")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Inotersen: new promise for the treatment of hereditary transthyretin amyloidosis
Received 11 September 2018
Accepted for publication 27 December 2018
Published 6 May 2019 Volume 2019:13 Pages 1515—1525
DOI https://doi.org/10.2147/DDDT.S162913
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Qiongyu Guo
Veena Mathew,1 Annabel K Wang1,2
1Department of Neurology, UCI ALS and Neuromuscular Center, University of California, Irvine, Orange, CA, USA; 2Neurology Section, Tibor Rubin VA Medical Center, Long Beach, CA, USA
Abstract: Hereditary transthyretin amyloidosis is a fatal autosomal dominant disorder characterized by deposition of transthyretin amyloid into the peripheral nervous system, heart, kidney, and gastrointestinal tract. Previous treatments using liver transplantation and small molecule stabilizers were not effective in stopping disease progression. Inotersen, a 2'-O-methyoxyethyl-modified antisense oligonucleotide, which acts by reducing the production of transthyretin, was recently demonstrated to improve disease course and quality of life in early hereditary transthyretin amyloidosis polyneuropathy in a 15-month Phase III study.
Keywords: antisense oligonucleotide, familial amyloid polyneuropathy, Inotersen, mRNA, RNase H
Introduction
Hereditary transthyretin amyloidosis
Familial amyloid polyneuropathy (FAP) was first described as a peculiar form of peripheral neuropathy with progressive lower extremity weakness, early loss of pain and temperature sensation, and gastrointestinal/genitourinary involvement by Corino Andrade in 1952.1 It is an autosomal dominant disorder characterized by deposition of amyloid in the peripheral nervous system, heart, kidney, and gastrointestinal tract. Amyloidosis occurs because of misfolded extracellular protein generating amyloid fibrils that are deposited into tissue. Deposition of transthyretin (TTR) amyloid fibrils in tissue is known as TTR amyloidosis (ATTR). Mutations in the TTR gene lead to hereditary forms of ATTR (hATTR), also referred to as ATTRm. The predominant feature is either polyneuropathy (hATTR-PN) or cardiomyopathy (hATTR-CM). hATTR can also affect the eyes and leptomeninges. Nonmutated or wild-type transthyretin amyloidosis (ATTRwt) typically presents as a cardiomyopathy in older adults and was previously referred to as senile systemic amyloidosis2 but deposition of ATTRwt in peripheral nerve has also been demonstrated.3
Disease onset occurs between the second and ninth decades of life but symptoms of hATTR usually manifest after age of 30 years. hATTR is classified as either early or late onset, where the cutoff is usually 50 years of age.4 Death occurs 3–15 years after the onset of symptoms, usually due to cardiac disease or malnutrition. Median survival is reduced in hATTR-CM compared with hATTR-PN.5 There are over 150 reported mutations of the TTR gene.6 The most common mutation is Val30Met, where valine is replaced by methionine at position 30 (148G→A). The Val30Met mutation is endemic in Portugal, Sweden, and Japan, with a large population in Brazil. The Val122Ile and Thr60Ala mutations are more common in the USA, and present with predominant cardiac symptoms.6 Penetrance is not complete, and there is a large variation in phenotype within genotypes. While polyneuropathy is a common presentation, initial symptoms range from cardiomyopathy, carpal tunnel syndrome, voice hoarseness due to recurrent laryngeal nerve palsies, vitreous opacities, and oculoleptomeningeal symptoms of seizure or cerebral hemorrhages.4 The global prevalence of ATTR is unknown, with estimates ranging from 5,000 to 40,000 affected individuals.7,8 This is likely an underestimate due to the lack of awareness among clinicians and the phenotypic variability of hATTR.
Background
Transthyretin
TTR is the primary transport protein for retinol-binding protein (RBP4)/Vitamin A and a redundant transport protein for thyroxine (T4). T4 binds to a central channel while RBP4 binds to surface receptors. There are four exons in the TTR gene, which is located on chromosome 18q11.2–12.1.9 A TTR monomer is derived from a single 127 amino acid polypeptide. Four identical monomers form TTR (homotetrameric structure). A single TTR gene mutation can replace one amino acid in a TTR monomer, leading to the formation of a mutant monomer which destabilizes of the homotetrameric structure of TTR.4,10–12 Tetramers can consist of 1–4 mutant monomers. The dissociation of the tetramer leads to misfolded monomers, which then form toxic oligomers and amyloid fibrils, which are deposited and lead to hATTR.
Treatment for hATTR
Initial therapy for hATTR-PN is symptomatic. The pain from the polyneuropathy is often severe and debilitating. Autonomic symptoms include profound orthostatic hypotension which limits the ability to stand and walk without fainting. Gastrointestinal symptoms can consist of bloating and alternating diarrhea and constipation, leading to dehydration and malnutrition. Assistance is required for self-care and activities of daily living due to significant hand weakness. Ankle foot orthoses, walking aids, and wheelchairs are eventually required. ATTR-CM may require pharmacological or electrophysiological interventions for congestive heart failure and arrhythmias.6,8
Therapeutic treatment of hATTR include removing the source of mutant TTR with liver transplantation (LTx), stabilizing the tetrameric structure of TTR using small molecules, reducing the amount of ATTR deposition with doxycycline plus tauroursodeoxycholic acid (TUDCA), or genetic therapy with TTR oligonucleotides to reduce the production of both mutated and wild-type TTR.
Liver transplantation
LTx was the first disease-modifying treatment for hATTR, initially performed in Sweden in 1990.13 Since the majority of TTR protein synthesis occurs in the liver,4 the rational was to remove the source and stop production of mutated TTR. LTx eliminates 90% of circulating mutant TTR in blood.12 Domino LTx has also been undertaken where the TTR liver is then transplanted in patients, such as elderly patients with liver cancer, who would otherwise never receive a new liver. Partial liver grafts from related living donors have also been utilized.14
Over 2,000 LTxs have been reported to the Familial Amyloid Polyneuropathy World Transplant Registry15 since 1990. The overall 20-year survival rate is >50%. In early onset Val30Met patients, the 15-year survival rate is >80%. In late-onset Val30Met patients or patients with non-Val30Met mutations, the 10-year survival rate is 50%. The 1-year mortality rate ranges from 7% to 24%, usually due to infection or cardiac disease. Posttransplant lymphoproliferative disease can also occur. Morbidity can be high with 20% of patients developing renal failure and diabetes. In addition to the age of onset and mutation type, prognosis also depends on the age at the time of LTx, duration of disease, and nutritional status.14,16
Progression of ATTR-PN and ATTR-CM occurs after LTx and is more common in patients with non Val30Met mutations.16,17 Progression after LTx is caused by wtTTR deposition, as mutant TTR is no longer being produced.14,16 Evaluation of nerve and cardiac amyloid deposits was found to contain 65%–75% mutant TTR with the remaining deposits consisting of wtTTR.18,19 Outcome of ATTR-CM can be improved with dual LTx and cardiac transplantation.16 Ocular and leptomeningeal ATTR continue to progress as mutant TTR is produced from the choroid plexus epithelium and retinal pigment epithelium, and not the liver.14 LTx slows but does not stop hATTR progression.
Although LTx improves survival in hATTR-PN, the limitations of LTx include the lack of organ improvement (nerve and heart), the shortage of available donor organs, the adverse effects of immunosuppressive agents, the cost of LTx and other additional treatments, and the difficulty in identifying LTx candidates early in the disease course.6,16 In non-Val30Met or late-onset hATTR patients, LTx is not recommended due to the poor response and lack of efficacy.16 De novo ATTR, referred to as iatrogenic ATTR, can also develop in the domino liver recipients.14,16 Due to the difficulties, complications, and disease progression associated with LTx, other treatment options were pursued for hATTR.
Small molecule stabilizers
Dissociation of the TTR homotetramer is naturally prevented by the binding of T4 in its central binding site. Stabilization of the mutant TTR tetramers stops the dissociation into misfolded monomers, thus preventing amyloid deposition and slowing disease progression. A small molecule can act as a kinetic stabilizer preventing the dissociation of TTR tetramers into monomers. NSAIDs, such as diflunisal, were demonstrated to bind to the T4 binding site. Tafamidis was developed to act as a small molecule stabilizer of TTR.20
Diflunisal
Diflunisal, a generic NSAID first discovered in the 1970s, binds to the central T4 binding complex preventing TTR tetramer dissociation into monomers.21 Diflunisal 250 mg twice a day was evaluated in a double-blind, placebo-controlled study with 130 patients with FAP.22 Inclusion criteria were symptoms and signs of sensorimotor or autonomic neuropathy in patients who routinely spent >50% of waking hours out of bed or out of chair (Eastern Cooperative Oncology Group Performance status of <3). Diflunisal reduced the rate of disease progression and preserved quality of life for 2 years. A twofold to threefold benefit was detected using the Neuropathy Impairment Score plus 7 nerve tests (NIS +7) as the primary end point of the study. The positive secondary end points included the Kumamoto Score (a qualitative polyneuropathy and end organ score for hATTR) and the modified body mass index (BMI, a predictor of survival in hATTR).
Diflunisal is not approved by the US Food and Drug Administration (FDA) and has been prescribed for off-label use. Continued progression on Diflunisal has been seen in late-onset hATTR and non-Val30Met patients.6 The use of Diflunisal is limited by renal insufficiency, congestive heart failure, and gastrointestinal adverse effects.
Tafamidis
Tafamidis (Vyndaqel®; Pfizer, New York, NY, USA), a 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid, was developed as a more potent kinetic stabilizer for TTR without the adverse effects of NSAIDs.20 In an 18-month Phase III trial, Tafamidis was evaluated in 128 Val30Met patients with early polyneuropathy.23 Based on intention to treat analysis, the primary end points of the NIS-Lower Limb score and the Norfolk Quality of Life Questionnaire-Diabetic Neuropathy (QoL-DN) were not met. Additional studies suggest that long-term therapy with Tafamidis is safe and delays disease progression in Val30Met patients. Data for non-Val30Met and late-onset Val30Met patients are less clear.6 Tafamidis was not approved for the treatment of hATTR-PN by the FDA in 2012 due to limited efficacy data. In 2011, Tafamidis was approved by the European Medicines Agency for treatment of stage 1 (walking without assistance) hATTR-PN in 2011. It is also approved in Japan and South America.6
In a recent 30-month Phase III study in 441 ATTR-CM patients, Tafamidis reduced all-cause mortality and cardiovascular-related hospitalizations compared with placebo.24
A new TTR stabilizer, AG10, is currently under evaluation for ATTR-CM.6,25
TTR stabilizers are generally safe and delay progression of the hATTR-PN but appear to be less efficacious in non-Val30Met patients and in those with more advanced disease.
Doxycycline and TUDCA
The combination of Doxycycline plus TUDCA6,26 was shown to reduce TTR deposition in transgenic Val30Met mice. These medications are well tolerated and were used in an open-label Phase III study using 100 mg twice a day of doxycycline and 250 mg of TUDCA daily. In the first 24 of 40 ATTR patients who completed the study (60% of patients enrolled), cardiac disease was stabilized in 75% and polyneuropathy was stabilized in 46%. Further studies are being undertaken.
The inability of LTx and current small molecule stabilizers to stop progression of hATTR led to the development of gene therapies to reduce the production of TTR.
TTR oligonucleotides
Individuals with ATTR are heterogeneous and produce both wt and mutant TTR. To prevent disease progression, all TTR production must be reduced or stopped. Because TTR was shown to be a nonessential protein transporter of T427 and vitamin A deficiency can be prevented using supplementation, hATTR was an ideal candidate disease for gene silencing. Oligonucleotides are short synthetic nucleic acid sequences containing 8–20 nucleotides that alter the complex intermediary metabolism of mRNA from transcription, translation, and degradation. They are complimentary to select sequences in the RNA strand that encode the amino acids required to make a protein. They reduce the production of mutant protein, thus modifying disease outcome.11,28–31 Two different oligonucleotides, siRNAs32 and antisense oligonucleotides (ASOs),33 have been studied in Phase III clinical trials in hATTR.
Protein synthesis of TTR begins with genetic coding in the nucleus from DNA, a double helix with two complimentary strands of nucleotides consisting of a phosphate group, a sugar (deoxyribose), and an organic base. Complimentary Watson–Crick base pairings of purines to pyrimidines are adenine to thymine and guanine to cytosine. In RNA, uracil replaces thymine (Figure 1). A sequence of three bases or a triplet codon codes for one amino acid and the sequence of amino acids determine the protein. Protein transcription involves duplicating the genetic code from DNA first to transcription of pre-mRNA which contains exons and introns. Pre-mRNA is then processed to form a mature mRNA. Protein translation occurs when mRNA is transported from the nucleus to ribosomes in the cytoplasm. Using complementary base pairing to the sense strand nucleic acid, siRNAs and ASOs affect gene expression of the TTR gene and thereby reduced the amount of total (mutant and wild-type) TTR that is produced by the liver.28–31,34,35
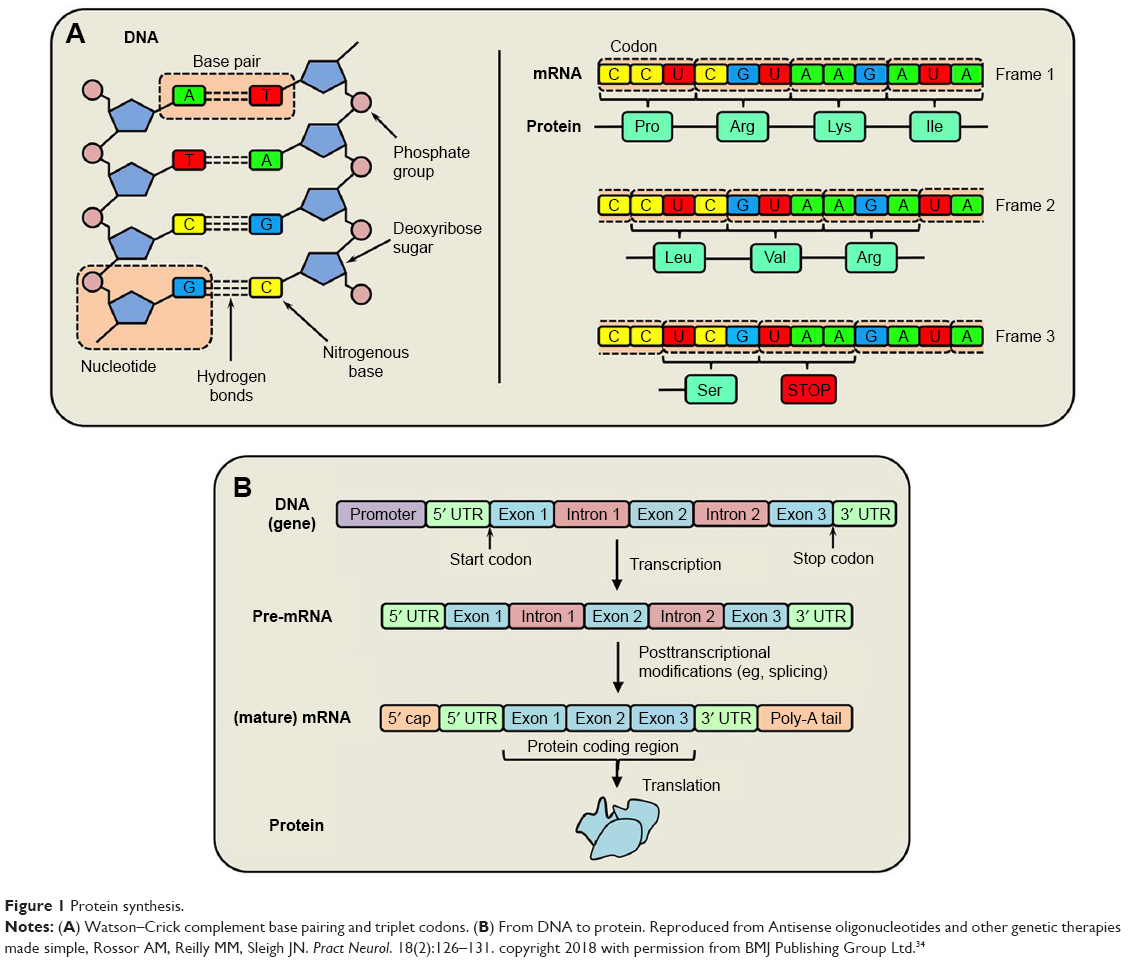 | Figure 1 Protein synthesis. |
RNAi/siRNA
RNAi or siRNA use dsRNAs, containing sense and antisense strands, which target both the mRNA and protein in the cytoplasm. RNAi is mediated by binding to mRNA/RNA-induced silencing complexes in the cytoplasm leading to degradation of mRNA by a cycloplasmic Dicer siRNA mechanism28–31,34 (Figure 2).
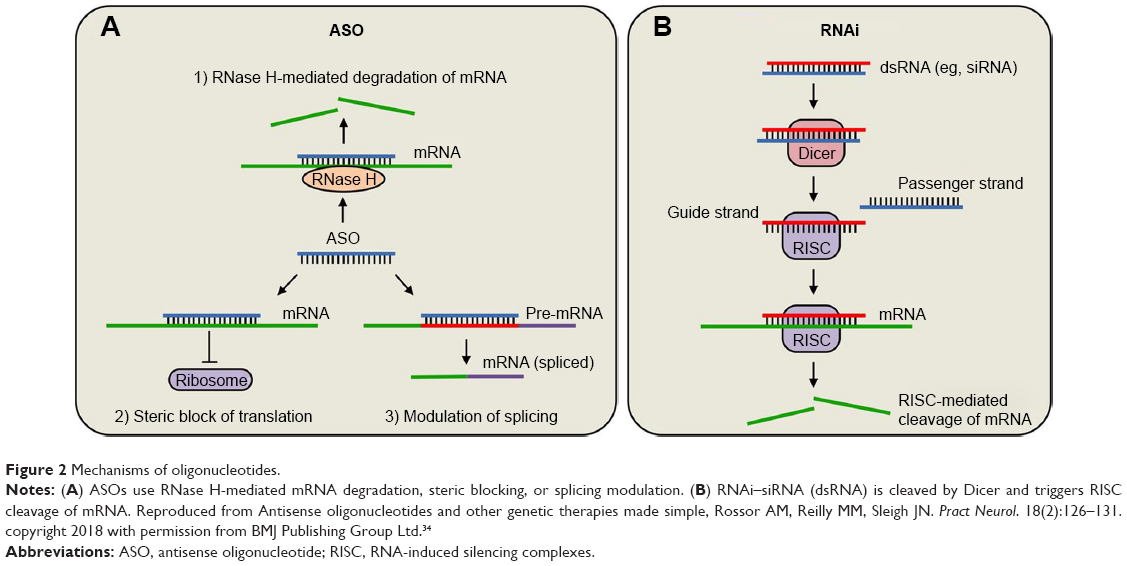 | Figure 2 Mechanisms of oligonucleotides. |
Patisiran
Patisiran (ALN-TTR02; Alnylam, Cambridge, MA, USA) is an siRNA encapsulated in a lipid nanoparticle delivered to intracellular compartments of hepatocytes using an intravenous infusion. It specifically binds to the 3′-UTRs of mutant and wt TTR mRNA.36
In a Phase III trial of Patisiran vs placebo, 0.3 mg per kilogram of body weight was intravenously administered over 80 minutes, every 3 weeks to patients for 18 months.32 The patients were randomized 2:1. One hundred forty-eight patients received Patisiran and 77 patients received placebo. To minimize infusion-related reactions, all patients were premedicated with intravenous dexamethasone (or equivalent), an H1 blocker, an H2 blocker, and oral acetaminophen 60 minutes before each infusion. Overall, 97% of patients in each trial group reported adverse events, most of which were mild or moderate in severity. Adverse events that occurred more often in Patisiran patients included peripheral edema and infusion-related reactions. Death, which was primarily cardiovascular related and consistent with expectations in hATTR, occurred in seven Patisiran patients (all cardiac events) and six placebo patients (one cardiac event). The primary end point (change from baseline to month 18 modified NIS +7 [mNIS +7]) and secondary end points (change from baseline to Month 18 Norfolk QoL-DN, 10-m walk test, and modified BMI) were met. Patisiran (Onpattro®) was approved by the FDA on August 10, 2018.37
Revusiran
Revusiran (Alnylam, Cambridge, MA, USA) is an siRNA conjugated with N-acetylgalactosamine and given subcutaneously (SC) in a Phase III clinical trial for hATTR-CM. The trial was halted due to an imbalance of mortality in the treatment arm compared with controls.29,35
Antisense oligonucleotides
Development of ASOs began in the late 1970s, but early ASOs were easily degraded by intracellular or serum nucleases.28–30 The unfavorable distribution and pharmacokinetics (PKs) with low affinity for binding to complementary sequences made it difficult to deliver ASOs to the appropriate intracellular compartments with adequate concentrations. Due to the large and strong negative polarity, ASOs were unable to cross cell membranes. ASOs also stimulated immune responses.
These challenges were addressed by chemically modifying the ribose backbone28–30 with phosphorothioate (PS) substitutes, which facilitated cell uptake and allowed broad distribution with systemic administration by binding to serum, cell surface, and intracellular proteins. PS substitutions improved PKs and optimal protein binding. Additional 2′-O-methoxyethyl (2′-MOE) modifications improved PKs by binding to plasma proteins, allowing for stable concentrations; reduced elimination half-life by increasing stability against nucleases; and increased potency by improving binding affinity of complementary hybridization. The MOE-modified nucleotides also ameliorated some of the high-dose toxicities via nonspecific protein binding and improved the safety profile compared with first-generation ASOs. The activation of the immune system was also dampened.
Protein production can be altered using one of two different mechanisms in the nucleus or cytoplasm.28,34,35 The first is by enzymatic RNA degradation using RNase H, which is an endonuclease within the nucleus. RNase H recognizes the RNA/DNA heteroduplex and catalyzes the cleavage of RNA, thus reducing the expression of mutant protein. RNase H also prevents RNA from attaching to the ribosome thereby blocking protein transcription. The second is by occupancy-only mechanism that pairs with target RNAs without triggering degradation and use alterations in RNA splicing: inhibiting translation to reduce production of toxic protein; enhancing translation to restore the deficient protein; and obstructing interactions that modify the functional effect of the protein or reduce the toxicity of the mutant protein (Figure 2).
Development of Inotersen
The most potent TTR inhibitors identified were ASOs to the TTR 3′-untranslated mRNA. They consisted of 20 base full-PS chimeric ASOs with five 2′-MOE-modified ribonucleotides at each terminus and a central region of ten 2′-deoxynucleotide residues (ie, 5-10-5 gapmer structure).38–40 Compounds were screened in vitro using mouse and monkey hepatocytes as well as human hepatoma HepG2 cells, and in vivo using TTR Ile84Ser transgenic mice, in a dose-dependent manner. Human mRNA and serum TTR levels were shown to be reduced in transgenic mice. No effect on activity, feeding, weight, or the liver was seen.
Four hundred TTR-specific ASO were screened, and from this, 36 ASOs were further evaluated for the ability to suppress the production of TTR. In ASO doses of 25 mg/kg, TTR RNA and protein levels were decreased by 90%. Eight of these ASOs were shown to suppress serum TTR levels in nonhuman primates.39,40 IONIS-TTRRX was found to reduce human TTR (mRNA and protein levels) up to 80% in human TTR Ile84Ser transgenic mouse model.38–40
Inotersen (Ionis, Carlsbad, CA, USA; previously IONIS-TTRRX/ISIS-420915) inhibits hepatic TTR production.40 It is a second-generation 2′MOE-modified ASO that is complimentary to a region in the 3′-UTR of the human TTR mRNA, which is not known to contain TTR mutations, and reduces all TTR production. It binds to mRNA with complimentary base pairing mimicking the DNA/RNA complex. This hybridization leads to an RNase H1-mediated degradation of TTR mRNA, thus preventing TTR production (Figure 2). Inotersen does not hybridize to any other known human gene. Unlike LTx, Inotersen reduces levels of both mutated and wild-type TTR. By decreasing the amount of liver-based serum TTR, the formation of TTR amyloid fibril deposits will be reduced, which effectively slows or halts disease progression.
Preclinical trials
Preclinical trials studied the PK and toxicology effects of repeat doses up to 6 months in mice and 9 months in cynomolgus monkey.38–40 Class effects occurred in a dose- and duration-dependent manner. In monkeys, they included transient effects on acute complement activation (at doses >10 mg/kg/week) and acute elevations in aPTT (at doses >12 mg/kg/week). Mice had inflammation that included presence of minimal to moderate mononuclear cell infiltrates in the sinusoids of liver lymph nodes and injection sites at doses >40 mg/kg/week after 3 and 6 months. The majority of Inotersen was cleared from plasma within hours, with the kidneys and liver having the highest concentrations. The half-life was 2–4 weeks. No effect was observed on TSH, total or free T4 or total or free T3 levels. RBP4 levels were reduced in parallel to TTR levels. Inotersen was well tolerated in mice and monkeys without adverse effects.
Phase I clinical trial
In 65 healthy volunteers, Inotersen demonstrated an average sustained, dose-dependent serum TTR reduction of 75% in a Phase I study (see Figure 3).40 There were no significant safety issues noted over 4 weeks. Of the four single dose levels and five multiple doses studied, the 300 mg dose was identified as appropriate for subsequent trials. The main safety findings, as with other 2′-MOE ASO profiles, included mild injection site reactions, primarily pain and erythema, typically lasting for 24 hours and occurred with some, but not all, injections. TTR binds to the retinol–RBP4 complex and prevents its excretion by the kidney. Retinol reductions below the lower limit of normal were found in about 50% of subjects and recovered about 1 month after the final dose. Treatment of Inotersen 400 mg SC for 4 weeks revealed unremarkable tolerability and safety without significant adverse effects. Data from the healthy volunteers led to the dose of 300 mg weekly SC dose of Inotersen in a Phase III trial.
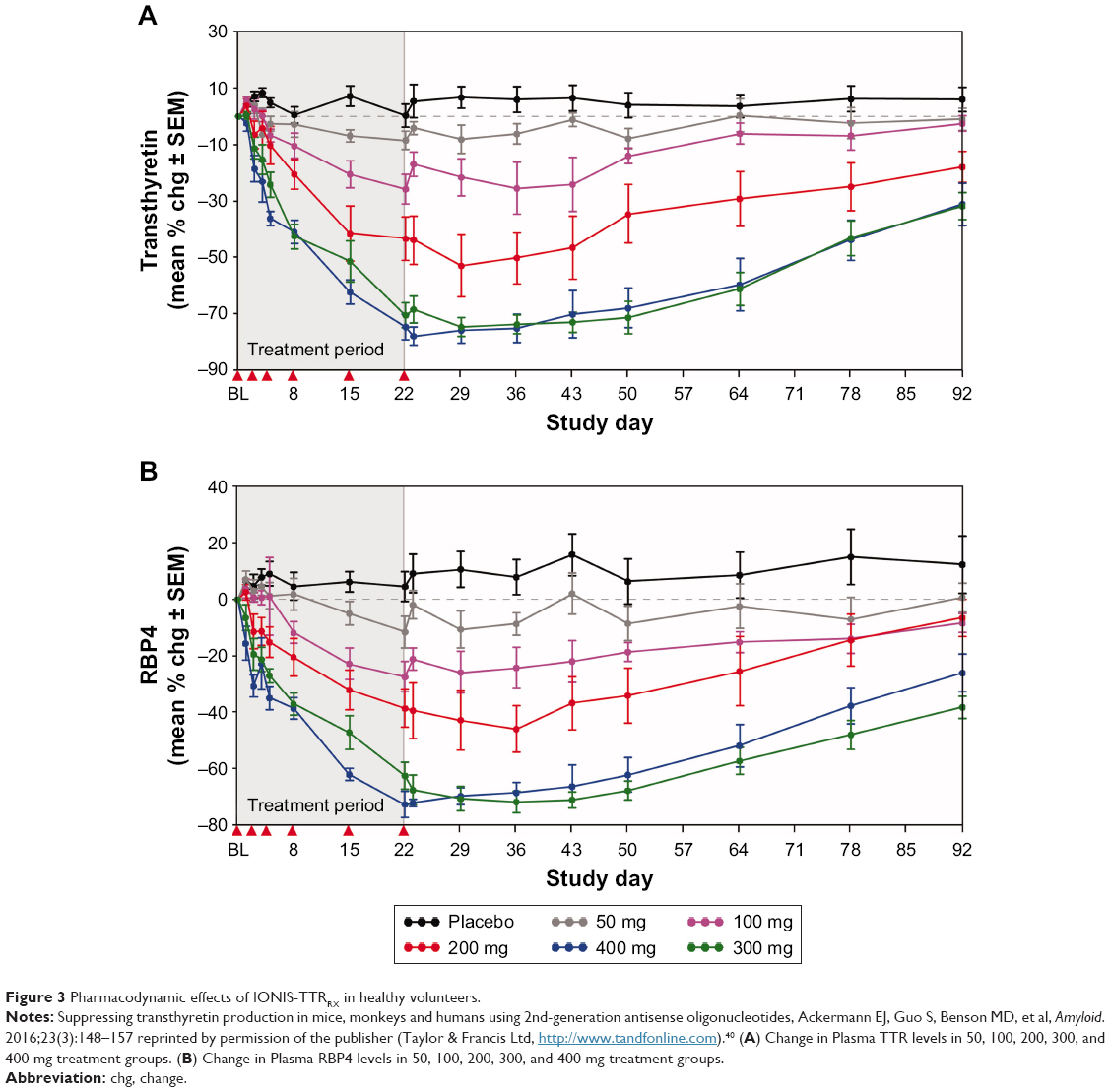 | Figure 3 Pharmacodynamic effects of IONIS-TTRRX in healthy volunteers. |
Phase III clinical trial
Given the limited and unequal standard of care options available internationally, an international, randomized, double-blind, placebo-controlled, 15-month Phase III study was undertaken with a 2:1 ratio of Inotersen (300 mg SC) to placebo.33 A total of 172 patients received at least one dose of Inotersen (112 patients) or placebo (60 patients). Approximately 50% of patients had the Val30Met mutation; 67% had stage 1 disease (ambulating without assistance); 33% had stage 2 disease (ambulating with assistance); 58% previously received Diflunisal or Tafamidis, and 63% had cardiomyopathy. At the 15-month intervention period, 139 patients had completed the study while 33 patients (25 Inotersen patients and 8 placebo patients) discontinued the study. The primary cause for discontinuation in the Inotersen group was adverse events, while the placebo group either discontinued for voluntarily reasons or disease progression.
The primary end points, the mNIS +7 and the Norfolk QoL-DN change from baseline, both showed statistically significant differences between the Inotersen and placebo groups after 15 months of intervention (Figure 4) and were independent of mutation type, disease stage, and baseline cardiomyopathy status. There are eight components of the mNIS +7 composite score (range −22.3 to 346.3) where a decrease in score indicates improvement and a 2-point change is defined as a minimally clinically meaningful change detectable. The Norfolk QoL-DN contains 5 domains with 35 questions (range −4 to 136) where a higher score indicates a poorer quality of life. Patients who received Inotersen had an average increase in mNIS +7 from baseline of 5.8 (95% CI, 1.6–10) points compared to placebo with an average increase of 25.5 (95% CI, 20.2–30.8) points. An average increase of 1 (95% CI, −3.2 to 5.2) points was seen in the Inotersen group compared to 12.7 (95% CI, 7.4–17.9) points in the placebo group in the Norfolk QoL-DN. An improvement in mNIS +7 (no increase from baseline) was seen in 36% of patients in the Inotersen group and 50% had improvement in the Norfolk QoL-DN. Serum TTR levels were reduced by a median nadir of 79% and mean nadir of 74%. Three patients (3%) developed glomerulonephritis. All had the Val30Met mutation. One was treated with glucocorticoids and another with glucocorticoids and cyclophosphamide. One was treated with hemodialysis. Three patients (3%) had thrombocytopenia. Two responded to glucocorticoids and dose reduction. There was one death due to grade 4 thrombocytopenia. Safety concerns of thrombocytopenia and renal dysfunction were managed with weekly platelet monitoring and bi-weekly serum creatinine collection. Affected patients were treated with split dosing or dose reduction of Inotersen. Disease course and quality of life were improved after treatment with Inotersen.
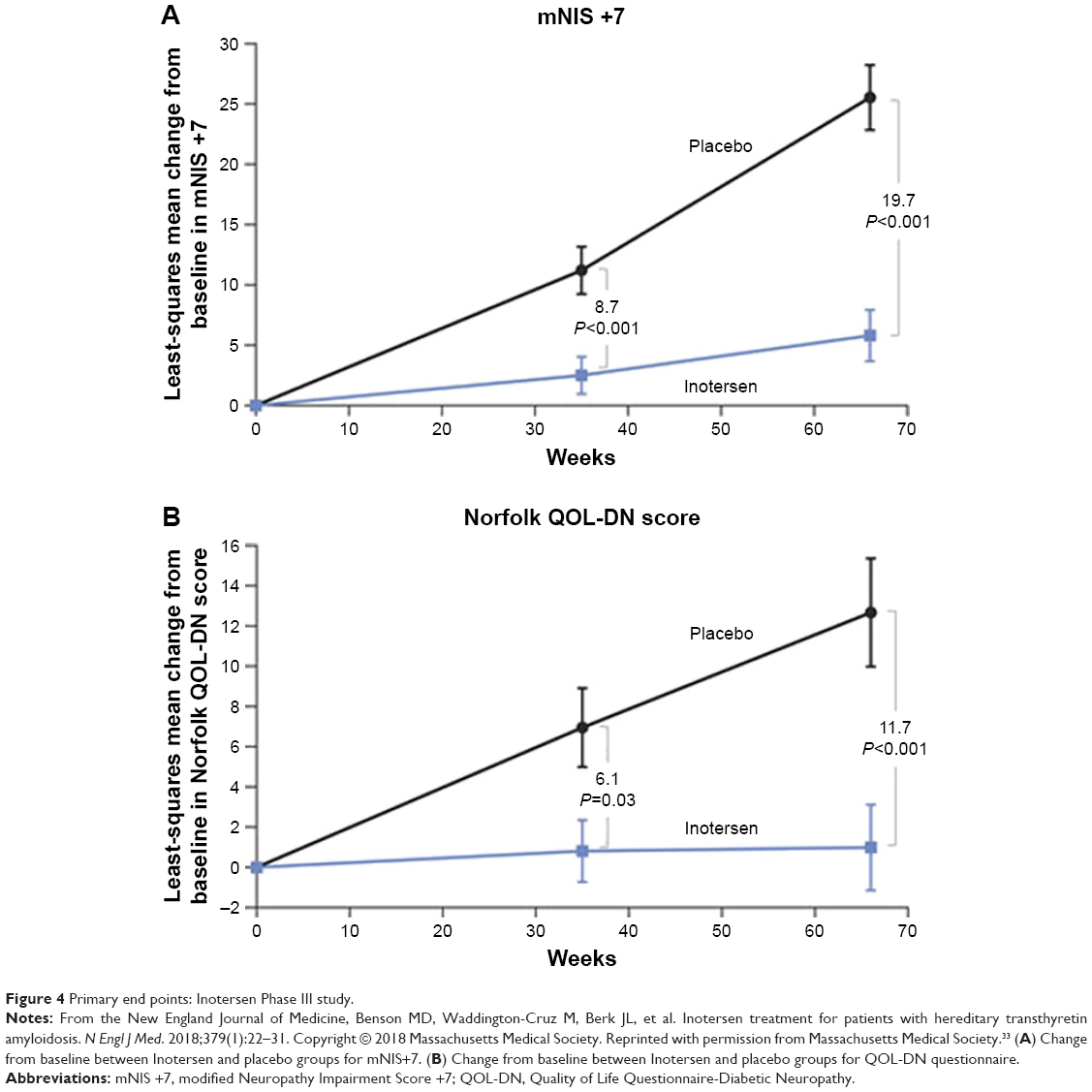 | Figure 4 Primary end points: Inotersen Phase III study. |
Patients who successfully completed this Phase III study were given the option to continue in the open-label extension study to receive Inotersen, 300 mg SC once weekly, for up to 5 years.41 One hundred twenty-eight eligible patients (95% of patients who completed the Phase III study) rolled over into the open-label extension study. Recent interim analysis findings demonstrated that the changes in the mNIS +7 and Norfolk-QOL in the Inotersen group continued to receive benefit after continuing treatment in the Phase III study. Similarly, the disease progression observed in the placebo group was reduced after initiating treatment with Inotersen in the open-label study. The average worsening in mNIS +7 and Norfolk QoL-DN changes from baseline to various time points in Phase III study was consistently higher in the placebo group compared with the Inotersen group, suggesting an increased therapeutic benefit with earlier treatment.
On August 20, 2018, Inotersen was approved in the European Union for the treatment of stage 1 or 2 adult patients with hATTR-PN (Figure 5).42
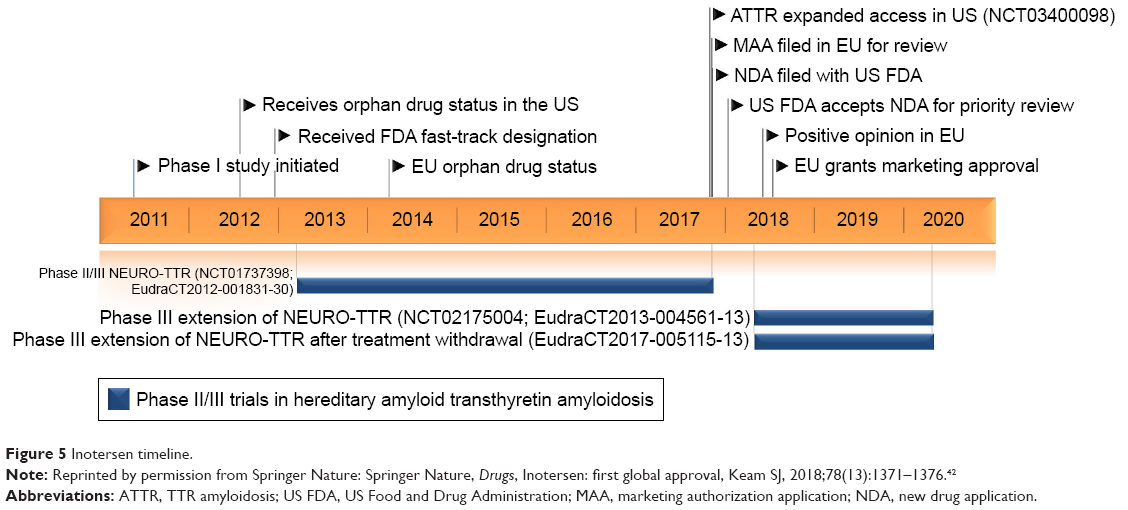 | Figure 5 Inotersen timeline. |
Inotersen in TTR-CM
LTx is less successful in the treatment of non Val30Met hATTR-CM as it does not stop cardiac amyloid deposition. wtATTR also leads to cardiomyopathy in patients who would not be candidates for cardiac transplantation due to their age and for whom LTx is of no benefit. Previously, there was no effective treatment to stop or slow the progression of ATTR-CM. The cardiomyopathy is characterized by congestive heart failure. Progression of disease can be monitored by changes in echocardiogram and cardiac magnetic resonance imaging (MRI). Twenty-two patients with biopsy-proven wtATTR or hATTR, signs of congestive heart failure, and left ventricular wall thickness ≥1.3 cm on echocardiogram were entered into a single-center, open-label study of Inotersen.44 Three patients left the study. One patient left the study to have a heart transplant, the second left the study after a fall leading to a head injury, and the third after developing pruritis which he felt was due to the study medication. Of the first 15 patients who completed the 12-month study, 8 patients had hATTR-CM (1 V30M, 6 T60A, 1 P24S) and 7 patients had wtATTR-CM. Mean reduction of TTR concentration was 72% (range 39%–91%). Lack of disease progression of hATTR-CM and wtATTR-CM was demonstrated by echocardiography and cardiac MRI over 12 months. There were no drug-related serious adverse events. Close monitoring revealed only mild decreases in platelet function and stable renal function. Inotersen appears to slow disease progression in ATTR-CM.
Conclusion
hATTR is a devastating disorder with death occurring within 15 years of disease onset, often beginning in patients around the age of 30. The inability of previous therapeutic treatments to prevent hATTR progression led to the discovery of Inotersen, an ASO that can be given once a week SC without pretreatment. It is one of the new treatments becoming available for hATTR that can prevent disease progression and improve quality of life. Inotersen (Tegsedi®; Akcea Therapeutics, Inc, Boston, MA, USA and Ionis Pharmaceuticals, Carlsbad, CA, USA) was approved for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults on October 05, 2018 by the FDA. Inotersen is now approved in the USA, European Union and Canada.43
As in all short-term studies, the long-term benefit and toxicities are yet to be answered. Sixty percent of patients treated with TTR oligonucleotides had a response over an 18-month period.10 Continued reduction of serum levels of TTR would presumably continue to prevent disease progression.
LTx patients who show signs of progression currently do not have any therapeutic options, and a trial with Inotersen could be considered.
Inotersen does not cross the blood–brain or blood–retinal barrier and thus would not be effective in hATTR patients with predominant retinal or leptomeningeal symptoms. ASOs can be injected intravitreally and intrathecally, and future studies may address these issues.28,30
As with all inherited disorders, the question of management and treatment of asymptomatic or pre-symptomatic gene carriers will need to be addressed.45 The efficacy of these medications for late onset or more severely affected patients, for whom there is no current adequate therapy, is still unknown. Given the heterogeneity of hATTR, however, response rates may be different. It may be that there is no one best treatment for all patients and combination therapy using the different mechanisms: TTR reduction, TTR stabilizers, and reduction of TTR deposits will be required.
With all new medications, cost is a concern. However, the cost of treatment of untreated hATTR patients and hATTR patients who have undergone LTx or cardiac transplantation are considerable.46 The cost of caregiver burden also needs to be considered.47 Reduced work productivity of the patient and caregiver, patient’s increasing need for assistance due to reduced ability for self-care and mobility, poor mental health, and poor quality of life are not easily quantified.
Disclosure
Dr Wang was the site Primary Investigator for the Phase III studies in Patisiran and Inotersen. She is currently involved in the Expanded Access Program for Inotersen. She also contributed to the Transthyretin-Associated Amyloidosis Outcomes Survey: A Global, Multi-center, Longitudinal, Observational Survey of Patients with Documented Transthyretin (TTR) Mutations or Wild-Type TTR Amyloidosis (THAOS). She has served on advisory boards for Ionis and Pfizer. The other author reports no conflicts of interest in this work.
References
Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75(3):408–427. | ||
Westermark P, Sletten K, Johansson B, Cornwell GG. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990;87(7):2843–2845. | ||
Wang AK, Gorevic PD. Neurologic improvement after cervical decompression in wild type transthyretin amyloidosis. In: Dyck PJ, editor. Companion to Peripheral Neurology. Philadelphia, PA, USA: Saunders; 2010:145–147. | ||
Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36(4):411–423. | ||
Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451–2466. | ||
Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265(4):976–983. | ||
Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–837. | ||
Hawkins PN, Ando Y, Dispenzeri A, Gonzalez-Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–638. | ||
Wallace MR, Naylor SL, Kluve-Beckerman B, et al. Localization of the human prealbumin gene to chromosome 18. Biochem Biophys Res Commun. 1985;129(3):753–758. | ||
Buxbaum JN. Oligonucleotide drugs for transthyretin amyloidosis. N Engl J Med. 2018;379(1):82–85. | ||
Niemietz C, Chandhok G, Schmidt H. Therapeutic oligonucleotides targeting liver disease: TTR amyloidosis. Molecules. 2015;20(10):17944–17975. | ||
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583–596. | ||
Holmgren G, Steen L, Suhr O, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–1116. | ||
Benson MD. Liver transplantation and transthyretin amyloidosis. Muscle Nerve. 2013;47(2):157–162. | ||
Familial Amyloid Polyneuropathy World Transplant Registry. Available from: http://fapwtr.org. Accessed September 11, 2018. | ||
Ericzon BG, Suhr O, Broomé U, et al. Liver transplantation halts the progress of familial amyloidotic polyneuropathy. Transplant Proc. 1995;27(1):1233. | ||
Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324–327. | ||
Yazaki M, Tokuda T, Nakamura A, et al. Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochem Biophys Res Commun. 2000;274(3):702–706. | ||
Yazaki M, Liepnieks JJ, Kincaid JC, Benson MD. Contribution of wild-type transthyretin to hereditary peripheral nerve amyloid. Muscle Nerve. 2003;28(4):438–442. | ||
Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421(2–3):185–203. | ||
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–249. | ||
Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–2667. | ||
Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. | ||
Maurer MS, Schwartz JH, Gundapaneni B, et al; ATTR-ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. | ||
Penchala SC, Connelly S, Wang Y, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013;110(24):9992–9997. | ||
Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19(Suppl 1):34–36. | ||
Palha JA, Hays MT, Morreale de Escobar G, Episkopou V, Gottesman ME, Saraiva MJ. Transthyretin is not essential for thyroxine to reach the brain and other tissues in transthyretin-null mice. Am J Physiol. 1997;272(3 Pt 1):E485–E493. | ||
Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-targeted therapeutics. Cell Metab. 2018;27(4):714–739. | ||
Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46(4):1584–1600. | ||
Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of antisense drugs. Annu Rev Pharmacol Toxicol. 2017;57(1):81–105. | ||
Goyal N, Narayanaswami P. Making sense of antisense oligonucleotides: a narrative review. Muscle Nerve. 2018;57(3):356–370. | ||
Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. | ||
Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. | ||
Rossor AM, Reilly MM, Sleigh JN. Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol. 2018;18(2):126–131. | ||
Crooke ST. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther. 2017;27(2):70–77. | ||
Suhr OB, Coelho T, Buades J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015;10(1):1–9. | ||
FDA approves first-of-its kind targeted RNA-based therapy to treat a rare disease. FDA news release August 10, 2018; 2018. Available from: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm616518.htm. Accessed September 11, 2018. | ||
Benson MD, Pandey S, Witchell D, et al. Antisense oligonucleotide therapy for TTR amyloidosis. Amyloid. 2011;18(Suppl 1):60. | ||
Ackermann EJ, Guo S, Booten S, et al. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid. 2012;19(Suppl 1):43–44. | ||
Ackermann EJ, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 2016;23(3):148–157. | ||
Brannagan T, Wang AK, Coehlo T, et al. Open-label Extension of the Phase 3 NEURO-TTR Study to assess the long-term efficacy and safety of Inotersen in patients with hereditary transthyretin amyloidosis. Neurology. 2018;90(15 Supplement):P1.234. | ||
Keam SJ. Inotersen: first global approval. Drugs. 2018;78(13):1371–1376. | ||
Press Release. Available from: https://ir.akceatx.com/news-releases/news-release-details/akcea-and-ionis-receive-fda-approval-tegseditm-inotersen. Accessed September 11, 2018. | ||
Benson MD, Dasgupta NR, Rissing SM, Smith J, Feigenbaum H. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid. 2017;24(4):217–223. | ||
Schmidt HH, Barroso F, González-Duarte A, et al. Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2016;54(3):353–360. | ||
Habka D, Mann D, Landes R, Soto-Gutierrez A. Future economics of liver transplantation: a 20-year cost modeling forecast and the prospect of bioengineering autologous liver grafts. PLoS One. 2015;10(7):e0131764–21. | ||
Stewart M, Shaffer S, Murphy B, et al. Characterizing the high disease burden of transthyretin amyloidosis for patients and caregivers. Neurol Ther. 2018;7(2):349–364. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.