Back to Journals » International Journal of General Medicine » Volume 14
Integrative Bioinformatics Analysis Revealed Mitochondrial Defects Underlying Hypoplastic Left Heart Syndrome
Authors Zhao D ![]() , Liu Y
, Liu Y ![]() , Xu Z, Shen H, Chen S, Zhang S, Li Y, Zhang H, Zou C, Ma X
, Xu Z, Shen H, Chen S, Zhang S, Li Y, Zhang H, Zou C, Ma X ![]()
Received 2 November 2021
Accepted for publication 2 December 2021
Published 14 December 2021 Volume 2021:14 Pages 9747—9760
DOI https://doi.org/10.2147/IJGM.S345921
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Diming Zhao,1 Yilin Liu,2 Zhenqiang Xu,3,4 Hechen Shen,1 Shanghao Chen,1 Shijie Zhang,1 Yi Li,1 Haizhou Zhang,3,4 Chengwei Zou,3,4 Xiaochun Ma3,4
1Department of Cardiovascular Surgery, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 2Department of Ophthalmology, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 3Department of Cardiovascular Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, People’s Republic of China; 4Department of Cardiovascular Surgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, People’s Republic of China
Correspondence: Xiaochun Ma
Department of Cardiovascular Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, No. 324, Jingwu Road, Jinan, 250021, People’s Republic of China
Tel +8615169196737
Email [email protected]
Background: Hypoplastic left heart syndrome (HLHS) is one of the most complex congenital cardiac malformations, and the molecular mechanism of heart failure (HF) in HLHS is still elusive.
Methods: Integrative bioinformatics analysis was performed to unravel the underlying genes and mechanisms involved in HF in HLHS. Microarray dataset GSE23959 was screened out for the differentially expressed genes (DEGs), after which the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses were carried out using the Metascape. The protein–protein interaction (PPI) network was generated, and the modules and hub genes were identified with the Cytoscape-plugin. And the integrated network of transcription factor (TF)-DEGs and miRNA-DEGs was constructed, respectively.
Results: A total of 210 DEGs were identified, including 135 up-regulated and 75 down-regulated genes. The functional enrichment analysis of DEGs pointed towards the mitochondrial-related biological processes, cellular components, molecular functions and signaling pathways. A PPI network was constructed including 155 nodes as well as 363 edges. And 15 hub genes, such as NDUFB6, UQCRQ, SDHD, ATP5H, were identified based on three topological analysis methods and mitochondrial components and functions were the most relevant. Furthermore, by integrating network interaction construction, 23 TFs (NFKB1, RELA, HIF1A, VHL, GATA1, PPAR-γ, etc.) as well as several miRNAs (hsa-miR-155-5p, hsa-miR-191-5p, hsa-mir-124-3p, hsa-miR-1-3p, etc.) were detected and indicated the possible involvement of NF-κB signaling pathways in mitochondrial dysfunction in HLHS.
Conclusion: The present study applied the integrative bioinformatics analysis and revealed the mitochondrial-related key genes, regulatory pathways, TFs and miRNAs underlying the HF in HLHS, which improved the understanding of disease mechanisms and the development of novel therapeutic targets.
Keywords: hypoplastic left heart syndrome, differential expression genes, transcription factors, microRNAs, regulatory networks, integrative bioinformatics analysis
Introduction
Hypoplastic left heart syndrome (HLHS) remains one of the most complex congenital cardiac diseases (CHD) and is characterized by the hypoplasia of the left ventricle, the stenosis or atresia of the mitral and aortic valve, and the underdevelopment of the aorta. Because the left ventricle and its components are incapable of supporting the systemic circulation, the disease is life-threatening without surgical intervention.1 A three-stage surgical palliation that includes the final Fontan surgery has revolutionized the surgical treatment with HLHS and made the survival with HLHS now possible.2
HLHS has been uniformly considered as a polygenic disease with a multifactorial inheritance pattern.1,3 Recent studies have identified a number of transcription factors (TFs) that play a fundamental role in the cardiac development, such as NK2 homeobox 5 (NKX2.5) and notch receptor 1 (Notch1).3 And somatic mutations in these TFs are also recognized as causal factors for HLHS.3,4 Dysregulation of microRNAs (miRNAs) has been proved as related to the occurrence of CHD in a variety of studies.5 The down-regulation of miR-592 has been demonstrated to inhibit the Notch signaling by up-regulating the potassium channel tetramerization domain containing 10 (KCTD10) expression, thereby protecting the mice from the hypoplastic heart and CHD.6 Liu et al found that the deletion of miR-133a resulted in the ectopic gene expression of the cardiac smooth muscle and abnormal cardiomyocyte proliferation.7 Through single-cell RNA profiling of hiPSC-derived endocardium and human fetal heart tissue with an underdeveloped left ventricle, Miao et al had identified a developmentally impaired endocardial population in HLHS.8 In addition, HLHS is also correlated with the Turner syndrome, trisomy 18 syndrome, DiGeorge syndrome, Jacobsen syndrome, Noonan syndrome, etc., suggesting the complexity of its underlying mechanisms.3
While HLHS patients have improved prognosis, 10-year transplant-free survival stands at only 39–50% for HLHS patients.9,10 The high morbidity/mortality is largely associated with ventricular dysfunction and acute heart failure (AHF).10 The Phase I TICAP (Transcoronary Infusion of Cardiac Progenitor Cells in Patients With Single-Ventricle Physiology) trial (NCT01273857)11,12 and Phase II PERSEUS randomized, controlled trial (NCT01829750) enrolled patients with various single-ventricle conditions-including HLHS and they were treated by intracoronary infusion of cardiosphere-derived cells (CDCs) after palliative operations. An improvement in ejection fraction of the RV, somatic growth, reduced heart failure status, and quality of life were observed in treated patients compared with those in controls.13 However, therapies that have been developed for HF in adults are still inefficient for treating HF in HLHS.14 The detailed molecular mechanisms underlying the HF in HLHS is to a large extent elusive despite recent advancements.15 A prior work focused on the peripheral blood mononuclear cells (PBMC) oxygen consumption rate (OCR) measured from single-ventricle congenital heart disease developing HF demonstrated the mitochondrial respiration defects including higher maximal respiratory capacity and respiratory reserve.16 Another study by Xu et al suggested that intrinsic mitochondrial dysfunction is linked with cardiac dysfunction and heart failure risk in HLHS. In this study, the induced pluripotent stem cells (iPSC) were established from HLHS patients and differentiated into cardiomyocytes (iPSC-CM). These HLHS patient iPSC-CM demonstrated the reduced mitochondrial membrane potential as well as diminished mitochondrial oxygen consumption rate.17 Hence, comprehensively exploring the novel therapeutic strategies is urgently warranted in order for effective treatments of HF in HLHS.3
Bioinformatics analysis provides important clues in understanding the molecular mechanisms of diseases, exploring the novel biomarkers related to the disease diagnosis and prognosis, and investigating the possible therapeutic targets in the early stages of diseases. A previous report by Liu and his colleagues on the HLHS mouse heart tissue suggested a mitochondrial maturation defect, and another study by Ricci identified the alternative mRNA splicing patterns in the pathogenesis of HLHS based on GSE23959. In HLHS, over 1800 mRNAs were differentially spliced and the most significant alterations in KEGG pathways involved the oxidative phosphorylation in mitochondria.16,18 To further unravel the molecular basis underlying the HF in HLHS, integrative bioinformatics analysis was performed to unravel the underlying genes and mechanisms involved in HLHS. A previous report by Liu and his colleagues on the HLHS mouse heart tissue suggested a mitochondrial maturation defect and another study by Xu et al demonstrated intrinsic mitochondrial dysfunction linked with cardiac dysfunction and heart failure risk in HLHS.16,17 Of note, our results revealed the mitochondrial-related key genes, regulatory pathways, TFs and miRNAs underlying HLHS, which added to deepen the understanding of mitochondrial maturation and function defects in HF in HLHS.
Materials and Methods
Microarray Data
The gene expression profile data GSE2395918 based on the platform of GPL5188 (Affymetrix Human Exon 1.0 ST Array) was obtained from the Gene Expression Omnibus (GEO) database, National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/geo/). The dataset available in this analysis was uploaded by Ricci et al, which includes 16 samples containing 10 healthy controls and 6 patients with HLHS. And the HLHS neonates were diagnosed based upon clinical features including hypoplasia/atresia of the ascending aorta, various degrees of underdevelopment of the aortic valve, mitral valve, and left ventricle (LV) cavity, and retrograde flow in the aortic arch as determined by conventional 2-D echocardiography. The myocardial samples were isolated from the right ventricles (RVs) of 6 HLHS neonates and all subjects underwent the stage 1 Norwood reconstruction. As for the control samples, the myocardial tissue of RVs and left ventricles (LVs) were obtained from 5 newborns each newborn with normal cardiac anatomy, who died due to non-cardiac causes. An overview of the detailed information on the samples analyzed in our study is shown in Table 1.
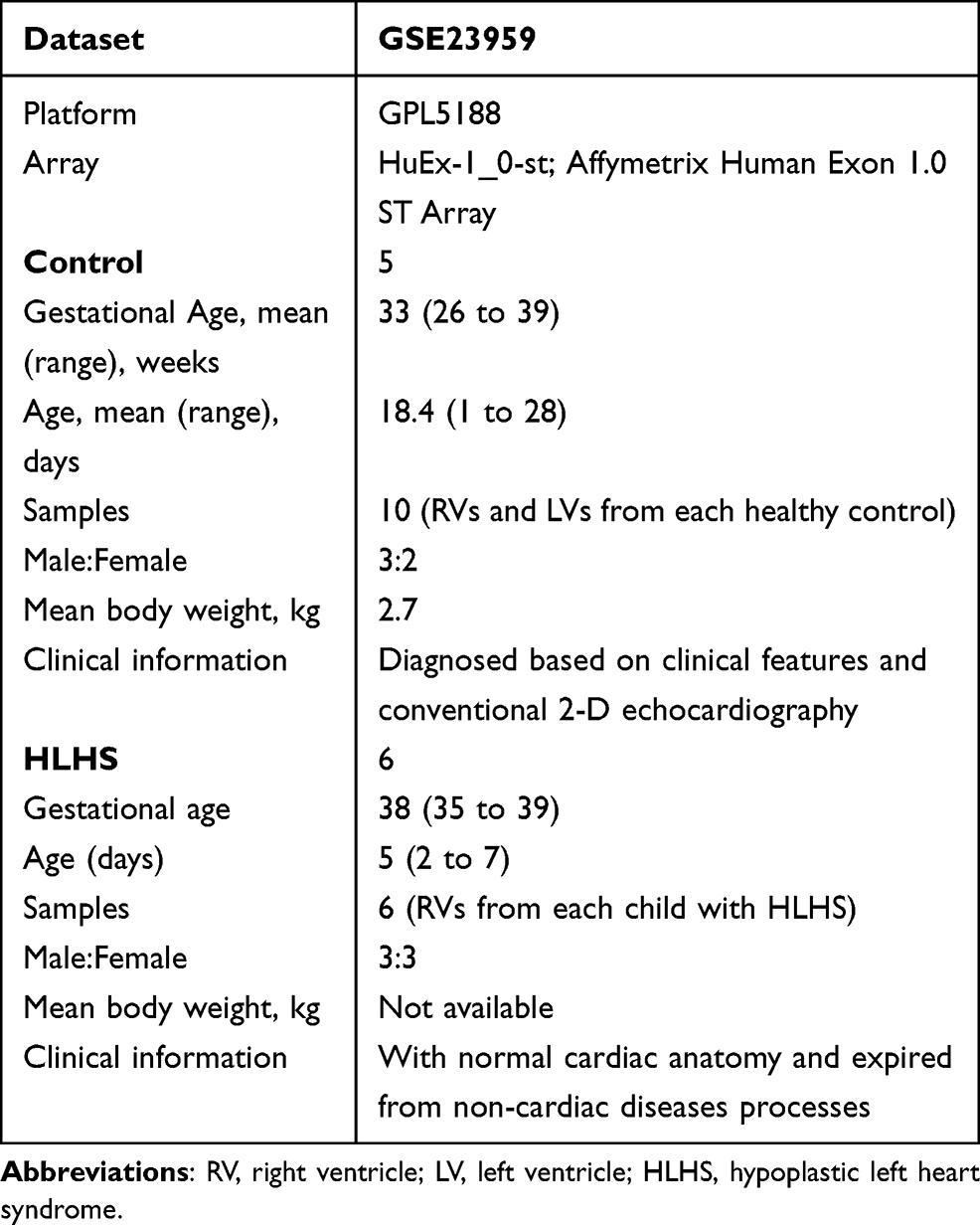 |
Table 1 Details of Clinical Samples from GSE23959 |
Identification of Differentially Expressed Genes (DEGs)
The Perl script was applied to convert the gene IDs to official gene symbols after downloading the probe expression matrix file GSE23959 series matrix.txt. If multiple probes match the same gene, the mean value of probes is calculated as the final value of this gene. The dataset was then normalized using the Normalized Between Arrays function of the Limma package in RStudio software (Version 1.2.5001).19 After processing, the Limma package was applied to screen the DEGs in this dataset.19 The DEGs were screened out following the standard of a corrected p value <0.05 and |log2 fold change (FC)| ≥1.00. Volcano and heatmap were constructed using the RStudio software with the ggplot2 package and pheatmap package, respectively.20,21 A list of DEGs including the up-regulated and down-regulated genes was saved for the following integration analysis.
Functional and Pathway Enrichment Analysis of DEGs
Metascape (http://metascape.org) is an integrated online tool providing comprehensive gene annotations and analyses for input list of genes.22 To explore the biological functions of DEGs in HLHS, Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed using Metascape with the criteria of minimum overlap >3, p value cut off <0.01, and minimum enrichment score >1.5. And the results were visualized by Hiplot, a comprehensive web platform for scientific data visualization (https://hiplot.com.cn).23
PPI Network Construction and Analysis of Modules
The Search Tool for the Retrieval of Interacting Genes (STRING) online database (https://string-db.org/) (version: 11.0), which provides the direct and indirect associations of proteins or genes, was utilized to detect further details of molecular interactions of DEGs.24 Medium confidence (minimum required interaction score >0.4) was chosen as a threshold and all positive interactions were included. Subsequently, the PPI network was visualized by Cytoscape software (vision 3.8.2) (https://cytoscape.org/).25 Then, a Cytoscape plugin known as the Molecular Complex Detection (MCODE) was applied to find out significant modules with degree cutoff = 2, node score cutoff = 0.2, k-Core = 2, max. Depth = 100 as a filter criterion.26 The modules with a MCODE score greater than 4 and containing more than five nodes were regarded as the key modules. Moreover, the overlapping genes were identified as hub genes according to the score calculated by three topological algorithms through the cytoHubba plugin of Cytoscape including Maximum Correlation Criterion (MCC), Density of Maximum Neighborhood Component (DMNC) and Maximum Neighborhood Component (MNC).27 The enrichment analysis was further performed for the key modules and hub genes through Metascape, respectively.22
TF-DEGs and miRNA-DEGs Regulatory Network Construction
The TFs fine-tune the downstream target genes at the pre-transcriptional stage and play a key role in HLHS. The TRRUST (version 2) is a manually curated database of human and mouse transcriptional regulatory networks, of which the TF-targeted regulatory interactions have been verified experimentally.28 The TRRUST was used to discover the TFs for DEGs and the interaction pairs whose FDR < 0.05 were visualized in Cytoscape. The target miRNAs of DEGs were predicted using a miRNA-centric network visual analytics platform named miRNet (version 2.0), which integrates data from 14 different miRNA databases.29 After obtaining the interactions between all DEGs and related microRNAs, the interaction pairs with the number of connected target genes in the top 10 were visualized in Cytoscape.
Results
The Identification of DEGs in HLHS
The gene expression profile of the GSE23959 was normalized and shown in Figure 1A. A total of 17,513 genes were detected in the myocardial samples, of which 135 up-regulated genes (adjusted p-value <0.05, log2 (Fold Change) >1.00) and 75 down-regulated genes (adjusted p-value <0.05, log2 (Fold Change) < −1.00) were identified as significant DEGs. Compared to the normal controls, the distribution of DEGs in HLHS patients was manifested in a volcano plot (Figure 1B). The hierarchical clustering analysis revealed that the DEGs were well clustered between HLHS tissues and normal tissues, as shown in the heatmap (Figure 1C). The details of 210 DEGs are presented in Supplementary Table S1.
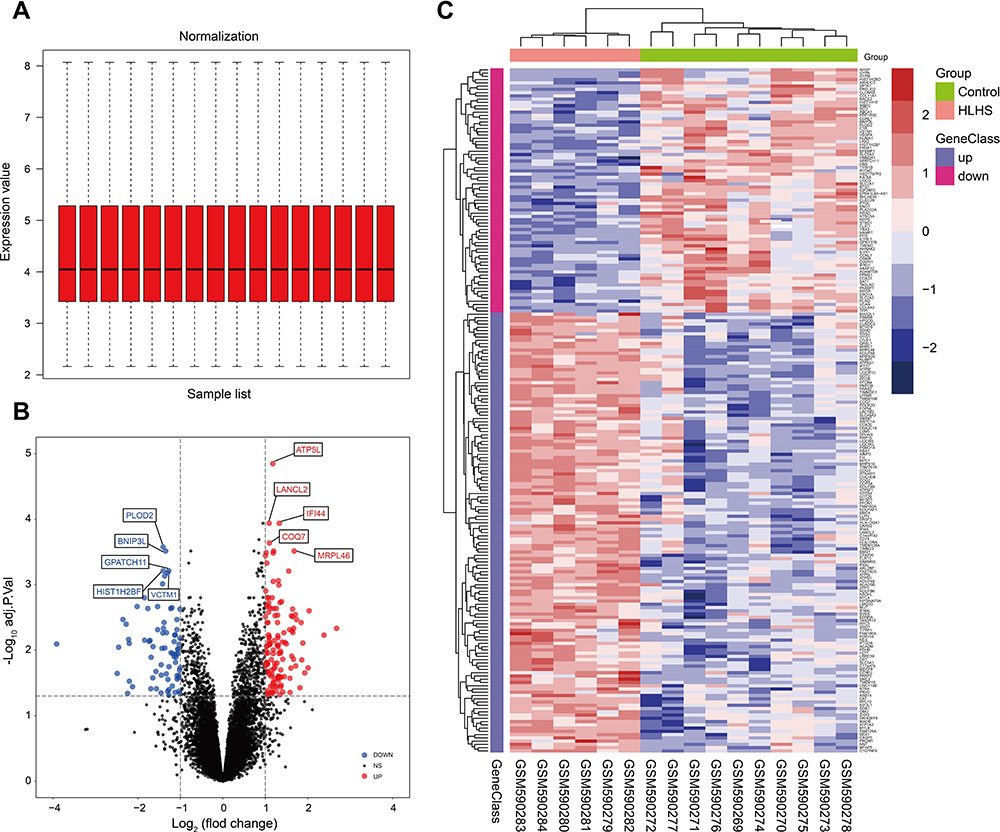 |
Figure 1 The identification of DEGs between the HLHS patients and normal controls. The box plot of gene expression data after normalization (A). The volcano plot of genes detected in HLHS in which the red dots represented the up-regulated genes and the blue dots represented the down-regulated genes (B). The heatmap of DEGs (adjusted p-value<0.05 and |log2(FC)|≥1.00) in which the up-regulated genes were in red color and the down-regulated genes were in blue color (C). Abbreviations: DEG, differentially expressed gene; HLHS, hypoplastic left heart syndrome; FC, fold change. |
The Functional and Pathway Enrichment Analysis
The up-regulated and down-regulated DEGs were uploaded to Metascape to further analyze their crucial biological functions, and the top ten enrichment results of each term were shown in Supplementary Table S2. The DEGs were mainly enriched in the generation of precursor metabolites and energy, cofactor metabolic process, monocarboxylic acid metabolic process, cellular amino acid metabolic process and response to decreased oxygen levels, etc., by the biological process (BP) analysis (Figure 2A). The results of cellular component (CC) in GO showed that the DEGs were mainly involved in mitochondrial envelope, mitochondrial matrix, ficolin-1-rich granule, sarcolemma and mitochondrial proton-transporting ATP synthase complex and etc., of which most were mitochondrial composition correlated (Figure 2B). For the molecular function (MF) group, oxidoreductase activity, cofactor binding, protein homodimerization activity, catalytic activity, acting on a tRNA, proton-transporting ATP synthase activity and rotational mechanism and etc., were the enriched terms (Figure 2C). Additionally, KEGG pathway analysis indicated that the DEGs were abundant in oxidative phosphorylation, carbon metabolism, valine, leucine and isoleucine degradation, cardiac muscle contraction, renal cell carcinoma, etc. (Figure 2D, Supplementary Table S3). The results above indicated that the DEGs were mainly enriched in the mitochondria-related biological processes, subcellular components, molecular functions and signaling pathways.
 |
Figure 2 The GO and KEGG pathway enrichment analysis of DEGs. The top ten biological process (A), cellular component (B), molecular function (C) and KEGG pathway (D) of DEGs. Abbreviations: GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEG, differentially expressed gene. |
The PPI Network Construction and Analysis of Modules
To assess the relationship between the DEGs, the STRING online database was utilized for further analysis. A total of 155 DEGs, including 104 up-regulated genes and 51 down-regulated genes, were finally filtered into the PPI network. The PPI network involved 155 nodes and 363 edges with a local clustering coefficient of 0.412 and the PPI enrichment p-value < 1.0 × 10−16 (Figure 3). And 15 hub genes, such as NDUFB6, UQCRQ, SDHD, ATP5H, were identified based on three topological analysis methods.
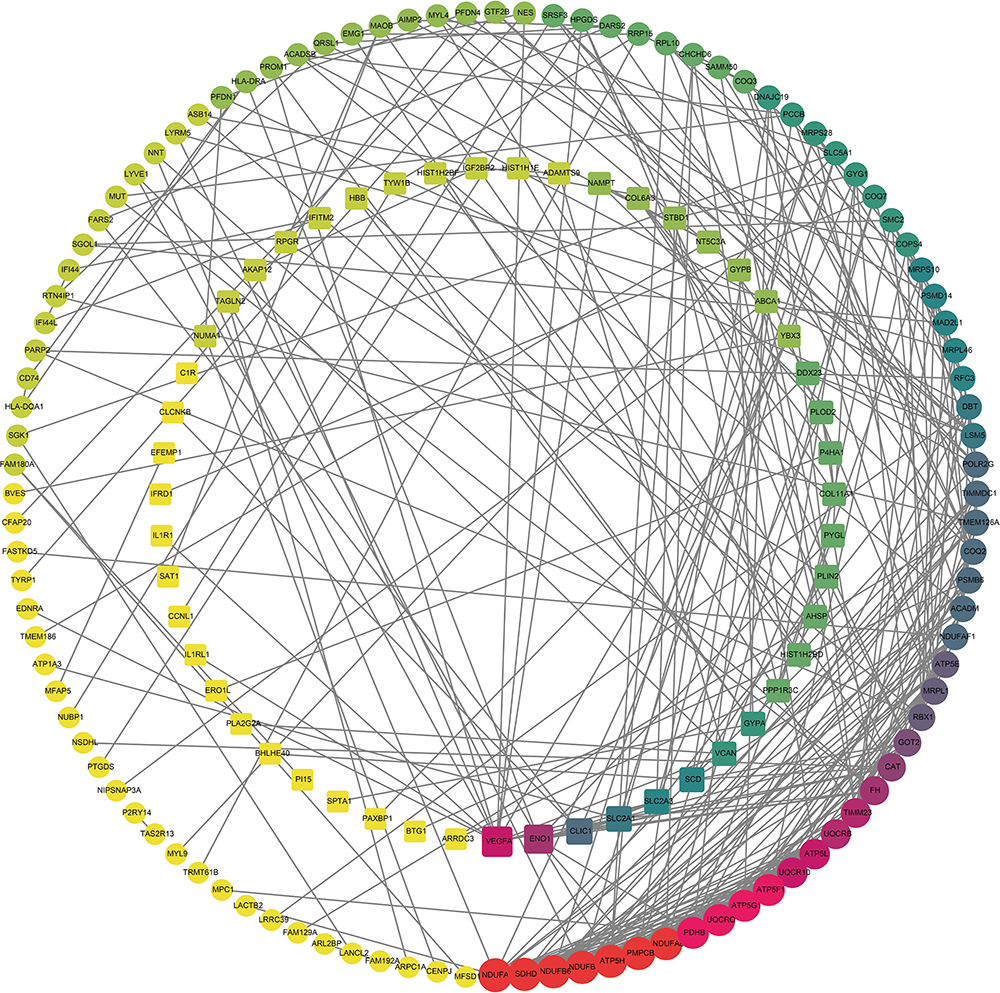 |
Figure 3 The PPI network of DEGs constructed using the Cytoscape, including 155 nodes and 363 edges. The up-regulated genes were depicted as the circles and the down-regulated genes as the squares. The red and large node showed a high degree, and the yellow and small node showed a low degree. Abbreviations: PPI, protein–protein interaction network; DEG, differentially expressed gene. |
Subsequently, the MCODE plugin of Cytoscape software, which detects densely connected regions in large PPI networks, was applied to explore significant modules. A total of ten modules were generated, and only two modules with a MCODE score greater than four and containing more than five nodes were screened out. The module 1 showed 13 nodes linked via 76 edges with an MCODE score of 12.667 and UQCR10 (ubiquinol-cytochrome c reductase, complex III subunit X) was regarded as seed gene. All DEGs of module 1 were up-regulated and GO and KEGG analysis showed that these genes projected the oxidative phosphorylation, mitochondrial inner membrane and respiratory chain complex, etc., as top terms, which were all mitochondrial components and functions related (Figure 4A and C and Supplementary Table S4). The module 2 was consisted of 5 nodes and 9 edges with an MCODE score of 4.500 and MRPS28 (mitochondrial ribosomal protein S28) was considered as seed gene. The enrichment analysis showed that these up-regulated genes were abundant in the ribosomal subunit, mitochondrial translational elongation and large ribosomal subunit, and mitochondrial components and functions were still the most relevant (Figure 4B and D and Supplementary Table S4).
 |
Figure 4 The network analysis of DEGs. The clustered module 1 identified from the whole PPI network (A). The enrichment analysis of the module 1 by the Metascape (C). The clustered module 2 identified from the whole PPI network (B). The enrichment analysis of the module 2 by the Metascape (D). The Venn diagram showing the number of overlapping genes among the three topological analysis methods (E). The interactions among the fifteen hub genes (F). The enrichment analysis of the hub genes by the Metascape (G). Abbreviations: DEG, differentially expressed gene; PPI, protein–protein interaction network. |
Furthermore, 3 topological analysis methods were adopted to identify the hub genes in PPI network. Fifteen hub genes were obtained after overlapping the genes according to these ranked means in cytoHubba plugin (Figure 4E and F). And the details of these hub genes are shown in Table 2. According to the enrichment results, the hub genes were mainly correlated with the oxidative phosphorylation, oxidoreductase complex, proton-transporting ATP synthase activity and mitochondrial respiratory chain complex III. Similar to the results of enrichment of modules, the hub genes were abundant in the biological processes and molecular functions of mitochondria (Figure 4G and Supplementary Table S5).
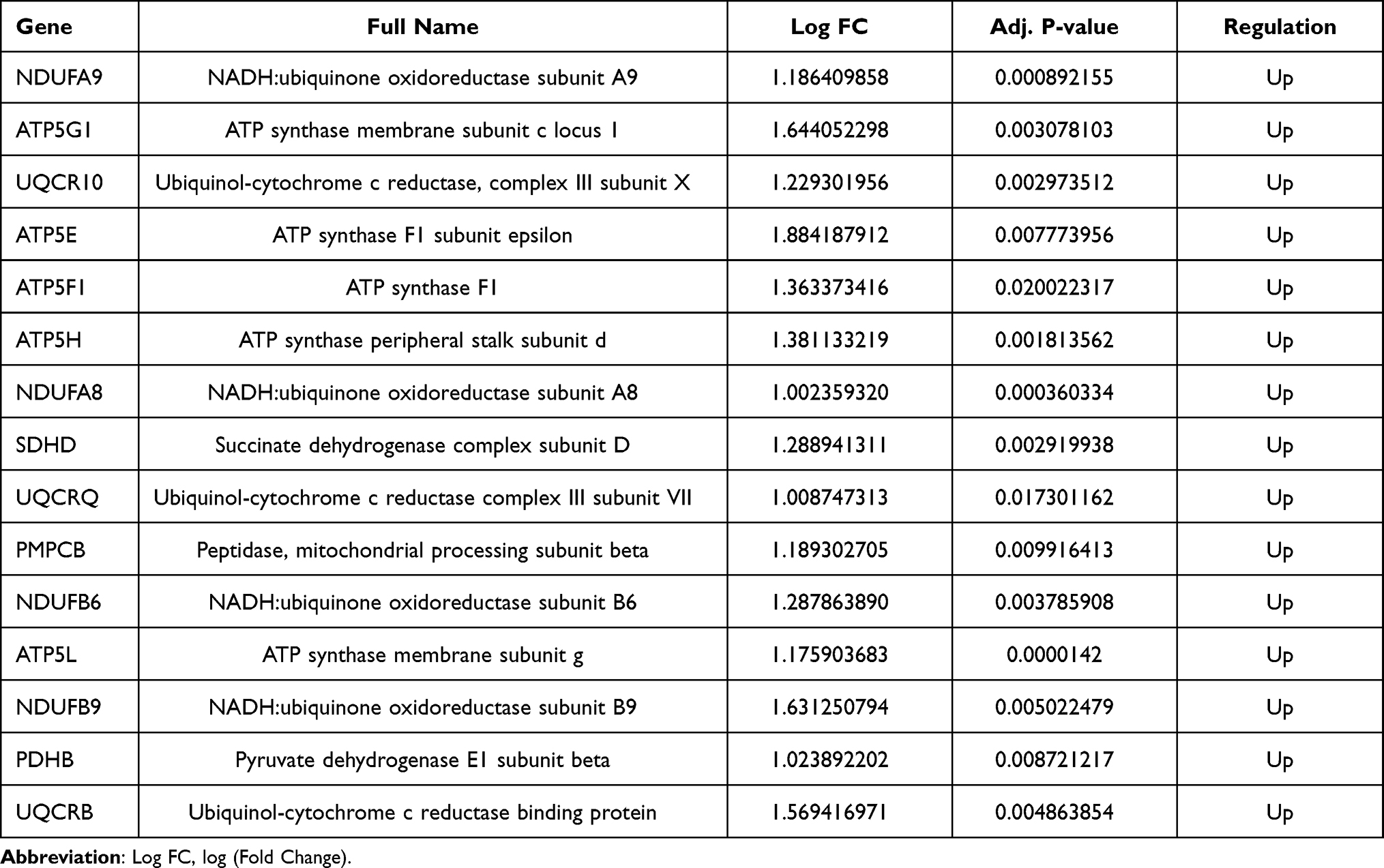 |
Table 2 Hub Genes Identified Through Three Topological Analysis Methods |
The TF-DEGs and miRNA-DEGs Regulating Network Analysis
TF-DEGs regulating network was constructed with the help of TRRUST exploring the relationships between TFs and DEGs (Figure 5). As a result, a total of 85 interactions among 23 TFs, 10 up-regulated DEGs and 19 down-regulated DEGs were identified. In this network, both NFKB1 and RELA possessed the similar maximum quantity of regulatory genes. NFKB1 regulated 8 DEGs such as VEGFA, NAMPT and IGF2BP2, while RELA regulated 8 DEGs such as ABCA1, CD74 and NPPB. Additionally, the top targeted DEGs for TFs was VEGFA that was regulated by 10 TFs and HLA-DRA that was regulated by 5 TFs. Interestingly, both NFKB1 and RELA belongs to the NF-κB family and this indicated the possible involvement of this canonical signaling pathways in mitochondrial dysfunction in HLHS.30–32
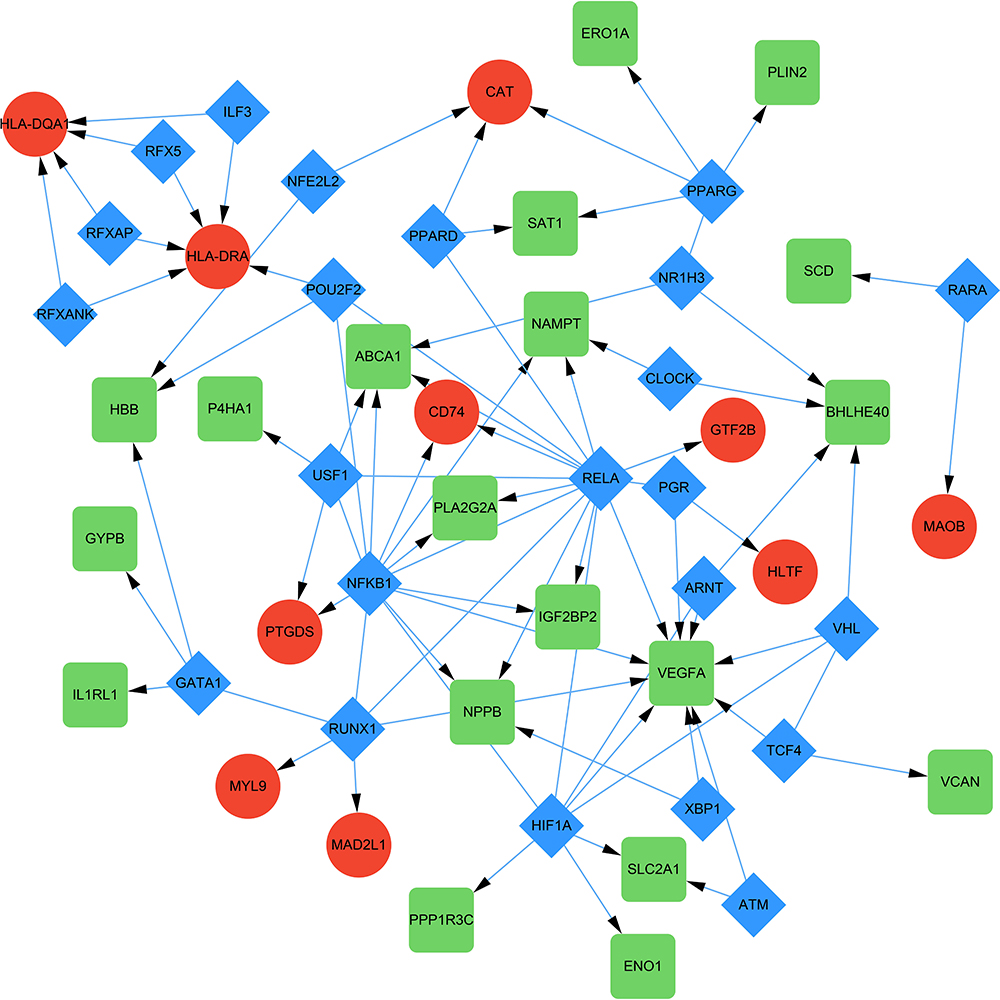 |
Figure 5 TF-target DEGs regulatory network. The green and red nodes for the DEGs, and the blue diamonds for the TFs. Abbreviations: TF, transcription factor; DEG, differentially expressed gene. |
Then, a total of 9062 interaction pairs were predicted, including 1949 miRNAs and 204 DEGs. The miRNAs were ranked according to the number of target genes and the top ten were screened out, and the relation pairs were finally visualized in Cytoscape (Figure 6). hsa-mir-1-3p had the highest connectivity with 119 target DEGs. Besides, hsa-mir-124-3p and hsa-mir-16-5p had 99 pairs and 87 pairs with DEGs, respectively. Of note, both hsa-mir-124 and hsa-mir-16 have been reported to mediate the NF-κB signaling and mitochondrial functions.33–35 Thus these above-mentioned results should promote further in-depth investigation of HLHS mechanism by which specific miRNAs dysregulates the mitochondrial dysfunction via NF-κB signaling.
 |
Figure 6 MicroRNA-target DEGs regulatory network. The green and red nodes for the DEGs, and the pink V-shapes for the miRNAs. Abbreviations: miRNA, microRNA; DEG, differentially expressed gene. |
Discussion
In this study, a total of 210 DEGs including 135 up-regulated genes and 75 down-regulated genes were identified. From the results of enrichment analysis of DEGs, multiple terms in BP, CC, MF and KEGG pathway were found to be associated with mitochondrial components and functions. Generation of precursor metabolites and energy, cofactor metabolic process, cristae formation, aerobic respiration, glucose import across plasma membrane, response to decreased oxygen levels, mitochondrial gene expression, mitochondrial electron transport and ubiquinol to cytochrome c were the top enriched terms in BP. The results of CC showed that the DEGs were mainly related to the mitochondrial envelope, mitochondrial matrix, mitochondrial proton-transporting ATP synthase complex, mitochondrial respiratory chain complex III, mitochondrial intermembrane space and intrinsic component of mitochondrial inner membrane. And the oxidoreductase activity, proton-transporting ATP synthase activity, rotational mechanism, cofactor binding and NADH dehydrogenase activity were found to be the most important MFs. The KEGG pathway analysis indicated that oxidative phosphorylation and carbon metabolism, ubiquinone biosynthesis, eukaryotes, 4-hydroxybenzoate ≤ ubiquinone were predominantly enriched.
Similar results were obtained in the subsequent analysis of modules and hub genes. The enrichment results of modules included the oxidative phosphorylation, mitochondrial inner membrane, respiratory chain complex, mitochondrion organization, mitochondrial proton-transporting ATP synthase complex, coupling factor F(o), mitochondrial respiratory chain complex III, and mitochondrial translational elongation. The oxidative phosphorylation, oxidoreductase complex, proton-transporting ATP synthase activity, rotational mechanism, mitochondrial respiratory chain complex III and mitochondrial matrix were found to be the main results of enrichment of hub genes.
Accumulating evidence has pointed towards the mitochondrial maturation and function defects in the pathogenesis of HF in HLHS. Ricci et al have previously identified the DEGs and alternative mRNA splicing patterns in the pathogenesis of HLHS. In HLHS, over 1800 mRNAs were differentially spliced and the most significant alterations in KEGG pathways involved the oxidative phosphorylation in mitochondria.18 A previous report by Liu and his colleagues on the HLHS mouse heart tissue suggested a mitochondrial maturation defect of fewer cristae, changed shape and decreased size. And the transcriptome profiling showed the metabolic and mitochondria-related pathways among the top impacted pathway.16 Another study by Xu et al demonstrated the intrinsic mitochondrial dysfunction is linked with cardiac dysfunction and heart failure risk in HLHS. In this study, the induced pluripotent stem cells (iPSC) were established from HLHS patients and differentiated into cardiomyocytes (iPSC-CM). These HLHS patient iPSC-CM demonstrated the reduced mitochondrial membrane potential as well as diminished mitochondrial oxygen consumption rate. Importantly, the patients with CM mitochondrial and differentiation defects had poorer clinical prognosis with severe ventricular dysfunction.17 A prior work focused on the peripheral blood mononuclear cell (PBMC) oxygen consumption rate (OCR) measured from single-ventricle congenital heart disease developing HF demonstrated the mitochondrial respiration defects including higher maximal respiratory capacity and respiratory reserve.16 A recent report using iPSC-CM generated from HLHS patients showed uncompensated mitochondrial-mediated oxidative stress underlying early HF in HLHS. Early-HF patient iPSC-CM showed the mitochondrial permeability transition pore (mPTP) opening, mitochondrial hyperfusion and respiration defects, which were associated with early-HF and poorer outcomes.36
Our results found that seed and hub genes were mostly involved in the mitochondrial functional and metabolic pathways. UQCR10 has been involved in the mitochondrial oxidative phosphorylation and myocardial contraction in cardiomyocytes.37 MrpS28 encodes a mitochondrial ribosomal protein, which is essential for mitochondrial ribosomal assembly, mitochondrial translation and oxidative phosphorylation.38 NDUFB6, a mitochondrial gene, plays a major role in reactive oxygen species (ROS) production.39 UQCRQ has been associated with the mitochondrial dysfunction and has been found to be up-regulated in mice with ischemic and dilated cardiomyopathy.40,41 As one of the four subunits of mitochondrial respiratory complex II, SDHD regulates reserve respiratory capacity and cell survival in cardiac myocytes.42,43 And the expression of ATP5H was apparently increased in the myocytes with mitochondrial dysfunction induced by fluoride.44
Among the predicted TFs in this study, some have been correlated with cardiac development and function. Both NFKB1 and RELA are members of the Rel/NFKB family of TFs and are involved in the regulation of immunity, inflammation, cell proliferation and apoptosis.45 Zhang et al found that the functional-94 insertion/deletion ATTG polymorphism in the promoter of NFKB1 is associated with the susceptibility to CHD.46 The RELA subunit of NF-κB is essential for the transition from the committed precursors into mature cardiomyocytes, and activation of TLR3/NF-κB pathway enhances the myocardial maturation.47 This indicated the possible involvement of these canonical signaling pathways in this complex CHD.48,49
MiRNAs are a class of small single-stranded RNA containing 19–24 nucleotides and fine-tunes the post-transcriptional gene expression. The dysregulation of miRNA expression has been found in a variety of cardiovascular diseases.50 The top 10 miRNAs were screened using the miRNet online tools, including hsa-let-7b-5p, hsa-mir-16-5p, hsa-mir-26a-5p, hsa-mir-27a-3p, hsa-mir-34a-5p, hsa-mir-1-3p, hsa-mir-124-3p, hsa-mir-191-5p, hsa-mir-155-5p, hsa-mir-129-2-3p. As a member of the miR-155 family, hsa-miR-155-5p have been used as a biomarker to distinguish the patients with hypertrophic cardiomyopathy from the normal controls.51,52 Moreover, hsa-miR-155-5p could regulate the mitochondrial biogenesis by regulating the expression level of TFAM.53,54 Hsa-miR-191-5p regulates the expression of PGC-1α, which is a key molecule in the process of mitochondrial production.51 Interestingly, both hsa-mir-124 and hsa-mir-16 have been demonstrated to regulate the NF-κB signaling and mitochondrial functions. In 2020, Chen et al revealed that long non-coding RNA NEAT1 could inhibit miR-124/NF-κB signaling pathway and regulate the expression of inflammatory factors to protect cardiomyocytes from As2O3 damage. It has also been proved that miR-124 was involved in the regulation of mitochondrial apoptosis in a variety of diseases.55–57 And Zhao et al found that miR-124 could inhibit the expression of CD151, leading to the aggravation of heart failure.58 In the apoptosis of neutrophils exposed to Pb, miR-16-5p activated the mitochondrial apoptotic pathway and death receptor pathway by down-regulating the expression of PiK3R1 and IGFR1.59 Besides, miR-16 could mediate the expression of CD40 and inhibit the NF-κB signaling pathway to alleviate LPS-induced myocardial cell injury.60 Thus, these above-mentioned results should promote further in-depth investigation of HLHS mechanism by which specific miRNAs dysregulate mitochondrial dysfunction via NF-κB signaling.
Limitations
There were several limitations that should be highlighted to interpret the results. First, confounding factors such as age, gender, BMI, and differences between the LV and RV of the samples were not carefully considered in our study. The results of this study must be interpreted with caution. Second, the HLHS group and the healthy group were not well matched to age, and some results may be due to differences in age. Third, the enrichment results for up- and down-regulated genes were not performed separately, and we might miss some of the valuable information. Fourth, due to the small number of clinical samples included in GSE23959, there may be selection bias in the study design and the sample size needs to be further expanded. Lastly, in further studies, HLHS patients with baseline HF should be considered to explore the mechanisms of HF.
Conclusion
In summary, a comprehensive analysis of DEGs was performed for HLHS, and the results demonstrated that the mitochondrial structure and function may be particularly relevant in the pathophysiological mechanisms of HF in HLHS. In addition, the network analysis detected 15 hub genes, 23 TFs and 10 miRNAs closely related to HLHS. These results have provided the evidence of novel mechanisms as well as promising therapeutic targets for HLHS and await in-depth investigation in the future.
Data Sharing Statement
The datasets presented in this study were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). And the acquisition and application methods complied with the corresponding database guidelines and policies.
Acknowledgments
The authors thank the openbiox community and Hiplot team (https://hiplot.com.cn) for providing the technical assistance and valuable tools for data analysis and visualization.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (81800255), the National Nature Science Foundation of Shandong Province (ZR2020MH044 to ZC) and Natural Science Foundation of Shandong Province (ZR2018BH002 to MX).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Barron D, Kilby M, Davies B, Wright J, Jones T, Brawn W. Hypoplastic left heart syndrome. Lancet. 2009;374(9689):551–564. doi:10.1016/S0140-6736(09)60563-8
2. Oster M, Lee K, Honein M, Riehle-Colarusso T, Shin M, Correa A. Temporal trends in survival among infants with critical congenital heart defects. Pediatrics. 2013;131(5):e1502–e1508. doi:10.1542/peds.2012-3435
3. Saraf A, Book W, Nelson T, Xu C. Hypoplastic left heart syndrome: from bedside to bench and back. J Mol Cell Cardiol. 2019;135:109–118. doi:10.1016/j.yjmcc.2019.08.005
4. Reamon-Buettner S, Ciribilli Y, Inga A, Borlak J. A loss-of-function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Hum Mol Genet. 2008;17(10):1397–1405. doi:10.1093/hmg/ddn027
5. You G, Zu B, Wang B, Fu Q, Li F. Identification of miRNA-mRNA-TFs regulatory network and crucial pathways involved in tetralogy of fallot. Front Genet. 2020;11:552. doi:10.3389/fgene.2020.00552
6. Pang X, Lin X, Du J, Zeng D. Downregulation of microRNA-592 protects mice from hypoplastic heart and congenital heart disease by inhibition of the Notch signaling pathway through upregulating KCTD10. J Cell Physiol. 2019;234(5):6033–6041. doi:10.1002/jcp.27190
7. Liu N, Bezprozvannaya S, Williams A, et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22(23):3242–3254. doi:10.1101/gad.1738708
8. Miao Y, Tian L, Martin M, et al. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell. 2020;27(4):574–589.e8. doi:10.1016/j.stem.2020.07.015
9. Driscoll D, Offord K, Feldt R, Schaff H, Puga F, Danielson G. Five- to fifteen-year follow-up after Fontan operation. Circulation. 1992;85(2):469–496. doi:10.1161/01.CIR.85.2.469
10. Garcia A, Beatty J, Nakano S. Heart failure in single right ventricle congenital heart disease: physiological and molecular considerations. Am J Physiol Heart Circ Physiol. 2020;318(4):H947–H965. doi:10.1152/ajpheart.00518.2019
11. Ishigami S, Ohtsuki S, Tarui S, et al. Intracoronary autologous cardiac progenitor cell transfer in patients with hypoplastic left heart syndrome: the TICAP prospective Phase 1 controlled trial. Circ Res. 2015;116(4):653–664. doi:10.1161/CIRCRESAHA.116.304671
12. Tarui S, Ishigami S, Ousaka D, et al. Transcoronary infusion of cardiac progenitor cells in hypoplastic left heart syndrome: three-year follow-up of the transcoronary infusion of cardiac progenitor cells in patients with single-ventricle physiology (TICAP) trial. J Thorac Cardiovasc Surg. 2015;150(5):1198–1208.e2. doi:10.1016/j.jtcvs.2015.06.076
13. Ishigami S, Ohtsuki S, Eitoku T, et al. Intracoronary cardiac progenitor cells in single ventricle physiology: the PERSEUS (cardiac progenitor cell infusion to treat univentricular heart disease) randomized phase 2 trial. Circ Res. 2017;120(7):1162–1173. doi:10.1161/CIRCRESAHA.116.310253
14. Shaddy R, Boucek M, Hsu D, et al. Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA. 2007;298(10):1171–1179. doi:10.1001/jama.298.10.1171
15. Bejjani AT, Wary N, Gu M. Hypoplastic left heart syndrome (HLHS): molecular pathogenesis and emerging drug targets for cardiac repair and regeneration. Expert Opin Ther Targets. 2021;25(8):621–632. doi:10.1080/14728222.2021.1978069
16. Liu X, Yagi H, Saeed S, et al. The complex genetics of hypoplastic left heart syndrome. Nat Genet. 2017;49(7):1152–1159. doi:10.1038/ng.3870
17. Xu X, Tan T, Lin J-HI, et al. Abstract 15746: intrinsic cardiomyocyte mitochondrial defects underlie cardiac dysfunction and heart failure risk associated with hypoplastic left heart syndrome. Circulation. 2018;138(Suppl_1):A15746–A15746.
18. Ricci M, Xu Y, Hammond H, et al. Myocardial alternative RNA splicing and gene expression profiling in early stage hypoplastic left heart syndrome. PLoS One. 2012;7(1):e29784. doi:10.1371/journal.pone.0029784
19. Diboun I, Wernisch L, Orengo C, Koltzenburg M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genom. 2006;7:252. doi:10.1186/1471-2164-7-252
20. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009.
21. Kolde R. Pheatmap: pretty heatmaps. R Package Version. 2012;61:617.
22. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
23. Hiplot: a free and comprehensive cloud platform for scientific computation and visualization. Openbiox Community; 2021.
24. Szklarczyk D, Gable A, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi:10.1093/nar/gky1131
25. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
26. Bader G, Hogue C. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi:10.1186/1471-2105-4-2
27. Chin C, Chen S, Wu H, Ho C, Ko M, Lin C. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(S4):S11. doi:10.1186/1752-0509-8-S4-S11
28. Han H, Cho J, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46:D380–D386. doi:10.1093/nar/gkx1013
29. Chang L, Zhou G, Soufan O, Xia J. miRNet 2.0: network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020;48:W244–W251. doi:10.1093/nar/gkaa467
30. Regula KM, Baetz D, Kirshenbaum LA. Nuclear factor-kappaB represses hypoxia-induced mitochondrial defects and cell death of ventricular myocytes. Circulation. 2004;110(25):3795–3802. doi:10.1161/01.CIR.0000150537.59754.55
31. Mauro C, Leow SC, Anso E, et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011;13(10):1272–1279. doi:10.1038/ncb2324
32. Johnson RF, Witzel II, Perkins ND. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-κB. Cancer Res. 2011;71(16):5588–5597. doi:10.1158/0008-5472.CAN-10-4252
33. Zhang G, Chen L, Khan AA, et al. miRNA-124-3p/neuropilin-1(NRP-1) axis plays an important role in mediating glioblastoma growth and angiogenesis. Int J Cancer. 2018;143(3):635–644. doi:10.1002/ijc.31329
34. Ye E-A, Liu L, Jiang Y, et al. miR-15a/16 reduces retinal leukostasis through decreased pro-inflammatory signaling. J Neuroinflammation. 2016;13(1):305. doi:10.1186/s12974-016-0771-8
35. Valentino A, Calarco A, Di Salle A, et al. Deregulation of MicroRNAs mediated control of carnitine cycle in prostate cancer: molecular basis and pathophysiological consequences. Oncogene. 2017;36(43):6030–6040. doi:10.1038/onc.2017.216
36. Xu X, Lin J, Bais A, et al. Mitochondrial respiration defects in single-ventricle congenital heart disease. Front Cardiovasc Med. 2021;8:734388. doi:10.3389/fcvm.2021.734388
37. Chang P, Niu Y, Zhang X, et al. Integrative proteomic and metabolomic analysis reveals metabolic phenotype in mice with cardiac-specific deletion of natriuretic peptide receptor A. Mol Cell Proteomics. 2021;20:100072. doi:10.1016/j.mcpro.2021.100072
38. Pulman J, Ruzzenente B, Bianchi L, et al. Mutations in the MRPS28 gene encoding the small mitoribosomal subunit protein bS1m in a patient with intrauterine growth retardation, craniofacial dysmorphism and multisystemic involvement. Hum Mol Genet. 2019;28(9):1445–1462. doi:10.1093/hmg/ddy441
39. Wang N, Maskomani S, Meenashisundaram G, Fuh J, Dheen S, Anantharajan S. A study of Titanium and Magnesium particle-induced oxidative stress and toxicity to human osteoblasts. Mater Sci Eng C Mater Biol Appl. 2020;117:111285. doi:10.1016/j.msec.2020.111285
40. Lu D, Xia Y, Chen Z, et al. Cardiac proteome profiling in ischemic and dilated cardiomyopathy mouse models. Front Physiol. 2019;10:750. doi:10.3389/fphys.2019.00750
41. Wen J, Garg N. Oxidative modification of mitochondrial respiratory complexes in response to the stress of Trypanosoma cruzi infection. Free Radic Biol Med. 2004;37(12):2072–2081. doi:10.1016/j.freeradbiomed.2004.09.011
42. Dhingra R, Kirshenbaum L. Succinate dehydrogenase/complex II activity obligatorily links mitochondrial reserve respiratory capacity to cell survival in cardiac myocytes. Cell Death Dis. 2015;6:e1956. doi:10.1038/cddis.2015.310
43. Jeon Y, He M, Austin J, Shin H, Pfleger J, Abdellatif M. Adiponectin enhances the bioenergetics of cardiac myocytes via an AMPK- and succinate dehydrogenase-dependent mechanism. Cell Signal. 2021;78:109866. doi:10.1016/j.cellsig.2020.109866
44. Wang H, Zhao W, Liu J, Tan P, Tian W, Zhou B. ATP5J and ATP5H proactive expression correlates with cardiomyocyte mitochondrial dysfunction induced by fluoride. Biol Trace Elem Res. 2017;180(1):63–69. doi:10.1007/s12011-017-0983-5
45. Herron B, Rao C, Liu S, et al. A mutation in NFkB interacting protein 1 results in cardiomyopathy and abnormal skin development in wa3 mice. Hum Mol Genet. 2005;14(5):667–677. doi:10.1093/hmg/ddi063
46. Zhang D, Li L, Zhu Y, et al. The NFKB1 -94 ATTG insertion/deletion polymorphism (rs28362491) contributes to the susceptibility of congenital heart disease in a Chinese population. Gene. 2013;516(2):307-310. doi:10.1016/j.gene.2012.12.078
47. Hodgkinson C, Pratt R, Kirste I, Dal-Pra S, Cooke J, Dzau V. Cardiomyocyte maturation requires TLR3 Activated nuclear factor kappa B. Stem Cells. 2018;36(8):1198–1209. doi:10.1002/stem.2833
48. Qing M, Schumacher K, Heise R, et al. Intramyocardial synthesis of pro- and anti-inflammatory cytokines in infants with congenital cardiac defects. J Am Coll Cardiol. 2003;41(12):2266–2274. doi:10.1016/S0735-1097(03)00477-7
49. Mirza H, Finkel MS, Johnson LA. Inflammatory mediators in congenital heart disease. Crit Care Med. 2002;30(4):941–942. doi:10.1097/00003246-200204000-00045
50. Kalayinia S, Arjmand F, Maleki M, Malakootian M, Singh C. MicroRNAs: roles in cardiovascular development and disease. Cardiovasc Pathol. 2021;50:107296. doi:10.1016/j.carpath.2020.107296
51. Shi H, Li J, Song Q, et al. Systematic identification and analysis of dysregulated miRNA and transcription factor feed-forward loops in hypertrophic cardiomyopathy. J Cell Mol Med. 2019;23(1):306–316. doi:10.1111/jcmm.13928
52. Derda A, Thum S, Lorenzen J, et al. Blood-based microRNA signatures differentiate various forms of cardiac hypertrophy. Int J Cardiol. 2015;196:115–122. doi:10.1016/j.ijcard.2015.05.185
53. Quiñones-Lombraña A, Blanco J. Chromosome 21-derived hsa-miR-155-5p regulates mitochondrial biogenesis by targeting mitochondrial transcription factor A (TFAM). Biochim Biophys Acta. 2015;1852(7):1420–1427. doi:10.1016/j.bbadis.2015.04.004
54. Zhao Q, Sun Q, Zhou L, Liu K, Jiao K. Complex regulation of mitochondrial function during cardiac development. J Am Heart Assoc. 2019;8(13):e012731. doi:10.1161/JAHA.119.012731
55. Jin L, Miao J, Liu Y, et al. Icaritin induces mitochondrial apoptosis by up-regulating miR-124 in human oral squamous cell carcinoma cells. Biomed Pharmacother. 2017;85:287–295. doi:10.1016/j.biopha.2016.11.023
56. Sun Y, Zhao X, Luo M, et al. The pro-apoptotic role of the regulatory feedback loop between miR-124 and PKM1/HNF4α in colorectal cancer cells. Int J Mol Sci. 2014;15(3):4318–4332. doi:10.3390/ijms15034318
57. Deng X, Chen Y, Liu Z, Xu J. MiR-124-3p.1 sensitizes ovarian cancer cells to mitochondrial apoptosis induced by carboplatin. Onco Targets Ther. 2020;13:5375–5386. doi:10.2147/OTT.S242342
58. Zhao Y, Yan M, Chen C, et al. MiR-124 aggravates failing hearts by suppressing CD151-facilitated angiogenesis in heart. Oncotarget. 2018;9(18):14382–14396. doi:10.18632/oncotarget.24205
59. Yin K, Cui Y, Sun T, Qi X, Zhang Y, Lin H. Antagonistic effect of selenium on lead-induced neutrophil apoptosis in chickens via miR-16-5p targeting of PiK3R1 and IGF1R. Chemosphere. 2020;246:125794. doi:10.1016/j.chemosphere.2019.125794
60. Li -Q-Q, Xi J, Li B-Q, Li N. MiR-16, as a potential NF-κB-related miRNA, exerts anti-inflammatory effects on LPS-induced myocarditis via mediating CD40 expression: a preliminary study. J Biochem Mol Toxicol. 2020;34(2):e22426. doi:10.1002/jbt.22426
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.