Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Inhibition of myeloid differentiation factor 88 signaling mediated by histidine-grafted poly(β-amino ester) ester nanovector induces donor-specific liver allograft tolerance
Authors Hu F, Wang H, Zhang S, Peng Y, Su L, Change J, Liu G
Received 23 January 2015
Accepted for publication 28 March 2015
Published 6 July 2015 Volume 2015:10(1) Pages 4367—4382
DOI https://doi.org/10.2147/IJN.S81413
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Fanguo Hu,1,* Hanjie Wang,2,* Shuangnan Zhang,2 Yao Peng,2 Lin Su,2 Jin Chang,2 Gang Liu1
1Department of General Surgery, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China; 2School of Life Sciences, Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering, Tianjin Engineering Center of Micro-Nano Biomaterials and Detection-Treatment Technology, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Abstract: Toll-like receptors (TLRs) activate biochemical pathways that evoke activation of innate immunity, which leads to dendritic cell maturation and initiation of adaptive immune responses that provoke allograft rejection. We aimed to prolong allograft survival by selectively inhibiting expression of myeloid differentiation factor 88 (MyD88), which is an essential adaptor in TLR signaling. We designed and synthesized a novel histidine-grafted poly(β-amino ester)(HGPAE) nanovector, which was shown to be safe and efficient both in vitro and in vivo for the delivery of a plasmid containing shRNA targeting MyD88 (pMyD88). We also demonstrated that the pMyD88/HGPAE complex mediated remarkable inhibition of MyD88 expression in rat liver in vivo. We transplanted Dark Agouti rat livers lacking MyD88 as result of transfection with the pMyD88/HGPAE complex into Lewis rats. The recipients survived longer and graft rejection of the donor liver as well as serum levels of IL-2 and IFN-γ in the recipient were significantly reduced.
Keywords: immune recognition, allograft rejection, MyD88, short hairpin RNA (shRNA), gene delivery, PAE
Introduction
Liver transplantation has achieved great success in patients with terminal liver diseases. Nevertheless, the success is limited by the requirement for lifelong use of immunosuppressants to prevent allograft rejection. Current immunosuppressants are not completely effective and result in complications, which limit graft and patient survival. Therefore, a novel therapeutic strategy for suppressing graft rejection with limited side effects is required. Immunosuppression targets the adaptive alloimmune response primarily; however, innate immunity is also important in allograft rejection because it both mediates inflammation and promotes adaptive alloimmune responses.1–3
Toll-like receptors (TLRs), which are innate immune receptors expressed by a variety of immune cells, recognize pathogen-associated molecular patterns present on microorganisms and also recognize endogenous ligands released from damaged tissue.4 All TLRs, except TLR3, signal through an adaptor molecule, myeloid differentiation factor 88 (MyD88), which leads to nuclear translocation of NF-κB and IRF7, with consequent upregulation of proinflammatory cytokines (Figure 1); this upregulation subsequently promotes the development of effective adaptive immunity through activation of antigen-presenting cells (APCs), via upregulation of major histocompatibility complex (MHC) class II antigens, costimulatory molecules, chemokines, and cytokines.5–7 It has been reported that skin allografts in mice with targeted deletion of the MyD88 adaptor protein are received without rejection.8 Therefore, MyD88 is implicated as an ideal target to inhibit innate immune responses by preventing TLR signal transduction.9,10
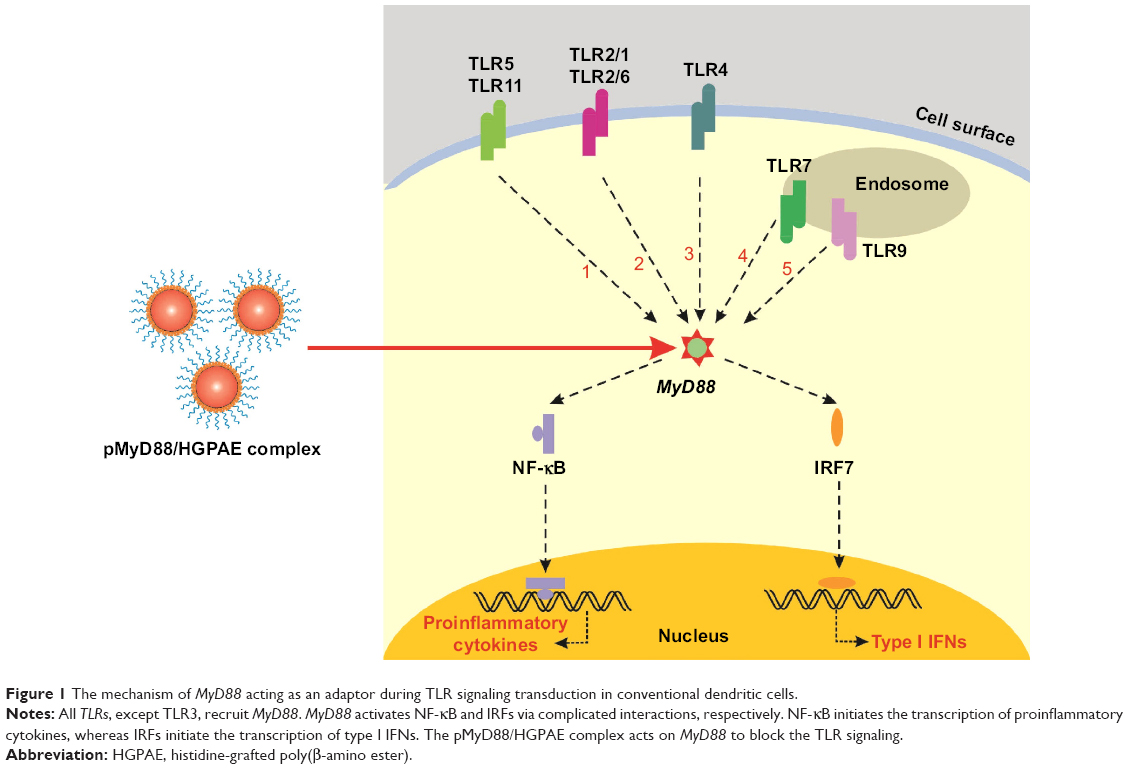 | Figure 1 The mechanism of MyD88 acting as an adaptor during TLR signaling transduction in conventional dendritic cells. |
The attenuation of allograft rejection by inhibiting MyD88 expression in liver transplantation has not yet been reported. Therefore, in this study, a plasmid expressing a short hairpin RNA (shRNA) targeting MyD88 (pMyD88) was designed, synthesized, and combined with a new histidine-grafted poly(β-amino ester) (HGPAE) nanovector to form the pMyD88/HGPAE complex. The complex was then used to attenuate graft rejection in a rat liver transplantation model by inhibiting the expression of MyD88 in vivo. To protect the recipient, we chose to inhibit MyD88 expression in the donor liver.
Materials and methods
Materials and animals
1,4-Butanediol diacrylate (90%), 4-amino-1-butanol (98%), 4-dimethylaminopyridine (99%), N,N′-dicyclohexylcarbodiimide (99%), N-cbz-L-histidine, 10% Pd-C, methylene chloride, and ethyl ether were purchased from Alfa Aesar (Ward Hill, MA, USA). pMyD88 and the negative control plasmid containing nonspecific shRNA sequence (pHK) were both designed and synthesized by Genesil Biotechnology (Wuhan, People’s Republic of China). Sprague Dawley rats and Lewis rats weighing approximately 250 g were obtained from the Experimental Animal Center of China’s Military Academy of Medical Sciences (Beijing, People’s Republic of China). DA rats were obtained from the Experimenta Animal Center of the Second Affiliated Hospital of Harbin Medical University (Harbin, People’s Republic of China).
Synthesis of poly(β-amino esters) and HGPAEs by Michael addition reaction
Poly(β-amino esters) (PAEs) containing degradable ester bonds were synthesized through the conjugation Michael addition reaction between 1,4-butanediol diacrylate and 4-amino-1-butanol. The details are as follows: 2.22 g 1,4-butanediol diacrylate powder and 2.50 g 4-amino-1-butanol were dissolved into 10 mL methylene chloride and both solutions were added into a flask with stirring. The mixed solution was heated to 60°C and the reaction was continued for 48 hours under argon. Ethyl ether was then added into the mixed solution to precipitate the polymers. The precipitates were centrifuged and washed with ethyl ether three times. Finally, the products were stored in a vacuum drying oven for subsequent experiments.
HGPAEs were synthesized by modification of the PAEs with histidine, which improves the protonation of PAEs. The details are as follows: 144.6 mg N-cbz-L-histidine, 6.1 mg 4-dimethylaminopyridine, and 158.2 mg PAEs were dissolved into 4 mL N,N-dimethyl formamide (Alfa Aesar). Then 113.4 mg N,N′-dicyclohexylcarbodiimide dissolved into 4 mL N,N-dimethyl formamide was added into the mixture and stirred at room temperature for 2 days under argon. Subsequently, the insoluble products were filtered out using oily membrane with aperture of 220 nm, and the remaining solution was precipitated with ethyl ether. The purified product was then dispersed into cyclohexene/ethanol (5/95% v/v) (Alfa Aesar) solution in the presence of 0.5 g 10% Pd-C. The solution was heated to 65°C and the reaction was continued for 8 hours under argon for the deprotection of carboxybenzyl groups of the conjugated N-cbz-L-histidine. Ethyl ether was added into the mixed solution to precipitate the polymers. The precipitates were centrifuged and washed with ethyl ether three times. Finally, the products were stored in a vacuum drying oven for subsequent experiments.
Structure and property characterization of PAEs and HGPAE
The chemical structure was characterized based on proton nuclear magnetic resonance spectra, which were recorded on a Varian UNITY Plus-400 nuclear magnetic resonance instrument (Palo Alto, CA, USA) using dimethyl sulfoxide as a solvent.
The buffering ability of PAEs and HGPAE was determined by acid/base titration. The details are as follows: the polymer solution was first adjusted to above pH 10 with 0.1 M NaOH and was then titrated with 0.1 M HCl. Titration profiles were plotted as changes in pH against the volume of HCl solution.
In addition, the pH sensitivity was tested by detecting the absorbance of HGPAE solutions at different pH values at 500 nm with UV spectrophotometry using a UV-2450 (Shimadzu, Kyoto, Japan).
Preparation and property characterization of the plasmid pHK/HGPAE complexes
The plasmid pHK was diluted in sodium acetate buffer and mixed with HGPAE to form the pHK/HGPAE complexes at a concentration of 1 mg/mL. After incubation at room temperature for 30 minutes, the complexes were used for further characterization.
Agarose gel retardation assays were used to test the gene combining ability and gene protection ability of HGPAE at different weight ratios of HGPAE to pHK. The size and potential changes of pHK/HGPAE complexes with different weight ratios were tested using a laser granulometer and a zeta potentiometer (BI-90Plus; Brookhaven, Brookhaven, NY, USA), respectively. The morphology of the complexes were observed with a JEOL-100 CXII transmission electron microscope (JEOL, Tokyo, Japan) at an acceleration voltage of 100 kV.
To verify the ability of the pHK/HGPAE complexes to expand and disassemble under acid conditions, the size changes were monitored kinetically by dynamic light scattering under different pH conditions using a Brookhaven BI-90Plus particle size analyzer.
Safety and transfection efficiency of HGPAE in vitro
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based assay was performed to test the cytotoxicity of HGPAE polymers in vitro. MIA PaCa-2 pancreatic cancer cells were seeded into a 96-well plate at a density of 4,000 cells per well and incubated at 37°C for 24 hours in Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and 5% CO2. Then, 100 μL of medium containing different concentrations of HGPAE was added. The cells were incubated for an additional 24 hours at 37°C and 20 μL of 0.5 mg/mL MTT solution was added to each well; cells were incubated for another 4 hours. The culture medium was subsequently removed and 200 μL of dimethyl sulfoxide was then added. Absorbance was measured at 570 nm in a microplate reader (model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA). Cell survival was then calculated as a percentage of the untreated cell number, which was designated as 100% survival.
The transfection efficiency of HGPAE nanovectors in vitro was evaluated by observing the EGFP expression in cells. Briefly, MIA PaCa-2s were seeded into a 24-well plate at a density of 3×104 cells per well and incubated at 37°C for 24 hours in 500 μL DMEM supplemented with 10% fetal bovine serum. The cells were treated with two different samples: pHK/Lipofectamine2000 (Thermo Fisher Scientific, Waltham, MA, USA) and pHK/HGPAE complexes. After 48 hours in culture, the expression of EGFP was observed by fluorescence microscopy.
Transfection safety and efficiency of HGPAE in vivo
To evaluate the transfection safety and efficiency of nanovector HGPAE in vivo, the following three groups were established: saline group, HGPAE vector group, and pHK/HGPAE group. Liver function and renal function were used to test the transfection safety of HGPAE in vivo. Serum aspartate transaminase (AST), alanine transaminase (ALT), and serum total bilirubin were used as liver function indexes and creatinine and blood urea nitrogen were used as renal function indexes. To evaluate the transfection efficiency of HGPAE in vivo, liver specimens were homogenized and EGFP expression was measured by fluorimetry (excitation wavelength of 485 nm and emission wavelength of 530 nm).
The details of transfections in vivo are as follows: Sprague Dawley rats (n=8 per group) were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (5 mg/kg), and maintained with isoflurane inhalation. A midline abdominal incision was made and the portal vein was gently exposed and clamped at a distal point. Then 2 mL of different samples of pHK/HGPAE containing 200 μg shRNA were injected into the proximal portal vein over approximately 20 seconds, and the portal vein was opened 2 minutes later. Tests were carried out on the first day and the third day after transfection.
Transfection of rat liver with pMyD88/HGPAE in vivo
Four groups of rats (saline control group, HGPAE vector control group, pHK/HGPAE control group, and pMyD88/HGPAE group) were used to evaluate the inhibitory effect of pMyD88/HGPAE on MyD88 gene expression in vivo. The details are as described in the section “Transfection safety and efficiency of HGPAE in vivo”. Liver specimens were harvested 72 hours after transfection and then assessed for MyD88 expression by real-time polymerase chain reaction (PCR) and Western blot analyses.
Real-time PCR
Total RNA was extracted from liver tissue using TRIzol reagent (Thermo Fisher Scientific). RNA was reverse-transcribed using oligo-(dT) primer and reverse transcriptase (Thermo Fisher Scientific). Primers used for the amplification of rat MyD88 and β-actin genes were as follows: MyD88, 5′-AGGACAAACGCCGGAACTTTT-3′ (forward) and 5′-GCC GATAGTCTGTCGTTCTAGT-3′ (reverse); and β-actin, 5′-GTCGTACCACTGGCATTGTG-3′ (forward) and 5′-CTCTCAGCTGTGGTGGTGAA-3′ (reverse). Real-time PCR reactions were performed in a PTC-200 PCR machine (Bio-Rad Laboratories Inc.) using SYBR green PCR Master Mix (Bio-Rad Laboratories Inc.) and 100 nM of forward and reverse primers. The PCR reaction conditions were 94°C for 2 minutes, 94°C for 15 seconds, 58°C for 45 seconds, and 72°C for 30 seconds (35 cycles), followed by 72°C for 10 minutes.
Western blot
Liver tissue was homogenized in RIPA lysis buffer and used for Western blot analysis. Briefly, 40 μg of protein extracts were boiled with sodium dodecyl sulfate sample buffer for 5 minutes before being electrophoretically resolved on sodium dodecyl sulfate polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Pierce Chemical Company, Rockford, Illinois, USA). The membranes were then blocked for 1 hour at room temperature in 5% skimmed milk containing 1× Tris-buffered saline and 0.1% Tween 20. After blocking, the membranes were incubated overnight at 4°C with rabbit-anti-rat monoclonal antibodies (1:500 dilution; Cell Signaling, Boston, MA, USA). The membranes were incubated for 2 hours with goat-anti-rabbit secondary antibodies (1:2,000 dilution; Cell Signaling). Finally, the membranes were washed and an electrochemiluminescence (ECL) signal detection kit (Amersham/GE Healthcare, Piscataway, NJ, USA) was used for visualization of the protein bands.
Rat orthotopic liver transplantation
DA and Lewis rats were used as donors and recipients, respectively. Rats were allocated to the following four groups: saline control group, HGPAE vector control group, pHK/HGPAE control group, and pMyD88/HGPAE group (n=16 per group). Transfection of the donor liver was performed as described in the section “Transfection of rat liver with pMyD88/HGPAE in vivo”. The liver transplantation was performed 3 days after transfection using the modified two-cuffed technique as reported previously.11 Five days after transplantation, eight recipients (Lewis rats) from each group were sacrificed humanely with cervical dislocation and both the liver and blood were harvested to evaluate the graft rejection and detect expression of MyD88, IL-2, and IFN-γ. The remaining rats in each group were used to observe the survival time.
Graft histology
Liver graft samples were fixed in 10% formaldehyde, embedded in paraffin, and sectioned for hematoxylin and eosin staining. The Banff pathological schema was used to evaluate pathological features such as portal area inflammation, bile duct inflammation injury, and venous endothelial cell inflammation.12 The severity of pathological changes were scored as none (0–3), mild (4–5), moderate (6–7), or severe (8–9).
Enzyme-linked immunosorbent assay analysis of serum IL-2 and IFN-γ concentrations
The blood samples of recipient rats were collected 5 days after liver transplantation, and the concentrations of IL-2 and IFN-γ analyzed using IL-2 and IFN-γ enzyme-linked immunosorbent assay kits (Boster Biological Engineering, Wuhan, People’s Republic of China), respectively, according to the manufacturer’s instructions. The absorbance was measured at a test wavelength of 570 nm with a microplate reader. The concentrations of IL-2 and IFN-γ were calculated according to a standard curve prepared using samples of known concentration.
Statistical analysis
The survival time of recipients is reported as median survival time and comparisons were made using the Kaplan–Meier cumulative survival method. Histological findings were analyzed using analysis of variance on rank tests. Statistical comparisons of gene expression (real-time PCR) were performed using one-way analyses of variance. Differences with P-values less than 0.05 were considered to indicate statistical significance.
Ethics statement
All animal experimental procedures were carried out according to the regulations and internal biosafety and bioethics guidelines of Tianjin Medical University (Tianjin, People’s Republic of China) and the Tianjin Municipal Science and Technology Commission (Tianjin, People’s Republic of China).
Results
Structure and properties of PAEs and HGPAE
PAEs as gene vectors can protect DNA under physiological pH and promote gene release by responding to the intracellular acid environment. In this study, a positively charged histidine residue was grafted onto HGPAE to further improve the gene loading efficiency. The synthesis process and proton nuclear magnetic resonance spectra are shown in Figures 2 and 3. As shown in Figure 3, the peaks corresponding to signals δ (ppm) 5.9, 6.5, and 2.9 that are characteristic of acrylate-terminated PAEs indicated successful copolymerization of PAEs. Some additional peaks at 7.80–7.83 ppm that are characteristic of histidine were detected on the HGPAE spectrum, which indicated the success of the conjugation of histidine and the chain of PAEs.
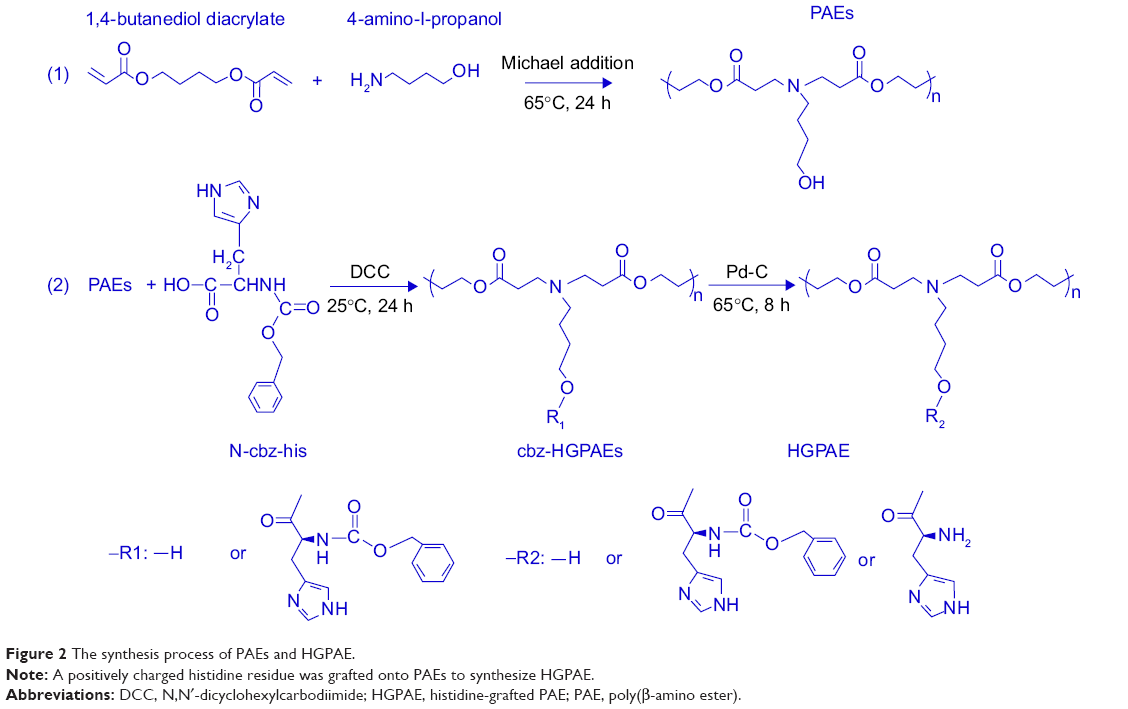 | Figure 2 The synthesis process of PAEs and HGPAE. |
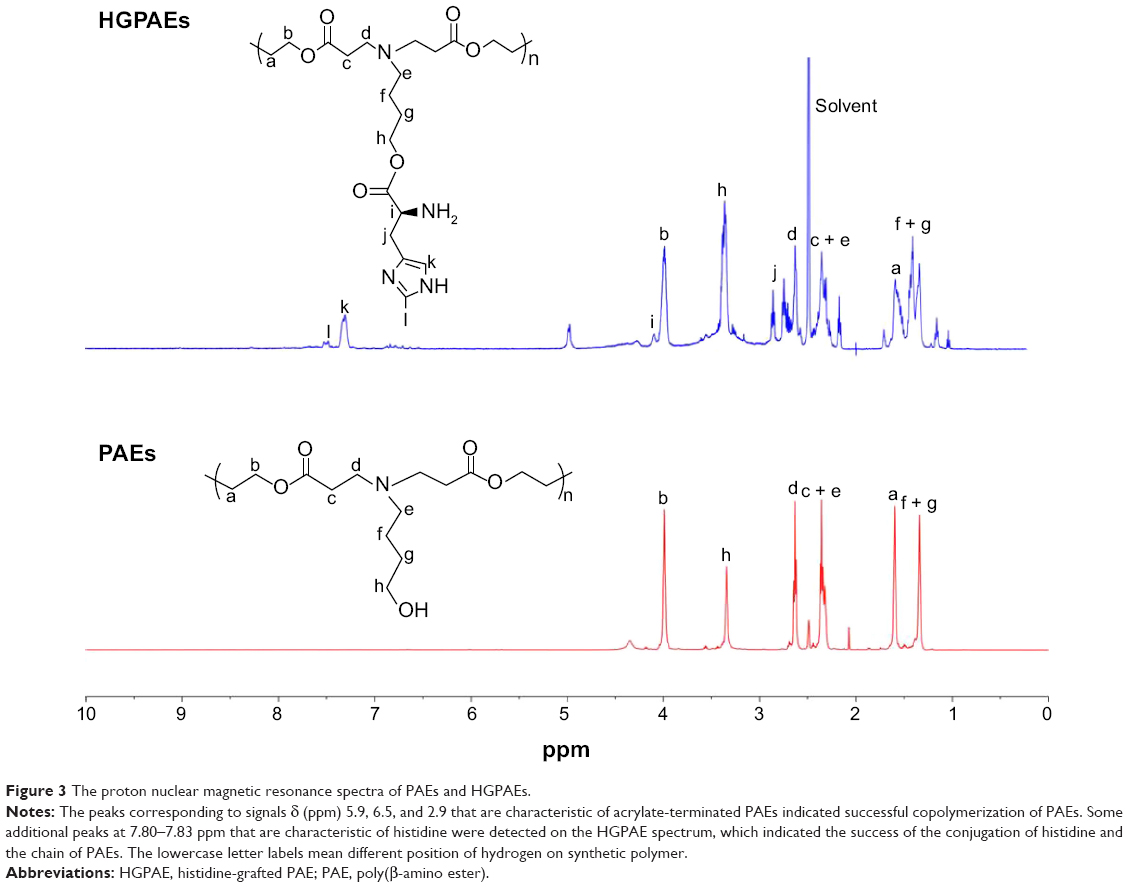 | Figure 3 The proton nuclear magnetic resonance spectra of PAEs and HGPAEs. |
The ion buffering capacity, or the potential for resistance to pH changes in different ionic environments, is one of the most important properties of the gene vector that is required for binding and release of the gene from endosome by the “proton sponge effect”. The buffering capacity of PAEs and HGPAE was investigated using the acid/base titration method. The titration curves are shown in Figure 4A. The NaCl solution profile showed a dramatic decrease in the pH range of 7.4 to 5.2, whereas that of the PAEs underwent a gradual decrease in the same pH range. Compared with the PAE profile, the decrease in the HGPAE profile was much more gradual. The ion buffering capacity according to the ratio between d[H] and d[pH] is shown in Figure 4B. Compared with PAEs, HGPAE slowed the change in pH much more significantly, indicating the larger buffering capacity of HGPAE. The reason for this may be that the imidazole and amine groups on the histidine moiety become protonated under acidic conditions.
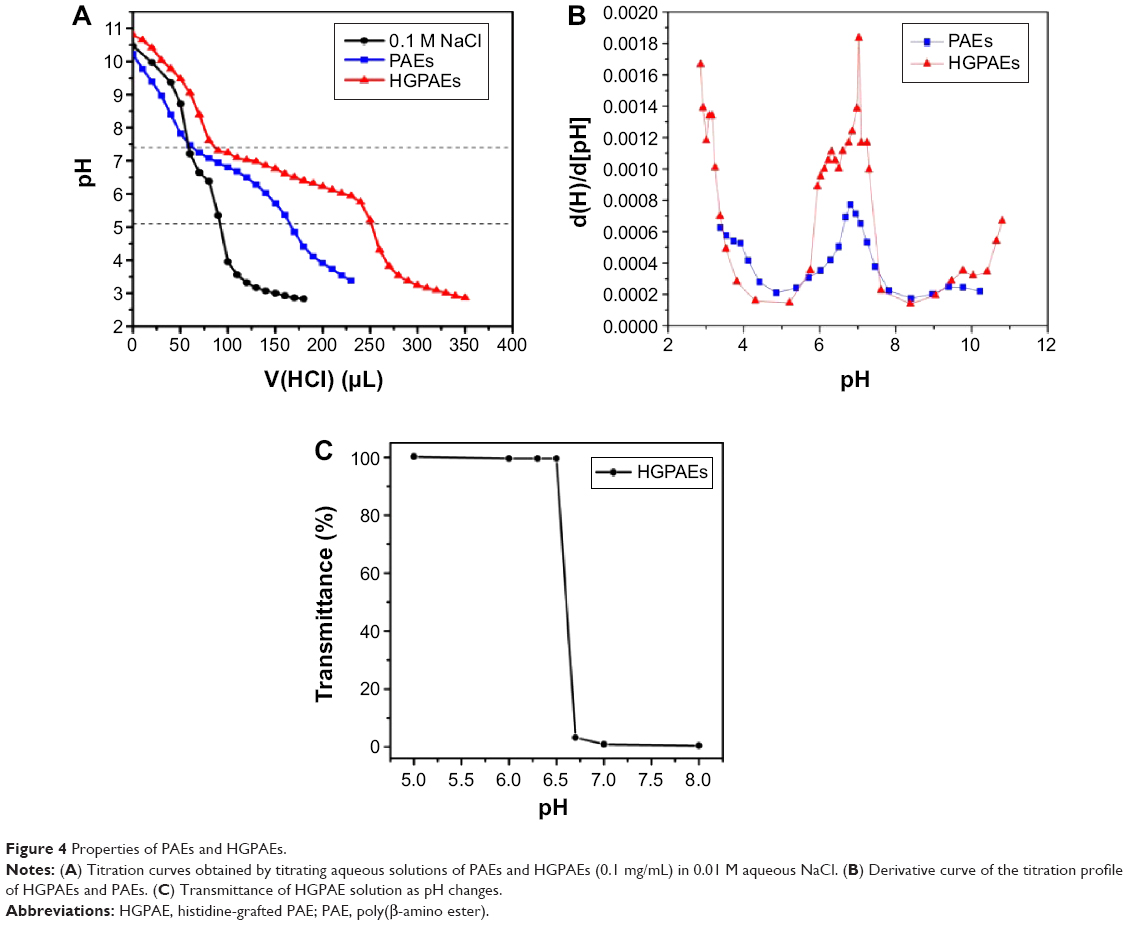 | Figure 4 Properties of PAEs and HGPAEs. |
As shown in Figure 4C, HGPAE showed a sensitive pH-dependent transmittance property. When exposed to basic conditions (pH >6.75), HGPAEs are prone to associate into stable nanoparticles and thus easily condense with the gene, while in acidic media (pH <6.5), they disintegrate, all of which indicates that HGPAEs are acid-sensitive and release the gene under acidic endosomal conditions.
Properties of pHK/HGPAE
As the weight ratio of HGPAE to pHK increases, the surface charge of the pHK/HGPAE complex decreases. As seen in Figure 5A, when the weight ratio of HGPAE to pHK reached 80:1, no DNA separation was detected, indicating complete association of the DNA with HGPAE. Therefore, the ratio 80:1 was selected as the ideal weight ratio for preparation of the pDNA/HGPAE complexes.
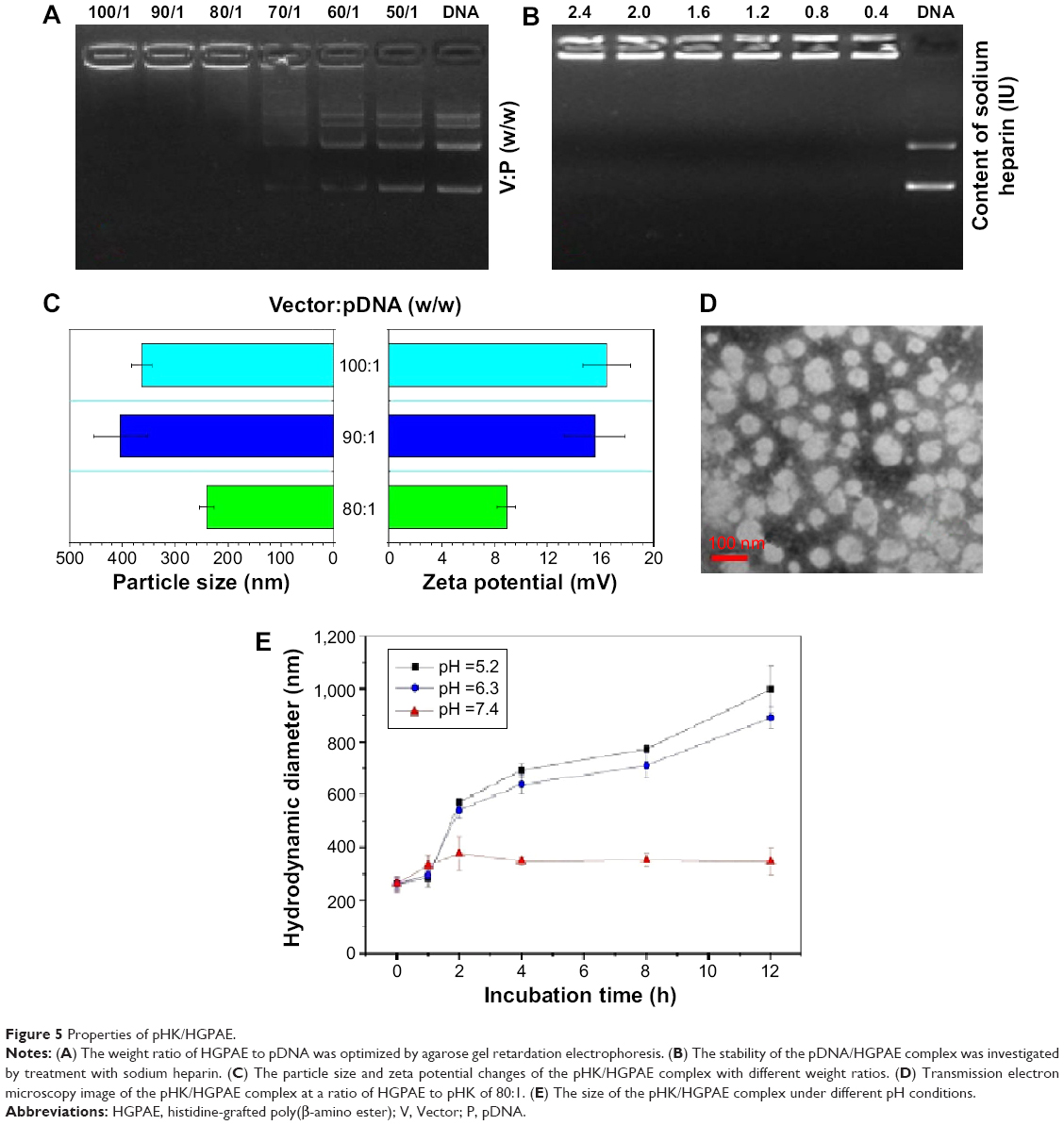 | Figure 5 Properties of pHK/HGPAE. |
Polyionic heparin sodium, which carries a strong negative charge, was used to imitate the in vivo environment for evaluation of the stability of the pHK/HGPAE complexes. As shown in Figure 5B, at heparin sodium concentrations ranging from 0.4 IU to 2.4 IU, no DNA was detected in the DNA electrophoresis samples, which showed the pHK/HGPAE complexes were sufficiently stable to avoid destruction of the polyanionic material.
As shown in Figure 5C, with increased incorporation of HGPAE, the particle size and the zeta potential of the pHK/HGPAE complexes became larger and more positive, respectively. As shown in Figure 5D, when HGPAE combined with pHK at a weight ratio of 80:1, the complexes displayed a spherical shape with a relatively homogeneous size distribution, which revealed the efficient condensation capability of HGPAE.
As shown in Figure 5E, after incubation in pH 7 buffer for 12 hours, the size of the pHK/HGPAE complexes remained in the range of 200–300 nm, indicating that HGPAE maintain the complex stability and protect pDNA sufficiently. In contrast, a rapid size increase was observed after incubation in pH 5.2 buffer and pH 6.3 buffer for 12 hours, which suggested the dissociation of the pHK/HGPAE complexes as a result of proton buffering capability and degradability. The pH response of pDNA/HGPAE complexes may be important in protecting pDNA from degradation by the endosome and allowing escape from the acidic endosome into cytoplasm.
Safety and transfection efficiency of HGPAE in vitro
The cytotoxicity of HGPAE in vitro was preliminarily estimated by MTT assays. As shown in Figure 6A, the cytotoxic activity was evaluated ranging from 60 μg/mL to 300 μg/mL. The HGPAE showed almost no significant cytotoxicity at 300 μg/mL. Therefore, in the cellular studies, HGPAE were used at 240 μg/mL to minimize the cytotoxic effects on cell viability.
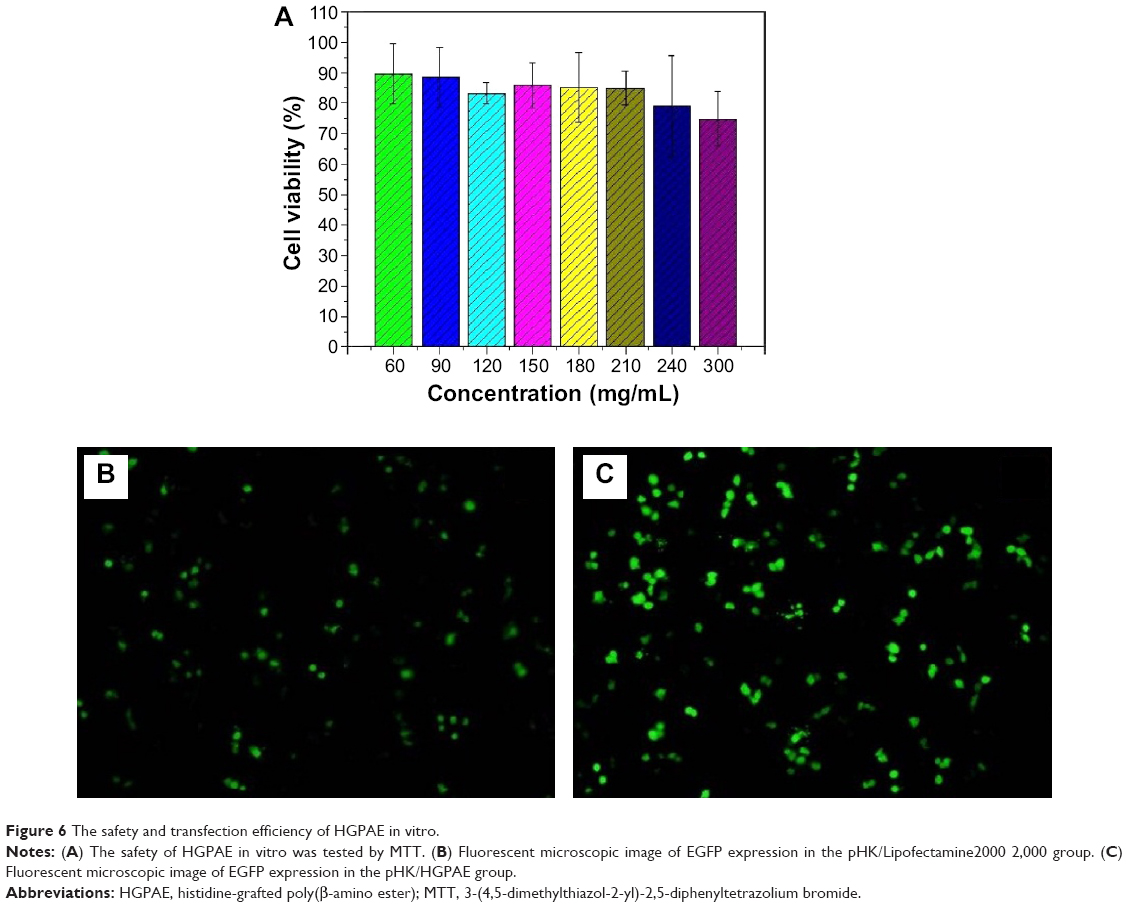 | Figure 6 The safety and transfection efficiency of HGPAE in vitro. |
As shown in Figure 6B and C, compared with the pHK/Lipofectamine2000 group, there was abundant expression of EGFP in the pHK/HGPAE group, which confirmed the transfection efficiency of the HGPAE vectors in vitro.
Safety and transfection efficiency of HGPAE in vivo
As shown in Figure 7C, both AST and ALT increased significantly in all the three groups (P<0.01) on the first day after transfection, although there was no significant difference among the three groups. On the third day after transfection, AST and ALT returned to normal levels in all three groups (Figure 7D). There was no significant increase in serum total bilirubin, creatinine, and blood urea nitrogen on either the first or third day after transfection.
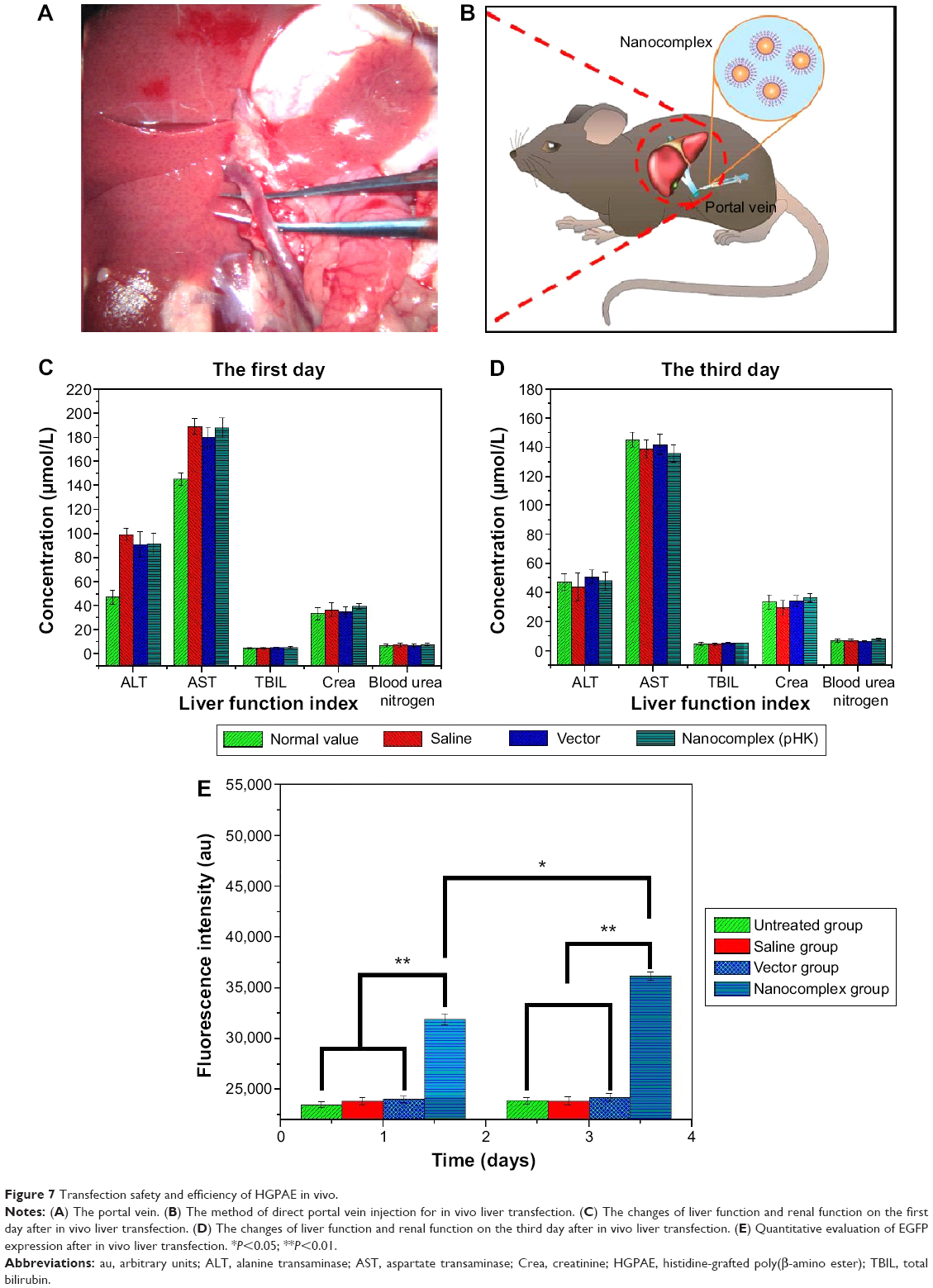 | Figure 7 Transfection safety and efficiency of HGPAE in vivo. |
As shown in Figure 7E, the amount of EGFP in the pHK/HGPAE group was significantly greater than that in the saline group and the HGPAE group on the first and third days after transfection in vivo (P<0.01), while there was no significant difference between the latter two groups (P>0.05). Moreover, comparisons of the amounts of EGFP detected on the first day and the third day after transfection revealed a significant increase in the pHK/HGPAE group (P<0.01), while there was still no significant difference in the saline group and the HGPAE group (P>0.05).
Gene expression of MyD88 after in vivo liver transfection
As shown in Figure 8A, 3 days after transfection, there were no significant differences in MyD88 mRNA expression among the saline control group, HGPAE vector control group, and pHK/HGPAE control group (P>0.05). However, compared with the three control groups, MyD88 mRNA expression in the pMyD88/HGPAE group was inhibited significantly (P<0.01). Similarly, there were no significant differences in MyD88 protein expression among the three control groups, while that in the pMyD88/HGPAE group was inhibited significantly (P<0.01) (Figure 8B–C).
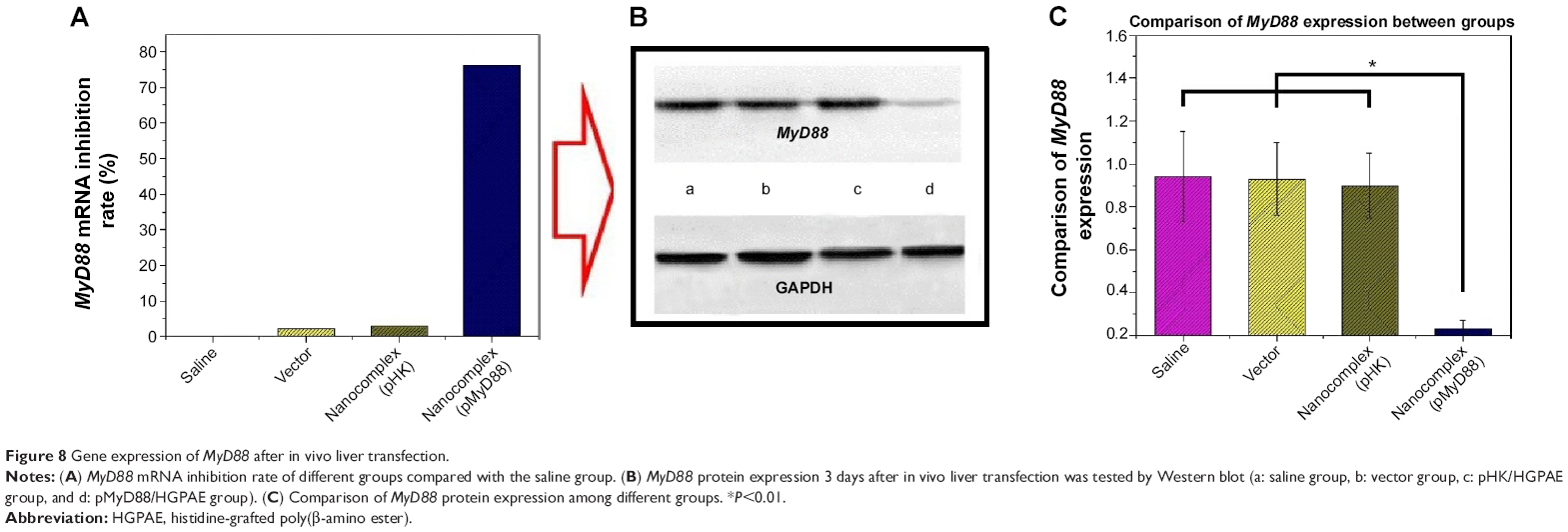 | Figure 8 Gene expression of MyD88 after in vivo liver transfection. |
Survival time of recipient rats after liver transplantation
As shown in Figure 9, compared with the three control groups, the median survival time of recipients in the pMyD88/HGPAE group was significantly longer (14 days versus 10 days in the saline group, 9 days in the vector group, and 8 days in the pHK/HGPAE group, P<0.05). However, there were no significant differences in the median survival time of recipient rats among the control groups (P>0.05).
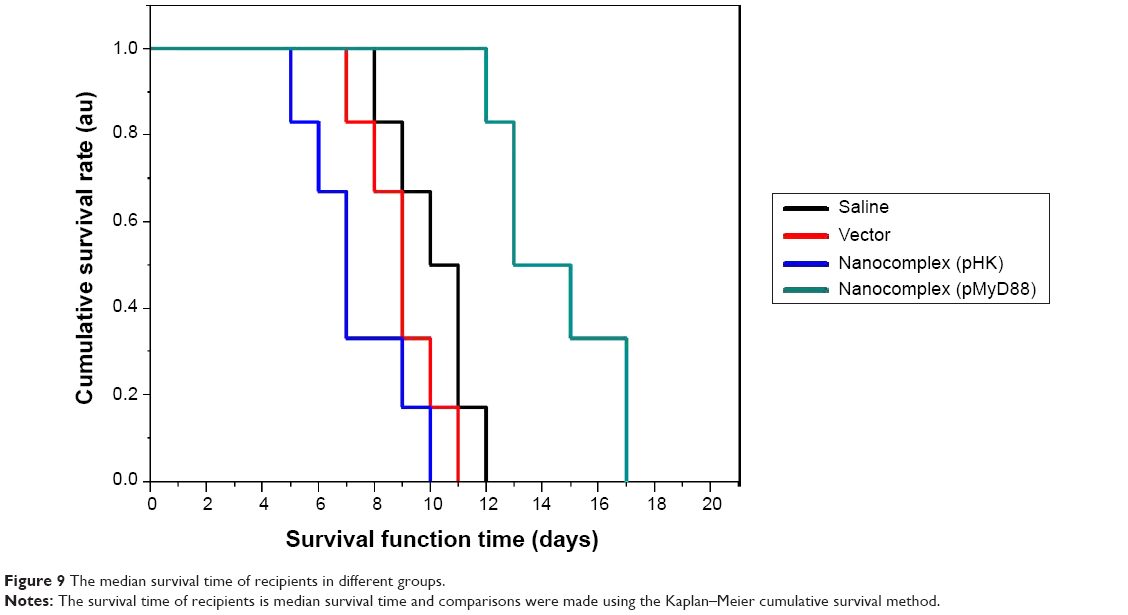 | Figure 9 The median survival time of recipients in different groups. |
Transplant rejection grades of the recipient rats
As shown in Figure 10, in the three control groups, liver grafts showed severe rejection-associated changes characterized by distinct inflammation in the portal areas with marked infiltration of neutrophils. In contrast, only mild histological changes were found in the pMyD88/HGPAE group. Significant differences in graft rejection evaluation scores based on the Banff pathological schema were found between the pMyD88/HGPAE group and the control groups (4.5±0.5 in the pMyD88/HGPAE group versus 8.62±0.6 in the saline group, 8.35±0.8 in the vector group, and 8.16±0.7 in the pHK/HGPAE group, P<0.05). However, there were no significant differences in the graft rejection evaluation scores among the three control groups (P>0.05).
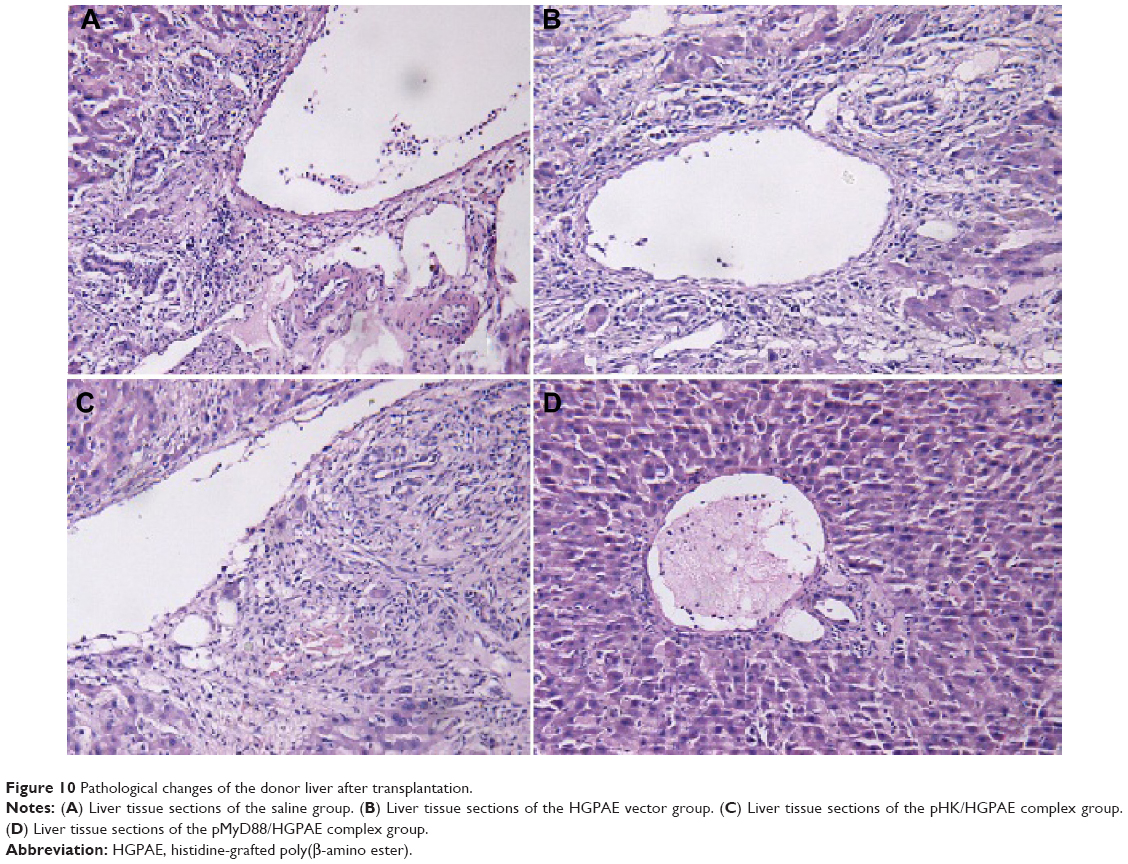 | Figure 10 Pathological changes of the donor liver after transplantation. |
Gene expression of MyD88 after liver transplantation
As shown in Figure 11, 5 days after liver transplantation, there were no significant differences in MyD88 mRNA and protein expression among the saline control, HGPAE vector control, and pHK/HGPAE control groups (P>0.05). However, compared with the three control groups, the expression of MyD88 mRNA and protein in the pMyD88/HGPAE group was suppressed significantly (P<0.01).
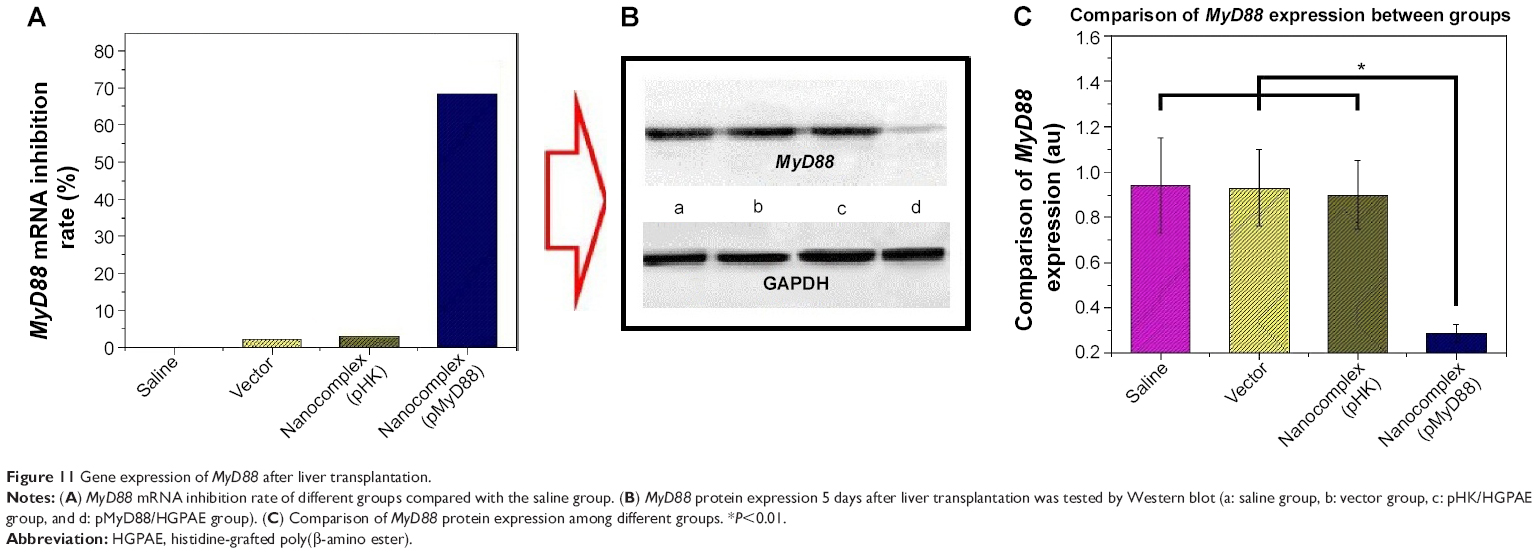 | Figure 11 Gene expression of MyD88 after liver transplantation. |
Expression of IL-2 and IFN-γ after liver transplantation
As shown in Figure 12, compared with the three control groups, there was an obvious reduction in IL-2 and IFN-γ concentrations in the pMyD88/HGPAE group (P<0.05). In contrast, there were no significant differences in the IL-2 and IFN-γ concentrations among the three control groups (P>0.05).
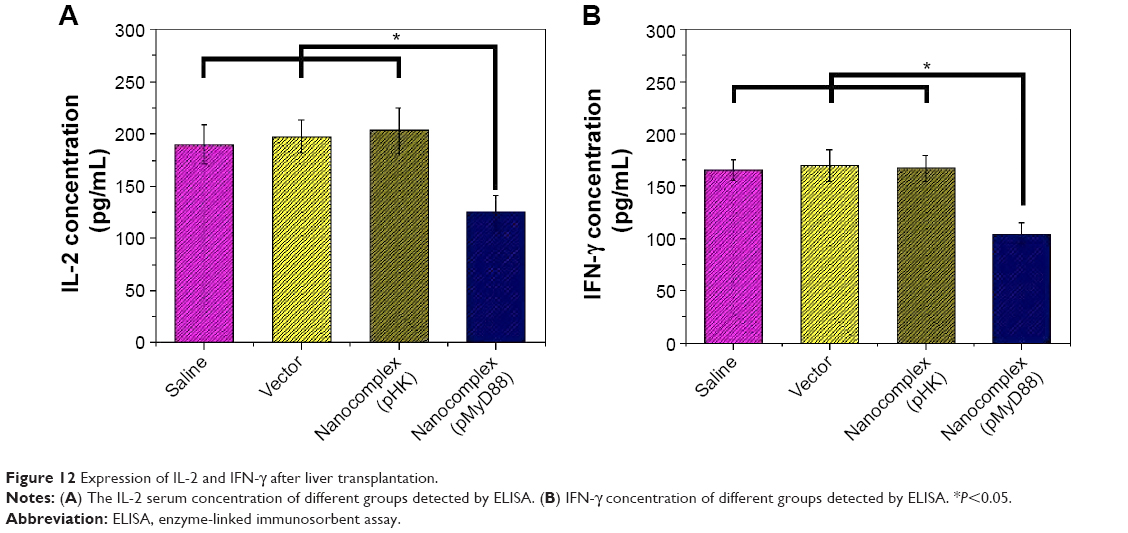 | Figure 12 Expression of IL-2 and IFN-γ after liver transplantation. |
Discussion
The availability of methods for efficient delivery of pDNA into cells in vivo limits the use of RNA interference technology both for research and clinical applications.13,14 In our study, we designed and synthesized a type of HGPAE nanovector, which was capable of complexing with an electronegative gene and promoting its release from the endosome and into the cytoplasm (Figure 13).15 Furthermore, we showed that at a weight ratio of HGPAE to DNA of 80:1, they formed electropositive spherical complexes, 50–150 nm in diameter, which were stable under electronegative conditions. The HGPAE nanovectors were shown to be efficient for transfection both in vitro and in vivo. In safety tests, both AST and ALT increased significantly in the saline group, HGPAE vector group, and pHK/HGPAE group on the first day after transfection, but returned to normal levels on the third day. We speculated that the increase in AST and ALT was due to surgical injury, such as anesthesia, incision, and injection via the portal vein. There were no significant differences in liver and renal function among the three groups, demonstrating the safety of HGPAE for use in vivo. Liver transfection in vivo is mainly performed either locally by injection through portal vein or systemically at sites such as the tail vein.16,17 As the systemic injection technique requires rapid injection of liquid at a rate equivalent to the circulation volume, it is mainly used in mice.18,19 The nanocomplex is absorbed by the reticuloendothelial system in vivo, so when injected systemically, part of the nanocomplex is absorbed by the spleen, while in delivery via the portal vein injection, the nanocomplex is absorbed by the liver.20,21 In our study, we achieved efficient transfection of the liver following delivery via the portal vein.
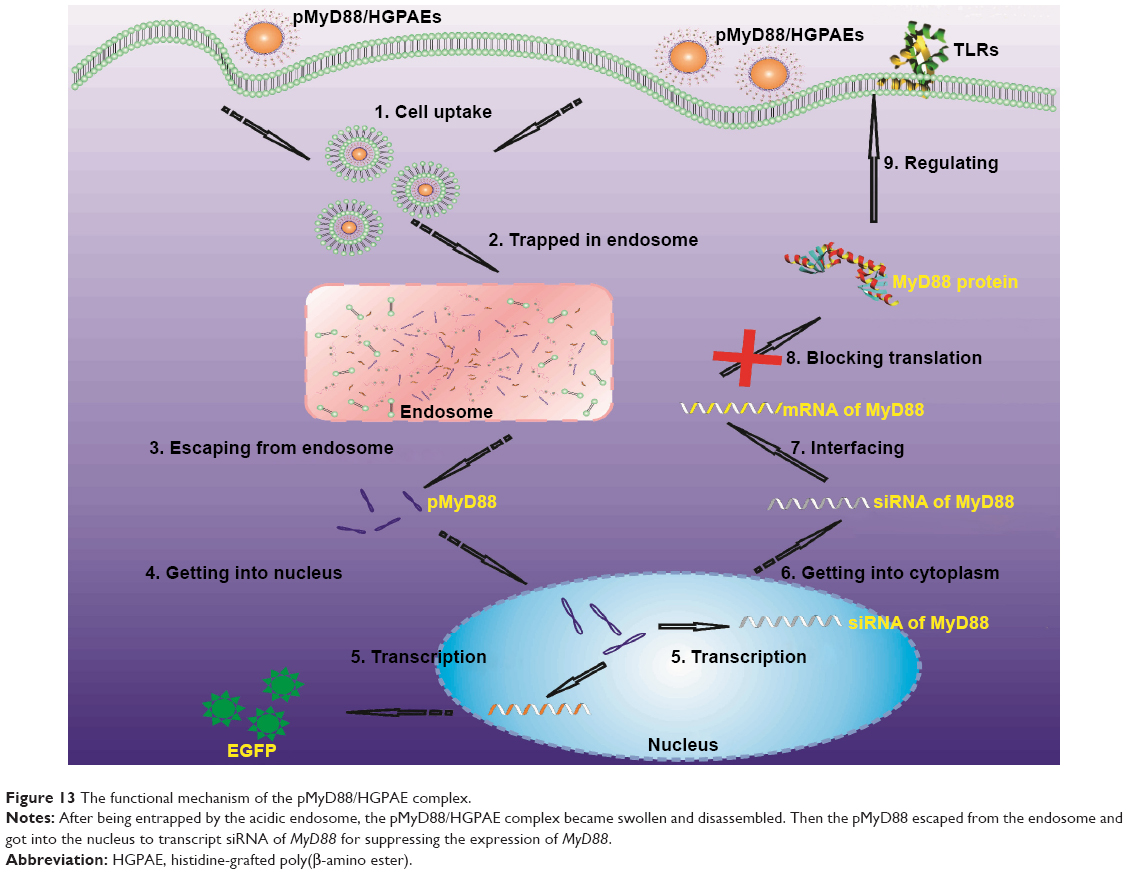 | Figure 13 The functional mechanism of the pMyD88/HGPAE complex. |
T-cells are necessary and sufficient for the rejection of almost all allogeneic tissues; therefore, transplantation research has focused on adaptive immunity in graft rejection.22,23 Only in recent years has the focus shifted to the role of innate immunity in promoting the adaptive response.24,25 TLRs are a family of molecules that activate innate immune responses and modulate adaptive immunity, and are actively involved in graft rejection in transplantation. TLRs play a critical role in the recognition of pathogen-associated molecular patterns as well as endogenous damage-associated molecular patterns.26 Following stimulation by signals, such as exogenous sources of ligands for TLRs during transplantation and endogenous molecules released during ischemia reperfusion injury, TLRs activate various downstream signals that induce the production of inflammatory cytokines and chemokines, which mediate innate immune attack on grafts, and also modulate alloantigen-specific adaptive immune rejection.27 All TLRs, except TLR3, are dependent on the adaptor molecule, MyD88. Hence, targeting the MyD88 could block TLR signaling to prevent graft injury and modulate immune rejection in organ transplantation. Based on these findings, several attempts to prevent graft rejection have been tested using TLR-deficient donors or recipients.10,25 For example, in a kidney transplantation trial, MyD88−/− allografts survived to 100 days when they were transplanted to MyD88−/− recipients. In contrast, wild-type (WT) allografts were rejected by WT mice with a mean graft survival of 40 days. When MyD88 was absent from recipients, five of five allografts survived to 100 days. When only the allograft was MyD88 deficient, survival was modestly prolonged compared with that of the WT allograft.28 To explore the role of MyD88-dependent TLR signaling in liver transplant rejection, we chose to knock down MyD88 expression in the donor liver using pMyD88/HGPAE, which was then used in MHC fully mismatched allogeneic liver transplantation. Our study demonstrated that knocking down MyD88 reduced graft rejection and prolonged the survival time of the recipient in a liver allograft model.
Graft rejection is initiated by recognition of donor graft antigens by the recipient’s T-cells; this recognition occurs via direct and indirect pathways. In the direct pathway, the recipient’s T-cells recognize intact allo-MHC molecules presented by donor APCs, while in the indirect pathway, recipient’s T-cells recognize processed alloantigen presented by the recipient’s APCs. TLRs are expressed primarily on macrophages and dendritic cells (DCs) and control the activation of these APCs. In DCs, TLR signaling triggers a maturation program that includes upregulation of MHC and costimulatory molecules and expression of proinflammatory cytokines, such as TNF-α, IL-1, and IL-6. This DC maturation significantly increases their ability to elicit the differentiation of naïve T-cells into mature effector and memory T-cells, which secrete cytokines such as IL-2 and IFN-γ to induce expression of class II MHC, adhesion molecules, and costimulatory molecules by endothelial cells.29,30 These molecules reinforce both the recognition pathways, thereby recruiting more T-cells and amplifying the rejection process. Therefore, blockade of donor TLR signaling by inhibition of MyD88 prevents maturation of the donor APCs. As a result, the direct pathway will be impaired, leading to reduced production of class II MHC and cytokines, with a consequent reduction in indirect pathway activity. Ultimately, the graft rejection will be alleviated. In support of this hypothesis, our data demonstrate that interruption of the TLR signaling in the donor liver by inhibition of MyD88 reduces cytokine production and alleviates graft rejection, leading to prolonged recipient survival.
Conclusion
In summary, pMyD88/HGPAE nanocomplexes, a new type of gene delivery system, were prepared successfully for inhibiting graft rejection and prolonging the survival time of liver transplant recipients in a high-responder rat liver transplantation model. We demonstrated that pMyD88/HGPAE nanovectors can be used to deliver and release the pMyD88 successfully both in vitro and in vivo. In liver transplantation, pMyD88/HGPAE nanocomplexes efficiently prevented MyD88 action with the therapeutic potential to prevent allograft rejection. This study demonstrates that pMyD88/HGPAE nanocomplexes with high transfection efficiency represent an alternative strategy for preventing allograft rejection in liver transplantation.
Acknowledgments
The authors gratefully acknowledge the National Natural Science Foundation of China (81102246, 51373117, and 51303126), Tianjin Natural Science Foundation (13JCZDJC33200 and 13JCQNJC11900), Doctoral Base Foundation of Educational Ministry of China (20120032110027), and Tianjin Municipal Education Commission Science Foundation (20090126).
Disclosure
The authors report no conflicts of interest in this work.
References
Benichou G, Tonsho M, Tocco G, Nadazdin O, Madsen JC. Innate immunity and resistance to tolerogenesis in allotransplantation. Front Immunol. 2012;3:73. | ||
Shin OS, Harris JB. Innate immunity and transplantation tolerance: the potential role of TLRs/NLRs in GVHD. Korean J Hematol. 2011;46(2):69–79. | ||
Miller DM, Rossini AA, Greiner DL. Role of innate immunity in transplantation tolerance. Crit Rev Immunol. 2008;28(5):403–439. | ||
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. | ||
Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111(10):1571–1578. | ||
Yamamoto M, Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010;2010:240365. | ||
Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17(1):1–14. | ||
Tesar BM, Zhang J, Li Q, Goldstein DR. Th1 immune responses to fully MHC mismatched allografts are diminished in the absence of MyD88, a toll-like receptor signal adaptor protein. Am J Transplant. 2004;4(9):1429–1439. | ||
Ro H, Lee EW, Hong JH, et al. Roles of islet Toll-like receptors in pig to mouse islet xenotransplantation. Cell Transplant. 2013;22(9):1709–1722. | ||
Ro H, Hong J, Kim BS, et al. Roles of Toll-like receptors in allogeneic islet transplantation. Transplantation. 2012;94(10):1005–1012. | ||
Kamada N, Calne RY. Orthotopic liver transplantation in the rat. Technique using cuff for portal vein anastomosis and biliary drainage. Transplantation. 1979;28(1):47–50. | ||
[No authors listed]. Banff schema for grading liver allograft rejection: an international consensus document. Hepatology. 1997;25(3):658–663. | ||
Borna H, Imani S, Iman M, Azimzadeh Jamalkandi S. Therapeutic face of RNAi: in vivo challenges. Expert Opin Biol Ther. 2015;15(2):269–285. | ||
Colombo S, Zeng X, Ragelle H, Foged C. Complexity in the therapeutic delivery of RNAi medicines: an analytical challenge. Expert Opin Drug Deliv. 2014;11(9):1481–1495. | ||
Cristiano RJ. Targeted, non-viral gene delivery for cancer gene therapy. Front Biosci. 1998;3:D1161–D1170. | ||
Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6(7):1258–1266. | ||
Budker V, Budker T, Zhang G, Subbotin V, Loomis A, Wolff JA. Hypothesis: naked plasmid DNA is taken up by cells in vivo by a receptor-mediated process. J Gene Med. 2000;2(2):76–88. | ||
Herrero MJ, Monleon D, Morales JM, Mata M, Serna E, Aliño SF. Analysis of metabolic and gene expression changes after hydrodynamic DNA injection into mouse liver. Biol Pharm Bull. 2011;34(1):167–172. | ||
Shashidharamurthy R, Machiah D, Bozeman EN, et al. Hydrodynamic delivery of plasmid DNA encoding human FcγR-Ig dimers blocks immune-complex mediated inflammation in mice. Gene Ther. 2012;19(9):877–885. | ||
Nakamura Y, Kominami A, Tsujimoto Y, et al. Actin and Vimentin proteins with N-terminal deletion detected in tumor-bearing rat livers induced by intraportal-vein injection of Ha-ras-transfected rat liver cells. Int J Cancer. 2009;124(11):2512–2519. | ||
Iimuro Y, Fujimoto J. Strategy of gene therapy for liver cirrohosis and hepatocellular carcinoma. J Hepatobiliary Pancreat Surg. 2003;10(1):45–47. | ||
Lo YC, Lee CF, Powell JD. Insight into the role of mTOR and metabolism in T cells reveals new potential approaches to preventing graft rejection. Curr Opin Organ Transplant. 2014;19(4):363–371. | ||
Mehrotra A, Leventhal J, Purroy C, Cravedi P. Monitoring T cell alloreactivity. Transplant Rev (Orlando). 2015;29(2):53–59. | ||
Ma Z, Zhang E, Yang D, Lu M. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell Mol Immunol. Epub 2014 Nov 24. | ||
Zhang X, Beduhn M, Zheng X, et al. Induction of alloimmune tolerance in heart transplantation through gene silencing of TLR adaptors. Am J Transplant. 2012;12(10):2675–2688. | ||
Leventhal JS, Schröppel B. Toll-like receptors in transplantation: sensing and reacting to injury. Kidney Int. 2012;81(9):826–832. | ||
Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291–295. | ||
Wu H, Noordmans GA, O’Brien MR, et al. Absence of MyD88 signaling induces donor-specific kidney allograft tolerance. J Am Soc Nephrol. 2012;23(10):1701–1716. | ||
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2(10):947–950. | ||
Ozato K, Tsujimura H, Tamura T. Toll-like receptor signaling and regulation of cytokine gene expression in the immune system. Biotechniques. 2002;Suppl:66–68, 70, 72. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.