")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 7
Inhibition of the methionine aminopeptidase 2 enzyme for the treatment of obesity
Authors Joharapurkar A, Dhanesha N, Jain M
Received 1 November 2013
Accepted for publication 9 January 2014
Published 28 February 2014 Volume 2014:7 Pages 73—84
DOI https://doi.org/10.2147/DMSO.S56924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Amit A Joharapurkar, Nirav A Dhanesha, Mukul R Jain
Department of Pharmacology and Toxicology, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India
Abstract: Worldwide prevalence of obesity has nearly doubled since 1980. Obesity is the result of interactions among the environmental factors, genetic predisposition, and human behavior. Even modest weight reduction in obese patients provides beneficial health outcomes. For effective weight reduction, a drug should either increase energy expenditure or decrease energy intake without causing serious adverse effects. To overcome lack of efficacy and central nervous system related side effects, exploitation of the peripheral mechanism of anti-obesity action is needed. Inhibition of pathological angiogenesis in adipose tissue is one such peripheral mechanism that has attracted the attention of researchers in this area. Although originally developed as anti-cancer agents, methionine aminopeptidase (MetAP2) inhibitors induce significant and sustained weight reduction. Here, we review preclinical and clinical pharmacology of MetAP2 inhibitors. Beloranib is a prototype MetAP2 inhibitor, and currently in advanced clinical trials for the treatment of obesity. Clinical data of beloranib indicate that MetAP2 inhibitors could be a future treatment option for weight reduction without serious adverse effects. Further clinical data from Phase III trials will add to our growing knowledge of MetAP2 inhibitor potential for anti-obesity therapy.
Keywords: angiogenesis, beloranib, body weight, MetAP2, anti-obesity
Introduction
Epidemic of obesity
The World Health Organization (WHO) has defined obesity as “abnormal or excessive fat accumulation that may impair health.”1 Obesity is commonly measured using body mass index (BMI, kg/m2). A BMI between 25.0 and 29.9 kg/m2 is categorized as overweight, and a BMI greater than 30.0 kg/m2 is defined as obese.1 Abdominal obesity, rather than total body fat, is also a valuable, independent predictor of several obesity related diseases.2 Obesity is one of the most important contributors to ill health in the current century. Worldwide prevalence of obesity has nearly doubled since 1980, with differential demographics; developed countries in North America and Europe show high rates of obesity, while the prevalence of obesity in Africa and Middle Eastern countries is variable, and Asian countries show low incidence.3 According to a WHO estimate in 2005, approximately 1.6 billion people worldwide were overweight and at least 400 million adults were obese. By 2015, approximately 2.3 billion adults will be overweight and at least 700 million will be obese.1
Obesity is the result of interactions between environmental factors, genetic predisposition, and human behavior.4 Lifestyle changes, like increasing the quantity and energy density of food consumed by adults, car ownership, television viewing, and proxy measures of physical inactivity are the main causes of increased prevalence of obesity in recent times.5–7 Genetic predisposition is another important driver of obesity. Genome wide association studies in European as well as Caucasian populations have identified single nucleotide polymorphisms (SNPs) associated with increased BMI and obesity.8 SNPs have little effect on an individual’s susceptibility to obesity. However, owing to their high frequency, they are a major contributor to obesity at the population level.9 Social networks also play an important role in development of obesity. Social networks of subjects from the Framingham Heart Study were examined for the effects of weight gain among friends, siblings, and spouses. It was observed that one’s risk of becoming obese increased by 57%, 40%, and 37% if a friend, sibling, or spouse became obese, respectively.10 Obesity is particularly harmful because it may cause a range of diseases like dyslipidemia, atherosclerosis, hypertension, and type 2 diabetes. Together, these diseases can increase the risk of stroke, angina, and myocardial infarction.11 Obesity also increases the risk of colon, breast, kidney, and digestive tract cancers. Other diseases associated with obesity include arthritis, sleep apnea, gallstones, and gout, as well as low self-esteem and affective disorders.11 More importantly, obesity is associated with an increased risk of death. A prospective study of more than 500,000 US men and women reports that among patients who had never smoked, the risk of death is increased by 2- to 3-fold in obese and by 20% to 40% in overweight patients, compared with non-obese people.12 Due to increased death risk and increased risk of morbidity, the obesity epidemic is a great financial liability on the economy.
Treatment options for obesity
If excess weight increases the risk of health problems, even modest weight reduction (5% to 10% of total body weight) in obese patients provides beneficial health outcomes.13 Weight loss to this extent reduces blood pressure and cholesterol levels, and improves blood glucose control along with a reduced risk of myocardial infarction, stroke and death. Lifestyle modification, ie, changes in diet and proper exercise, is the best and cheapest obesity treatment. However, it is ineffective in most individuals due to lack of long-term compliance.14 Pharmaceutical industries and universities have therefore hunted to discover novel therapies that could serve as adjunctive treatments to diet, exercise, and lifestyle modification regimes. Thyroid preparations were used for weight reduction in the eighteenth century. 2,4-Dinitrophenol (DNP) was introduced as an anti-obesity drug in the early twentieth century. Amphetamine was introduced in the 1930s and phentermine was approved by the US Food and Drug Administration (FDA) in 1959. Sibutramine and rimonabant are two recent additions to obesity therapy, but both of these agents were withdrawn owing to serious adverse events.15 Currently available treatment options for obesity include Orlistat (Xenical®; F. Hoffman-La Roche Ltd., Basel, Switzerland; Alli, GlaxoSmithKline plc, Brentford, England), a gastric and pancreatic lipase inhibitor, lorcaserin (Belviq®; Arena Pharmaceuticals Inc., San Diego, CA, USA), a selective small molecule agonist of the 5-hydroxytryptamine 2C (5-HT2C)receptor, and a combination of phentermine plus topiramate (Qsymia®; Vivus Inc., Mountain View, CA, USA).15 Table 1 shows investigational and approved drugs for obesity treatment.
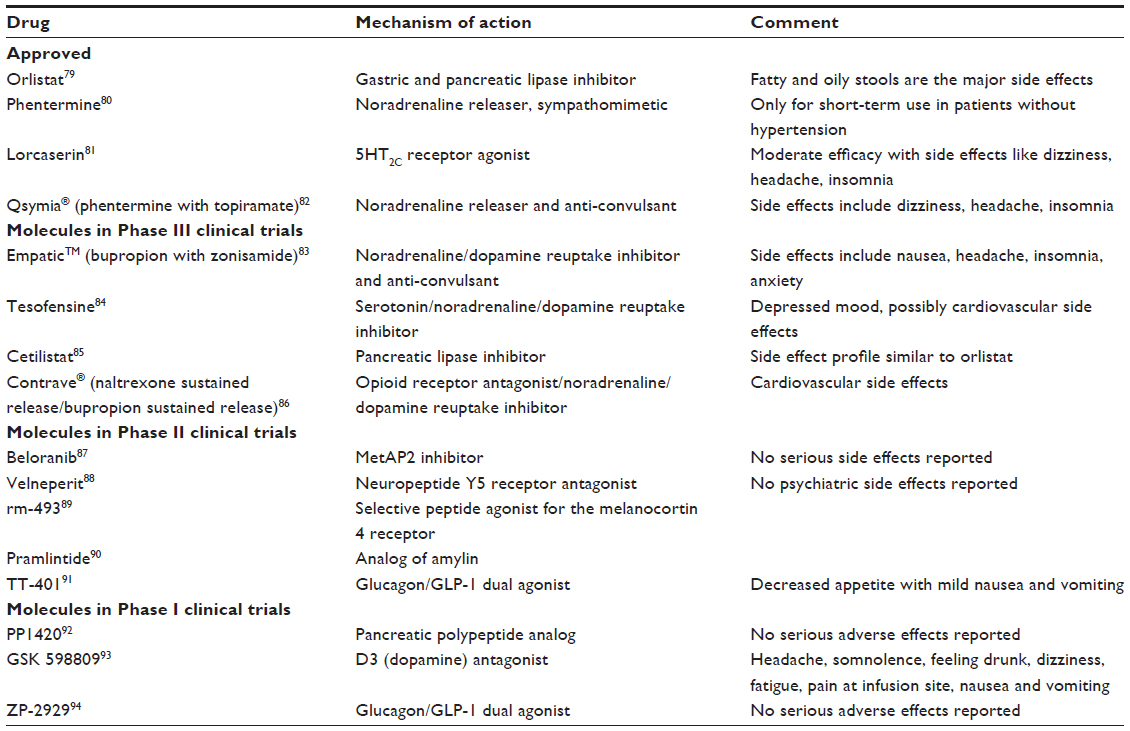 | Table 1 Anti-obesity drugs that are approved or in clinical trials |
For effective weight management therapy a drug should either increase energy expenditure or decrease energy intake without causing serious adverse effects. Drugs that increase energy expenditure, such as DNP, thyroid hormone, and thyromimetics, effectively reduce body weight, but are associated with potential side effects. On the other hand, drugs that decrease food intake, such as lorcaserin, phentermine with topiramate, or liraglutide are modestly efficacious anti-obesity agents with lesser side effects.16 The major mechanism of these agents is central nervous system (CNS) control. There are many peripheral pathways that can affect mitochondrial metabolism, nutrient absorption, vagal feedback pathway or lipolysis, and nutrient absorption or the vagal feedback pathway, such that these peripherally acting agents can potentially be combined with CNS agents to achieve maximal efficacy. It is expected that newer generations of anti-obesity therapeutics will be superior to existing agents and will facilitate lifestyle modification. To fulfill this expectation we can exploit the peripheral mechanism of anti-obesity action, such that these peripherally acting agents can be complimentary to the centrally acting agents. This will pave the way to truly effective and safer anti-obesity treatment. Inhibition of pathological angiogenesis is one such peripheral mechanism that has attracted the attention of researchers in this area.
Targeting angiogenesis in obesity
Angiogenesis is the process of new blood vessel formation from existing blood vessels. It differs from vasculogenesis, which is the differentiation of mesoderm cell precursors. Angiogenesis is mainly involved in growth and development; however, it is also involved in tissue repair, as in during wound healing, and in the formation of granulation tissue. Pathologically, however, it also drives tumors from a benign state to malignant. Based on these observations, the concept of anti-angiogenic therapy was proposed, which has successfully been translated into the development of anti-angiogenic drugs for the treatment of cancer and ophthalmological disorders.17 Similar to cancer cells, brown adipose tissue (BAT) is highly vascularized tissue, and white adipose tissue (WAT) undergoes expansion and shrinkage throughout the life.18 Owing to this nature, continuous remodeling of capillary networks, ie, dynamic angiogenesis, is required in both BAT and WAT.19 Hence, modulation of adipose tissue function by controlling angiogenesis can be targeted for the treatment of obesity. It has been demonstrated that cells located in the vessel wall contain precursor cells, which differentiate into adipocytes.20 These cells express growth factors and transcription factors. Either through these special cells or through endothelial cells, adipogenesis is controlled by angiogenesis.
On the other hand, adipose-derived stem cells produce growth factors like vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), hepatocyte growth factor (HGF), placental growth factor (PlGF), secreted protein acidic and rich in cysteine (SPARC)/osteonectin, angiopoietins, matrix metalloproteinases (MMPs), leptin, resistin, and visfatin, which boost angiogenesis.21 Adipose tissues, especially in pathological states, suffer from hypoxia, and hence express hypoxia-inducible factor 1-alpha (HIF-1α). Hypoxia induced HIF-1α and subsequent VEGF production is an important driver of angiogenesis in WAT.22 VEGF induction can also take place by HIF-1α independent mechanisms to facilitate angiogenesis. VEGF-A stimulates physiological as well as pathological angiogenesis by signaling through VEGF receptor (VEGFR)-2.21,22 Certain adipocyte lipids, such as monobutyrin, can also induce angiogenesis.23 On the other hand, several endogenous angiogenesis inhibitors like thrombospondin 1 and plasminogen activator inhibitor prevent further neovascularization.21,22 Adipose tissue releases adipokines like leptin that synergize with known angiogenetic factors, such as VEGF. Conversely, adiponectin is an adipokine that significantly suppresses angiogenesis.24
PlGF is a 25 kDa homologue of VEGF-A that enhances angiogenesis only in pathological conditions. Angiogenesis is impaired in the ischemic retina, limb, and heart, in wounded skin, and in tumors after loss of PlGF.25 Differentiation, migration, and proliferation of endothelial cells are also stimulated by FGF-2 and enhance adipocyte differentiation in vivo.26 Synthesis of proteinases such as collagenase and urokinase-type plasminogen activator (u-PA), and of integrins was stimulated by FGF-2 in order to form new capillary cord structures.21,22
Maintenance, growth, and stabilization of blood vessels is also controlled by the tyrosine kinase with immunoglobulin (Ig) and epidermal growth factor homology domains (TIE)-1 and -2 receptors.27 TIE-2 binds the angiopoietins (Ang-1 and Ang-2), whereas the ligands of TIE-1 are less well characterized. Ang-2 activates TIE-2 on some cells but blocks it on others while Ang-1 consistently activates TIE-2.28,29 Dysfunction of either endothelial cells or adipocytes results in substantial impact on the other system due to mutual interplay between these compartments. For example, type 2 diabetes may occur in obese individuals owing to endothelial dysfunction.30
Adipose circulatory vessels have many functions, namely: 1) supplying oxygen and nutrients to and removal of waste from adipose tissue; 2) transporting growth factors, cytokines, and hormones to and from other tissues; 3) supplying precursor cells that mature into adipocytes; 4) supplying stem cells and inflammatory cells that affect adipocyte functions; and 5) controlling oxygen concentration and pH in the adipose tissue. Though these functions are common for both BAT and WAT, their implications are very different. The main function of WAT is to store energy in the form of lipids, whereas the primary function of BAT is to transfer energy from nutrients into heat and it serves as a site for non-shivering thermogenesis. In WAT, promotion of angiogenesis results in adipose tissue expansion, and promotes obesity. Conversely, activation of angiogenesis in BAT improves oxygen supply and energy expenditure and thus reduces obesity.19 BAT is largely accountable for energy metabolism, and efficient blood perfusion is required for its function in order to export heat. BAT hyperplasia is significantly dependent on angiogenesis, as it requires rapid development of capillaries. The existence and significance of active BAT in infants has been well reported. However, in adult humans, the tissue regresses and functionally relevant BAT is considered to be essentially absent. Hence, therapeutic utility of BAT enhancers as a target of obesity is limited.31
Recent studies indicate that adult humans show the presence of typically inducible (from WAT) brown-like adipose tissue, while infants have actual BAT.32–34 The prevalence and activity of this adult human BAT are inversely related to body fat content and decrease with age.34 This type of BAT mediates thermogenesis via uncoupling protein-1 (UCP-1). The VEGF-VEGFR-2 system regulates angiogenesis in this adult BAT. Another mechanism which may stimulate adipose angiogenesis is the sympathetic release of neuropeptide Y (NPY). NPY stimulates NPY-Y2 receptors, which activate adipose angiogenesis in BAT.35 To conclude, activation of BAT and transition of WAT into BAT may require different mechanisms. Though the precise mechanism is not clear, it has been shown that adipose VEGF expression protects mice from high fat diet-induced obesity, glucose intolerance, insulin resistance, and adipose tissue inflammation. Conversely, genetic deletion of VEGF in mouse adipose tissue produces an obese and insulin resistant phenotype. It has also been shown that specific pharmacological inhibition of VEGFR-2 results in arresting adipose tissue expansion, mostly because of angiogenesis inhibition.36 Imbalanced production of VEGF, FGF-2, adiponectin, and thrombospondin 1 results in the formation of angiogenic phenotypes in both healthy and pathological organs;37 VEGF-A inhibitors are effective treatment options for various human cancers38,39 and non-malignant diseases, such as choroidal neovascularization in age-related macular degeneration.39,40
Taken together, the aforementioned studies indicate that inhibiting angiogenesis in WAT may be a better option for the treatment of obesity and metabolic diseases though a thorough understanding of its clinical implications is needed.41 Since obesity and other obesity-associated disorders, like diabetic complications, cardiovascular disorders, and malignancies, are associated with pathological angiogenesis42,43 inhibiting angiogenesis could be a novel and effective treatment method.25
Methionine aminopeptidases (MetAP2) in angiogenesis
MetAPs are enzymes responsible for the removal of methionine from the amino-terminus of newly synthesized proteins.44,45 Removal of methionine is crucial for further amino terminal modifications (eg, acetylation by N-alpha-acetyltransferase and myristoylation of glycine by N-myristoyltransferase; NMT) and for protein stability.46,47 MetAP2, a member of the dimetallohydrolase family, is encoded by the METAP2 gene in humans.48 Increased expression of this gene is associated with various forms of cancer.49 This gene is a member of the methionyl aminopeptidase family and encodes a protein that binds to cobalt or manganese ions.48 Literature suggested that MetAP2 plays a critical role in the growth of different types of tumors via promoting angiogenesis. Malignant mesothelioma cells expressed higher MetAP2 messenger ribonucleic acid (mRNA) levels compared to normal mesothelioma cells.50 Transfection of mesothelioma cells with an MetAP2 anti-sense oligonucleotide revealed a time-dependent inhibition of cell survival and induced nucleosome formation. MetAP2 is a main regulator of the proliferative and apoptotic pathways in mesothelioma cells and MetAP2 inhibition may represent a potential target for therapeutic intervention in human mesothelioma.50 MetAPs are inhibited by the natural product fumagillin and its derivatives by irreversibly binding to the active site of the enzyme. These compounds are potent anti-angiogenic agents that prevent tumor vascularization and metastasis.51
MetAP2 inhibitors for the treatment of obesity
Although originally developed as anti-cancer agents,52 MetAP2 inhibitors induce significant and sustained weight reduction. The anti-obesity efficacy of MetAP2 inhibitors has been demonstrated in animal models of obesity and in humans at low doses that do not affect angiogenesis.53–56 Figure 1 demonstrates the chemical structures of MetAp2 inhibitors.
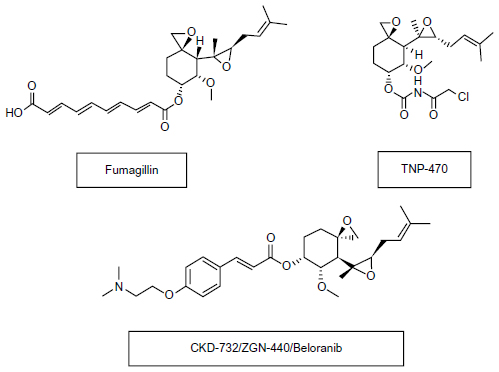 | Figure 1 Chemical structures of MetAP2 inhibitors. |
Mechanism of action
The precise mechanism for the anti-obesity effect of MetAP2 inhibitors is not well elucidated. However, non-enzymatic actions of MetAP2 to suppress activity of extracellular signal regulated kinases 1 and 2 (ERK1/2) represent one of the key mechanisms for the observed anti-obesity effect.57 MetAP2 inhibition results in to suppression of sterol regulatory element binding protein (SREBP) activity, leading to reduced lipid and cholesterol biosynthesis via ERK-related pathways.58,59 Extended fumagillin exposure results in changes in the expression patterns of hepatic and adipose tissue genes suggesting that MetAP2 inhibition also alters the relative abundance of factors involved in inflammation, consistent with reduced ERK-dependent cellular processes.60
Recent clinical trials with beloranib (MetAP2 inhibitor) demonstrated an increase in the levels of key catabolic hormones adiponectin and FGF-21. Coupled with the appearance of ketone bodies (beta-hydroxybutyrate), this suggests that MetAP2 inhibition stimulates energy expenditure, fat utilization, and lipid excretion.60 The reduction in leptin levels was also consistent with a decrease in total adipose tissue and negative energy balance.61 Figure 2 demonstrates the mode of action of MetAP2 inhibitors and its impact on obesity.
The active site of MetAP2 has a structural motif characteristic of many metalloenzymes, including the dioxygen carrier protein, hemerythrin; the dinuclear non-heme iron protein, ribonucleotide reductase; leucine aminopeptidase; urease; arginase; several phosphatases and phosphoesterases, which include two bridging carboxylate ligands and a bridging water or hydroxide ligand.62
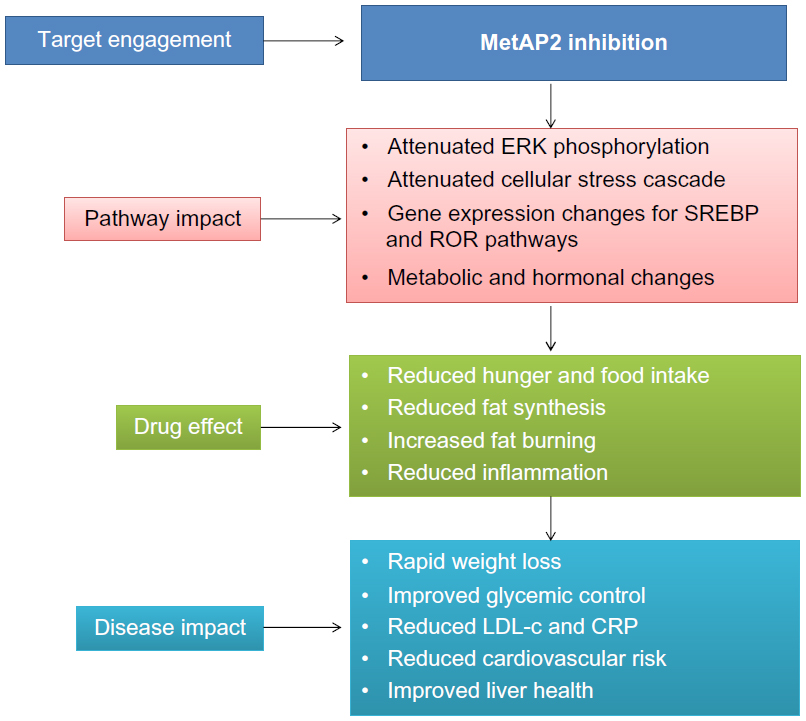 | Figure 2 Mode of action for MetAP2 inhibitors and impact on obesity. |
Fumagillin
Fumagillin, a prototype MetAP2 inhibitor, is a natural product isolated from Aspergillus fumigatus.64 Fumagillin covalently binds and modifies histidine 231, the active site of MetAP2 while it does not inhibit MetAP1.65 In previous clinical trials, it was evaluated mainly for anticancer indication and as an antiparasitic agent. It was well tolerated, and no serious adverse events were noted.65,66
The anti-obesity effect of fumagillin was evaluated in 11 week old C57BL/6J mice previously kept on a high fat diet for 6 weeks.64 Fumagillin was orally dosed at 1 mg/kg/day for 4 weeks. After completion of the treatment, body weight of fumagillin treated mice was significantly lower than that of pair fed mice and of mice fed ad libitum.64 The intra-abdominal (gonadal [GON]) and inguinal subcutaneous (SC) fat mass were significantly lower in the fumagillin treatment group. The weights of some other organs, including the spleen, kidney, pancreas, and heart were also lower in the treatment group. Body temperature and physical activity remained constant throughout the 4-week experimental period and were not different between groups. Insulin levels at the end of the treatment were lower in the pair fed group compared to the treatment group while leptin levels were decreased in the treatment and pair fed group compared to the control group. Total and high-density lipoprotein cholesterol as well as triglyceride levels at the end of the study were elevated in pair fed control animals compared to treated animals. Histological analysis revealed smaller adipocyte size in SC and GON adipose tissues of treated mice, compared to pair fed control groups, associated with increased adipocyte density. Fumagillin treatment induced marked adipocyte hypotrophy associated with enhanced density compared to mice fed a high fat diet ad libitum, but less marked compared to pair fed control mice.64 The number of blood vessels surrounding each adipocyte was lowest in the fumagillin treated group, and for GON fat, it was significantly lower than for pair fed or ad libitum fed controls.64
Short-term effects of fumagillin on established obesity were evaluated in 20-week-old obese C57BL/6J mice at a dose of 1 mg/kg/day over 4 days.64 Despite a significantly higher food intake, a similar weight loss was observed in fumagillin treated mice compared to control mice and was not associated with effects on other metabolic parameters. Histological analysis of SC fat pads revealed a significantly smaller adipocyte size and higher density in fumagillin treated samples compared to controls.64 Expression levels of MetAP2, TIE-2, angiopoietin-1, and angiopoietin-2 mRNA in GON adipose tissues were significantly upregulated in the fumagillin treatment group.64
TNP-470 or AGM-1470
TNP-470 or AGM-1470, a synthetic analog of fumagillin, selectively inhibits endothelial cell growth and angiogenesis.67 The angiostatic mechanism of TNP-470 involves suppression of MetAP2 in endothelial cells, thereby preventing cell proliferation.65,68 Anti-obesity efficacy of TNP-470 was evaluated in genetically (ob/ob) and diet induced obese mice. Four to five week-old ob/ob mice were treated with TNP-470 at a dose of 20 mg/kg on alternate days. TNP-470 treatment for 8 weeks resulted in significant weight reduction, which was associated with reduced food intake compared to pair fed and ad libitum fed controls.65,69 Significant reduction was observed in subcutaneous and omental fat depots, liver weight, and total body fat after 8 weeks of TNP-470-treatment in ob/ob mice compared with both ad libitum fed and pair fed control mice.69 The ratio of lean body mass/body weight was relatively increased in the TNP-470-treated group and thus the reduction of body weight with TNP-470 was well correlated with reduction of adipose tissue mass.69 Significant reduction in adipose tissue vascularization and average size of adipocytes in subcutaneous fat deposits was observed in the TNP-470-treated mice compared with non-treated, obese animals. Serum cholesterol, serum glucose, and serum insulin levels were decreased while serum triglycerides and free fatty acid (FFA) levels remain unchanged in TNP-470-treated ob/ob mice compared to control mice.69
Another study demonstrated that TNP-470 treatment at 2.5, 5, 7.5, and 10 mg/kg/day, delivered subcutaneously, resulted in a dose dependent decrease in body weight gain in ob/ob mice.70 To test reversibility, the authors intermittently treated ob/ob mice with TNP-470 until they were reduced to the weight of age-matched lean C57BL/6J mice, and then suspended treatment until mice regained the weight; mice reduced weight while on TNP-470 and regained weight off treatment. At the end of the fourth cycle, control ob/ob mice were morbidly obese, whereas treated mice were similar in weight to lean mice.70 Decreased food intake with TNP-470 treatment was transient and matched with controls as treatment continued. In the study, TNP-470 and leptin similarly reduced fat mass relative to controls resulting in similar decreases in body fat percentage, which was supported by significantly lower adipose tissue bed weights in both treated groups.70 Basal metabolic rates increased in TNP-470-treated ob/ob mice by day 4 and in C57BL/6J mice by day 8. Respiratory exchange ratio decreased with TNP-470 by day 8 in both strains, suggesting the preferred energy substrate shifted from carbohydrates to fat during weight loss.70
Anti-obesity efficacy of TNP-470 was also evaluated in mice fed a high fat diet (45% kcal fat in food) and treated for 12 weeks.69 A significant reduction in body weight gain was observed in treated animals compared with control animals. Histological examination of high fat diet fed mice after 16 weeks of treatment with TNP-470 revealed that the depots of subcutaneous, perigonadal, and omental fat were dramatically decreased compared with control mice.69 As observed in ob/ob mice, lean body mass was increased in TNP-470-treated mice compared to control mice as a result of a decrease in total body fat.69 There was no change in energy expenditure in the TNP-470-treated mice that might have contributed to the reduction in body weight gain. Analyses of BAT capacity, such as noradrenaline-induced increase in oxygen consumption and protein levels of UCP-1 in BAT tissue, also showed no increase in thermogenic capacity in TNP-470-treated mice. Similar to the ob/ob mice data, TNP-470 treatment resulted in significant reduction in serum cholesterol, serum glucose, serum insulin levels, adipose tissue vascularization, and average size of adipocytes in subcutaneous fat deposits in high fat fed C57BL/6J mice.69 The authors noted that although TNP-470 is a selective angiogenesis inhibitor in clinical trials for the treatment of cancer, high dosages of this agent may affect other systems in the body and hence TNP-470 may prevent obesity through mechanisms other than by affecting antiangiogenesis.69
The effect of TNP-470 on caloric intake and energy expenditure in high fat fed C57BL/6J mice was thoroughly assessed recently by White et al. The authors speculated that previous studies69,70 possibly overlooked the effects of TNP-470 on increased energy expenditure.71 As observed in the White et al study, TNP-470 at 20 mg/kg/day also reduced body weight gain and epididymal adipose tissue weight in diet induced obese mice. Similarly, liver weight as a percentage of body weight, serum triglycerides, serum glucose, and liver lipid accumulation was decreased in TNP-470-treated animals compared to control animals.71 By day 5, TNP-470-treated mice consumed significantly less grams of high fat food than vehicle treated high fat fed mice. By treatment day 15, TNP-470 mice were consuming an equivalent number of calories to that of chow fed mice, despite the provision of high fat diet. In contrast to previous studies, over the entire 24-hour period, energy expenditure in TNP-470-treated mice was greater than both in high fat fed and chow fed mice while locomotion did not differ between treatment and control groups.71 The authors propose that a novel signal from the adipose tissue vasculature acts centrally to regulate energy balance as a possible mechanism for the increased energy expenditure after TNP-470 treatment.71
CKD-732
CKD-732 is a synthetic derivative of TNP-470. It has higher affinity for MetAP2 and higher efficacy for suppressing of tumor growth compared to TNP-470.72 Anti-obesity efficacy of CKD-732 was evaluated in various animal models at different doses (C57BL/6J mice, 5 mg/kg per day; arcuate nucleus lesion mice, 1 mg/kg per day; Sprague Dawley rats, 5 mg/kg per day; Long–Evans Tokushima Otsuka (LETO) rats, 1 mg/kg per day; and Otsuka Long–Evans Tokushima fatty (OLETF) rats, 1 mg/kg per day). Seven days of CKD-732 subcutaneous injection decreased cumulative food intake and body weight in all animals, but to a greater extent in obese models.73 The core temperature of CKD-732-treated mice was greater than control mice, which suggests an increase in energy expenditure following CKD-732 treatment. In CKD-732-treated LETO and OLETF rats, regional fat pad weights, especially epididymal and mesenteric fat pads, and the size of adipocytes were significantly decreased.73
To evaluate the central anorexic effect, intracerebroventricular (ICV) injection of CKD-732 was administered in ARC lesion mice and ob/ob mice. Although anorexic effects were not found in normal littermates, the 24-hour body weight and 4- and 24-hour food intake were significantly decreased in a dose-dependent manner.73 On the other hand, the core body temperature was not altered in ARC lesion mice and normal littermates by a single central injection of CKD-732. Interestingly, expression of pro-opiomelanocortin (POMC), NPY, Agouti-related peptide (AgRP), and melanin-concentrating hormone (MCH) mRNA were not significantly altered by CKD-732.73 The authors checked conditioned taste aversion (CTA) following a single peripheral injection of CKD-732: LETO and OLETF rats with saline injection exhibited no preference for saccharin flavor. In contrast, the rats in both the lithium chloride and CKD-732-treated groups significantly preferred the neutral flavor, indicating the development of CTA.73 CKD-732 acutely causes CTA, which may contribute to decreased food intake caused by CKD-732, especially in obese models, and suggested that the CKD-732 has a multifunctional action that induces direct shrinkage of adipose tissue and anorexia caused by direct hypothalamic stimuli or CTA.73
ZGN-201
Researchers at Zafgen Inc., (Cambridge, MA, USA) evaluated the effect of 9 months of treatment with ZGN-201 on body weight and metabolic parameters in obese mice.74 During the first 4 weeks of treatment with 1 mg/kg ZGN-201 per oral treatment, mice lost all excess body weight, driven by a 30% reduction in food intake during the initial 2 weeks of treatment. This decrease in body weight was stable by day 28, food intake returned to 13% below high fat fed control animals, and was stable for the following 8 months, during which time the ZGN-201-treated animals had stable weights.74 Following 9 months of treatment, body weights of ZGN-201-treated mice were 43% lower than high fat fed control animals. Fasting plasma glucose and insulin were reduced by ZGN-201 treatment and plasma beta hydroxybutyrate was increased relative to high fat fed control animals, a consistent feature of MetAP2 treatment. Gene expression analysis revealed a downregulation of key lipid synthesis genes in the livers of ZGN-201-treated animals. The insulin and carbohydrate responsive genes, acetyl CoA carboxylase 1 and 2, fatty acid synthase, stearoyl CoA desaturase 1, and sterol regulatory element-binding protein (SREBP)-1c were downregulated in ZGN-201-treated animals when compared to chow fed controls.74
The effect of ZGN-201 was also evaluated in high fat, high fructose fed obese and chow fed overweight dogs for 8 weeks.75 In high fat fed dogs, daily oral treatment with ZGN-201 promoted loss of 81% of excess body weight, whereas treatment in overweight dogs inhibited weight gain. ZGN-201 reduced feed intake in obese dogs by 29%, but had minimal effect on overweight dogs.75 Glycerol levels increased in both obese and overweight dogs, reflecting enhanced lipolysis, while beta hydroxybutyrate levels, reflecting fat oxidation, were only increased in obese dogs.75
Clinical studies on MetAP2 modulators
Beloranib (CKD-732, ZGN-440, or ZGN-433) is an investigational drug candidate for the treatment of obesity. It was discovered by Chong Kun Dang Pharmaceutical Corp. (Seoul, South Korea) and is currently being developed by Zafgen Inc. Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme MetAP2.73 It was originally designed as an angiogenesis inhibitor for the treatment of cancer.73 However, once the potential anti-obesity effects of MetAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.
The first Phase I study showed that beloranib (ZGN-433) at a dose of 0.9 mg/m2 was well tolerated and reduced body weight by a median value of 1 kg per week and 3.1% over 26 days relative to placebo in severely obese subjects.76 The results of the study also demonstrated a decline in hunger as well as meaningful changes in lipid parameters following treatment at 0.9 mg/m2. These changes included a 38% reduction in triglyceride levels and a 23% reduction in low-density lipoprotein (LDL) cholesterol levels. Additionally, beta-hydroxybutyrate, an indicator of fat oxidation, increased to levels seen with very low-energy diets. No treatment-related serious adverse events were observed.76
The second Phase I trial was a randomized, double-blind, placebo controlled study to evaluate the safety, tolerability, and efficacy of twice-weekly intravenously administered beloranib in severely obese women with a BMI of 39.1±3.7 kg/m2.76 Individuals received 0.9 mg/m2, 3 mg/m2, or 6 mg/m2 beloranib (n=17) or placebo (n=11) twice weekly over a 25-day period (seven doses). Patients were allowed to eat normally and were not advised to change their exercise habits. The trial enrolled 34 subjects of whom 28 completed the study.76
Beloranib treatment for 25 days resulted in significantly more weight reduction versus the placebo-controlled group (4.3±0.4 kg versus –0.6±0.5 kg in placebo). Additionally, subjects treated with beloranib showed improvements in cardiometabolic risk factors, including reduced triglycerides, LDL cholesterol, waist circumference, diastolic blood pressure, and C-reactive protein, whereas there was no change with placebo.76 There was a trend for beloranib to be associated with a reduction in the percentage of subjects with metabolic syndrome that did not reach statistical significance. The most common adverse events were nausea, infusion site injury, and headache. Most events were of mild/moderate intensity and tended to be self-limiting.76
The third Phase I trial was a randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and efficacy of intravenously administered ZGN-433 in severely obese women with a BMI of 37.8±0.6 kg/m2.76 Individuals received ZGN-433 at 0.1 mg/m2, 0.3 mg/m2, or 0.9 mg/m2 or placebo twice weekly by intravenous administration over a 4-week period. Patients were allowed to eat normally and were not counseled to change their exercise habits. The trial enrolled 31 subjects and 26 people completed the study.76
After 26 days of treatment, subjects had decreased LDL cholesterol levels by 22% in the group that received 0.9 mg/m2 of ZGN-433 versus a 2% increase in the placebo group. Consistent with results seen in obese mouse models, treatment with ZGN-433 at a dose of 0.9 mg/m2 increased beta-hydroxybutyrate, an indicator of fat oxidation, increased plasma adiponectin concentrations, and increased the ratio of adiponectin/leptin.76
On November 15, 2013, Zafgen Inc., announced final body weight loss and cardiometabolic data from a Phase II study of beloranib for obesity. The 12-week study results showed significant weight loss and improvements in cardiometabolic risk markers in 147 obese individuals over 12 weeks of treatment, the largest and longest trial to date for the beloranib program.76
In this double blind, placebo-controlled study, Zafgen Inc., investigated the safety, tolerability, pharmacokinetics, and metabolic effects of beloranib in obese men and women. The trial enrolled 147 patients, of whom 122 completed the study. Subjects were mostly obese women with a mean age of 48.4 years, mean body weight of 100.9 kg, and a mean BMI of 37.6 kg/m2, who were enrolled into one of the four arms of the trial: n=37 for 0.6 mg; 37 for 1.2 mg; 35 for 2.4 mg; and n=38 in the placebo arm. Patients were allowed to eat normally and were not counseled to change their diet or exercise habits.76
Results indicate that after 12 weeks of treatment, subjects on 0.6 mg, 1.2 mg, or 2.4 mg of beloranib lost an average (± standard error of the mean) of 5.5±0.5 kg, 6.9±0.6 kg, and 10.9±1.1 kg, respectively, versus losses of 0.4±0.4 kg for those on placebo (all P<0.0001 versus placebo). The results have also demonstrated that weight loss with beloranib was progressive and continuing at week 12, and included a reduced sense of hunger, improved cardiometabolic risk markers, and was generally well tolerated. As observed in previous trials, this study also showed improvements in LDL-cholesterol, high density lipoprotein-cholesterol, triglycerides, and reduction in blood pressure. The most common adverse events with a higher incidence rate in those taking beloranib were nausea, diarrhea, headache, injection site bruising, and insomnia.76
Conclusion and place in therapy
Although MetAP2 inhibitors were originally developed as anticancer therapies, research has demonstrated their efficacy for effective and sustained weight reduction along with increased energy expenditure and reduced calorie consumption. Inhibition of MetAP2, and hence angiogenesis, is a novel mechanism for the treatment of obesity and related complications. MetAP2 inhibitors target angiogenic vessels to arrest their growth, which would prevent further development of adipose tissue and thus obesity. As opposed to anti-angiogenic therapy for cancer, MetAP2 inhibition for obesity treatment might not lead to drug resistance because of the genomic stability of adipocytes and endothelial cells.77 Nevertheless, long-term activation of different signaling pathways may result in reciprocal switching of angiogenic responses. Additional advantages of such treatments is that the timescale is not designed for a lifetime; when target body weight is achieved, the treatment can be stopped.77 Although no meaningful adverse effects are noted in clinical trials of beloranib, theoretically, patients who are obese and have already developed cardiometabolic complications, such as hypertension, might not be appropriate for this type of therapy. Another possible adverse effect can include delayed wound healing in obese subjects and patients with type 2 diabetes.77
Robust clinical data of beloranib indicate that it has very good potential for weight reduction in moderate to severe obese patients without serious adverse effects. The reduction in cardiometabolic risk factors is an added advantage of such treatment. The absolute weight loss achieved by the beloranib treatment is similar or greater than approved anti-obesity therapies. For example, absolute weight loss achieved by the highest dose of orlistat, lorcaserin, or phentermine/topiramate combination is 3.0, 4.8, and 12.2 kg, respectively versus 10.5 kg for beloranib treatment.76,78 Further clinical data from Phase III trials will add to our growing knowledge of MetAP2 inhibitor potential for anti-obesity therapy.
Acknowledgment
The authors are grateful to Mr Pankaj R Patel for his guidance. This is ZRC communication number 462.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization [webpage on the Internet]. Media centre fact sheets. Obesity and overweight. Geneva: World Health Organization; 2013. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/index.html. Accessed October 22, 2013. | |
Kumanyika SK, Obarzanek E, Stettler N, et al; American Heart Association Council on Epidemiology and Prevention, Interdisciplinary Committee for Prevention. Population-based prevention of obesity: the need for comprehensive promotion of healthful eating, physical activity, and energy balance: a scientific statement from American Heart Association Council on Epidemiology and Prevention, Interdisciplinary Committee for Prevention (formerly the expert panel on population and prevention science). Circulation. 2008;118(4):428–464. | |
Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132(6):2087–2102. | |
Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin North Am. 2010;39(1):1–7. | |
Dietz WH, Gortmaker SL. Do we fatten our children at the television set? Obesity and television viewing in children and adolescents. Pediatrics. 1985;75(5):807–812. | |
Holsten JE. Obesity and the community food environment: a systematic review. Public Health Nutr. 2009;12(3):397–405. | |
Prentice AM, Jebb SA. Obesity in Britain: gluttony or sloth? BMJ. 1995;311(7002):437–439. | |
Andreasen CH, Andersen G. Gene-environment interactions and obesity – further aspects of genomewide association studies. Nutrition. 2009;25(10):998–1003. | |
Andreasen CH, Stender-Petersen KL, Mogensen MS, et al. Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes. 2008;57(1):95–101. | |
Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007;357(4):370–379. | |
Heal DJ, Gosden J, Smith SL. Regulatory challenges for new drugs to treat obesity and comorbid metabolic disorders. Br J Clin Pharmacol. 2009;68(6):861–874. | |
Adams KF, Schatzkin A, Harris TB, et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. N Engl J Med. 2006;355(8):763–778. | |
Oster G, Thompson D, Edelsberg J, Bird AP, Colditz GA. Lifetime health and economic benefits of weight loss among obese persons. Am J Public Health. 1999;89(10):1536–1542. | |
Wu T, Gao X, Chen M, van Dam RM. Long-term effectiveness of diet-plus-exercise interventions vs diet-only interventions for weight loss: a meta-analysis. Obes Rev. 2009;10(3):313–323. | |
Adan RA. Mechanisms underlying current and future anti-obesity drugs. Trends Neurosci. 2013;36(2):133–140. | |
Adan RA. Mechanisms underlying current and future anti-obesity drugs. Trends Neurosci. 2013;36(2):133–140. | |
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–31. | |
Cao Y. Angiogenesis modulates adipogenesis and obesity. J Clin Invest. 2007;117(9):2362–2368. | |
Bukowiecki L, Lupien J, Follea N, Paradis A, Richard D, LeBlanc J. Mechanism of enhanced lipolysis in adipose tissue of exercise-trained rats. Am J Physiol. 1980;239(6):E422–E429. | |
Gupta RK, Mepani RJ, Kleiner S, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15(2):230–239. | |
Cao Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol. 2001;33(4):357–369. | |
Sun K, Wernstedt Asterholm I, Kusminski CM, et al. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A. 2012;109(15):5874–5879. | |
Dobson DE, Kambe A, Block E, et al. 1-Butyryl-glycerol: a novel angiogenesis factor secreted by differentiating adipocytes. Cell. 1990;61(2):223–230. | |
Mahadev K, Wu X, Donnelly S, Ouedraogo R, Eckhart AD, Goldstein BJ. Adiponectin inhibits vascular endothelial growth factor-induced migration of human coronary artery endothelial cells. Cardiovasc Res. 2008;78(2):376–384. | |
Lijnen HR. Angiogenesis and obesity. Cardiovasc Res. 2008;78(2):286–293. | |
Kawaguchi N, Toriyama K, Nicodemou-Lena E, Inou K, Torii S, Kitagawa Y. De novo adipogenesis in mice at the site of injection of basement membrane and basic fibroblast growth factor. Proc Natl Acad Sci U S A. 1998;95(3):1062–1066. | |
Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87(7):1161–1169. | |
Christiaens V, Lijnen HR. Angiogenesis and development of adipose tissue. Mol Cell Endocrinol. 2010;318(1–2):2–9. | |
Davis S, Papadopoulos N, Aldrich TH, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nat Struct Biol. 2003;10(1):38–44. | |
Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag. 2007;3(6):853–876. | |
Zafrir B. Brown adipose tissue: research milestones of a potential player in human energy balance and obesity. Horm Metab Res. 2013;45(11):774–785. | |
Yoneshiro T, Aita S, Matsushita M, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. 2013;123(8):3404–3408. | |
Virtanen KA, Lidell ME, Orava J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360(15):1518–1525. | |
Saito M, Okamatsu-Ogura Y, Matsushita M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58(7):1526–1531. | |
Kuo LE, Kitlinska JB, Tilan JU, et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med. 2007;13(7):803–811. | |
Tam J, Duda DG, Perentes JY, Quadri RS, Fukumura D, Jain RK. Blockade of VEGFR2 and not VEGFR1 can limit diet-induced fat tissue expansion: role of local versus bone marrow-derived endothelial cells. PLoS One. 2009;4(3):e4974. | |
Cao R, Brakenhielm E, Wahlestedt C, Thyberg J, Cao Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc Natl Acad Sci U S A. 2001;98(11):6390–6395. | |
Escudier B, Eisen T, Stadler WM, et al; TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. | |
Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–2049. | |
Steinbrook R. The price of sight – ranibizumab, bevacizumab, and the treatment of macular degeneration. N Engl J Med. 2006;355(14):1409–1412. | |
Rupnick MA, Panigrahy D, Zhang CY, et al. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A. 2002;99(16):10730–10735. | |
Duh E, Aiello LP. Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes. 1999;48(10):1899–1906. | |
Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438(7070):960–966. | |
Jackson R, Hunter T. Role of methionine in the initiation of haemoglobin synthesis. Nature. 1970;227(5259):672–676. | |
Solbiati J, Chapman-Smith A, Miller JL, Miller CG, Cronan JE. Processing of the N termini of nascent polypeptide chains requires deformylation prior to methionine removal. J Mol Biol. 1999;290(3):607–614. | |
Selvakumar P, Pasha MK, Ashakumary L, Dimmock JR, Sharma RK. Myristoyl-CoA:protein N-myristoyltransferase: a novel molecular approach for cancer therapy (Review). Int J Mol Med. 2002;10(4):493–500. | |
Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem Sci. 1998;23(7):263–267. | |
Arfin SM, Kendall RL, Hall L, et al. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc Natl Acad Sci U S A. 1995;92(17):7714–7718. | |
Selvakumar P, Lakshmikuttyamma A, Das U, Pati HN, Dimmock JR, Sharma RK. NC2213: a novel methionine aminopeptidase 2 inhibitor in human colon cancer HT29 cells. Mol Cancer. 2009;8:65. | |
Catalano A, Romano M, Robuffo I, Strizzi L, Procopio A. Methionine aminopeptidase-2 regulates human mesothelioma cell survival: role of Bcl-2 expression and telomerase activity. Am J Pathol. 2001;159(2):721–731. | |
Ingber D, Fujita T, Kishimoto S, et al. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature. 1990;348(6301):555–557. | |
Yeh JR, Ju R, Brdlik CM, et al. Targeted gene disruption of methionine aminopeptidase 2 results in an embryonic gastrulation defect and endothelial cell growth arrest. Proc Natl Acad Sci U S A. 2006;103(27):10379–10384. | |
White HM, Acton AJ, Considine RV. The angiogenic inhibitor TNP-470 decreases caloric intake and weight gain in high-fat fed mice. Obesity (Silver Spring). 2012;20(10):2003–2009. | |
Brakenhielm E, Cao Y. Angiogenesis in adipose tissue. Methods Mol Biol. 2008;456:65–81. | |
Kim YM, An JJ, Jin YJ, et al. Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732. J Mol Endocrinol. 2007;38(4):455–465. | |
Hughes TE, Kim DD, Marjason J, Proietto J, Whitehead JP, Vath JE. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (Silver Spring). 2013;21(9):1782–1788. | |
Datta B, Majumdar A, Datta R, Balusu R. Treatment of cells with the angiogenic inhibitor fumagillin results in increased stability of eukaryotic initiation factor 2-associated glycoprotein, p67, and reduced phosphorylation of extracellular signal-regulated kinases. Biochemistry. 2004;43(46):14821–14831. | |
Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab. 2008;19(2):65–73. | |
Kotzka J, Knebel B, Avci H, et al. Phosphorylation of sterol regulatory element-binding protein (SREBP)-1a links growth hormone action to lipid metabolism in hepatocytes. Atherosclerosis. 2010;213(1):156–165. | |
Hughes TE, Kim DD, Marjason J, Proietto J, Whitehead JP, Vath JE. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (Silver Spring). 2013;21(9):1782–1788. | |
Baratta R, Amato S, Degano C, et al. Adiponectin relationship with lipid metabolism is independent of body fat mass: evidence from both cross-sectional and intervention studies. J Clin Endocrinol Metab. 2004;89(6):2665–2671. | |
Zhang P, Nicholson DE, Bujnicki JM, et al. Angiogenesis inhibitors specific for methionine aminopeptidase 2 as drugs for malaria and leishmaniasis. J Biomed Sci. 2002;9(1):34–40. | |
Zafgen [webpage on the Internet]. Cambridge: Zafgen; 2013. Available from: http://zafgen.com/zafgen/our-approach/the-science. Accessed October 22, 2013. | |
Lijnen HR, Frederix L, Van Hoef B. Fumagillin reduces adipose tissue formation in murine models of nutritionally induced obesity. Obesity (Silver Spring). 2010;18(12):2241–2246. | |
Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94(12):6099–6103. | |
Killough JH, Magill GB. A comparative study of fumagillin and oxytetracycline in amebiasis. Am J Trop Med Hyg. 1954;3(6):999–1007. | |
Kusaka M, Sudo K, Fujita T, et al. Potent anti-angiogenic action of AGM-1470: comparison to the fumagillin parent. Biochem Biophys Res Commun. 1991;174(3):1070–1076. | |
Garrabrant T, Tuman RW, Ludovici D, et al. Small molecule inhibitors of methionine aminopeptidase type 2 (MetAP-2). Angiogenesis. 2004;7(2):91–96. | |
Bråkenhielm E, Cao R, Gao B, et al. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ Res. 2004;94(12):1579–1588. | |
Rupnick MA, Panigrahy D, Zhang CY, et al. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A. 2002;99(16):10730–10735. | |
White HM, Acton AJ, Considine RV. The angiogenic inhibitor TNP-470 decreases caloric intake and weight gain in high-fat fed mice. Obesity (Silver Spring). 2012;20(10):2003–2009. | |
Chun E, Han CK, Yoon JH, Sim TB, Kim YK, Lee KY. Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model. Int J Cancer. 2005;114(1):124–130. | |
Kim YM, An JJ, Jin YJ, et al. Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732. J Mol Endocrinol. 2007;38(4):455–465. | |
Hughes TE, Nichols AJ, Vath JE. ZGN-201 (ZGN), a methionine aminopeptidase 2 (MetAP2) inhibitor, durably eliminates excess body fat in obese mice through regulation of fat metabolism and food intake. Poster presented at: European Association for the Study of Diabetes; September 20–24, 2010; Stockholm, Sweden. Presentation 244. | |
Hughes TE, Vath JE, Scott MF, et al. ZGN -201, a methionine aminopeptidase 2 inhibitor, normalizes glucose tolerance in overweight dogs. Abstract presented at: American Diabetes Association’s 70th Scientific Sessions; June 25–29, 2010. Orlando, FL. Abstract number 24-LB. | |
Zafgen [website on the Internet]. Press releases. Cambridge: Zafgen; 2013. Available from: http://zafgen.com/zafgen/newsroom/press-releases. Accessed October 22, 2013. | |
Cao Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov. 2010;9(2):107–115. | |
Powell AG, Apovian CM, Aronne LJ. New drug targets for the treatment of obesity. Clin Pharmacol Ther. 2011;90(1):40–51. | |
US Food and Drug Administration. Orlistat. City: Publisher; Year. Available from: http://www.fda.gov/downloads/drugs/development approvalprocess/developmentresources/ucm163348.pdf. Accessed October 22, 2013. | |
Drugs Information Online [website on the Internet]. Phentermine. City: Drugs.com; 2014. Available from: http://www.drugs.com/monograph/phentermine html. Accessed on October 22, 2013. | |
US Food and Drug Administration [website on the Internet]. FDA approves Belviq to treat some overweight or obese adults. Silver Spring: US Food and Drug Administration:2012. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm309993.htm. Accessed on October 22, 2013. | |
US Food and Drug Administration [website on the Internet]. FDA approves weight-management drug Qysmia. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Accessed October 22, 2013. | |
Gadde KM, Yonish GM, Foust MS, Wagner HR. Combination therapy of zonisamide and bupropion for weight reduction in obese women: a preliminary, randomized, open-label study. J Clin Psychiatry. 2007;68(8):1226–1229. | |
Appel L, Bergström M, Buus Lassen J, Långström B. Tesofensine, a novel triple monoamine re-uptake inhibitor with anti-obesity effects: Dopamine transporter occupancy as measured by PET. Eur Neuropsychopharmacol. 2013. | |
Kopelman P, Groot Gde H, Rissanen A, et al. Weight loss, HbA1c reduction, and tolerability of cetilistat in a randomized, placebo-controlled phase 2 trial in obese diabetics: comparison with orlistat (Xenical). Obesity (Silver Spring). 2010;18(1):108–115. | |
Naltrexone/bupropion: Contrave(R); naltrexone SR/bupropion SR. Drugs R D. 2010;10(1):25–32. | |
Hughes TE, Kim DD, Marjason J, Proietto J, Whitehead JP, Vath JE. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (Silver Spring). 2013;21(9):1782–1788. | |
Drugs Information Online [website on the Internet]. Shionogi announces positive top-line efficacy results from year-long studies of Velneperit, a novel NPY Y5 receptor antagonist being investigated for the treatment of obesity. City: Drugs.com; 2014. Available from: http://www.drugs.com/clinical_trials/shionogi-announces-positive-top-line-efficacy-results-year-long-studies-velneperit-novel-npy-y5-6742.html. Accessed October 22, 2013. | |
Rhythm Pharmaceuticals, Inc. Phase 2 study to evaluate safety and efficacy of rm-493 in obese patients. Available from: http://clinicaltrials.gov/show/NCT01749137. NLM Identifier: NCT01749137. Accessed October 22, 2013. | |
Bristol-Myers Squibb. A study to examine the long term effect of Pramlintide on body weight in obese subjects. Available from: http://clinicaltrials.gov/ct2/show/NCT00189514?term=Pramlintide&rank=4. NLM Identifier:NCT00189514. Accessed October 22, 2013. | |
Transitiontherapeutics.com TT-401. Available from http://www.transitiontherapeutics.com/media/news.php. Accessed on October 22, 2013. | |
Tan TM, Field BC, Minnion JS, et al. Pharmacokinetics, adverse effects and tolerability of a novel analogue of human pancreatic polypeptide, PP 1420. Br J Clin Pharmacol. 2012;73(2):232–239. | |
Dodds CM, O’Neill B, Beaver J, et al. Effect of the dopamine D3 receptor antagonist GSK598809 on brain responses to rewarding food images in overweight and obese binge eaters. Appetite. 2012;59(1):27–33. | |
ZEAL& [webpage on the Internet]. ZP-2929. Glostrup: Zealand Pharma A/s; 2012. Available from: http://www.zealandpharma.com/product-pipeline/cardio-metabolic-diseases/drug-candidates-for-diabetes-and-metabolic-diseases/zp2929. Accessed October 22, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.