")
Back to Journals » OncoTargets and Therapy » Volume 11
Inhibition of MUC1-C entering nuclear suppresses MYC expression and attenuates malignant growth in esophageal squamous cell carcinoma
Authors Xin Z, Xin G, Shi M, Song L, Wang Q , Jiang B , Liu X
Received 21 March 2018
Accepted for publication 1 May 2018
Published 19 July 2018 Volume 2018:11 Pages 4125—4136
DOI https://doi.org/10.2147/OTT.S168813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Zhongwei Xin,* Gongsun Xin,* Mo Shi, Liang Song, Qiang Wang, Bin Jiang, Xiangyan Liu
Department of Thoracic Surgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, People’s Republic of China
*These authors contributed equally to this work
Background: The mucin 1 (MUC1) heterodimeric protein (N-terminal subunit and C-terminal subunit) is aberrantly overexpressed in esophageal squamous cell carcinoma (ESCC) and has been linked to poor outcomes in this disease. The detailed mechanism(s), however, remains unclear. In this article, we investigate the effects of the MUC1 C-terminal transmembrane subunit (MUC1-C) through the inhibitor GO-201, which inhibits MUC1-C targeting to nuclear.
Patients and methods: The expression of MUC1-C and MYC in the ESCC samples and cell lines was detected by immunohistochemistry, immunofluorescence and western blotting. MYC mRNA level was determined by using quantitative real-time polymerase chain reaction. In addition, Cell Counting Kit-8, clonogenic assay, transwell assay and tumor xenograft in nude mice assay were utilized to determine the role of MUC1-C in proliferation, invasion and migration of ESCC cells.
Results: The level of MUC1-C in nuclear and MYC in whole cells in the ESCC tissue is significantly higher than that in the noncancerous tissue. Treatment of MUC1-C-overexpressing ESCC cells with GO-201 was associated with downregulation of MYC expression and induction of apoptosis. Besides, in vitro and in vivo assays have both shown that inhibiting MUC1-C targeting to the nucleus by the GO-201 significantly decreased the abilities of proliferation, invasion and migration in ESCC cells.
Conclusion: Our findings suggest that MUC1-C targeting to the nucleus plays an important role in suppressing the malignant growth of ESCC and indicate that MUC1-C is a potential target for the treatment of ESCC.
Keywords: MUC1 C-terminal subunit (MUC1-C), MYC, esophageal squamous cell carcinoma (ESCC), GO-201, malignant growth
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the most common digestive tumors worldwide.1 Surgery still remains to be the first choice of treatment for resecting ESCC. Even though there are different advanced methodologies in surgery, chemotherapy and nutritional aid therapies at present, the long-term ESCC survival rate, which is only approximately 30%–50%, has only been slightly improved in recent years.2,3 It is therefore a formidable task to find novel biological molecules related to the malignant behavior of the carcinoma and elucidate the underlying therapeutic targets for ESCC patients.
Mucin 1 (MUC1) is a transmembrane heterodimer protein, which has been aberrantly overexpressed in ESCC.4 Its overexpression is correlated with tumor growth, lymph node metastasis and resistance to apoptosis.5 MUC1 consists of an extracellular MUC1 N-terminal subunit (MUC1-N) and transmembrane MUC1 C-terminal transmembrane subunit (MUC1-C). These two subunits form a complex, which is one of the characteristics of the mucin family.6 MUC1-C includes an intrinsically disordered 72 amino acid cytoplasmic domain that is phosphorylated by diverse kinases and interacts with various effectors that have been linked to transformation.7 In addition MUC1-C is also mainly targeted to the nucleus, where it interacts with diverse activators and repressors of transcription.8 The MUC1-C cytoplasmic domain contains a CQC motif that is necessary for MUC1-C homodimerization and entering the nucleus.9,10 Accordingly, cell-penetrating peptides, such as GO-201, have been developed to target the MUC1-C CQC motif and block the formation of the homodimerization that inhibits the MUC1-C entering the nucleus.11
MYC functions as a DNA-binding transcription factor that activates a cellular program of genes contributing to the control of cell growth, metabolism, protein synthesis and survival.12,13 Dysregulation of MYC expression occurs in diverse human cancers14 and is sufficient to confer tumorigenesis in transgenic mouse models.15 MYC is also of utmost importance for tumor maintenance as evidenced by tumor regressions in response to MYC downregulation16 or treatment with an inhibitor of MYC heterodimerization.17
However, previous researches have paid little attention to the location of the MUC1. There are a few reports of MUC1 expression in the nucleus of ESCC and no available information on the cross-talk between MUC1 and MYC in the malignant behavior of the ESCC. In this study, we paid more attention to the location of the MUC1, and explored the function of MUC1-C entering nucleus in MYC expression and malignant growth of ESCC.
Patients and methods
Patients
Forty pairs of ESCC and paracancerous tissue (more than 5 cm from the margin of tumor) from patients who underwent esophagectomy in the Department of Thoracic Surgery at Shandong Provincial Hospital Affiliated to Shandong University were collected and collated from January 2015 to June 2016. The inclusion criteria of the patient were as follows:
- Squamous cell carcinoma of the middle thoracic esophagus diagnosed by postoperative pathology with staging I–III.
- No preoperative chemotherapy or radiotherapy.
- No contraindications to surgery.
All patients underwent esophagectomy with complete resection: no residual tumor cells were present on upper or lower cutting edge as verified by pathology, lateral margins with no residual focus to the naked eye. TNM staging was determined by the criteria established by the International Union Against Cancer in 2009. Written informed consent was obtained from all patients, and the study protocol was approved by the institutional ethics committee of Shandong Provincial Hospital Affiliated to Shandong University according to Guide for Chinese Ethics Review Committees.
Cell lines and culture
The human ESCC cell lines KYSE450 and TE-1 were purchased from the Cell Bank of the Shanghai Institute in People’s Republic of China. The cells were cultured in Roswell Park Memorial Institute 1640 enriched with 1% penicillin/streptomycin (Sigma-Aldrich Co., St Louis, MO, USA), 10% fetal bovine serum (Gibco, Waltham, MA, USA) and 1% L-glutamine. Cell culture plates were maintained in humidified incubators at 37°C in a 5% CO2 incubator.
Immunohistochemistry and immunofluorescence
The immunohistochemistry studies of MUC1-C and MYC were performed by the streptavidin peroxidase method. The formalin-fixed, paraffin-embedded tissues were cut into sections, which then were deparaffinized and incubated with hydrogen peroxide. Rabbit anti-MUC1-C/anti-MYC antibodies (Abcam, Cambridge, MA, USA) were diluted to 1:100 and incubated at 4°C overnight. The subsequent steps were followed according to the instructions of the secondary biotinylated antibody kit (Zhongshan Biotech, Guangzhou, People’s Republic of China). Stained slides were scored by two blinded, independent observers. Integrated optical density was measured in three random fields from each patient using Image-Pro Plus 6.0. Values were represented as mean ± SD. Immunofluorescence process was also performed to verify the expression of MUC1-C and MYC protein in frozen sections. Following fixation with 4% paraformaldehyde, the frozen sections were treated with 0.25% Triton and blocked in 10% goat serum. Rabbit anti-MUC1-C/anti-MYC antibodies (Abcam) were diluted to 1:150, and the goat anti-rabbit DyLight 594/488 antibody (Abcam) was diluted to 1:300 for 1 h followed by counter staining of DAPI for 5 min. The images were examined using a fluorescence microscope (Olympus BX51; Olympus, Tokyo, Japan).
Protein extraction and western blotting
Protein was extracted both from tissues and cells by using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA). The extracted proteins were electrophoresed on SDS polyacrylamide gels and transferred onto PVDF membrane filters (Millipore Corporation, Billerica, MA, USA). Membranes were blocked with non-fat dried milk and incubated overnight with antibodies (anti-MUC1-C, anti-MYC and anti-PCNA were obtained from Abcam; anti-β-actin was obtained from Boster Biological Technology Co. Ltd., CA, USA) in blocking buffer. After being washed, the membranes were incubated with secondary antibody conjugated with horseradish peroxidase anti-mouse/rabbit IgG (Boster). The bands were visualized by an enhanced chemiluminescence detection system (Amersham imager 600; General Electric, Fairfield, CT, USA).
Colony formation assays
Cells were seeded in 6-well plates for 24 h and then treated with GO-201 (NJpeptide, Nanjing, People’s Republic of China), CP-1 (NJpeptide) or pure culture media every 3 days. The plates were incubated at 37°C in a 5% CO2 incubator for 10 days. Then, colonies were stained with crystal violet, and those with >50 cells were scored as surviving colonies. The cloning efficiency was calculated by dividing the average number of colonies/dish by the number of plated cells. This experiment was performed in triplicates.
CCK-8 assay
Cell proliferation was detected by using Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan). The logarithmically growing cells were seeded in 96-well plates and incubated with GO-201, CP-1 or pure culture media, respectively, for 24, 48 and 72 h. At different time intervals, the cells were incubated with CCK-8 reagent for 1 h at 37°C. The absorbance of each well was measured using Thermo Scientific Varioskan Flash (Thermo Fisher Scientific, Vantaa, Finland). This experiment was performed in triplicates.
Wound-healing assay
Cells were treated with GO-201, CP-1 or pure culture media in plates separately until 95% confluence. A straight “wound” was made by scratching with a plastic pipette tip. Having cultured in FBS-free medium for 12 and 24 h, cell migration was analyzed by using light microscopy. The residual area of the scratch at the same position was measured by using ImageJ software. The migration rate was calculated by the following formula:
|

The area of initial wound.
Cell apoptosis detection
Flow cytometric assays were conducted using the Annexin V-FITC apoptosis detection kit I (BD Biosciences, San Jose, CA, USA), according to the manufacturer’s instructions. Cells from the three groups (treated with GO-201, or treated with CP-1, or left untreated) were harvested by trypsinization and resuspended in 1X binding buffer. After double staining with P-phycoerythrin (PE) and 7-Amino-actinomycin D (7-AAD), the samples were analyzed by flow cytometry and the data were analyzed by using FlowJo software.
Real-time PCR
After being treated with GO-201, CP-1 or left untreated, the whole-cell RNA was extracted using the RNAiso Plus (TaKaRa, Shiga Japan). Then quantitative polymerase chain reaction (PCR) was carried out with SYBR Green PCR kit according to the manufacturer’s instructions (TaKaRa). Amplifications were performed in ABI PRISM 7500 Sequence Detection System (Applied Biosystems). Relative transcript quantities were calculated by using the DDCt method with β-actin as the endogenous reference gene. PCR primers were as follows: MYC, 5′-GGAGGCTATTCTGCCCATTTG-3′ (forward primer) and 5′-CGAGGTCATAGTTCCTGTTGGTG-3′ (reverse primer); β-actin, 5′-AGAGCCTCGCCTTTGCCGATCC-3′ (forward primer) and 5′-ATACACCCGCTGCTCCGGGTC-3′ (reverse primer). This experiment was performed in triplicates.
Cell migration and invasion assay
For both migration and invasion assays, the cells treated with GO-201, CP-1 or left untreated were precultured in FBS-free medium for 24 h. The uncoated transwells were used in the migration assay, while Matrigel (1:4; BD Biosciences) was pre-coated to the upper surface of transwells in the invasion assay. Then, starved cells were seeded into the upper chamber of 24-well transwells (8 mm pore size polycarbonate membrane; Merck KGaA, Darmstadt, Germany) with FBS-free medium, and medium with FBS was added to the low chamber. The migration assay was allowed to incubate for 24 h, and the invasion assay was allowed to proceed for 48 h. Then, cells on the lower surface of membrane were fixed and stained. The cell numbers were calculated from three independent visual fields under light microscopy.
ESCC tumor xenograft treatment studies
Six-week-old female BALB/c nude mice were injected with 2×106 ESCC cells subcutaneously in the flank. When the tumors reached a size of ~150 mm3, the mice were pair matched into 3 groups of 5 mice each, and treated by 1) control vehicle, 2) 15 mg/kg GO-201 administered intraperitoneally (IP) each day or 3) 15 mg/kg CP-1 administered IP daily. We measured the size of tumors every 4 days and weighed the tumors after 20 days. Tumor volumes were calculated by using the formula V=L×W2/2, where L and W are the larger and smaller diameters, respectively. All procedures relating to animal handling, care and treatment were performed in strict accordance with the recommendations of regulations on the management of experimental animals, which were approved by State Council of the People’s Republic of China on October 31, 1988, and promulgated by Decree No. 2 of the State Science and Technology Commission on November 14, 1988. The protocol was approved by the ethics committee of Provincial Hospital affiliated to Shandong University.
Statistical analysis
The quantitative data were expressed as mean ± SD. p-values were generated using the Student’s t-test (2 groups) and one-way analysis of variance (ANOVA) (≥3 groups). Pearson correlation analysis was used for the correlation between MUC1-C and MYC in immunohistochemistry. The significant effects from ANOVA were further examined with least significant difference-t and Student-Newman-Keuls test. Values were considered significant if p < 0.05. All statistical procedures were performed using SPSS 20.0 (IBM Corp, Armonk, NY, USA).
Results
The level of MUC1-C and MYC in the ESCC tissues is significantly higher than that in the noncancerous tissues
Results of immunohistochemistry and immunofluorescence staining for the 40 sample pairs showed that MUC1-C was weakly expressed in the nucleus of normal esophageal epithelium, meanwhile little MYC was detectable (Figure 1A and C). On the contrary, MUC1-C and MYC were moderately or strongly expressed in most ESCC specimens (Figure 1A and C). Moreover, quantitative analysis of MUC1-C and MYC expression in all immunohistochemistry staining showed a significant correlation (p < 0.001, Figure 1B). To be consistent with these immunohistochemistry and immunofluorescence staining results, western blot analysis on tumor tissues and the matched normal tissues revealed that MUC1-C and MYC proteins were present at a considerably higher level in tumor tissues when compared with normal tissues (Figure 1D–G).
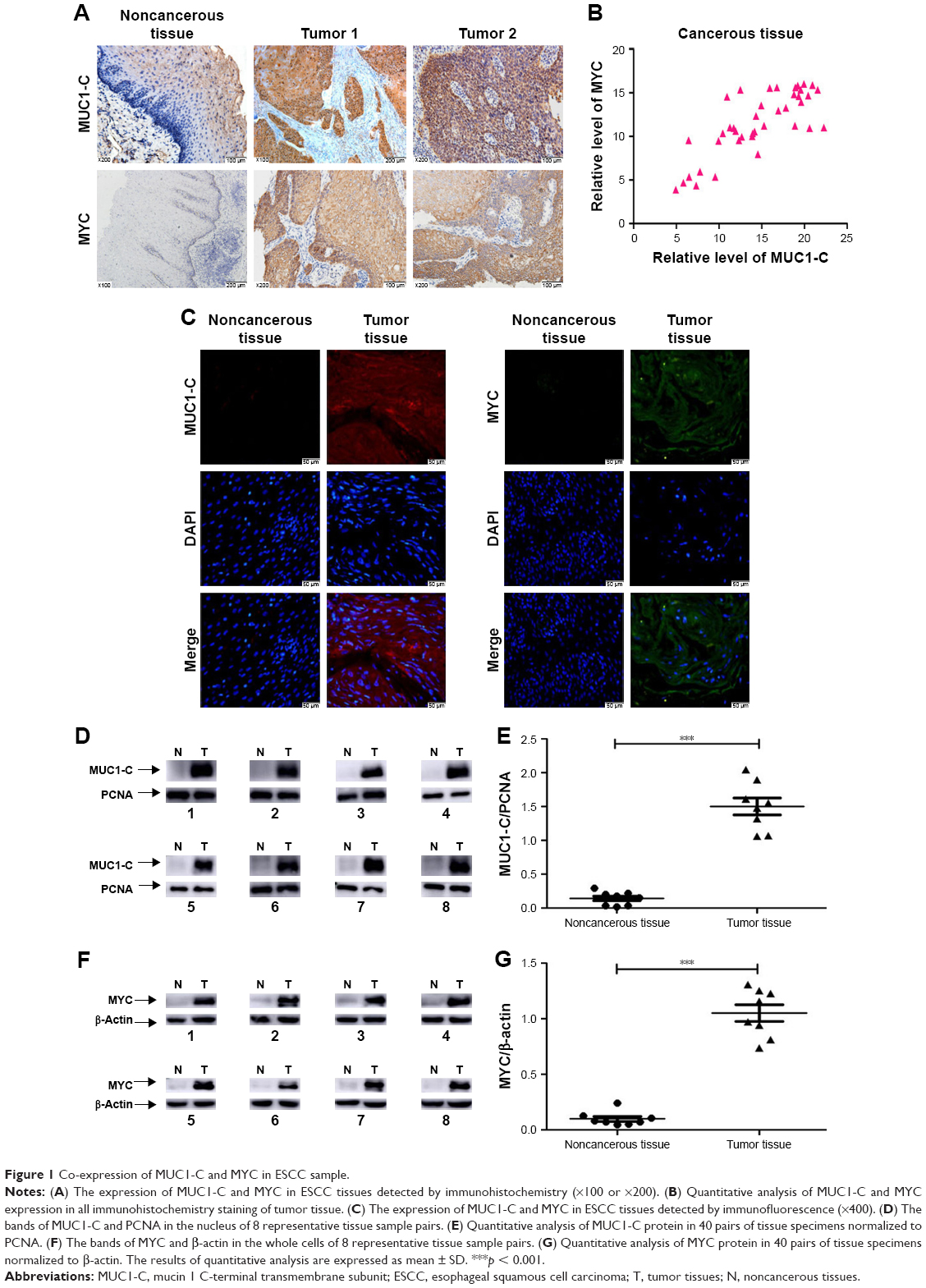 | Figure 1 Co-expression of MUC1-C and MYC in ESCC sample. |
Targeting MUC1-C inhibits MYC transcription and thereby downregulates the level of MYC in ESCC cells
The co-expression of MUC1-C in the nucleus and MYC in ESCC suggests a potential interaction of these two proteins, which has been studied by targeting MUC1-C. The MUC1-C cytoplasmic domain contains a CQC motif that is necessary for MUC1-C homodimerization and function9–11 (Figure 2A). Accordingly, the cell penetrating peptide, designated as GO-201, was developed to block the MUC1-C CQC motif in the ESCC and other carcinoma cells7,11 (Figure 2A). Notably, treatment of KYSE450 and TE-1 cells with GO-201, but not the control peptide CP-1, was obviously inhibiting the entry of MUC1-C into the nucleus, which is confirmed by western blot analysis (Figure 2B and C). However, there was no significant difference in the level of MUC1-C in the cytoplasm (Figure 2D and E). Meanwhile, the downregulation of MYC (Figure 2F and G) was also detected by western blot analysis. The same result is further approved by the quantitative real-time PCR. Targeting MUC1-C by using GO-201 in the cells resulted in downregulation of MYC mRNA levels (Figure 2H); thereby, it has obviously suppressed the MYC transcription.
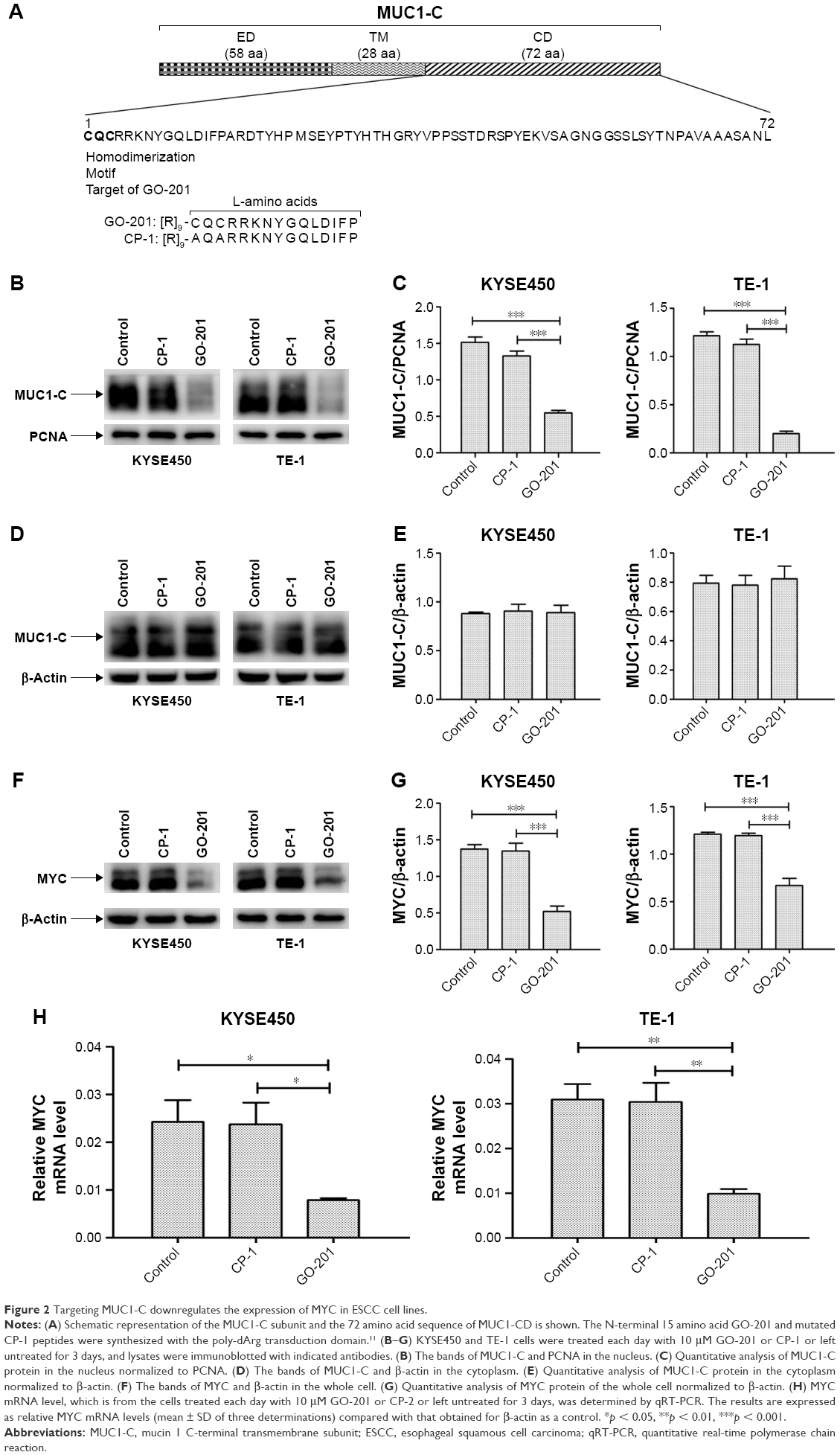 | Figure 2 Targeting MUC1-C downregulates the expression of MYC in ESCC cell lines. |
Targeting MUC1-C decreases ESCC cell lines’ proliferation ability
To determine if inhibiting the MUC1-C into the nucleus could suppress the proliferation of ESCC cells, clonogenic assay and CCK-8 were conducted. The colony formation assays showed that treatment of KYSE450 and TE-1 cells with GO-201 was apparently suppressed in clonogenic survival compared to that obtained with CP-1 or left untreated (Figure 3A and B). The similar result was obtained from the CCK-8 assay. The OD values (450 nm) of the KYSE450 and TE-1 cells with GO-201 showed significant decrease at 24, 48 and 72 h, compared with those cells with CP-1 or without penetrating peptide (Figure 3C).
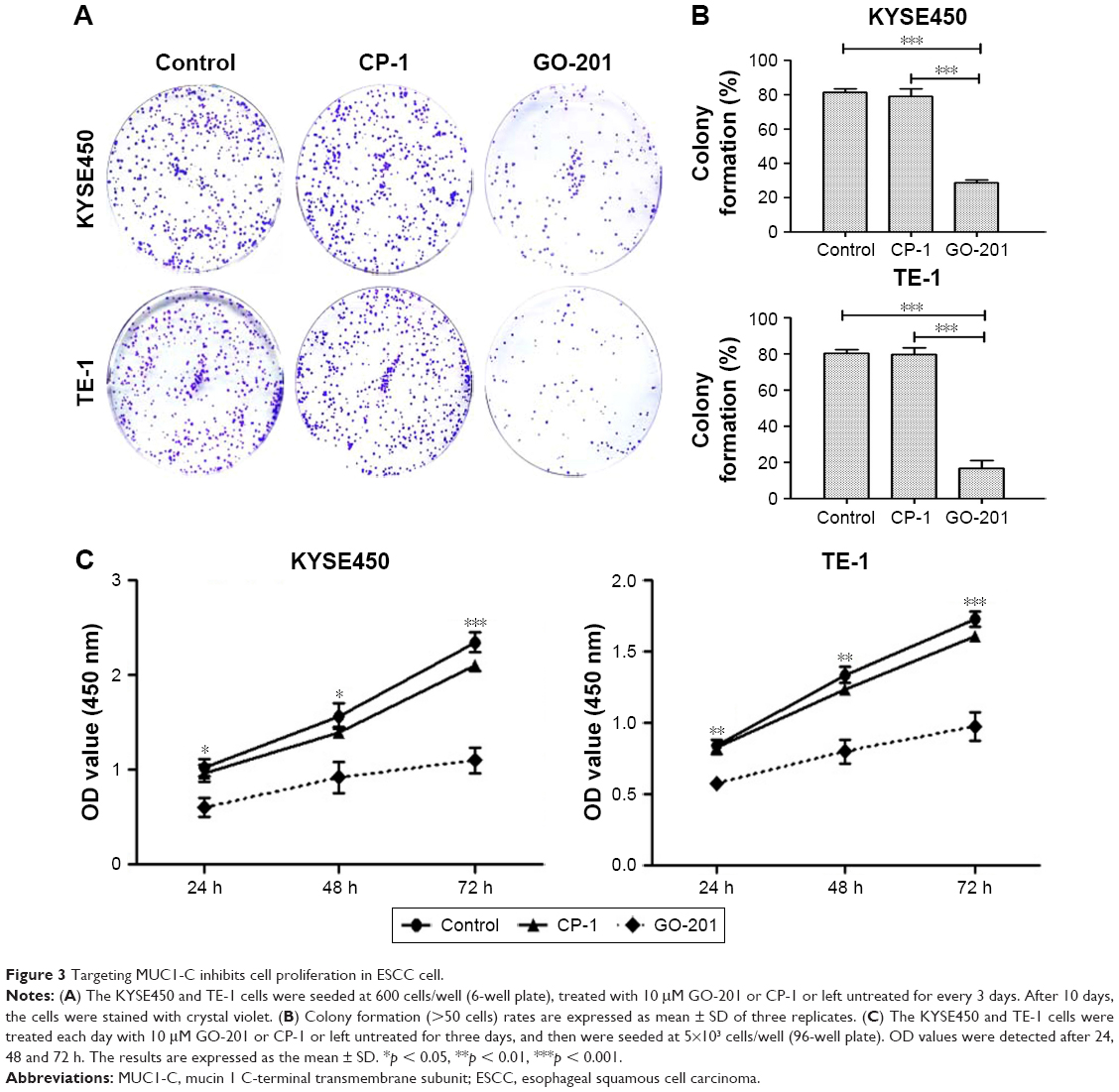 | Figure 3 Targeting MUC1-C inhibits cell proliferation in ESCC cell. |
Targeting MUC1-C inhibits the migration and invasion of ESCC cell lines
To evaluate the migration and invasion abilities of ESCC cells, which are the signs of a malignant degree, the wound-healing and transwell assays were performed. With respect to wound-healing assay, the closure of the wounds was apparently slower in ESCC cells treated by GO-201, in which most wounds remained open, whereas most space along the migrating fronts fused in the control group (Figure 4A and B). In the transwell assay, compared to the control group, the number of ESCC cells treated with GO-201 that penetrated the transwell membrane was significantly lower (Figure 4C–F). These results indicated that inhibiting MUC1-C into the nucleus by GO-201 could remarkably suppress the migration and invasion of ESCC cells.
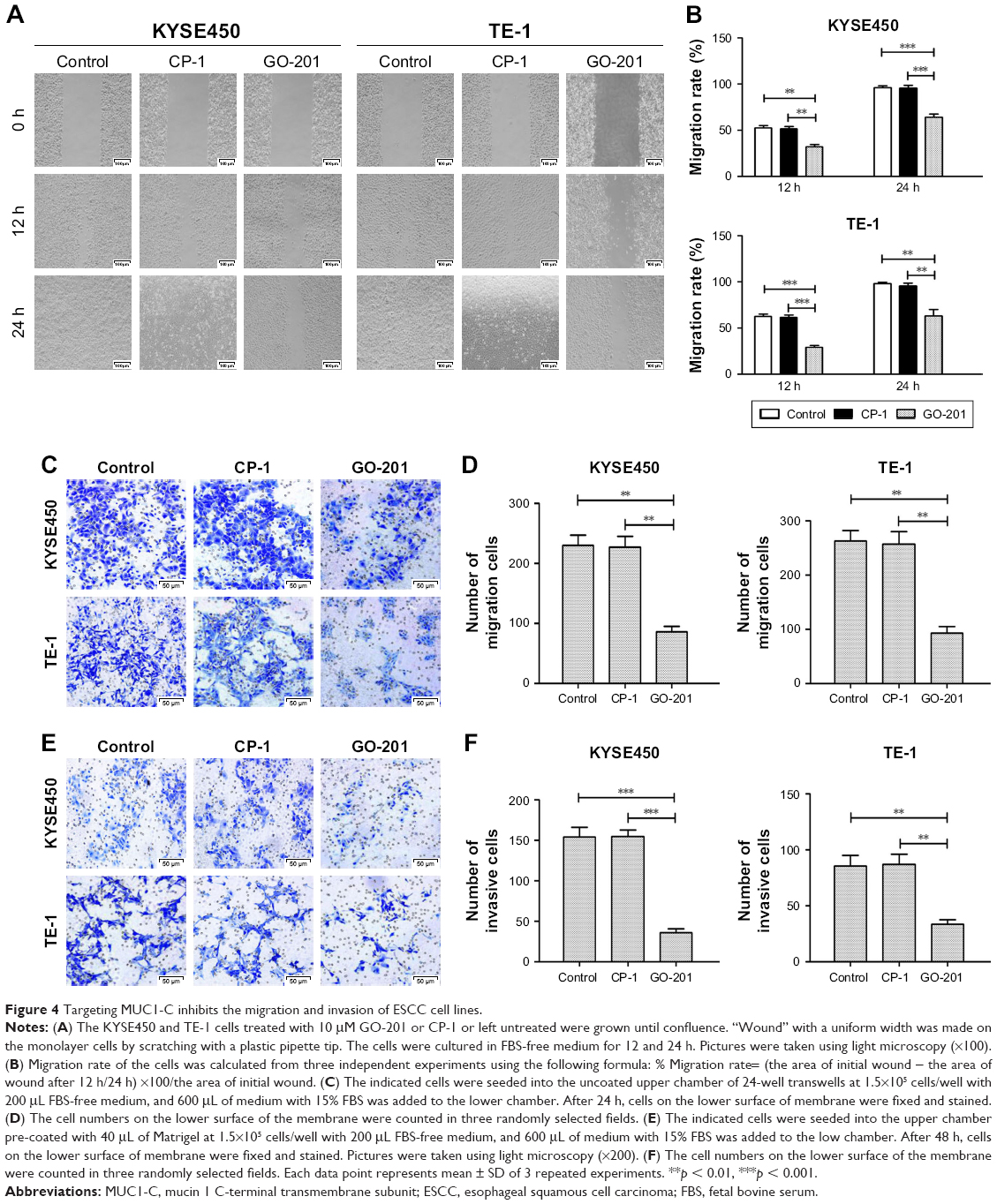 | Figure 4 Targeting MUC1-C inhibits the migration and invasion of ESCC cell lines. |
Targeting MUC1-C promotes apoptosis in ESCC cells
In order to determine whether apoptosis resulted in proliferation inhibition, a flow cytometric apoptosis assay was performed. The data showed that in contrast to control groups the number of apoptotic KYSE450 and TE-1 cells treated with GO-201 for 4 days increased significantly (Figure 5A and B). Based on these results, we concluded that inhibiting MUC1-C into the nucleus promotes apoptosis of KYSE450 and TE-1 cells.
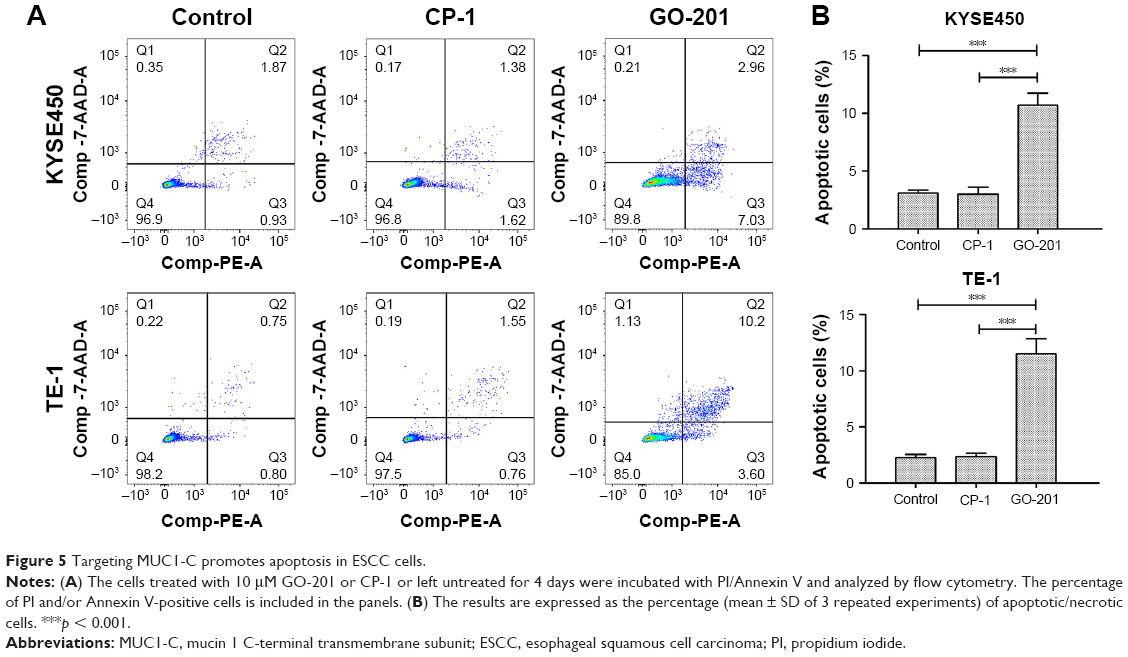 | Figure 5 Targeting MUC1-C promotes apoptosis in ESCC cells. |
GO-201 could inhibit tumor growth in mice
The effects of GO-201 on inhibiting tumor growth in vivo were also evaluated by zoopery. Compared to the control group, the average volumes of tumors in the mice treated by GO-201 were significantly decreased (Figure 6A and B). That is to say, the growth velocity of experimental group was obviously decreased (Figure 6C). In contrast to the activity of GO-201, treatment with CP-1 had no effect on ESCC tumor growth (Figure 6A–C). Importantly, GO-201 was not associated with loss of body weight or other apparent toxicities. The results illustrate that GO-201 could inhibit tumor growth in vivo.
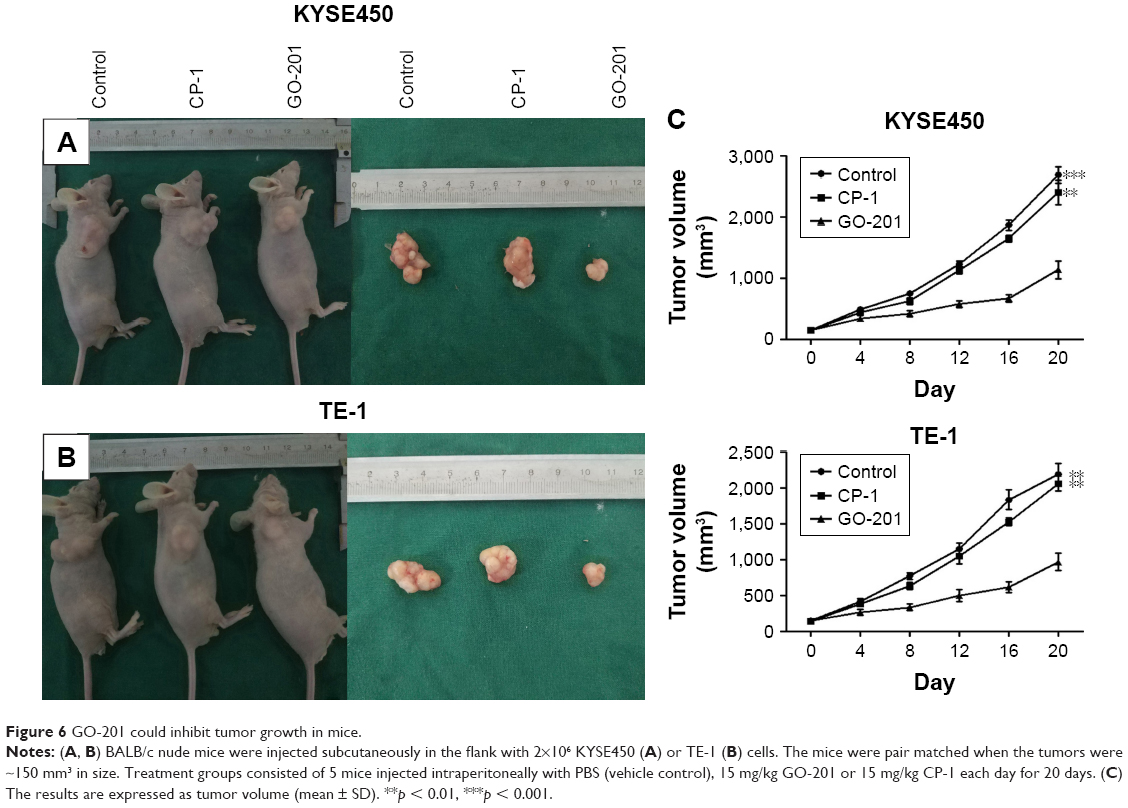 | Figure 6 GO-201 could inhibit tumor growth in mice. |
Discussion
ESCC is one of the most common fatal diseases and it has a high incidence in some developing countries.1,18,19 Despite of the significant improvements in therapeutic regimens, surgical resection is still considered to be the first-line treatment for early stage and localized ESCC.20 However, ESCC is often resistant to conventional therapeutic agents.3 In addition, inhibitors of ESCC have been clinically ineffective up-to-date. Hence, some biological targets that may be helpful in treatments are becoming the hot spot of improving the prognosis of patients with ESCC.
The aberrant overexpression of MUC1 has been detected in ESCC and is correlated with poor prognosis.21,22 Our previous work showed that silencing MUC1 in ESCC cells is associated with decreases in self-renewal and tumorigenicity;23 however, the detailed mechanism(s) responsible for these responses remains unclear. MUC1 consists of two sub-units: MUC1-N and MUC1-C that functions as an oncoprotein.6,7 The MUC1-C 72-amino-acid cytoplasmic domain is an intrinsically disordered structure,24 which has the plasticity to interact with multiple kinases and effectors that have been linked to transformation.6,7 Therefore, the focus of this experiment was on the MUC1-C.
MUC1-C is overexpressed in most ESCC, which has been confirmed in our study. The MUC1-C transforming function is dependent on homodimer formation, which is mediated by a CQC motif present in the cytoplasmic domain of MUC1-C.9 GO-201 is an inhibitor of MUC1-C, blocks MUC1-C oligomerization and inhibits the function of MUC1-C as an intracellular signaling protein.11 This inhibitor has been reported to be effective in inhibiting cell proliferation in vitro and in xenografts models of breast,11 prostate,25 lung26 and certain hematologic malignancy.27 Here, we show that treatment of MUC1-C-positive ESCC cells with GO-201 is associated with growth inhibition. Meanwhile, we further suppose that the inhibitory effects of GO-201 may relate to the level of MUC1-C in the nucleus. The specificity of GO-201 on MUC1-C-positive ESCC cells is significant, because GO-201 binds to the CQC motif of MUC1-C and directly blocks MUC1-C function. This is where we can control the variables properly. On the contrary, the control peptide CP-1, in which the CQC motif has been changed to an AQA motif,11 does not bind to MUC1-C and therefore has exerted no effect on the growth of MUC1-positive ESCC cells.
In the study, it was also found that MYC was overexpressed in ESCC cells. Furthermore, the correlation between the two is obvious in the immunohistochemistry staining. Therefore, the present studies provide new insights on the function of MUC1-C on ESCC cells by demonstrating that targeting MUC1-C is associated with downregulation of MYC expression. MYC is of functional importance in orchestrating transcriptional pathways that regulate cell cycle progression, metabolism, survival and stemness.12,28,29 Therefore, the effects of targeting MUC1-C on self-renewal of ESCC cells can be attributable, at least in partial, to suppression of MYC signaling. Notably, we have used GO-201 inhibitor for targeting MUC1-C. Under these experimental conditions, ESCC cells respond with MYC downregulation by providing convincing support for a MUC1-C/MYC pathway.
What is more, treatment of ESCC tumors in nude mice responded to GO-201 treatment with an apparent slowing of growth. At 20 days of GO-201 treatment, the speed of ESCC tumor growth was reduced and the tumors volume was significantly smaller. On the contrary, the control group, with CP-1, had no obvious inhibitory effects. These findings indicate that inhibition of MUC1-C entering nucleus is effective in inhibiting growth and survival of ESCC cancer cells in xenograft models. What is more, we found that the inhibitory effect of GO-201 was not associated with loss of body weight or other apparent toxicities, which is crucial for whether it can be applied clinically. We suppose that there may be GO-201 dose−response effect with regression of tumors at higher doses administered for longer periods (20 days). The effectiveness of GO-201 in ESCC tumor xenografts may be linked to MUC1-C entering nucleus and MYC expression in human ESCC cancers. Therefore, the MUC1-C may become a potential target and GO-201 may be useful for the ESCC cancer patients who overexpress MUC1-C.
Although the works have proved that targeting MUC1-C, which is associated with downregulation of MYC expression, could attenuate the biological characteristics of ESCC cells, the more specific signaling pathway is still unclear. Importantly, MUC1-C is intrinsically disordered and thereby has the capacity to serve as a substrate for multiple kinases and as a binding partner for diverse effectors of gene transcription. For instance, the MUC1-C cytoplasmic domain is phosphorylated by GSK3b and binds directly to β-catenin, linking MUC1-C to the WNT path-way.30 Moreover, MYC is regulated by a whole host of transcription factors, including TCF that is downstream of the WNT pathway. TCF is a transcription factor that plays a role in deregulated MYC expression downstream of the WNT pathway, such as with the loss of the tumor suppressor APC that results in constitutive nuclear localization of the TCF co-factor β-catenin.12 Therefore, it is possible that MUC1-C drives MYC gene transcription by activation of the WNT/β-catenin pathway, and hence targeting MUC1-C with GO-201 could also inhibit these pathways in ESCC cancer cells.
Conclusion
Based on the aforesaid experiments and results, a conclusion can be reached that targeting MUC1-C, which is associated with downregulation of MYC expression, can block the cellular proliferation and survival of ESCC cells. In addition targeting MUC1-C function by GO-201 can lead to tumor regression in esophageal xenografts models. Hence, MUC1-C may become a potential target for treating ESCC cancer, and MUC1-C inhibitor may become a new agent for treating ESCC patients.
Acknowledgments
This work was supported by grants from Natural Science Foundation of Shandong Province (2014ZRB14125). The authors appreciate the help from the Department of Thoracic Surgery, Pathology Department and Central Laboratory of Shandong Provincial Hospital affiliated to Shandong University.
Author contributions
ZWX and GSX participated in the design of the study, performed the experiments and drafted the paper. QW and BJ performed the experiments and analyzed data. XYL conceived the study, participated in its design and coordination and helped to draft the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All the authors read and approved the final paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Mizoguchi K, Ishiguro H, Kimura M, et al. Induction of apoptosis by eicosapentaenoic acid in esophageal squamous cell carcinoma. Anticancer Res. 2014;34(12):7145–7149. | ||
Mariette C, Piessen G, Triboulet JP. Therapeutic strategies in oesophageal carcinoma: role of surgery and other modalities. Lancet Oncol. 2007;8(6):545–553. | ||
Ye Q, Yan Z, Liao X, et al. MUC1 induces metastasis in esophageal squamous cell carcinoma by upregulating matrix metalloproteinase 13. Lab Invest. 2011;91(5):778–787. | ||
Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. 2014;20(6):332–342. | ||
Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9(12):874–885. | ||
Kufe DW. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2012;32(9):1073–1081. | ||
Rajabi H, Kufe D. MUC1-C oncoprotein integrates a program of EMT, epigenetic reprogramming and immune evasion in human carcinomas. Biochim Biophys Acta. 2017;1868(1):117–122. | ||
Leng Y, Cao C, Ren J, et al. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J Biol Chem. 2007;282(27):19321–19330. | ||
Raina D, Ahmad R, Rajabi H, Panchamoorthy G, Kharbanda S, Kufe D. Targeting cysteine-mediated dimerization of the MUC1-C oncoprotein in human cancer cells. Int J Oncol. 2012;40(5):1643–1649. | ||
Raina D, Ahmad R, Joshi MD, et al. Direct targeting of the mucin 1 oncoprotein blocks survival and tumorigenicity of human breast carcinoma cells. Cancer Res. 2009;69(12):5133–5141. | ||
Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. | ||
Li B, Simon MC. Molecular pathways: targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin Cancer Res. 2013;19(21):5835–5841. | ||
Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. | ||
Leder A, Pattengale PK, Kuo A, Stewart TA, Leder P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: multiple neoplasms and normal development. Cell. 1986;45(4):485–495. | ||
Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16(4):313–317. | ||
Soucek L, Whitfield J, Martins CP, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–683. | ||
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. | ||
Napier KJ, Scheerer M, Misra S. Esophageal cancer: a review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6(5):112–120. | ||
Stahl M, Mariette C, Haustermans K, Cervantes A, Arnold D; ESMO Guidelines Working Group. Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi51–vi56. | ||
Li J, Xiang S, Zhang Q, et al. Combination of curcumin and bicalutamide enhanced the growth inhibition of androgen-independent prostate cancer cells through SAPK/JNK and MEK/ERK1/2-mediated targeting NF-κB/p65 and MUC1-C. J Exp Clin Cancer Res. 2015;34(1):46. | ||
Sagara M, Yonezawa S, Nagata K, et al. Expression of mucin 1 (MUC1) in esophageal squamous-cell carcinoma: its relationship with prognosis. Int J Cancer. 1999;84(3):251–257. | ||
Shi M, Chen D, Yang D, Liu XY. CCL21-CCR7 promotes the lymph node metastasis of esophageal squamous cell carcinoma by up-regulating MUC1. J Exp Clin Cancer Res. 2015;34(1):149. | ||
Raina D, Agarwal P, Lee J, et al. Characterization of the MUC1-C cytoplasmic domain as a cancer target. PLoS One. 2015;10(8):e0135156. | ||
Joshi MD, Ahmad R, Yin L, et al. MUC1 oncoprotein is a druggable target in human prostate cancer cells. Mol Cancer Ther. 2009;8(11):3056–3065. | ||
Raina D, Kosugi M, Ahmad R, et al. Dependence on the MUC1-C oncoprotein in non-small cell lung cancer cells. Mol Cancer Ther. 2011;10(5):806–816. | ||
Yin L, Ahmad R, Kosugi M, et al. Terminal differentiation of chronic myelogenous leukemia cells is induced by targeting of the MUC1-C oncoprotein. Cancer Biol Ther. 2014;10(5):483–491. | ||
Bretones G, Delgado MD, León J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849(5):506–516. | ||
Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4(1):a014357. | ||
Yin L, Tagde A, Gali R, et al. MUC1-C is a target in lenalidomide resistant multiple myeloma. Br J Haematol. 2017;178(6):914–926. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.