")
Back to Journals » Cancer Management and Research » Volume 12
Inhibition of Skp2 Sensitizes Chronic Myeloid Leukemia Cells to Imatinib
Authors Chen X, Huang Z, Wu W, Xia R
Received 10 March 2020
Accepted for publication 13 May 2020
Published 22 June 2020 Volume 2020:12 Pages 4777—4787
DOI https://doi.org/10.2147/CMAR.S253367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Xiaowen Chen, Zhenqi Huang, Wei Wu, Ruixiang Xia
Department of Hematology, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui 230022, People’s Republic of China
Correspondence: Ruixiang Xia Email [email protected]
Introduction: Skp2 is an E3 ubiquitin ligase that plays an important role in modulating tumor progression. The mechanisms underlying Skp2 in the promotion of proliferation and its function in the primary resistance to tyrosine kinase inhibitors (TKIs) in human CML remain to be determined. This study aimed to investigate the function of Skp2 in CML progression as well as its effects on TKI sensitivity.
Methods: Expression of Skp2 in leukocytes from patients with CML and normal blood samples was analyzed by qRT-PCR. Cell proliferation was analyzed by EdU incorporation and cell counting assays. Luciferase reporter and chromatin immunoprecipitation assays were used for examination of the effects of CREB on Skp2 expression. The apoptosis in vitro of K562 cells was analyzed by MTT and caspase 3/7 activity assays.
Results: The present study demonstrates that Skp2 was expressed at a higher level in patients with CML compared with healthy donors, and the elevated expression of Skp2 is critical for CML cell proliferation. Mechanistically, Skp2 was transcriptionally upregulated by CREB responsive to the PI3K/Akt signaling pathway. Furthermore, inhibition of Skp2 expression by shRNAs or blocking the PI3K/Akt/CREB pathway greatly enhances the sensitivity of CML cells to Imatinib treatment.
Conclusion: We conclude that the PI3K/Akt/CREB axis regulates the sensitivity of K562 cells to Imatinib via mediating Skp2 expression. The present study revealed an unknown role of Skp2 in CML progression and provided new aspects on the Skp2-modulated TKI sensitivity in CML, contributing to the development of potential therapeutic anticancer drugs.
Keywords: Skp2, PI3K/Akt, CREB, Imatinib
Introduction
Chronic myeloid leukemia (CML) is a malignant myeloproliferative disorder, which is characterized by the production of large amounts of immature white blood cells1. The BCR-ABL fusion gene produces a chimeric protein known as BCR-ABL with constitutive tyrosine kinase activity, which is considered to be the most critical cause of and another feature of CML.2,3 CML is a common hematologic neoplasm and accounts for ~20% of newly diagnosed cases of adult leukemia.4
In clinical therapy, tyrosine kinase inhibitors (TKIs), which are specific inhibitors targeting BCR-ABL, have been widely applied in the clinical treatment of CML and significantly improved the survival rate of patients.5,6 Imatinib is the first TKI used as a CML treatment strategy that inhibits tyrosine kinase activity through blocking the inactive conformation of the BCR-ABL protein. Imatinib treatments exhibited high efficacy and low toxicity in patients with CML,7 and the primary TKIs resistance is not commonly observed in clinical practice. However, there remains a certain proportion of patients with CML who are not sensitive or develop drug resistance to Imatinib by a variety of cellular mechanisms.1,7-9 As a result, screening for effective therapeutic targets in the progression of CML is essential, and will be useful to improve the chemotherapeutic effect of CML under Imatinib treatment.
Human S-phase kinase-associated protein 2 (Skp2) is an E3 ligase encoded by the SKP2 gene.10,11 Skp2 is frequently upregulated in cancer and plays an important role in controlling cell cycle progression. In serum-starved cells, Skp2 overexpression promotes S-phase entry by inducing the accumulation of cyclin A and phosphorylation-dependent degradation of p27.12,13 Skp2 is also involved in the ubiquitination of other cell-cycle regulatory proteins, such as cyclin E and the transcription factor E2F1.12,14 Furthermore, accumulating evidence has indicated the function of Skp2 in anticancer drug resistance. For instance, Skp2 positively regulates MAD2 expression through the p27-CDKs-E2F1 pathway and inhibition of Skp2 sensitizes lung cancer cells to paclitaxel.10
The molecular mechanism of deregulated-Skp2 in CML is, and remains to be an intensive area of research. Emerging evidence has demonstrated that amplification of the SKP2 gene or upregulation of Skp2 is observed in CML and other hematopoietic malignancies.15–17 More recently, Skp2 has been shown to mediate Myc-dependent transformation, and it was identified to be a direct Myc targeted-gene in human leukemia cells.18 In addition, Skp2 was demonstrated to be transcriptionally activated by BCR-ABL responsive to the PI3K pathway to promote p27 degradation and proliferation of chronic myelogenous leukemia cells.19–21 Given the complexity of these identified signaling pathways that are associated with Skp2 dysregulation, fully investigating its potential underlying mechanisms remains a priority in order to further clarify the underlying pathogenesis of CML.
In the present study, we reported that Skp2 expression is significantly upregulated in patients with CML, which leads to the abnormal proliferation of CML cells. Furthermore, Skp2 is transcriptionally activated by CREB responsive to PI3K/Akt signaling, and it was revealed that inhibition of the PI3K/Akt pathway by specific inhibitors or by direct knockdown of Skp2 could sensitize the CML cell line K562 to Imatinib. Collectively, these data indicate that Skp2 is critical to the proliferation of K562 cells and inhibition of Skp2 sensitizes K562 cells to Imatinib treatment.
Materials and Methods
Ethical Statement
This study was conducted in accordance with the Declaration of Helsinki and written informed consents were obtained from all patients and healthy donors.
Patient Samples Collection
Blood samples from 24 patients with newly diagnosed CML and 7 healthy donors were collected in this study with approval from the Ethics Committee of First Affiliated Hospital of Anhui Medical University. All methods were performed in accordance with the relevant guidelines and regulations.
Cell Lines
HEK293T cells and the human chronic myeloid leukemia cell line K562 were purchased from Cell Bank of Chinese Academy of Sciences and cultured in RPMI 1640 medium containing 10% fetal bovine serum at 37°C under an atmosphere with 5% CO2.
Reagents and Antibodies
The PI3K inhibitor LY294002 was obtained from Sigma Aldrich (L9908, USA). The following antibodies for Western blot analysis were used in the present study: anti-CREB (Cell Signaling Technology; catalog no. 9197S); anti-Skp2 (Proteintech; catalog no. 15010-1-AP), anti-ACTIN (Proteintech; catalog no. 20536-1-AP), anti-PARP (Proteintech; catalog no. 13371-1-AP), anti-Flag (Sigma Aldrich; catalog no. F3165), anti-caspase-3 (Stressgene; catalog no. AAP-113).
Leukocyte Isolation
Peripheral blood samples from healthy volunteers and patients with CML were treated with red blood cell lysis buffer (Sigma Aldrich; Merck KGaA) for 10 min with a gyratory shaker. Blood samples were then centrifuged at 500 x g at 4°C for another 10 min. Leukocyte pellets were washed and centrifuged again. The remaining leukocytes were collected for further experiments.
Luciferase Reporter Assay
In order to determine the effect of CREB on the Skp2 promoter, K562 cells were co-transfected with the pGL3-based luciferase reporters containing wildtype CREB binding site (WT) or mutant CREB binding site (MUT) and Renilla plasmids using lipofectamine2000. After 24 h transfection, the luciferase activities were measured using a Dual-Luciferase Reporter Assay System (Promega Corp.) and the firefly activity was normalized to the Renilla enzyme activity.
EdU Incorporation Assay
K562 cells with shRNA-control or shRNA-Skp2 were seeded in 24-wells plate for 72 h. According to the manufacturer’s protocol, the EdU incorporation analysis was performed using an EdU assay kit (RibBio). Briefly, K562 cells were incubated in the medium containing 50 uM EdU for 2 h and then the nuclei were stained with Hoechst33342. The proportion of EdU positive cells was determined by fluorescence microscope (Olympus DP80).
ChIP Assays
ChIP assays were performed using the Millipore ChIP kit (17-371RF) and anti-CREB antibody as previously described.22 The specific primers (F: 5ʹ-TGTAATCTCAGCACTTTAGGA-3ʹ; R: 5ʹ-GAAATTTCACTCTTATTGCCC −3ʹ) were used to detect the bound DNA fragment using real-time PCR assays.
RNA Isolation and Reverse Transcription-Quantitative PCR (RT–qPCR) Analysis
The total RNA was isolated using Trizol® (Invitrogen; Thermo Fisher Scientific, Inc.) reagent. A total of 1μg of RNA was used to synthesize cDNA using the PrimeScriptTM RT reagent kit (TaKaRa). RT-qPCR was performed using SYBR Green RT-PCR kit (TaKaRa) with the specific primers and analyzed by the Stratagene Mx3000p (Agilent Technology). The primers used in this assay were as follows: Skp2, Forward: 5ʹ-AGTCTCTATGGCAGACCTTAGACC-3ʹ and Reverse: 5ʹ-TTTCTGGAGATTCTTTCTGTAGCC-3ʹ.
RNA Interference
In order to knockdown the expression of Skp2, PLKO.1-based Skp2 shRNA plasmids were constructed. PLKO.1 based shRNA-Skp2 plasmids were co-transfected with pREV, pGag and pVSVG at the ratio of 2:2:2:1 into HEK293T cells. 48 h after transfection, lentiviral supernatant were collected and used for the target cells infection. The sequences used to construct shRNAs were as follows: Skp2: 5ʹ-GCCTAAGCTAAATCGAGAGAA-3ʹ and 5ʹ-CCATTGTCAATACTCTCGCAA-3ʹ.CREB:5ʹ-GCTCGATAAATCTAACAGTTA-3ʹ and 5ʹ-GCAAACATTAACCATGACCAA-3ʹ; Akt:5ʹ-GCATCGCTTCTTTGCCGGTAT-3ʹ and 5ʹ-CGCGTGACCATGAACGAGTTT-3ʹ
Lentiviral Overexpression of Skp2
HEK293T cells were co-transfected with pCDH-Flag-Skp2, pSPAx2 and pMD2.g at the ratio of 2:1.5:1. 48 h after transfection, lentiviral supernatant was used for K562 cells infection.
Western Blot Analysis
Protein samples were extracted using RIPA lysis buffer (Sangon) containing protease inhibitor cocktail (Beyotime). Then, protein lysates were subjected to SDS-PAGE and transferred onto nitrocellulose membrane (EMD, Millipore). The membrane was blocked with 5% milk and probed with the indicated antibodies, and then visualized using HRP substrate kit (EMD, Millipore).
The Caspase 3/7 Activity Assay
The caspase3/7 activity assay was performed using a caspase3/7 assay kit (Promega Corp. G8090) according to the manufacturer’s protocol. The activity of caspase3/7 was normalized to protein concentration.
Cell Viability Assay
A total of 1 × 103 K562 cells with different treatment were incubated in 96-wells plates for 24 h, and following exposure to 1μM Imatinib for another 24 h, cell viability was evaluated via MTT assay and analyzed with a microplate reader at a wavelength of 490 nm.
Statistical Analysis
Statistical analysis was performed using Microsoft Excel software (Microsoft) and GraphPad Prism Software (GraphPad). Differences between experimental groups were assessed using Student’s t-test. P< 0.05 was considered to indicate a statistically significant difference.
Results
Aberrant Expression of Skp2 in Patients with CML
Previous studies have revealed that aberrant expression of Skp2 may induce multiple hematological malignancies.23,24 With the objective of deciphering the regulatory networks of Skp2 in CML, the present study first compared the expression levels of Skp2 in leukocytes from patients with CML and normal blood samples. Leukocytes from 24 patients with CML and 7 healthy donor blood samples were collected and analyzed by RT-qPCR. As presented in Figure 1A, CML samples exhibited significantly upregulated Skp2. In order to further investigate this observation, the expression levels of Skp2 protein were analyzed via Western blotting, and the CML samples exhibited relatively higher levels of Skp2 than the healthy controls (Figure 1B and C). Thus, these data demonstrated that the expression levels of Skp2 were aberrantly upregulated in patients with CML.
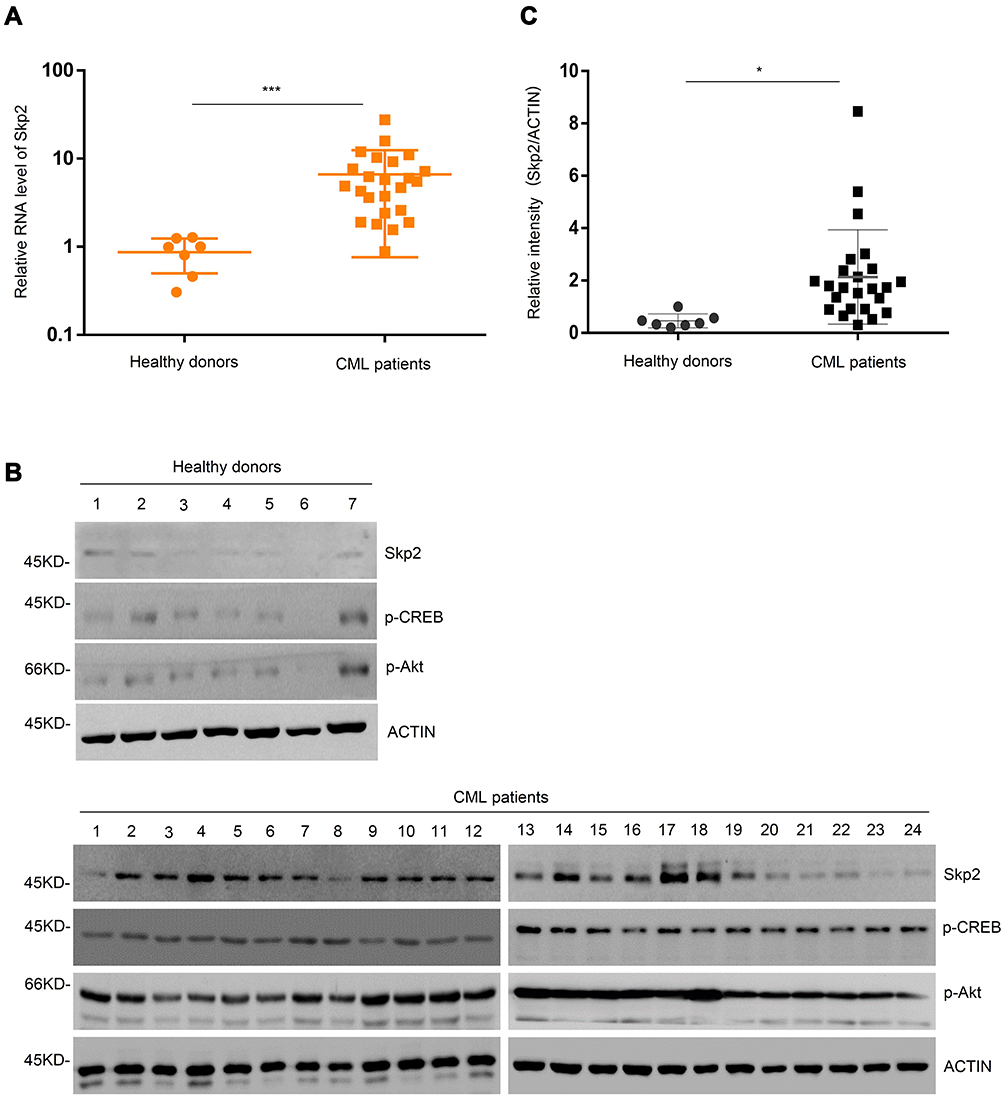 |
Figure 1 Skp2 is upregulated in patients with CML. (A) Real-time-PCR analysis of Skp2 mRNA expression in the healthy donors and patients. Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. ***P < 0.001 versus healthy donors. (B) Skp2, phosphorylated-CREB and phosphorylated-Akt protein levels were evaluated by Western blot analysis in the healthy donors (1–7) and patients with CML (1–24). (C) The relative intensity of Skp2 was quantified using ImageJ software. Data are shown as mean ± SD; two-tailed paired Student’s t-test. *P<0.05 versus healthy donors. |
Skp2 Is Critical for CML Cell Proliferation
The present study next investigated the role of Skp2 in K562 cells, a cell line frequently used as a model of CML. Skp2 was knocked-down in K562 cells (Figure 2A), and when compared with scrambled, Skp2 shRNA induced a dramatic decrease in the cell proliferation rate of K562 cells (Figure 2B). Meanwhile, the EdU assay also demonstrated that the proliferation capacity of K562 cells was significantly lower following Skp2 knockdown (Figure 2C). The present study next evaluated the effect of Skp2 overexpression on cell proliferation in K562 cells. In contrast with Skp2 deficiency, cell numbers were elevated by ectopic expression of Skp2 (Figure 2D and E). Consistently, Skp2 overexpression also resulted in a significantly increased proportion of EdU-positive cells in K562 cells (Figure 2F). Taken together, these results strongly suggested that Skp2 stimulates the proliferation of K562 cells.
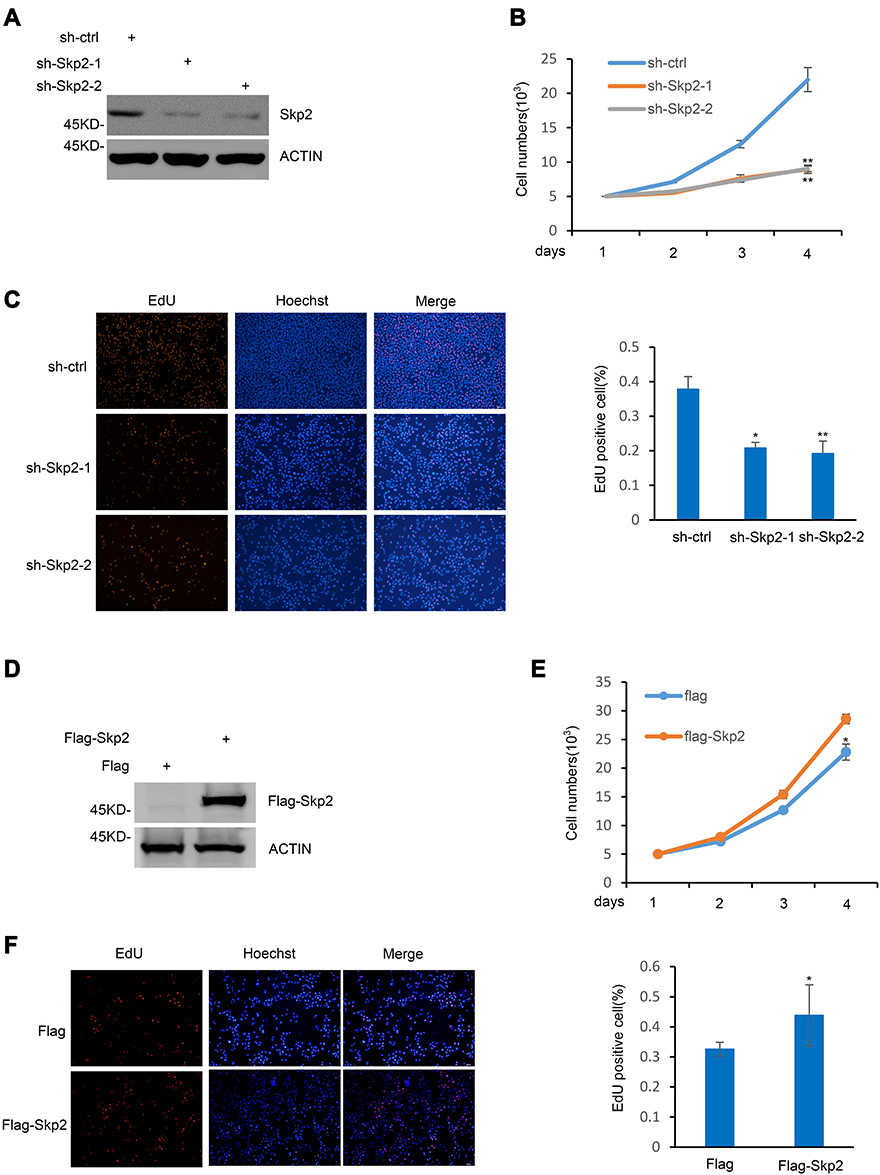 |
Figure 2 Skp2 regulates CML cell proliferation. (A) The protein level of Skp2 in K562 cells expressing shRNA-ctrl, shRNA-Skp2-1 or shRNA-Skp2-2 were examined by Western blot analysis. (B) Cell proliferation rates were evaluated in K562 cells with and without Skp2 stable knockdown by cell count analysis. Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. **P<0.01 versus shRNA-ctrl group. (C) K562 cells stably expressing shRNA-ctrl, shRNA-Skp2-1 or shRNA-Skp2-2 were stained with EdU and the nuclei were visualized using Hoechst 33342 staining (left). The ratio of EdU-positive cells to total Hoechst 33342-positive cells was presented from three independent experiments (right). Data are shown as mean ± SD; two-tailed paired Student’s t-test.*P<0.05; **P<0.01 versus shRNA-ctrl group. (D) Cell lysates from K562 cells with ectopic expression of Skp2 were subjected to Western blot analysis with the indicated antibodies. (E) Cell numbers of K562 cells with ectopic expression of Skp2 were determined by cell count analysis. Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05 versus the group with flag overexpression. (F) K562 cells with ectopic expression of Flag or Flag-Skp2 were stained with EdU and the nuclei were visualized using Hoechst 33342 staining (left). The ratio of EdU-positive cells to total Hoechst 33342-positive cells was presented from three independent experiments (right). Data are shown as mean ± SD; two-tailed paired Student’s t-test. *P<0.05 versus the group with flag overexpression. |
Skp2 Is Transcriptionally Regulated by CREB
The present study investigated the molecular mechanism by which Skp2 is elevated in CML cells. The genomic sequence upstream of the Skp2 gene was analyzed using the JASPAR software.25 One putative CREB-binding region (BR) was identified within the promoter of Skp2 (Figure 3A). The subsequent ChIP assays indicated that CREB was associated with the chromatin fragment, including the putative CREB-BR of Skp2 promoter (Figure 3B). Furthermore, to test whether CREB indeed regulates Skp2 expression through binding with this potential CREB-BR, a series of pGL3-basic vector based luciferase reporter plasmids including the wild-type CREB-BR (WT) or a deletion of CREB-BR (MUT) were constructed in the present study (Figure 3A). These plasmids were transfected into K562 cells with or without CREB overexpression, and their transcriptional activities were measured using luciferase reporter assays. As presented in Figure 3C, the pGL3-based wild-type CREB-BR luciferase reporter construct, rather than the mutant construct or the pGL3 empty vector, exhibited CREB-responsive transcriptional activity, whereas knockdown of CREB diminished the wild-type CREB-BR reporter activity (Figure 3D).
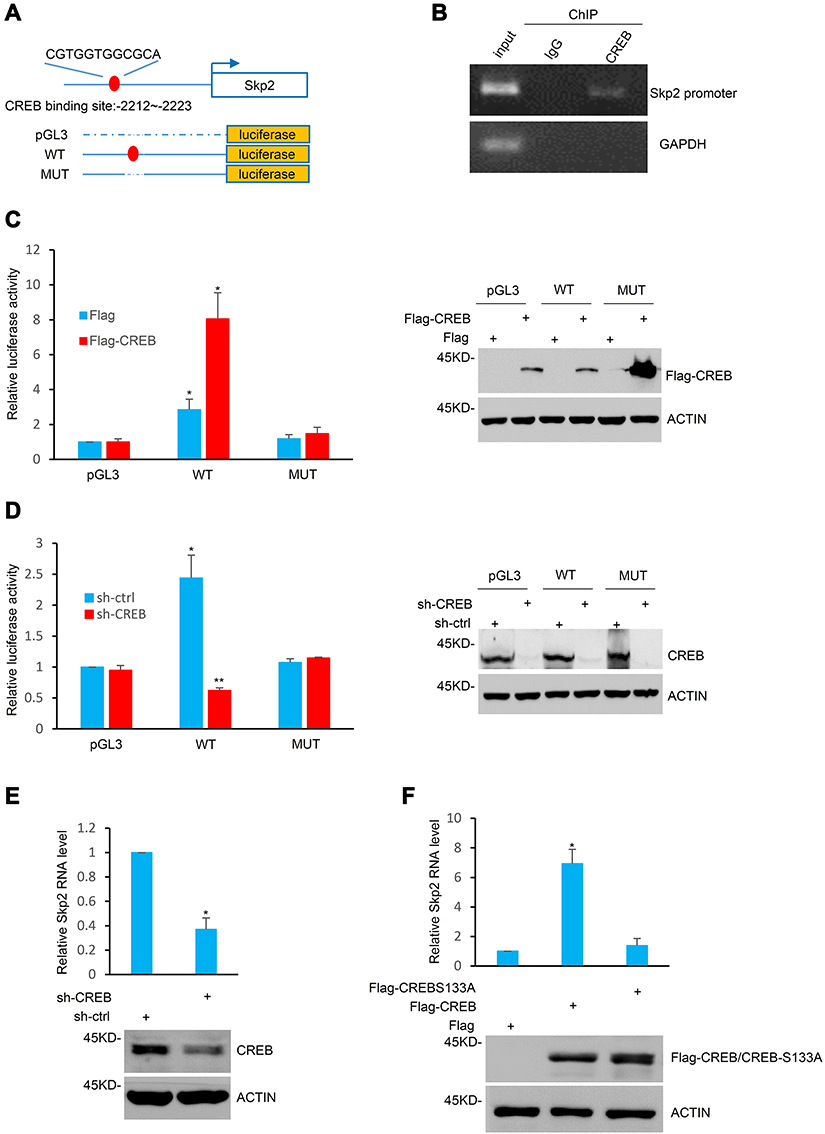 |
Figure 3 Skp2 is a transcriptional target of CREB. (A) Schematic diagram of the putative CREB binding region located 2212–2223 bp upstream of the Skp2 transcriptional start site and the pGL3-basic based Skp2 promoter reporter constructs. (B) ChIP assays were performed in K562 cells with anti-CREB antibodies or rabbit IgG as a control. ChIP products for Skp2 along with GAPDH as a negative control were amplified by semi-quantitative reverse transcription-PCR. (C) K562 cells were co-transfected with the indicated pGL3-based luciferase reporter constructs containing the wild-type or mutant Skp2 promoter regions together with either Flag or Flag-CREB. After 24 h transfection, transcriptional activity was determined via luciferase assays (left). The successful ectopic expression of CREB was confirmed via Western blotting (right). Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05 versus the group with pGL3 vectors. (D) K562 cells with and without CREB knockdown were co-transfected with the indicated reporter constructs. After 24 h transfection, cell lysates were subjected to luciferase analysis (left) and Western blot analysis (right). Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05;**P<0.01 versus the group with pGL3 vectors. (E) Skp2 mRNA levels of K562 cells with and without CREB knockdown were determined via Real-time-PCR analysis (top) and the CREB protein level was examined via Western blot analysis with CREB antibody (bottom). Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05 versus the group with shRNA-ctrl. (F) K562 cells were transfected with Flag, Flag-CREB or Flag-CREB S133A. After 24 h transfection, Skp2 mRNA levels were determined via Real-time-PCR analysis (top) and the successful ectopic expression of Flag-CREB and Flag-CREB S133A were confirmed via Western blotting (bottom). Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05 versus the group with flag overexpression. |
In addition, knockdown of CREB led to a dramatic decrease in the mRNA level of Skp2 in K562 cells (Figure 3E). Due to the transcriptional activity of CREB being enhanced following ser133 phosphorylation, the present study examined the effects of wild-type CREB and mutant S133A-CREB on Skp2 expression. As presented in Figure 3F, wild-type CREB, but not S133A-CREB, increased the mRNA level of Skp2.
To further examine the expression of Skp2 was correlated with the PI3K-CREB axis in CML, we analyzed the activation of Akt and CREB in patients with CML and healthy donors. As presented in Figure 1B, CML samples displayed relatively high levels of Skp2, pAkt and pCREB, whereas healthy controls associated with less Skp2, pAkt and pCREB. These results indicate that Skp2 expression is transcriptionally regulated by CREB and is positively correlated with PI3K-CREB axis.
Inhibition of PI3K/Akt-CREB-Skp2 Axis Sensitizes CML Cells to Imatinib
In order to investigate the functions of Skp2 in the chemosensitivity of CML cells, the present study analyzed whether inhibition of Skp2 sensitize CML cell lines to Imatinib, a classic tyrosine kinase inhibitor for treating CML.26,27 Wild-type, CREB and Skp2 knockdown K562 cells were first treated with Imatinib individually to assess the cell survival rates. As anticipated, cell viability of K562 cells with CREB or Skp2 stable knockdown significantly decreased with Imatinib treatment (Figure 4A). Furthermore, knockdown of either CREB or Skp2 also resulted in a dramatic increase in caspase-3/7 activity in response to treatment with Imatinib (Figure 4C). And higher level of cleaved PARP and caspase-3 were observed in CREB or Skp2 stable knockdown cells following Imatinib treatment (Figure 4D), indicating that CREB- or Skp2-deficient cells are more vulnerable to apoptosis.
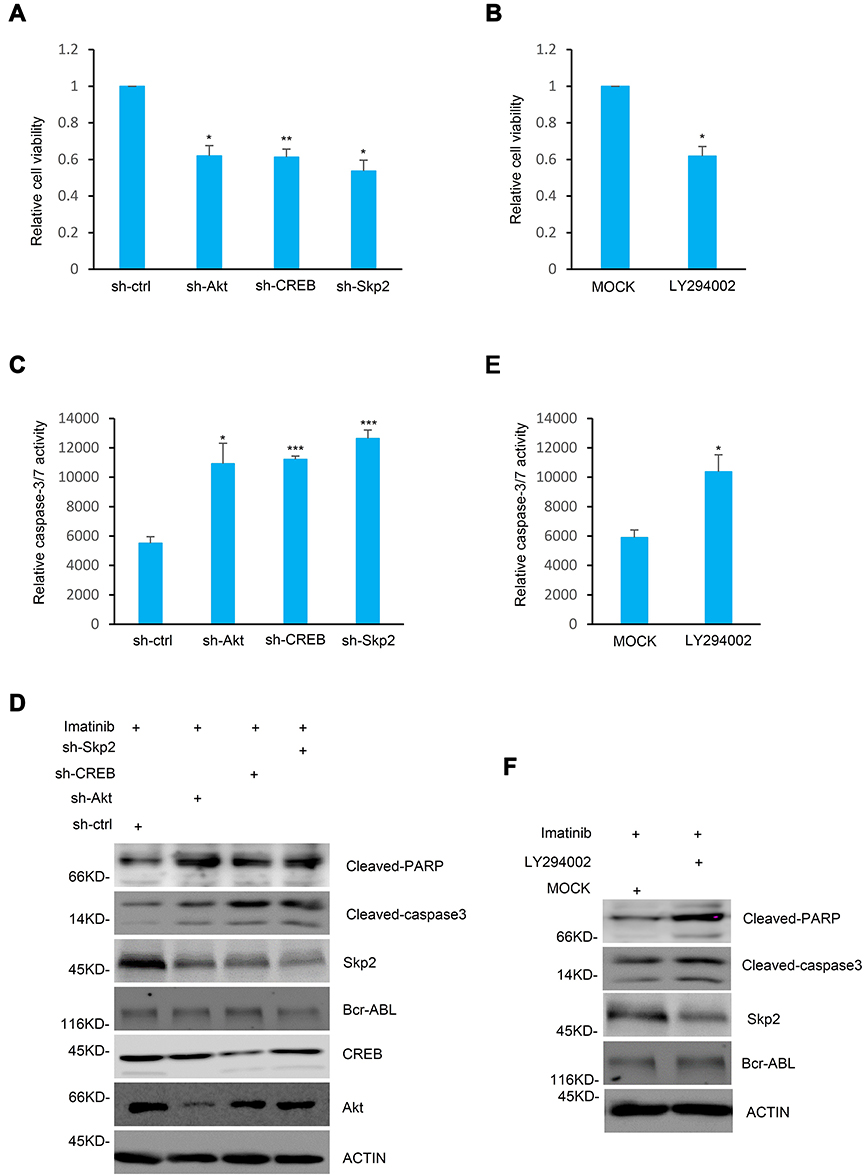 |
Figure 4 Inhibition of the PI3K/Akt/CREB/Skp2 axis sensitizes CML cells to Imatinib. (A) K562 cells stably expressing shRNA-ctrl, shRNA-Akt, shRNA-CREB or shRNA-Skp2 were treated with 1 μM Imatinib for 24 h. The cell viability was determined by MTT analysis and the relative cell viability was presented from triplicate experiments compared to the shRNA-ctrl group. Data are shown as mean ± SD; two-tailed paired Student’s t-test. *P<0.05; **P<0.01 versus the group with the group with shRNA-ctrl. (B) K562 cells were pre-treated with DMSO or LY294002 for 4 h, followed by treatment with 1 μM Imatinib for another 24 h. MTT assays were used to determine the cell viability and the relative cell viability was presented by comparison with the control group. Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05 versus the mock group. (C and D) K562cells with shRNA-ctrl, shRNA-Akt, shRNA-CREB or shRNA-Skp2 expression were treated with 1 μM Imatinib for 24h. Cell lysates were analyzed by (C) caspase3/7 activity and (D) Western blot analysis. Data are shown as mean ± SD; two-tailed paired Student’s t-test. *P<0.05; ***P<0.001 versus the group with the group with shRNA-ctrl. (E and F) K562 cells with and without LY294002 pre-treatment were also treated with 1 μM Imatinib for 24 h. Cell lysates were then subjected to (E) caspase3/7 activity assay and (F) Western blot analysis. Data are shown as mean ± SD; n = 3 independent experiments, two-tailed paired Student’s t-test. *P<0.05 versus the mock group. |
It has been reported that CREB is phosphorylated at Ser133 via the PI3K/Akt pathway and then activated.28 In order to determine the specific function of the PI3K/Akt pathway in inducing CREB activation and Skp2 expression, the K562 cells were treated with Akt shRNA or LY294002 to inhibit the PI3K-Akt pathway. Indeed, inhibition of the PI3K-Akt pathway significantly decreased Skp2 expression (Figure 4D and F). In addition, silencing of Akt or LY294002 treatment led to a noticeable decrease in cell viability with Imatinib treatment (Figure 4A and B). Furthermore, either treatment with LY294002 or stable knockdown of Akt in K562 cells strongly enhanced caspase-3/7 activity in response to treatment with Imatinib (Figure 4C and E). Consistently, higher levels of caspase-3 activation and PARP cleavage were observed following Imatinib treatment in K562 cells with Akt knockdown or treatment with LY294002 (Figure 4D and F). Taken together, these results illustrate an important role of the PI3K/Akt/CREB axis in regulating the sensitivity of K562 cells to Imatinib via mediating Skp2 expression.
Discussion
In the early 1970s, BCR-ABL kinase has been identified as the main cause of CML development.1 Specific inhibitors targeting BCR-ABL kinase, such as TKIs, have been in development for a number of years. Imatinib, one of the most widely used TKIs, inhibits the BCR-ABL kinase activity specifically and efficiently and has been listed as the preferred choice of drug in clinical CML therapy.5,6 However, oncogenic BCR-ABL has been discovered to be an effective drug target for decades, but the treatment of CML remains challenging. Not all the patients with CML are typical BCR-ABL kinase carriers, and some other dysregulated molecules, such as DNMT3A, may also promote the development of CML.29 In addition, the primary TKIs resistance is also commonly observed among patients with CML. The over-activated pathways in CML, including PI3K/AKT and MAPK/ERK signaling are potential targets for solving the issue surrounding TKIs resistance.30,31 The present study discovered that Skp2 was aberrantly upregulated in patients with CML compared with the healthy donors which was responsive to the PI3K/Akt signaling pathway and mediated Imatinib resistance in patients with CML. As mentioned in previous studies, the protein level of BCR-ABL was associated with the sensitivity to Imatinib.32,33 The present study also investigated the effect of the inhibition of PI3K/Akt/CREB pathway on BCR-ABL expression, and failed to detect any change in BCR-ABL under these conditions (Figure 4D and F). In agreement with the data in the present study, an increasing number of studies have demonstrated that BCR-ABL-independent resistance to Imatinib was associated with transcription factor NF-κB, HDACs and protein kinase C in CML, but not BCR-ABL9,34,35. These data indicate that Skp2 is specifically responsive to PI3K/Akt/CREB pathway and contributed a BCR-ABL independent Imatinib resistance.
Skp2 was originally identified as an F-box protein and a member of the ubiquitin ligase SCF-Skp2 complex which also contains three other subunits RBX1, CUL1, and Skp1.24,36 Skp2 expression was regulated in a cell-cycle dependent manner and several proteins that participate in cell cycle control (such as cyclin E, E2F1, and p21) have been identified as substrates for Skp2.24 Skp2 functions as an oncogenic regulator in transformation assays and is frequently aberrantly expressed in human cancer23,24. However, the role of Skp2 in CML remains largely unclear. A recent study reported that Skp2 is an MYC-direct target gene, and Skp2 mediates the downregulation of MYC on p27 in leukemia cells.18 The present study provided evidence that Skp2 is highly upregulated in patients with CML, and knockdown of Skp2 significantly inhibits the proliferation of CML cells. The data from the present study uncover a previously unidentified role of Skp2 in association with CML cell proliferation.
The present study also demonstrated that the increase of Skp2 in CML is due to increased transcription mediated by the transcription factor CREB. It has been reported that CREB transcriptional activity was activated following its phosphorylation at Ser133 by various signaling pathways, such as PI3K/Akt and MAPK/ERK.37,38 The results of the present study demonstrated that CREB inactivation resulted in a significant decline of the mRNA level of Skp2, exhibiting a specific role of CREB in the upregulation of Skp2 in CML cells.
This study established that inhibition of Skp2 or the PI3K/Akt/CREB pathway significantly increased the sensitivity of K562 cells to Imatinib treatment. These results suggested that the PI3K/Akt/CREB/Skp2 axis may be involved in the primary sensitivity of K562 cells to Imatinib. Consistent with the results of the present study, a recent study reported that the constitutive activation of the PI3K/Akt signaling pathway contributes to the pathogenesis and survival of mantle cell lymphoma.39 Despite the detailed mechanism underlying this phenomenon still requiring further investigation, the results of the present study provide insights into potential therapeutic strategies for the treatment of CML that have primary resistance to the tyrosine kinase inhibitors.
Conclusion
In conclusion, we found that Skp2 was transcriptionally upregulated by CREB responsive to the PI3K/Akt signaling pathway in CML and elevated expression of Skp2 was critical for CML cell proliferation. Moreover, our study confirmed that inhibition of Skp2 expression by shRNAs or blocking the PI3K/Akt/CREB pathway greatly enhanced the sensitivity of CML cells to Imatinib treatment. Our findings revealed an unknown role of Skp2 in CML progression and provides new aspects on the Skp2-modulated TKI sensitivity in CML.
Abbreviations
CREB, cAMP response element binding protein; TKI, tyrosine kinase inhibitor; CML, chronic myeloid leukemia; Skp2, S-phase kinase-associated protein 2; PI3K, phosphatidylinositol 3-kinase; ChIP, chromatin immunoprecipitation; shRNA, short hairpin RNA.
Data Sharing Statement
Further information and resources in this study are available from the corresponding author on reasonable request.
Acknowledgment
This work was supported by Natural Science Research Project for Universities of Anhui Province (KJ2018ZD020).
Disclosure
The authors have declared that no competing interest exists.
References
1. Jiang MJ, Dai JJ, Gu DN, Huang Q, Tian L. MicroRNA-7 inhibits cell proliferation of chronic myeloid leukemia and sensitizes it to imatinib in vitro. Biochem Biophys Res Commun. 2017;494(1–2):372–378. doi:10.1016/j.bbrc.2017.10.001
2. Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. doi:10.1038/243290a0
3. Deng Y, Li X, Feng J, Zhang X. Overexpression of miR-202 resensitizes imatinib resistant chronic myeloid leukemia cells through targetting Hexokinase 2. Biosci Rep. 2018;38(3). doi:10.1042/BSR20171383
4. Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract. 2014;2014:357027.
5. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. doi:10.1182/blood-2013-05-501569
6. O’Brien S, Radich JP, Abboud CN, et al. Chronic myelogenous leukemia, version 1.2014. J Natl Compr Canc Netw. 2013;11(11):1327–1340. doi:10.6004/jnccn.2013.0157
7. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. doi:10.1056/NEJM200104053441401
8. Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J Biol Chem. 2004;279(33):34227–34239. doi:10.1074/jbc.M402290200
9. Donato NJ, Wu JY, Stapley J, et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res. 2004;64(2):672–677. doi:10.1158/0008-5472.CAN-03-1484
10. Huang T, Yang L, Wang G, et al. Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel. Onco Targets Ther. 2017;10:439–446. doi:10.2147/OTT.S125789
11. Lim MS, Adamson A, Lin Z, et al. Expression of Skp2, a p27(Kip1) ubiquitin ligase, in malignant lymphoma: correlation with p27(Kip1) and proliferation index. Blood. 2002;100(8):2950–2956. doi:10.1182/blood.V100.8.2950
12. Nakayama K, Hatakeyama S, Nakayama K. Regulation of the cell cycle at the G(1)-S transition by proteolysis of cyclin E and p27(Kip1). Biochem Biophys Res Commun. 2001;282(4):853–860. doi:10.1006/bbrc.2001.4627
13. Zhang H, Kobayashi R, Galaktionov K, Beach D. p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell. 1995;82(6):915–925. doi:10.1016/0092-8674(95)90271-6
14. Marti A, Wirbelauer C, Scheffner M, Krek W. Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nat Cell Biol. 1999;1(1):14–19. doi:10.1038/8984
15. Pene F, Claessens YE, Muller O, et al. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002;21(43):6587–6597. doi:10.1038/sj.onc.1205923
16. Min YH, Cheong JW, Lee MH, et al. Elevated S-phase kinase-associated protein 2 protein expression in acute myelogenous leukemia: its association with constitutive phosphorylation of phosphatase and tensin homologue protein and poor prognosis. Clin Cancer Res. 2004;10(15):5123–5130. doi:10.1158/1078-0432.CCR-04-0136
17. Latres E, Chiarle R, Schulman BA, et al. Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci U S A. 2001;98(5):2515–2520. doi:10.1073/pnas.041475098
18. Bretones G, Acosta JC, Caraballo JM, et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. J Biol Chem. 2011;286(11):9815–9825. doi:10.1074/jbc.M110.165977
19. Andreu EJ, Lledo E, Poch E, et al. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27 (Kip1) degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res. 2005;65(8):3264–3272. doi:10.1158/0008-5472.CAN-04-1357
20. Chen JY, Wang MC, Hung WC. Transcriptional activation of Skp2 by BCR-ABL in K562 chronic myeloid leukemia cells. Leuk Res. 2009;33(11):1520–1524. doi:10.1016/j.leukres.2009.03.007
21. Yang G, Ayala G, De Marzo A, et al. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res. 2002;8(11):3419–3426.
22. Hu WL, Jin L, Xu A, et al. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat Cell Biol. 2018;20(4):
23. Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112(7):1415–1424. doi:10.1002/cncr.23317
24. Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8(6):438–449. doi:10.1038/nrc2396
25. Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32(Database issue):D91–D94. doi:10.1093/nar/gkh012
26. Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566. doi:10.1038/nm0596-561
27. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–2653. doi:10.1182/blood-2004-08-3097
28. Du KY, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273(49):32377–32379. doi:10.1074/jbc.273.49.32377
29. Schmidt M, Rinke J, Schafer V, et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia. 2014;28(12):2292–2299. doi:10.1038/leu.2014.272
30. Burchert A, Wang Y, Cai D, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774–1782. doi:10.1038/sj.leu.2403898
31. Aceves-Luquero CI, Agarwal A, Callejas-Valera JL, et al. ERK2, but not ERK1, mediates acquired and “de novo” resistance to imatinib mesylate: implication for CML therapy. PLoS One. 2009;4(7):e6124. doi:10.1371/journal.pone.0006124
32. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880. doi:10.1126/science.1062538
33. le Coutre P, Tassi E, Varella-Garcia M, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95(5):1758–1766. doi:10.1182/blood.V95.5.1758.005a41_1758_1766
34. Ma LY, Shan Y, Bai R, et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6(252):252ra121. doi:10.1126/scitranslmed.3009073
35. Roychowdhury S, Talpaz M. Managing resistance in chronic myeloid leukemia. Blood Rev. 2011;25(6):279–290. doi:10.1016/j.blre.2011.09.001
36. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–381. doi:10.1038/nrc1881
37. Ozgen N, Obreztchikova M, Guo J, et al. Protein kinase D links Gq-coupled receptors to cAMP response element-binding protein (CREB)-Ser133 phosphorylation in the heart. J Biol Chem. 2008;283(25):17009–17019. doi:10.1074/jbc.M709851200
38. Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18(4):1946–1955. doi:10.1128/MCB.18.4.1946
39. Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108(5):1668–1676. doi:10.1182/blood-2006-04-015586
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.