")
Back to Journals » OncoTargets and Therapy » Volume 10
Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel
Authors Huang T, Yang L, Wang G, Ding G, Peng B, Wen Y, Wang Z
Received 26 October 2016
Accepted for publication 14 December 2016
Published 18 January 2017 Volume 2017:10 Pages 439—446
DOI https://doi.org/10.2147/OTT.S125789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Tonghai Huang, Lin Yang, Guangsuo Wang, Guanggui Ding, Bin Peng, Yuxin Wen, Zheng Wang
Department of Thoracic Surgery, Shenzhen People’s Hospital, Shenzhen, Guangdong Province, People’s Republic of China
Abstract: S-phase kinase-associated protein 2 (Skp2) is an E3 ubiquitin ligase and plays an important role in the control of cell cycle progression. Skp2 is upregulated in several cancers, including lung cancers, but the role of Skp2 in the tumorigenesis and anticancer drug resistance in human lung cancer remains to be determined. We report here that Skp2 positively regulated mitotic arrest deficient 2 (MAD2) expression and that inhibition of Skp2 sensitizes human lung cancer cells to paclitaxel. Knockdown of Skp2 by small interfering RNA (siRNA) decreased Mad2 messenger RNA (mRNA) and protein levels in A549 and NCI-H1975 cells, accompanied with upregulation of p27 but decrease of the phosphorylation of retinoblastoma (Rb). In contrast, ectopic overexpression of Skp2 increased Mad2 mRNA and protein levels and phosphorylation of Rb, while it decreased p27. Pharmacological inhibition of CDK1/2 by flavopiridol or E2F1 with HLM006474 led to downregulation of Mad2 expression and prevented the increase of Mad2 expression by Skp2. Most importantly, pharmacological inhibition of Skp2 sensitized A549 and NCI-H1299 cells to paclitaxel. Our results demonstrated that SKP2 positively regulates the gene expression of MAD2 through p27-CDKs-E2F1 signaling pathway and that inhibition of Skp2 sensitizes A549 and NCI-H1299 cells to paclitaxel, suggesting that small molecule inhibitors of Skp2 are potential agents for the treatment of lung cancer with upregulation of Skp2.
Keywords: SKP2, MAD2, spindle assembly checkpoint, lung cancer, paclitaxel
Introduction
Spindle assembly checkpoint (SAC) controls the accurate and complete separation of sister chromatins during mitosis and thereby plays a pivotal role in the maintenance of chromosome stability in all eukaryotes.1 Chromosome instability is now recognized as a hallmark of human cancer cell, highlighting the important contribution of the deregulation of SAC during the multistep processes of tumorigenesis.2 However, the molecular mechanism by which SAC dysregulation promotes tumorigenesis remains to be determined.
Deregulation of the components of SAC is a frequent characteristic of cancer, especially solid tumors.3 Mitotic arrest deficient 2 (Mad2) is an essential component of SAC and has been found highly expressed in a variety of human malignancies.4–7 In most lung cancers, Mad2 messenger RNA (mRNA) was found to be elevated. High-level Mad2 expression in human non-small-cell lung cancer (NSCLC) correlates with tumor progression, and patients with tumors with elevated Mad2 expression demonstrate significantly shorter survival time.8 Similarly, overexpression of Mad2 in transgenic mice results in a wide variety of tumors.9 It was suggested that high-level Mad2 expression might be an independent prognostic factor for NSCLC.8 However, the mechanism by which Mad2 is deregulated in lung cancer is largely unknown.
S-phase kinase-associated protein 2 (Skp2) is an F-box protein of SCF ubiquitin ligase complex, which plays an important role in the regulation of cell cycle progression.10 One of the main targets of Skp2 is p27, an inhibitor of cyclin-dependent kinases (CDKs).11 It was reported that Skp2 is upregulated in NSCLC, and overexpression of Skp2 is correlated with a decrease of p27.12 Moreover, it was found that Skp2 expression was significantly associated with tumor status, lymph node metastasis, stage and vascular invasion.13 Skp2 was also found to be an independent prognostic factor for survival in NSCLC.14 These findings clearly indicate that Skp2 plays an important role in the oncogenesis and development of NSCLC.
Thus, both Mad2 and Skp2 are upregulated in lung cancers, and high-level expression of either Mad2 or Skp2 is associated with tumor progression and predicts poor survival of NSCLC patients, suggesting that there might be a functional link between Mad2 and Skp2 in the promotion of tumorigenesis of lung cancer. It has been reported that Skp2 is an E2F target gene and retinoblastoma (Rb) directly binds Skp2 to repress its ability to mediate p27 degradation and to bring it to anaphase promoting complex/cyclosome for ubiquitination and degradation.15 Recently, it was found that Mad2 is positively regulated by Rb-E2F1 at the transcriptional level.16,17 Rb-E2F1 is controlled by CDKs, while Skp2 negatively regulates p27, an inhibitor of CDKs. In addition, it has been reported that inhibition of Skp2 sensitizes human prolactinoma cells to bromocriptine and Skp2 is related to paclitaxel resistance in prostate cancer cells.18,19 The simultaneous upregulation of Skp2 and Mad2 and regulation of Mad2 by Skp2 suggest that inhibition of Skp2 may sensitize human lung cancer cells to paclitaxel.
In this study, we investigated the expression of Mad2 by silencing Skp2 with small interfering RNA (siRNA) and ectopic overexpression of Skp2 in human lung cancer A549 and NCI-H1975 cells. We further assessed the cell viability following pharmacological inhibition of Skp2 in the presence or absence of paclitaxel.
Experimental section
Drugs
CDK inhibitor flavopiridol was purchased from Selleck Chemicals (Houston, TX, USA). E2F inhibitor HLM006474 and SKP2 inhibitor SMIP004 were from Millipore.
Cells and cell transfection
Human NSCLC cell lines A549 and NCI-H1975 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 100 units/mL penicillin, 100 mg/mL streptomycin and 2 mmol/L L-glutamine; the cells were incubated at 37°C with a 5% CO2 atmosphere. Control and ON-TARGETplusSMARTpool siRNAs of SKP2 and MAD2 were purchased from Dharmacon (Chicago, IL, USA). Plasmid pcDNA-SKP2 was from Addgene (Cambridge, MA, USA). Plasmid pcDNA-MAD2 was constructed by cloning the open-reading frame of MAD2 gene from A549 cells into vector pcDNA3.1. Lipofectamine 2000 was from Invitrogen (Carlsbad, CA, USA), and transfection of siRNA or plasmids in A549 or NCI-H1975 cells was performed according to the manufacturer’s instructions.
Cell proliferation assay
Cell proliferation was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded at 4×103/well in 96-well plates the day before transfection. Following transfection of siRNAs for 24 h, cells were treated with paclitaxel or SMIP004 for 24 h. Then, 20 μL of 5 mg/mL MTT solution was added to the wells, and the cells were cultured for additional 4 h. The culture medium was removed, and 150 μL dimethylsulfoxide was added to dissolve formazan. Cell viability was quantified by measuring absorbance at 490 nm using a microplate spectrophotometer to calculate the optical density values.
Colony formation assay
Following treatment, 500 cells were seeded in each well of 6-well plates. After 2 weeks of culture, the cells were fixed in 4% formaldehyde and stained with crystal violet for 20 min. The number of cell clones of a diameter >1.5 mm was counted. This assay was repeated at least three times.
Western blotting
Total protein was extracted from cells using lysis buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% DTT and 1% protease inhibitor cocktail (Roche, Basel, Switzerland). Equal amounts of protein extracts (40 μg) were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred on to a PVDF membrane. Membranes were blocked with 5% w/v nonfat dry milk dissolved in Tris-buffered saline plus Tween-20 (0.1% Tween-20; pH 8.3) at room temperature for 1 h, then incubated with primary antibodies at 4°C overnight. The primary antibodies used were rabbit anti-Mad2 (Abcam, Cambridge, MA, USA), rabbit anti-Skp2 and GADPH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit anti-Rb, pRb-Ser807/811 and pRb-S780 (Cell Signaling Biotechnology, Boston, MA, USA). After washing with Tris-buffered saline plus Tween-20, membranes were incubated with horseradish peroxidase-labeled secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Immunobands were visualized using enhanced chemiluminescence (ECL) kit (GE Healthcare, Waukesha, WI, USA) according to the manufacturer’s instructions and exposed to X-ray films.
RNA isolation, cDNA synthesis and RT-PCR
Total RNA was extracted from cells using Trizol reagent (Invitrogen). Reverse transcription was performed using the First-Strand cDNA Synthesis System (Invitrogen). Real-time PCR was performed on the 7900HT Fast Real-Time PCR System using the TaqMan® Universal Mastermix II. Human MAD2 and SKP2 expression was quantified in real time with MAD2 and SKP2-specific FAM dye-labeled minor groove binder probes and normalized to GAPDH.GAPDH was used as an internal control. Each experiment was repeated twice in triplicate. The relative expression of target genes was calculated using the 2−ΔΔCT method.
Statistical analysis
All data were analyzed using SPSS19.0 statistical software. Measurement data are expressed as mean ± standard error of the mean. Comparison was made by t-test between two groups. A P-value of <0.05 was considered statistically significant.
Results
Skp2 positively regulates Mad2 expression in human lung cancer cells
The upregulation of both MAD2 and SKP2 in human lung cancer suggests that MAD2 might be regulated by SKP2. To test this hypothesis, we knocked down SKP2 by siRNA in human lung cancer A549 and NCI-H1975 cells and determined the mRNA and protein levels of MAD2 by RT-quantitative PCR (QPCR) and Western blotting, respectively. In comparison with control siRNA, SKP2 siRNA decreased Skp2 protein levels 48 h after transfection in both A549 and NCI-H1975 cells (Figure 1A). As expected, the Mad2 protein levels were drastically decreased by Skp2 siRNA (Figure 1A). Consistent with the decrease of Mad2 protein, the mRNA levels of Mad2 were also significantly downregulated by Skp2 siRNA in both A549 and NCI-H1975 cells (Figure 1B). To further support the above observation, we transfected A549 and NCI-H1975 cells with SKP2 plasmid to ectopically overexpress SKP2 and determined the mRNA and protein levels of MAD2 by RT-QPCR and Western blotting, respectively. In comparison to control vector pcDNA3.1, transfection of pcDNA-SKP2 obviously increased Skp2 protein levels 24 h after transfection and apparently after 48 and 72 h in both A549 (Figure 1C) and NCI-H1975 cells (Figure 1D). The mRNA levels of Mad2 were also significantly increased by pcDNA-SKP2 in both A549 and NCI-H1975 cells (Figure 1E). Together, these results clearly demonstrated that Skp2 signaling controls Mad2 expression at the transcriptional level in A549 and NCI-H1975 cells.
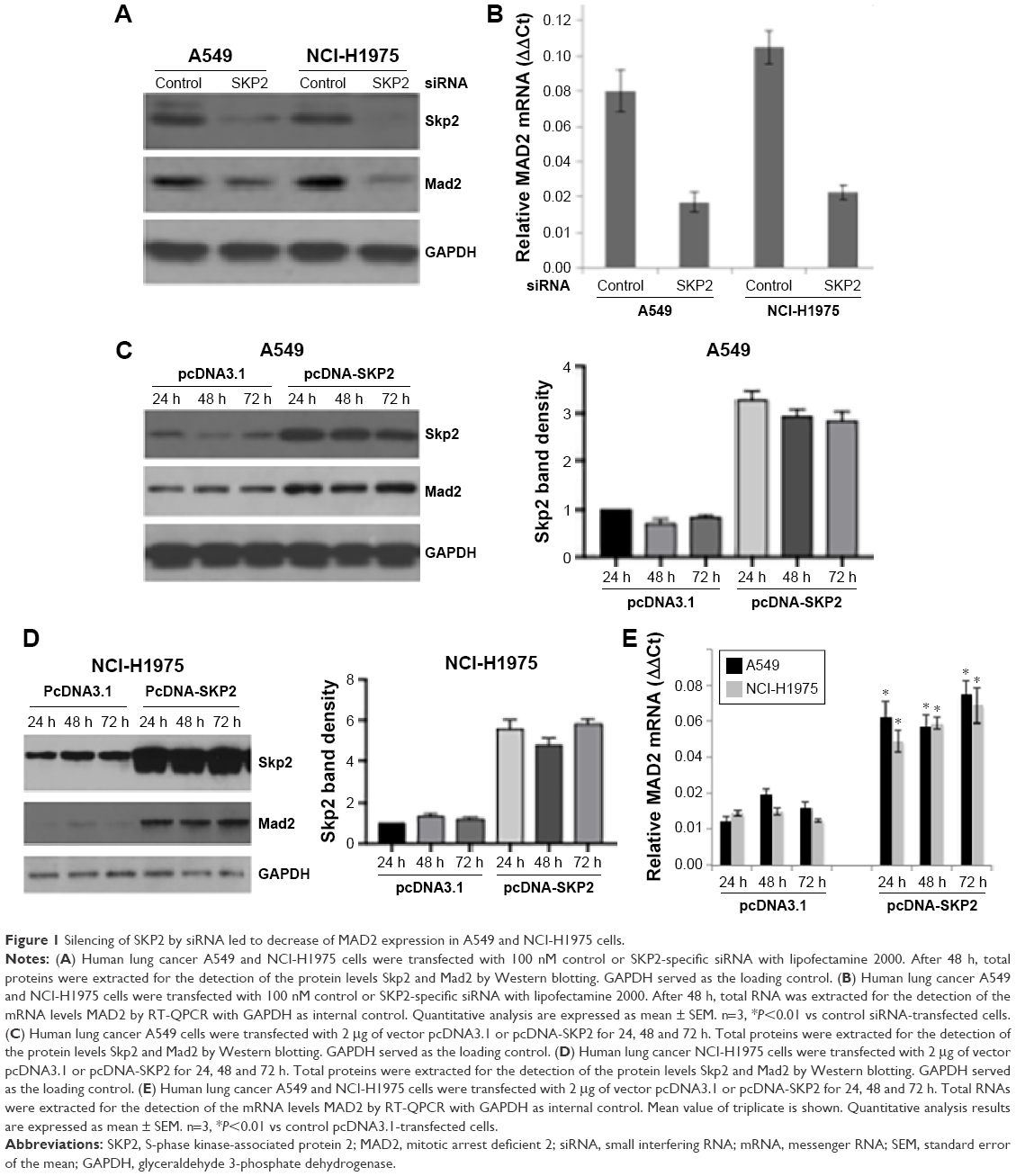 | Figure 1 Silencing of SKP2 by siRNA led to decrease of MAD2 expression in A549 and NCI-H1975 cells. |
Skp2 controls the phosphorylation of Rb in human lung cancer cells
To investigate the underlying mechanism by which Skp2 regulates Mad2 expression, we first assessed the protein level of p27, a well-known downstream target of Skp2, after silencing or overexpression of Skp2 in A549 and NCI-H1975 cells by immunoblotting. As shown in Figure 2A, knockdown of Skp2 by siRNA led to an increase of p27 in both A549 and NCI-H1975 cells. p27 is a potent inhibitor of CDKs. We further tested the phosphorylation of Rb at Ser780 and Ser807/811, a marker of the activation of CDKs. Consistent with the upregulation of p27, pRb-S780 and pRb-S807/811 signals were apparently decreased following SKP2 siRNA transfection in A549 and NCI-H1975 cells (Figure 2A). In contrast, ectopic overexpression of SKP2 resulted in decrease of p27 but increase of the phosphorylation of Rb at Ser780 and Ser807/811 in these cells (Figure 2B). These results showed that Skp2 signaling positively regulates the activity of CDKs by decreasing p27 in A549 and NCI-H1975 cells, supporting the conclusion that Skp2 promotes cell cycle progression by degrading p27.
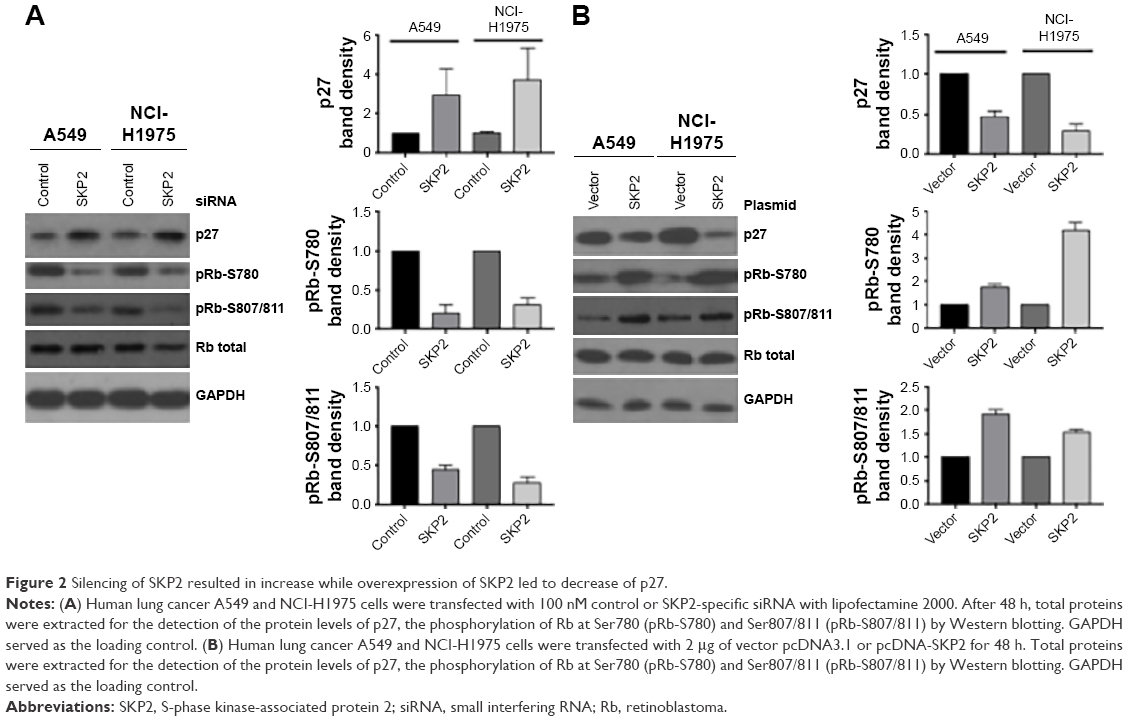 | Figure 2 Silencing of SKP2 resulted in increase while overexpression of SKP2 led to decrease of p27. |
Pharmacological inhibition of CDK1/2 or E2F1 abolishes the promotion of the expression of Mad2 by Skp2
MAD2 gene transcription is regulated by Rb-E2F1, which is controlled by CDKs. Our observation that Skp2 promotes the activity of CDKs by downregulating p27 suggests that Skp2 may positively regulate MAD2 expression via CDKs-E2F1 axis. To test this hypothesis, we treated A549 cells transfected with pcDNA-SKP2 with CDK1/2 inhibitor flavopiridol or E2F1 inhibitor HLM006474, which is a small molecule of pan-E2F inhibitor and has been shown to specifically inhibit E2F target genes in melanoma cells and synergies with paclitaxel lung cancer cells.20,21 Then, we determined MAD2 expression by RT-QPCR and immunoblotting. In comparison to untreated control, flavopiridol or HLM006474 alone decreased the mRNA levels of MAD2. Transient transfection of SKP2 plasmid resulted in elevation of the mRNA level of MAD2, whereas either flavopiridol or HLM006474 significantly abolished the increase of MAD2 mRNA by SKP2 overexpression (Figure 3A). In agreement with the alteration of MAD2 mRNA levels, flavopiridol or HLM006474 alone decreased the protein levels of Mad2 compared with control; ectopic overexpression of SKP2 led to increase of the protein levels of Skp2 and Mad2 and phosphorylation of Rb at Ser780; however, either flavopiridol or HLM006474 apparently prevented the increase of Mad2 protein and pRb-S780 signal but not the level of Skp2 protein (Figure 3B). In addition, CDK or E2F1 inhibitor treatment did not further decrease Mad2 expression in Skp2 knockdown cells (Figure 3C). Thus, Skp2 promotes the gene transcription of MAD2 via p27-CDKs-E2F1 signaling.
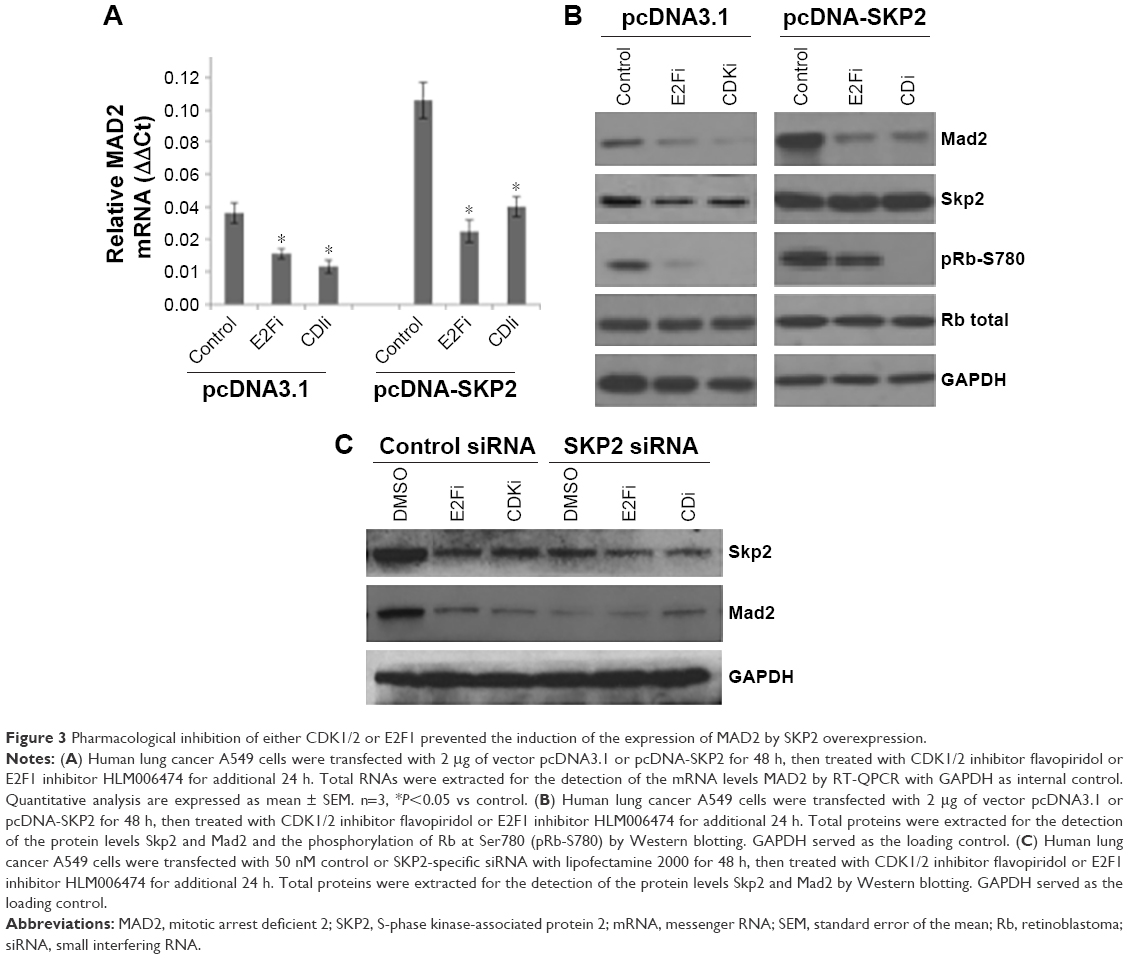 | Figure 3 Pharmacological inhibition of either CDK1/2 or E2F1 prevented the induction of the expression of MAD2 by SKP2 overexpression. |
Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel
Lots of mitotic spindle-damaging agents such as paclitaxel are chemotherapy drugs for various types of cancers. Mitotic checkpoint arrests cells in response to mitotic spindle damage, and increased expression of MAD2 enhances mitotic checkpoint.7 The positive regulation of MAD2 by SKP2 suggests that inhibition of Skp2 may sensitize lung cancer cells to mitotic spindle-damaging agents. To assess this possibility, we transfected A549 cells with different concentrations of SKP2 siRNA or treated A549 cells with different concentrations of paclitaxel or SKP2 inhibitor SMIP004 and then determined the cell viability by MTT assay. Paclitaxel concentration dependently inhibited the viability of A549 cells (IC50 of 1 μM; Figure 4A). SKP2 siRNA decreased A549 cell viability with IC50 of 200 nM (Figure 4B). SMIP004 also decreased the viability of A549 cells in a concentration-dependent manner with IC50 of 500 nM (Figure 4C). Then, A549 cells were treated with 100 nM paclitaxel alone or in combination with 50 nM SKP2 siRNA or 50 nM SMIP004, all of which did not significantly reduce A549 cell viability at these concentrations. MTT assay showed that 100 nM paclitaxel, 50 nM SKP2 siRNA or 50 nM SMIP004 alone did not lead to significant loss of cell viability; however, the combination of 100 nM paclitaxel and 50 nM SKP2 siRNA or 50 nM SMIP004 significantly resulted in loss of cell viability (Figure 4D). We then performed colony formation assay and found that either 50 nM SKP2 siRNA or 50 nM SMIP004 alone did not significantly inhibit the colony formation when compared to the untreated control. Though 100 nM paclitaxel obviously reduced the number of colonies, the combination of 100 nM paclitaxel and 50 nM SKP2 siRNA or 50 nM SMIP004 significantly reduced the colony numbers compared to that by 100 nM paclitaxel alone (Figure 4E).
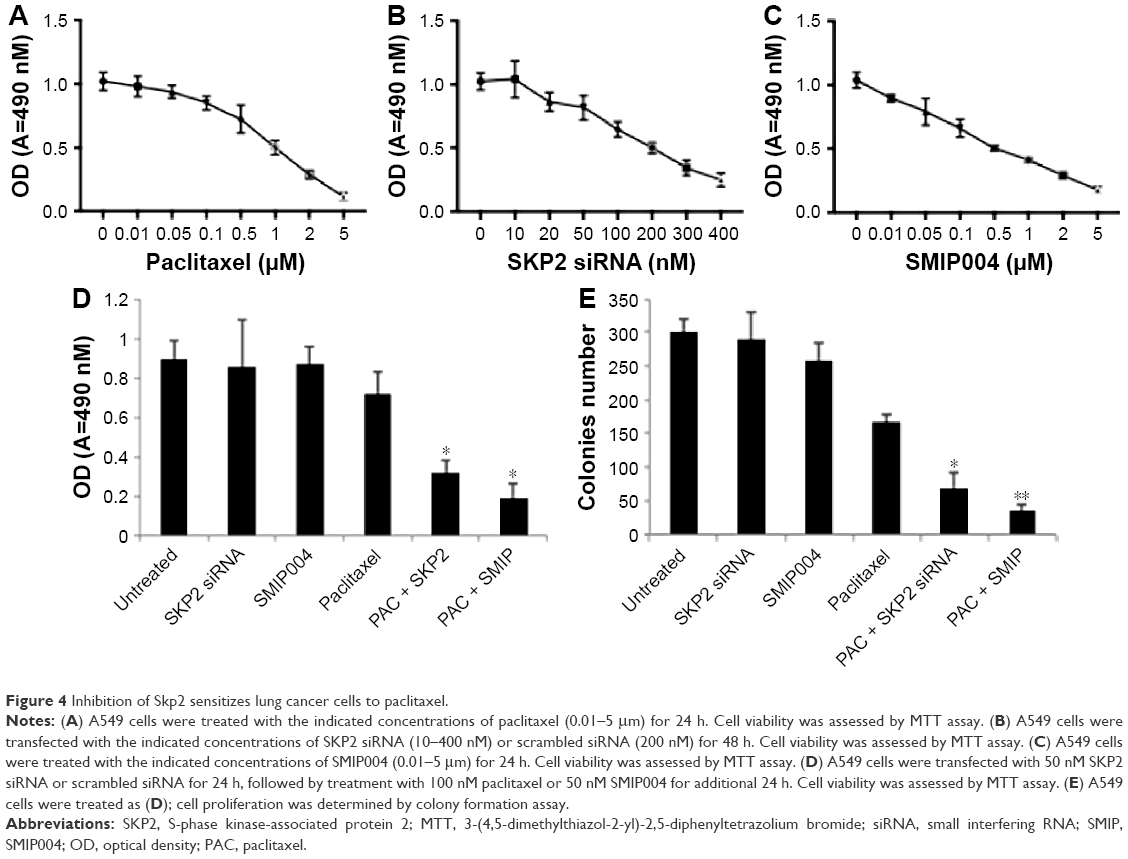 | Figure 4 Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel. |
Discussion
In this study, we demonstrated that silencing of SKP2 by siRNA led to decrease of MAD2 expression, while ectopic overexpression of SKP2 resulted in increase of MAD2 expression in A549 and NCI-H1975 cells. Moreover, knockdown of SKP2 resulted in elevation of p27 and downregulation of phosphorylation of Rb, whereas overexpression of SKP2 led to downregulation of p27 and upregulation of phosphorylation of Rb. Furthermore, pharmacological inhibition of either CDK1/2 or E2F1 reduced the increase of MAD2 expression by SKP2 overexpression. Finally, silencing of SKP2 by siRNA or pharmacologic inhibition of Skp2 sensitized A549 cells to paclitaxel. Our findings indicate that Skp2 positively regulates Mad2 via p27-CDKs-E2F1 signaling axis, suggesting that small molecule inhibitors of Skp2 are potential agents in combination with paclitaxel for the treatment of lung cancer patients with upregulation of Skp2.
Chromosomal instability is a hallmark of cancer cell and may promote chromosome translocations, aneuploidy, gene dosage change and other chromosomal chaos of cancer cells.1 More than 100 years ago, Theodor Boveri stated that chromosomal instability drives tumorigenesis, and recent studies demonstrated that chromosomal instability drives a mutation phenotype both in yeast and in human cancers.22,23 It was once believed that inactivation of SAC (also called mitotic checkpoint) promotes chromosomal instability. However, genetic inactivation mutation of the components of SAC was rarely found in human cancers.1 In contrast, increasing evidence implies that overexpression but not downregulation of the components of SAC results in missegregation of chromosomes and hence genome instability.1,6,16,24,25 Oncogene activation leads to chromosomal instability but the underlying mechanism is largely unknown.
It was well documented that MAD2 overexpression promotes tumorigenesis.25,26 MAD2 is frequently overexpressed in chromosomally unstable tumors.27 Moreover, MAD2 overexpression is frequently observed in various tumors including liver cancer,28 breast cancer,29 soft-tissue sarcoma4 and NSCLC.8 However, the mechanism by which Mad2 is upregulated in lung cancer is largely unknown. Our observation that Skp2 positively regulates Mad2 via the p27-CDKs-E2F1 signaling pathway suggests that oncogene activation such as SKP2 may promote chromosome instability through deregulating MAD2.
Lung cancer is one of the leading causes of cancer-related death in the world. In the past decades, great advances have been made in the diagnosis, systemic treatment and prognosis of lung cancers, especially the development of molecular targeted therapies including small molecule inhibitors of EGFR and ALK.30–33 With a long history of clinical application, mitotic spindle-targeting agents including vinca alkaloids, taxanes and epothilones are the most classical and reliable anticancer drugs.34 We showed that silencing of SKP2 by siRNA or pharmacologic inhibition of Skp2 sensitized A549 cells to paclitaxel, implying that Skp2 inhibition and paclitaxel could be synergistic, or paclitaxel could sensitize cells to Skp2 inhibition. Recently, several small molecule inhibitors of Skp2 kinase and Skp2–Cks1 interaction have been identified and shown to inhibit tumor growth in vitro.35–37 Importantly, Skp2 inhibitors have also been shown to inhibit the growth of tumor xenografts of prostate and lung cancers in mice.37 As SKP2 is highly expressed in NSCLC, our data suggest that small molecule inhibitors of Skp2 are potential agents for the treatment of lung cancer patients with upregulation of Skp2 in combination with mitotic spindle-targeting agents.
Conclusion
We found that SKP2 positively regulated the gene expression of MAD2 through p27-CDKs-E2F1 signaling pathway and inhibition of Skp2 sensitized lung cancer cells to paclitaxel. Our findings may provide an explanation for the simultaneous upregulation of MAD2 and SKP2 and the basis for the development of combinatorial therapy strategies of small molecule inhibitors of Skp2 with mitotic spindle-targeting agents in lung cancers.
Acknowledgments
We thank all other members of Zheng Wang’s laboratory for their critical comments and discussion.
Disclosure
The authors report no conflicts of interest in this work.
References
Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10(7):478–487. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5(10):773–785. | ||
Hisaoka M, Matsuyama A, Hashimoto H. Aberrant MAD2 expression in soft-tissue sarcoma. Pathol Int. 2008;58(6):329–333. | ||
Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–180. | ||
Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. MAD2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19(6):701–714. | ||
Schuyler SC, Wu YF, Kuan VJ. The Mad1-Mad2 balancing act – a damaged spindle checkpoint in chromosome instability and cancer. J Cell Sci. 2012;125(pt 18):4197–4206. | ||
Kato T, Daigo Y, Aragaki M, et al. Overexpression of MAD2 predicts clinical outcome in primary lung cancer patients. Lung Cancer. 2011;74(1):124–131. | ||
Yu L, Liu S, Guo W, Zhang B, Liang Y, Feng Q. Upregulation of Mad2 facilitates in vivo and in vitro osteosarcoma progression. Oncol Rep. 2012;28(6):2170–2176. | ||
Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428(6979):190–193. | ||
Muth D, Ghazaryan S, Eckerle I, et al. Transcriptional repression of SKP2 is impaired in MYCN-amplified neuroblastoma. Cancer Res. 2010;70(9):3791–3802. | ||
Hu X, Liu F, Jiang B, Wang Y. The expression of Skp2 in human non-small cell lung cancer and its correlation with expression of p27 protein. Zhongguo Fei Ai Za Zhi. 2008;11(4):547–550. | ||
Takanami I. The prognostic value of overexpression of Skp2 mRNA in non-small cell lung cancer. Oncol Rep. 2005;13(4):727–731. | ||
Osoegawa A, Yoshino I, Tanaka S, et al. Regulation of p27 by S-phase kinase-associated protein 2 is associated with aggressiveness in non-small-cell lung cancer. J Clin Oncol. 2004;22(20):4165–4173. | ||
Zhang L, Wang C. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 2006;25(18):2615–2627. | ||
Sotillo R, Hernando E, Diaz-Rodriguez E, et al. MAD2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11(1):9–23. | ||
Hao Z, Huang S. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Front Biosci (Landmark Ed). 2015;20:474–490. | ||
Huang J, Zhang F, Jiang L, et al. Inhibition of SKP2 sensitizes bromocriptine induced apoptosis in human prolactinoma cells. Cancer Res Treat. Epub 2016 Jul 28. doi:10.4143/crt.2016.017. | ||
Yang Y, Lu Y, Wang L, et al. Skp2 is associated with paclitaxel resistance in prostate cancer cells. Oncol Rep. 2016;36(1):559–566. | ||
Ma Y, Kurtyka CA, Boyapalle S, et al. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 2008;68(15):6292–6299. | ||
Kurtyka CA, Chen L, Cress WD. E2F inhibition synergizes with paclitaxel in lung cancer cell lines. PLoS One. 2014;9(5):e96357. | ||
Sheltzer JM, Blank HM, Pfau SJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333(6045):1026–1030. | ||
Solomon DA, Kim T, Diaz-Martinez LA, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333(6045):1039–1043. | ||
Sotillo R, Schvartzman JM, Socci ND, Benezra R. MAD2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464(7287):436–440. | ||
van Deursen JM. Rb loss causes cancer by driving mitosis mad. Cancer Cell. 2007;11(1):1–3. | ||
Malumbres M. Oncogene-induced mitotic stress: p53 and pRb get mad too. Cancer Cell. 2011;19(6):691–692. | ||
Pérez de Castro I, de Cárcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28(5):899–912. | ||
Zhang SH, Xu AM, Chen XF, Li DH, Sun MP, Wang YJ. Clinicopathologic significance of mitotic arrest defective protein 2 overexpression in hepatocellular carcinoma. Hum Pathol. 2008;39(12):1827–1834. | ||
Scintu M, Vitale R, Prencipe M, et al. Genome instability and increased expression of BUB1B and MAD2L1 genes in ductal breast cancer carcinoma. Cancer Lett. 2007;254(2):298–307. | ||
Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol. 2016;9(1):59. | ||
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34. | ||
Iragavarapu C, Mustafa M, Akinleye A, et al. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17. | ||
Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9(1):19. | ||
Chan KS, Koh CG, Li HY. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 2012;3:e411. | ||
Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19(12):1515–1524. | ||
Ungermannova D, Lee J, Zhang G, Dallmann HG, McHenry CS, Liu X. High-throughput screening AlphaScreen assay for identification of small-molecule inhibitors of ubiquitin E3 ligase SCFSkp2-Cks1. J Biomol Screen. 2013;18(8):910–920. | ||
Chan CH, Morrow JK, Li CF, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154(3):556–568. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.