")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Inhibition of miR-448-3p Attenuates Cerebral Ischemic Injury by Upregulating Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)
Authors Xu M, Xiang D, Wang W, Chen L, Lu W, Cheng F
Received 10 March 2021
Accepted for publication 23 September 2021
Published 19 October 2021 Volume 2021:17 Pages 3147—3158
DOI https://doi.org/10.2147/NDT.S310495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jun Chen
Min Xu,1,* Dingchao Xiang,2,* Wenhua Wang,1 Long Chen,1 Wei Lu,1 Feng Cheng3
1Department of Neurosurgery, Kunshan Hospital of Traditional Chinese Medicine, Kunshan Affiliated Hospital of Nanjing University of Chinese Medicine, Kunshan City, Jiangsu Province, 215300, People’s Republic of China; 2Department of Neurosurgery, Wuxi clinical medical school of Anhui Medical University, 904th Hospital of PLA(Taihu Hospital of Wuxi), Wuxi, 214000, People’s Republic of China; 3Department of Neurosurgery, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, 215300, Jiangsu Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Cheng
Department of Neurosurgery, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, 215300, Jiangsu Province, People’s Republic of China
Tel +86-13913208695
Email [email protected]
Background: Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key regulator responsible for oxidative stress in brain injury. This study aimed to investigate the potential mechanism of miR-448-3p and Nrf2 in cerebral ischemia/reperfusion (I/R) injury.
Methods: In vitro and in vivo cerebral I/R injury models were constructed, and Nrf2 expression levels were detected by qRT-PCR and Western blot. The potential miRNAs for Nrf2 were predicted by bioinformatic analysis. The binding interaction between miR-448-3p and Nrf2 was determined by luciferase reporter assay. The effects of miR-448-3p on neurological deficit, infarct volume, and brain water content in mice were tested. The effects of miR-448-3p on oxidative stress indicators (SOD activity, MDA content, and ROS production) were detected by commercial assay kits. The levels of HO-1 and cleaved caspase-3 were evaluated by Western blot. Cell viability was evaluated by MTT assay, and cell apoptosis was evaluated by TUNEL staining and flow cytometry.
Results: Nrf2 was significantly downregulated and miR-448-3p was upregulated in cerebral I/R injury both in vivo and in vitro. MiR-448-3p downregulation efficiently attenuated brain injury and reduced oxidative stress and apoptosis. MiR-448-3p was identified to act as ceRNA of Nrf2 and negatively regulated Nrf2 expression, which was consistent with the animal studies. In addition, Nrf2 silencing obviously attenuated the neuroprotective effects of miR-448-3p inhibitor in vitro.
Conclusion: MiR-448-3p participated in the regulation of cerebral I/R injury via inhibiting Nrf2.
Keywords: ischemia/reperfusion, miR-448-3p, Nrf2, oxidative stress, apoptosis
Introduction
Stroke is one of the major leading causes of disability and death in the world.1 Cerebral ischemia-reperfusion (I/R) injury is one type of brain injuries during treating ischemic stroke by vascular recanalization.2,3 One of the most crucial components of cerebral I/R injury is reactive oxygen species (ROS)-induced tissue damages and apoptosis.4 Therefore, better understanding the regulation involved in oxidative injury contributes to identifying potential therapeutic targets.
Increasing evidences confirmed the important roles of oxidative stress in the pathogenesis of cerebral I/R injury.5 Wu et al found that ROS and malondialdehyde (MDA) were dramatically elevated in brain tissues of mice subjected to middle cerebral artery occlusion (MCAO), and the activity of antioxidants, including SOD, was significantly reduced.6 Nrf2 has been identified to be a key regulator in response to antioxidant stress.7 Previous studies demonstrated that Nrf2 promotes the activity of antioxidants to reduce ROS production in brain and attenuates cerebral I/R injury.8 However, the regulatory mechanisms of Nrf2 in cerebral I/R injury have not been well elucidated.
MicroRNAs (miRNAs) are emerged as a class of small endogenous molecules with approximately 22 nucleotides in length.9 It has been reported that miRNAs function as competing endogenous RNAs (ceRNAs) of mRNAs to regulate a series of pathogenic processes.10 Along with the development of ischemic stroke studies, more and more miRNAs involved in cerebral I/R injury have been identified. For example, miR-132 attenuates cerebral injury via protecting blood–brain barrier disruption.11 In addition, many miRNAs participate in cerebral I/R injury, including miR-34b,12 miR-124-5p,13 miR-19a-3p,14 and miR-7-5p.15 Despite identification of many potential miRNA biomarkers in cerebral I/R injury, more effective miRNA indicators are still urgent.
In this study, we constructed I/R injury models both in vitro and in vivo and confirmed Nrf2 downregulation during I/R injury. Moreover, we identified that miR-448-3p was an upstream miRNA of Nrf2, and this axis closely participated in the regulation of oxidative stress injury and apoptosis during I/R injury. Our study demonstrated that miR-448-3p inhibition efficiently attenuated cerebral I/R injury by regulating Nrf2, suggesting that miR-448-3p might be a potential diagnostic and therapeutic target for cerebral I/R injury.
Materials and Methods
Animal Model
C57BL/6 J mice (adult, male, weighing 22–25 g) were maintained in standard environment. These mice were randomly divided into four groups with 6 mice per group, namely sham group, I/R group, miR-448-3p inhibitor group, and NC inhibitor group. 100 nM miR-448-3p inhibitor and 100 nM NC inhibitor were injected into the left lateral ventricle of mice (depth: 3.0 mm dorsal) through intracerebroventricular injection into mice in the miR-448-3p inhibitor group and in the NC inhibitor group, respectively. At 24 h after injection, all mice except those in the shame group were subjected to transient cerebral artery occlusion (tMCAO) to construct the cerebral ischemia model as previously described.16 Briefly, mice were subjected to middle cerebral artery occlusion (MCAO) for 2 h under anesthesia. Then, the right internal carotids, internal carotid arteries and common carotid artery (CCA) were separated. Next, the right external cerebral artery (CA) and the CCA were ligated to the distal and proximal ends, and a piece of monofilament (0.26-mm, round) was inserted from the external CA to internal CA until it reached the middle CA to block the blood supply of the middle CA, leading to occlusion of the middle CA. After 2 h of ischemia, the filaments were retracted a few inches to complete reperfusion. Mice in the sham group were subjected to the same operation without using monofilaments. At 24 h after operation, all mice were anesthetized with 3% sodium pentobarbital (30 mg/kg body weight, Sigma, USA) by intraperitoneal injection and sacrificed by cervical spine dislocation. During the operation, the body temperature of all rats was maintained at 37.0°C. Neurological deficits were evaluated as previously reported.17 All animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals and approved by the Affiliated Kunshan Hospital of Jiangsu University.
Infarct Volume
The brain tissues were cut into 1–2 mm coronal sections, and then incubated with 2% 2, 3,5-triphenyltetrazolium chloride (TTC; Sigma) for 30 min at 37°C. TTC stained sections were photographed using a digital camera successively. The infarct volume was calculated as 100% × (infarcted area/total brain area).
Brain Water Content
After 24 h of reperfusion, infarct brain hemispheres were weighted using an electronic scale and presented as wet weight. After drying overnight at 105°C, dry weight was measured. The brain water content was calculated as 100% × (wet weight-dry weight)/wet weight.
Hematoxylin and Eosin (H&E) Staining
The brain tissues were embedded in paraffin, sliced into 4 μm serial sections, and subjected to hematoxylin and eosin (H&E) staining as previously described.18 Histopathologic changes in mouse hippocampal CA1 or cerebral cortex regions were observed using an optical microscope (DM2500; Leica Microsystems, Germany).
Oxidative Stress Measurement
ROS production in brain tissues and cells was detected by Reactive Oxygen Species Assay Kits (Beyotime, Shanghai, China). Malondialdehyde (MDA) and superoxide dismutase (SOD) levels in brain tissues and cells were detected using commercial assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
TUNEL Staining
Brain tissue sections or cultured cells were fixed with paraformaldehyde for 10 min. According to commercially available TUNEL assay kit (Beyotime, Shanghai, China), the sections were incubated with 50 μL of TUNEL reaction buffer for 50 min. Afterwards, the section was incubated with diaminobenzidine (DAB) for 3 min, photographed using an epifluorescence microscope at ×400 magnification (Nikon Eclipse 80i).
Cell Culture and OGD/R Model
The mouse neuronal cell line HT22 was purchased from ATCC and cultured in DMEM media containing 10% FBS in an incubator with 5% CO2 at 37°C. To mimic I/R injury, HT22 cells were transferred into glucose-free DMEM medium and maintained in hypoxic incubator chamber (95% N2 and 5% CO2) for 2 h and recultured in glucose-containing DMEM and maintained in normoxic conditions for 24 h for re-oxygenation. Control cells were cultured in glucose-containing DMEM under normoxic conditions.
Cell Transfection
MiR-448-3p mimics, NC mimics, miR-448-3p inhibitor, and NC inhibitor were synthesized by GenePharma (Shanghai, China). Specific siRNA-Nrf2 (si-Nrf2) (sc-37049) and siRNA-NC (si-NC) (sc-37007) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cell transfection was performed by using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, transfection efficiency was confirmed by qRT-PCR and cells were subjected to OGD/R treatment.
MTT Assay
Cell viability was detected using MTT assay. In brief, 1 × 104 HT22 cells were seeded into each well of 96-well plates and cultured for 24 h. Then, cells were incubated in media with 50 μL of MTT (5 mg/mL) for another 4 h at 37°C. The absorbance at 570 nm was detected using a microplate reader.
QRT-PCR
Total RNA was extracted from brain tissues, plasma, and cultured cells using Trizol reagent. After reverse transcription, real-time PCR was performed on the ABI 7300 Real-Time PCR System with β-actin and U6 as the internal reference for mRNAs and miRNAs, respectively. The expression levels of targets were calculated using the 2−ΔΔCt method. The primers used for qRT-PCR were listed in Table 1.
 |
Table 1 Sequences of Primers Used in qRT-PCR |
Western Blot
Total proteins were extracted from brain tissues and cultured cells using RIPA lysis buffer, separated by SDS-PAGE, and transferred onto membranes. After blocking, the membranes were incubated with primary antibodies against Nrf2 (67 kDa) (ab89443, 1:500), cleaved Caspase 3 (32 kDa) (ab49822, 1:500), pro-Caspase 3 (35 kDa) (ab32499, 1:500), HO-1 (33 kDa) (ab13243, 1:500), Lamin B (66 kDa) (ab122919, 1:1000), and β-actin (42 kDa) (ab6276, 1:1000). On the next day, the membranes were subjected to HRP-conjugated secondary antibody for 1 h. Protein bands were visualized using enhanced chemiluminescence substrates, and the gray value of targets were analyzed using Image Lab 5.0.
Bioinformatic Analysis
To determine the upstream miRNAs of Nrf2, Targetscan (http://www.targetscan.org/vert_72/) and microrna.org (http://www.microrna.org/microrna/home.do) were used to predict the potential target miRNAs of Nrf2.
Dual Luciferase Reporter Assay
The 3′-UTR fragments of Nrf2 containing wild type (WT) and mutant type (MT) binding site of miR-448-3p were cloned into pmirGLO vector (Promega, Madison, WI, USA) to generate recombinant vectors Nrf2 3′-UTR WT/MUT. HT22 cells were co-transfected with recombinant vectors and miR-448-3p mimics or NC mimics. After 48 h of transfection, luciferase activity was measured by the Dual-Luciferase Reporter Assay System (Promega).
Statistical Analysis
Statistical analysis was performed using SPSS 21.0. The data were presented as means ± standard deviation (SD). Differences between two groups and among multiple groups were tested by Student’s t-test and one-way ANOVA (Tukey’s post hoc), respectively. P < 0.05 was considered as the significant threshold.
Results
MiR-448-3p/Nrf2 Axis Might Be Involved in Cerebral I/R Injury
It has been reported that Nrf2 plays a crucial role in antioxidant stress system and significantly decreased in cerebral I/R injury.19 In this study, we established a mouse model with cerebral I/R injury and confirmed that Nrf2 was significantly downregulated in brain tissues of mice subjected to I/R injury (p < 0.01, Figure 1A–C). Considering that miRNAs are a class of regulator of genes such as Nrf2,20 we performed bioinformatic analysis to predict the potential miRNAs of Nrf2 and found that there were seven probable miRNAs (Figure 1D). Meanwhile, qRT-PCR results showed that only miR-448-3p was significantly upregulated in brain tissues of mice subjected to I/R injury (p < 0.01, Figure 1D). In addition, miR-448-3p level was also obviously increased in plasma of mice subjected to I/R injury (p < 0.01, Figure 1E). These results indicated that miR-448-3p/Nrf2 axis may be associated with cerebral I/R injury.
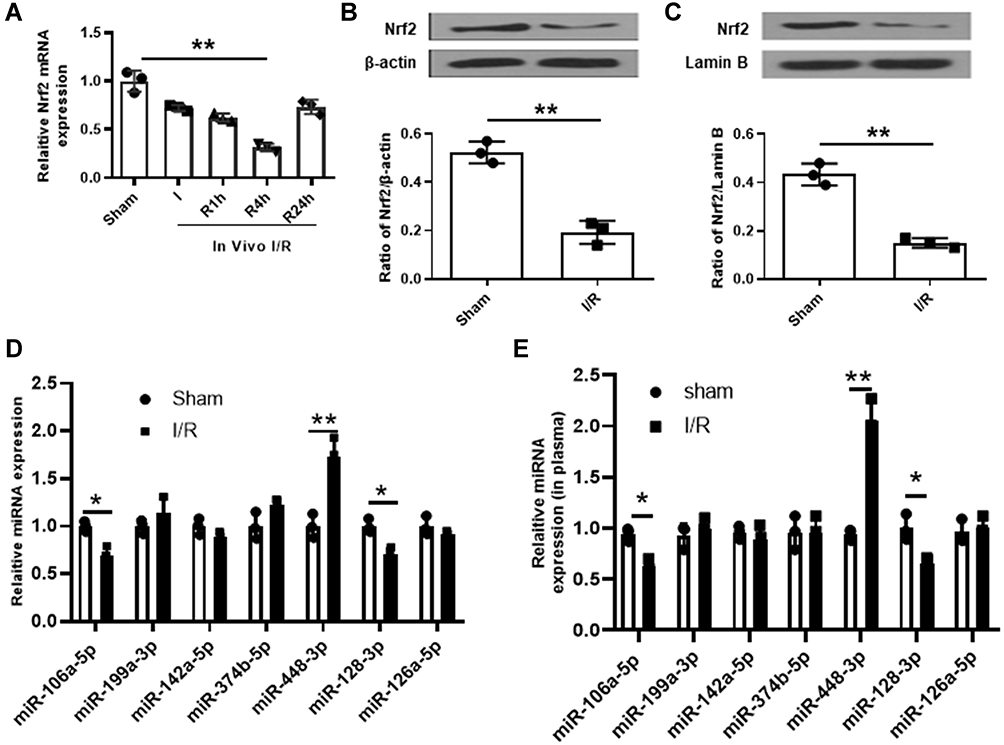 |
Figure 1 MiR-448-3p might serve as a ceRNA of Nrf2 in cerebral I/R injury. (A) Nrf2 expression in brain tissues was evaluated by qRT-PCR. (B and C) Total Nrf2 protein (B) and nuclear Nrf2 protein (C) levels in brain tissues were evaluated by Western blot. (D) The seven miRNAs potential upstream of Nrf2 in brain tissues were evaluated by qRT-PCR. (E) MiR-448-3p expression in plasma was evaluated by qRT-PCR. *p < 0.05 and **p < 0.01. |
MiR-448-3p Downregulation Attenuated Cerebral I/R Injury in vivo
MiR-448-3p expression in brain tissues was upregulated after 24h reperfusion (p < 0.01, Figure 2A). To determine the role of miR-448-3p in cerebral I/R injury, miR-448-3p inhibitor was injected into mice followed by MCAO/R treatment. The injection efficiency was confirmed by qRT-PCR (p < 0.01, Figure 2B). MCAO/R treatment significantly increased the neurological scores (p < 0.01), infarct volumes (p < 0.01), and brain water content (p < 0.01) of mice compared with sham-operation, while miR-448-3p downregulation obviously reduced MCAO/R-induced elevation of neurological scores (p < 0.01), infarct volumes (p < 0.01), and brain water content (p < 0.01) in mice (Figure 2C–E). As shown in Figure 2F, H&E staining in hippocampal CA1 regions of brain tissues showed that MCAO/R treatment significantly induced hippocampal CA1 neuron death (p < 0.01), while miR-448-3p downregulation attenuated I/R injury-induced neuron death (p < 0.01, Figure 2F). H&E staining of sections of cerebral cortex hippocampal CA1 regions also showed that MCAO/R treatment significantly induced hippocampal CA1 neuron death (p < 0.01, Figure 2G). These results indicated that miR-448-3p downregulation attenuated cerebral I/R injury.
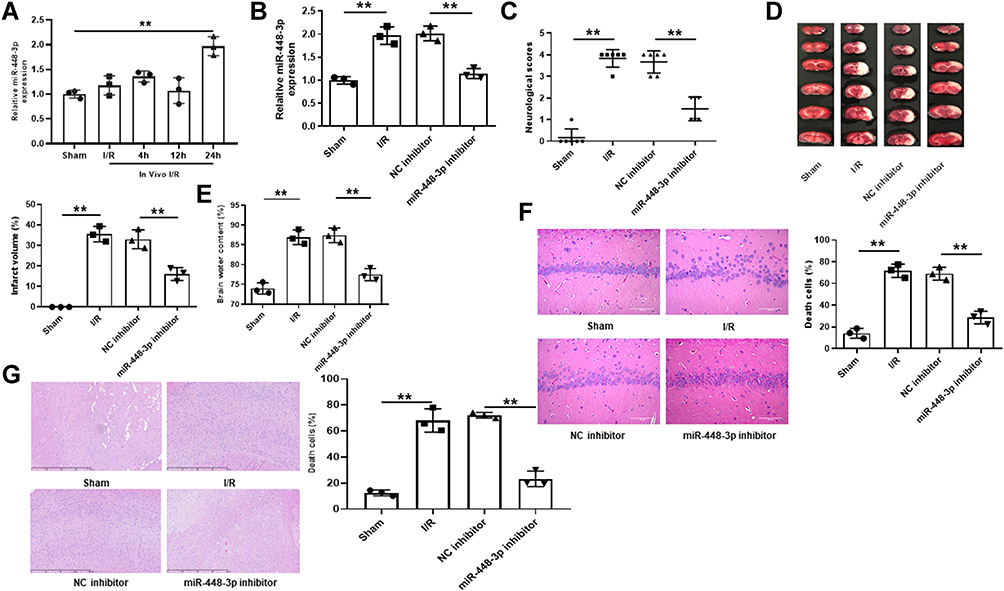 |
Figure 2 MiR-448-3p downregulation attenuated cerebral I/R injury in vivo. (A) MiR-448-3p expression in brain tissues was evaluated by qRT-PCR at 4 h, 12h, and 24h of post-reperfusion. (B) MiR-448-3p expression in brain tissues was evaluated by qRT-PCR. (C) Neurobehavioral deficits. (D) Infarct volume. (E) Brain water content. (F) H&E-stained sections of hippocampal CA1 regions (magnification, ×200, scale bar = 100μm) and the percentage of necrotic cells. (G) H&E-stained sections of cerebral cortex regions (magnification, ×200, scale bar = 100μm) and the percentage of necrotic cells. **p < 0.01. |
MiR-448-3p Downregulation Reduced I/R Injury-Induced Oxidative Stress and Neuron Apoptosis in Mouse Brains
MCAO/R treatment significantly reduced Nrf2 expression in brain tissues of mice subjected to I/R injury (p < 0.01), and miR-448-3p inhibitor obviously increased Nrf2 expression compared with NC inhibitor (p < 0.01, Figure 3A–C). Further, the effects of miR-448-3p on oxidative stress and apoptosis of brain tissues were investigated. MCAO/R treatment significantly reduced HO-1 expression (p < 0.01) and SOD activity (p < 0.01) and increased MDA content (p < 0.01) and ROS production (p < 0.01) in brain tissues of mice compared with sham operation, while miR-448-3p downregulation reversed MCAO/R-induced changes in brain tissues compared with NC inhibitor (HO-1, p < 0.01; SOD activity, p < 0.05; MDA content, p < 0.01; ROS production, p < 0.05; Figure 3D–G). Meanwhile, we found that MCAO/R treatment significantly increased cleaved-caspase-3 level in brain tissues of mice compared with sham operation (p < 0.01), and miR-448-3p downregulation obviously reduced its expression compared with NC inhibitor (p < 0.01, Figure 3H). In addition, TUNEL staining of brain tissue sections showed that MCAO/R treatment significantly promoted neuron apoptosis compared with sham operation (p < 0.01), and miR-448-3p downregulation reduced I/R injury-induced neuron apoptosis compared with NC inhibitor (p < 0.01, Figure 3I). These results indicated that miR-448-3p downregulation reduced I/R injury-induced oxidative stress and neuron apoptosis in mice brains.
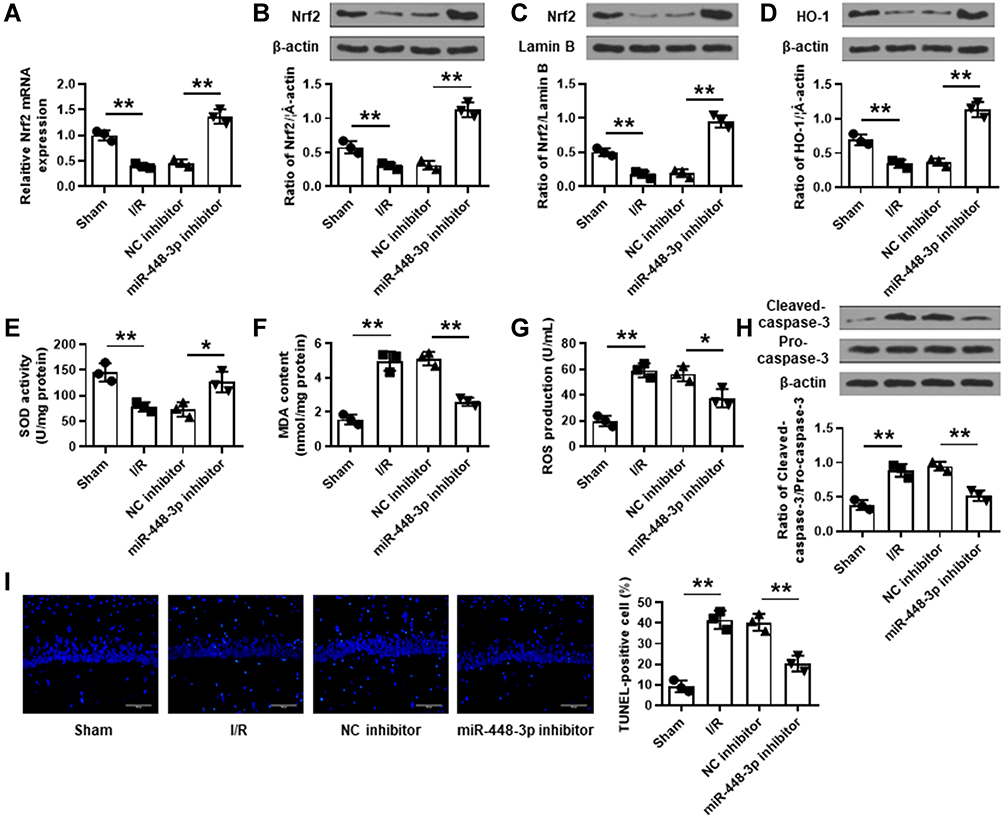 |
Figure 3 MiR-448-3p downregulation reduced I/R injury-induced oxidative stress and neuron apoptosis in mice brains. (A) Nrf2 expression in brain tissues was evaluated by qRT-PCR. (B–D) The levels of total Nrf2 protein (B), nuclear Nrf2 protein (C), and HO-1 protein (D) in brain tissues were evaluated by Western blot. (E–G) SOD activity (E), MDA content (F), and ROS production (G) in brain were detected by commercial assay kits. (H) Cleaved caspase-3 levels in brain tissues were evaluated by Western blot. (I) Neuron apoptosis in brain tissues was evaluated by TUNEL staining. Magnification: ×200, scale bar = 100 μm. *p < 0.05 and **p < 0.01. |
Nrf2 Was a Target of miR-448-3p
To further determine the relationship between miR-448-3p and Nrf2, their putative binding site was predicted, and the results showed that Nrf2 might be a target of miR-448-3p (Figure 4A). In addition, luciferase reporter assay was performed in HT22 cells, and the results showed that miR-448-3p overexpression significantly reduced the relative luciferase activity of Nrf2 3ʹ-UTR WT compared with NC mimics (p < 0.01) but did not change that of Nrf2 3ʹ-UTR MT (Figure 4B). These results indicated that Nrf2 was a target of miR-448-3p.
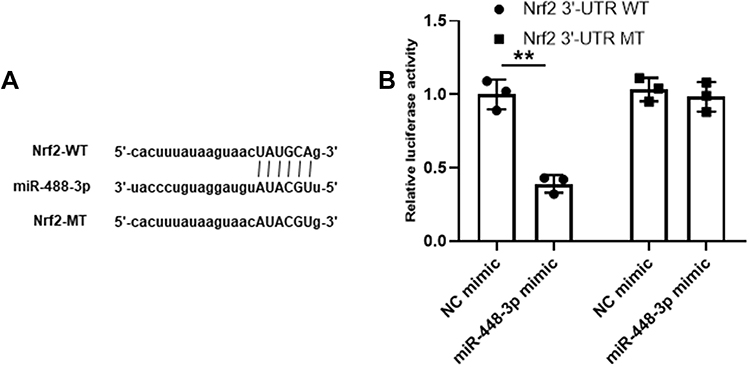 |
Figure 4 Nrf2 was a target of miR-448-3p. (A) The putative binding site between miR-448-3p and Nrf2. (B) Dual luciferase reporter assay. **p < 0.01. |
MiR-448-3p Downregulation Reduced OGD/R-Induced Oxidative Stress in HT22 Cells
To confirm the role of miR-448-3p, HT22 cells were transfected with miR-448-3p, and subjected to 2 h of OGD followed by 24 h of re-oxygenation. The transfection efficiency was confirmed by qRT-PCR (p < 0.01, Figure 5A). MTT assay showed that OGD/R treatment significantly reduced HT22 cell viability compared with control (p < 0.01), and miR-448-3p downregulation obviously enhanced OGD/R-induced growth defect compared with NC inhibitor (p < 0.05, Figure 5B). OGD/R treatment significantly reduced Nrf2 expression in HT22 cells (p < 0.01), and miR-448-3p inhibitor obviously increased Nrf2 expression in OGD/R-treated HT22 cells compared with NC inhibitor (p < 0.01, Figure 5C–F). In addition, OGD/R treatment significantly reduced HO-1 expression (p < 0.01) and SOD activity (p < 0.01) and increased MDA content (p < 0.01) and ROS production (p < 0.01) in HT22 cells compared with the control, while miR-448-3p downregulation reversed OGD/R-induced changes in HT22 cells compared with NC inhibitor (HO-1, p < 0.01; SOD activity, p < 0.05; MDA content, p < 0.01; ROS production, p < 0.05; Figure 5G–J). These results indicated that miR-448-3p downregulation reduced OGD/R induced oxidative stress in HT22 cells.
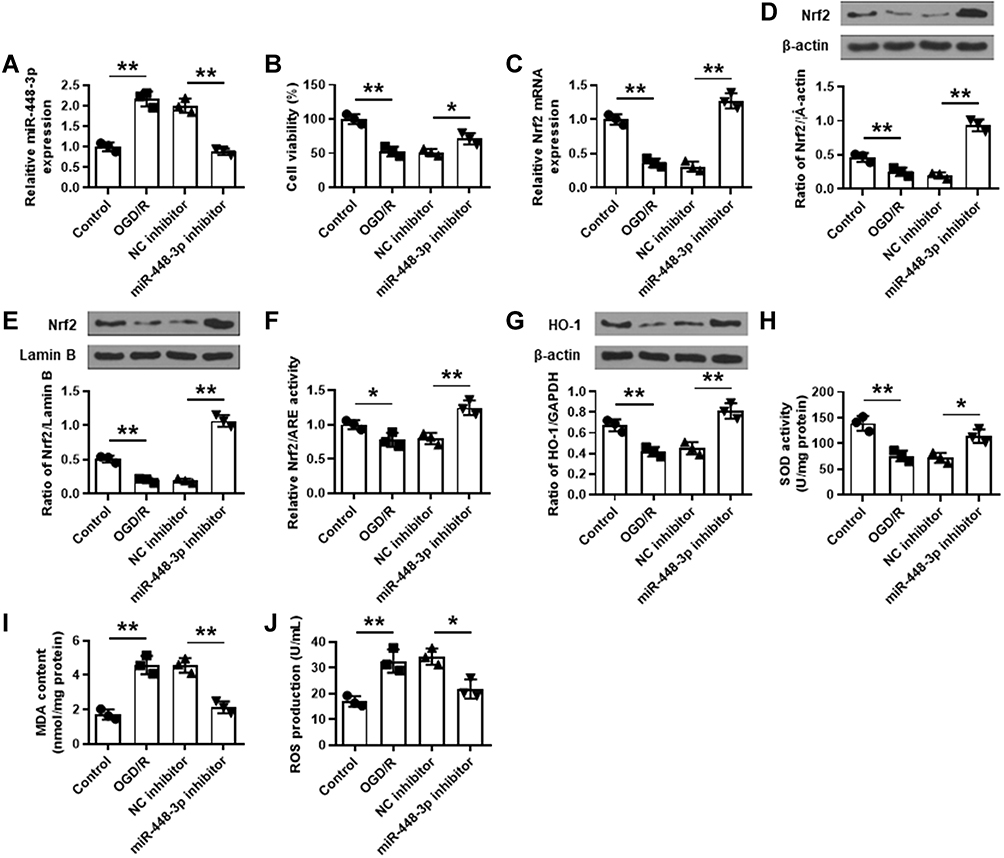 |
Figure 5 MiR-448-3p downregulation reduced OGD/R-induced oxidative stress in HT22 cells by regulating Nrf2. (A) The transfection efficiency was confirmed by qRT-PCR. (B) Cell viability was evaluated by MTT assay. (C–E) Nrf2 expression was detected by qRT-PCR (C) and Western blot (D and E). (F) The luciferase reporter assay of Nrf2/ARE activity. (G–J) HO-1 expression (G), SOD activity (H), MDA content (I), and ROS production (J) in HT22 cells were detected by commercial assay kits. *p < 0.05 and **p < 0.01. |
MiR-448-3p Downregulation Reduced OGD/R-Induced Apoptosis in HT22 Cells
Similarly, OGD/R treatment significantly increased cleaved caspase-3 level in HT22 cells (p < 0.01), and miR-448-3p downregulation obviously reduced OGD/R- induced elevation of cleaved caspase-3 in HT22 cells (p < 0.01, Figure 6A). Meanwhile, TUNEL staining (Figure 6B) and flow cytometry (Figure 6C) showed that OGD/R treatment significantly promoted HT22 cell apoptosis (p < 0.01), and miR-448-3p downregulation obviously inhibited OGD/R-treated HT22 cell apoptosis compared with NC inhibitor (TUNEL staining, p < 0.01; flow cytometry, p < 0.05). These results indicated that miR-448-3p downregulation reduced OGD/R-induced HT22 cell apoptosis.
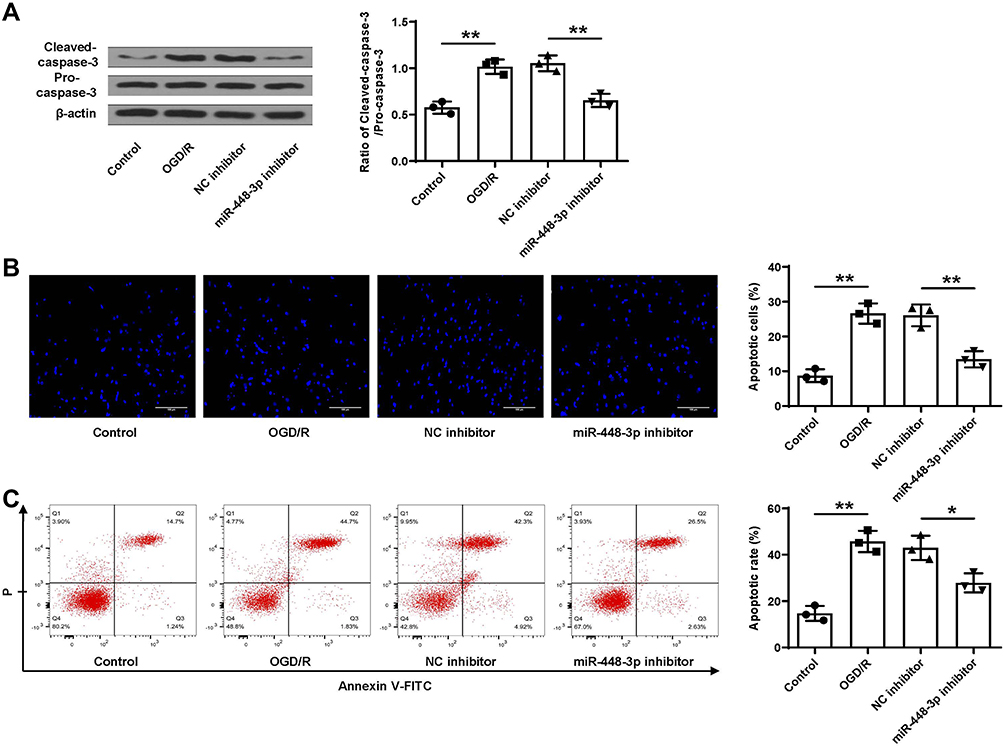 |
Figure 6 MiR-448-3p downregulation reduced OGD/R-induced apoptosis in HT22 cells. (A) Cleaved caspase-3 level was evaluated by Western blot. (B and C) Cell apoptosis was evaluated by TUNEL staining (B, magnification, ×200, scale bar = 100μm) and flow cytometry (C). *p < 0.05 and **p < 0.01. |
Nrf2 Silencing Attenuated the Protective Effects of miR-448-3p Inhibitor on Oxidative Stress and Apoptosis in vitro
To further determine whether Nrf2 mediated the neuroprotective effects of miR-448-3p inhibitor, si-Nrf2 was transfected into HT22 cells, and the transfection efficiency was confirmed by qRT-PCR (p < 0.01, Figure 7A). HT22 cells were transfected with miR-448-3p inhibitor, or co-transfected with miR-448-3p inhibitor and si-Nrf2, and then subjected to 2 h of OGD followed by 24 h of re-oxygenation. We found that the neuroprotective effects of miR-448-3p inhibitor including cell viability, oxidative stress indicators (HO-1 expression, SOD activity, MDA content and ROS production), and cleaved caspase-3 level in OGD/R treated HT22 cells were all reversed by co-transfection of miR-448-3p inhibitor and si-Nrf2 (all p < 0.05, Figure 7B–G). These results indicated that the neuroprotective effects of miR-448-3p inhibitor were partially mediated by Nrf2.
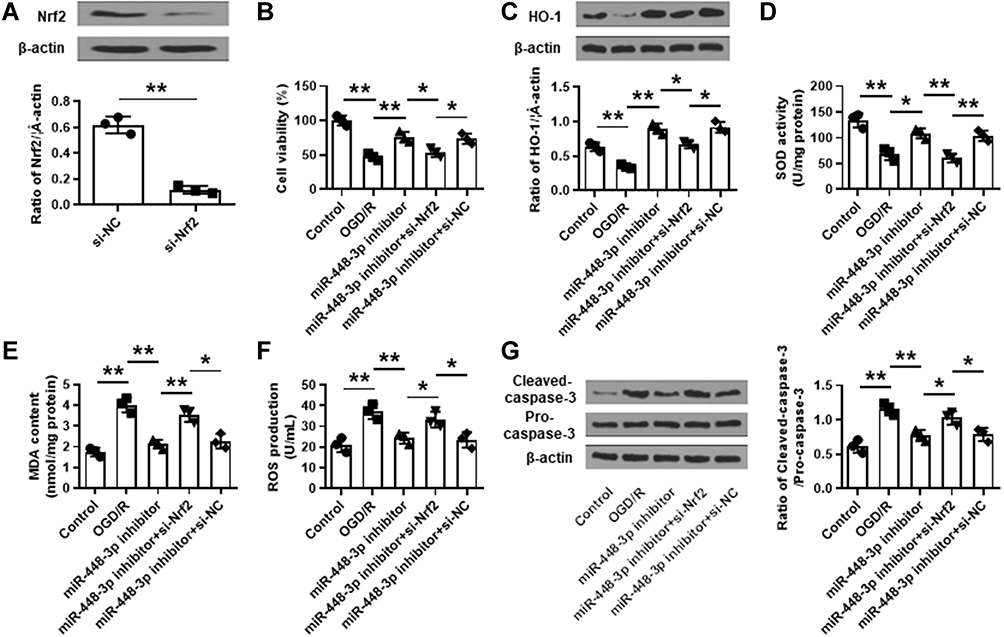 |
Figure 7 Nrf2 silencing attenuated the protective effects of miR-448-3p inhibitor on oxidative stress and apoptosis in vitro. (A) The transfection efficiency of si-Nrf2 in HT22 cells was confirmed by qRT-PCR. (B–G) HT22 cells were transfected with miR-448-3p inhibitor, or co-transfected with miR-448-3p inhibitor and si-Nrf2, and then subjected to 2 h of OGD followed by 24 h of re-oxygenation. (B) Cell viability was evaluated by MTT assay. (C) HO-1 expression was detected by Western blot. (D–F) SOD activity (D), MDA content (F), and ROS production (F) were detected by commercial assay kits. (G) Cleaved caspase-3 levels in brain tissues were evaluated by Western blot. *p < 0.05 and **p < 0.01. |
Discussion
In brain tissues, oxidative stress is the main pathophysiological mechanism, and tissues further damaged when there are no enough antioxidant defenses to remove the ROS.21 Previous studies have reported that ROS production is increased in brain tissues of mice subjected to I/R injury during the reperfusion process.22 Our study demonstrated that cerebral I/R injury significantly increased ROS production and reduced SOD activity in brain tissues. Moreover, miR-448-3p downregulation efficiently reduced oxidative stress and apoptosis in brain tissues of MCAO/R-treated mice and OGD/R-treated HT22 cells. These findings suggested that miR-448-3p served as a ceRNA of Nrf2 and might be a potential biomarker and efficient target for cerebral I/R injury.
Nrf2/HO-1-mediated ROS elimination plays an important protective role in cerebral I/R injury.23 Our results confirmed that Nrf2 was significantly downregulated during I/R injury both in vitro and in vivo. In addition, I/R injury reduced HO-1 expression and increased MDA content in MCAO/R-treated mouse brains and OGD/R-treated HT22 cells, consistent with previous studies. MiRNAs have emerged as promising regulators in cerebral I/R injury.24 Previous studies reviewed that many miRNAs mediated the regulation of the major redox homeostasis switch, Nrf2, in cerebral I/R injury.25 MiR-93 downregulation attenuates cerebral I/R injury by regulating Nrf2/HO-1 defense pathway.26 To further identify one or more potential miRNAs of Nrf2, the potential miRNAs against Nrf2 were predicted, and there were seven probable miRNAs (miR-106a-5p, miR-199a-3p, miR-142a-5p, miR-374b-5p, miR-448-3p, miR-128-3p and miR-126a-5p), suggesting that these seven miRNAs might be upstream regulators of Nrf2. Of these seven miRNAs, only miR-448-3p was significantly upregulated, while miR-106a-5p and miR-128-3p were downregulated in brain tissues of mice subjected to I/R injury, and miR-106a-5p and miR-128-3p downregulation did not show a very significant difference. Therefore, we paid more attention to the function of miR-448-3p. Considering the negative correlation between miRNAs and mRNAs, miR-448-3p might be the ceRNA of Nrf2. Luciferase reporter assay further determined their binding interaction. MiR-448-3p is a recently identified miRNA, and its roles have not been well explored. MiR-448-3p improves diabetic vascular dysfunction by suppressing epithelial–mesenchymal transition.27 In addition, one previous study revealed that miR-448 showed a pivotal role in the development of ROS-induced cardiomyopathy,28 indicating that miR-448 might be involved in ROS-mediated physiological metabolism. In this study, we demonstrated that miR-448-3p downregulation attenuated cerebral I/R injury, and Nrf2 silencing obviously reversed the neuroprotective effects of miR-448-3p inhibitor. These further confirmed the regulation of miR-448-3p and Nrf2 in cerebral I/R injury.
Except for miR-448-3p, there were also two significantly downregulated miRNAs (miR-106a-5p and miR-128-3p). Although the role of miR-106a-5p in cerebral I/R injury remains unclear, its important function in ROS-mediated cell injury has been studied. MiR‑106a‑5p downregulation reduces ox‑LDL‑induced endothelial cell injury and ROS production by regulating STAT3 signaling pathway.29 In addition, recent studies have revealed that miR-106a-5p acts as a tumor suppressor in different human cancers such as renal cell carcinoma,30 osteosarcoma,31 prostate cancer,32 and ovarian cancer.33 MiR-128-3p has also been reported to be associated with oxidative stress in various pathogenic processes including doxorubicin-induced liver injury34 and mitochondrial dysfunction in glioma cells.35 Considering the negative regulation of miRNA and Nrf2, both miR-106a-5p and miR-128-3p are not ceRNA of Nrf2 and might be involved in oxidative stress during cerebral I/R injury. These need to be determined in the subsequent experiments.
In addition, some miRNAs for Nrf2 have been identified in previous studies such as miR-153,36 miR-129-3p,37 miR-144,38 and miR-140-5p.39 Although this study did not predict more potential miRNAs, whether these identified miRNAs are also involved in Nrf2 regulation in cerebral I/R injury should be investigated in the future.
Conclusion
Our results demonstrated that miR-448-3p downregulation protected brain against I/R injury through upregulating Nrf2, resulting in the reduction of oxidative stress and apoptosis. Our study suggested that miR-448-3p might be a potential diagnostic and therapeutic target for cerebral I/R injury.
Availability of Supporting Data
The data are not publicly available due to their containing information that could compromise the privacy of research participants, but are available on request from the corresponding author.
Ethical Approval and Consent to Participate
All animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals and approved by the Affiliated Kunshan Hospital of Jiangsu University. Procedures operated in this research were completed in keeping with the standards set out in the principles on ethical animal research outlined in the Basel Declaration.
Funding
This study was supported by grant from the Suzhou Science and Technology Plan Project (People's Livelihood Science and Technology) (Grant No. SYS2020067).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Miranda B, Fonseca AC, Ferro JM. Patent foramen ovale and stroke. J Neurol. 2018;265(8):1943–1949. doi:10.1007/s00415-018-8865-0
2. Rabinstein AA. Update on treatment of acute ischemic stroke. Continuum. 2020;26(2):268–286.
3. Silva GS, Nogueira RG. Endovascular treatment of acute ischemic stroke. Continuum. 2020;26(2):310–331.
4. Degracia DJ. Towards a dynamical network view of brain ischemia and reperfusion. Part I: background and preliminaries. J Exp Stroke Transl Med. 2010;3(1):59–71. doi:10.6030/1939-067X-3.1.59
5. Wu J, Chen Y, Yu S, et al. Neuroprotective effects of sulfiredoxin-1 during cerebral ischemia/reperfusion oxidative stress injury in rats. Brain Res Bull. 2017;132:99–108. doi:10.1016/j.brainresbull.2017.05.012
6. Li R, Li X, Wu H, Yang Z, Fei L, Zhu J. Theaflavin attenuates cerebral ischemia/reperfusion injury by abolishing miRNA‑128‑3p‑mediated Nrf2 inhibition and reducing oxidative stress. Mol Med Rep. 2019;20(6):4893–4904.
7. Khajevand-Khazaei MR, Azimi S, Sedighnejad L, et al. S-allyl cysteine protects against lipopolysaccharide-induced acute kidney injury in the C57BL/6 mouse strain: involvement of oxidative stress and inflammation. Int Immunopharmacol. 2019;69:19–26. doi:10.1016/j.intimp.2019.01.026
8. Chen M, Dai LH, Fei A, Pan SM, Wang HR. Isoquercetin activates the ERK1/2-Nrf2 pathway and protects against cerebral ischemia-reperfusion injury in vivo and in vitro. Exp Ther Med. 2017;13(4):1353–1359. doi:10.3892/etm.2017.4093
9. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–1207. doi:10.1016/j.jaci.2017.08.034
10. Mohr AM, Mott JL. Overview of microRNA biology. Semin Liver Dis. 2015;35(1):3–11. doi:10.1055/s-0034-1397344
11. Zuo X, Lu J, Manaenko A, et al. MicroRNA-132 attenuates cerebral injury by protecting blood-brain-barrier in MCAO mice. Exp Neurol. 2019;316:12–19. doi:10.1016/j.expneurol.2019.03.017
12. Huang R, Ma J, Niu B, et al. MiR-34b protects against focal cerebral ischemia-reperfusion (I/R) injury in rat by targeting Keap1. J Stroke Cerebrovasc Dis. 2019;28(1):1–9. doi:10.1016/j.jstrokecerebrovasdis.2018.08.023
13. Wu Y, Yao J, Feng K. miR-124-5p/NOX2 axis modulates the ROS production and the inflammatory microenvironment to protect against the cerebral I/R injury. Neurochem Res. 2020;45(2):404–417. doi:10.1007/s11064-019-02931-0
14. Chai Z, Gong J, Zheng P, Zheng J. Inhibition of miR-19a-3p decreases cerebral ischemia/reperfusion injury by targeting IGFBP3 in vivo and in vitro. Biol Res. 2020;53(1):17. doi:10.1186/s40659-020-00280-9
15. Zhao J, Wang B. MiR-7-5p enhances cerebral ischemia-reperfusion injury by degrading sirt1 mRNA. J Cardiovasc Pharmacol. 2020;76(2):227–236. doi:10.1097/FJC.0000000000000852
16. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi:10.1161/01.STR.20.1.84
17. Pagani IS, Spinelli O, Mattarucchi E, et al. Genomic quantitative real-time PCR proves residual disease positivity in more than 30% samples with negative mRNA-based qRT-PCR in Chronic Myeloid Leukemia. Oncoscience. 2014;1(7):510–521. doi:10.18632/oncoscience.65
18. Zhang B, Wei K, Li X, et al. Upregulation of Cdh1 signaling in the hippocampus attenuates brain damage after transient global cerebral ischemia in rats. Neurochem Int. 2018;112:166–178. doi:10.1016/j.neuint.2017.07.005
19. Hou Y, Wang Y, He Q, et al. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res. 2018;336:32–39. doi:10.1016/j.bbr.2017.06.027
20. Ayers D, Baron B, Hunter T. miRNA influences in NRF2 pathway interactions within cancer models. J Nucleic Acids. 2015;2015:143636. doi:10.1155/2015/143636
21. Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4(6):461–470. doi:10.1111/j.1747-4949.2009.00387.x
22. Awooda HA, Lutfi MF, Sharara GG, Saeed AM. Oxidative/nitrosative stress in rats subjected to focal cerebral ischemia/reperfusion. Int J Health Sci. 2015;9(1):17–24. doi:10.12816/0024679
23. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88(Pt B):179–188. doi:10.1016/j.freeradbiomed.2015.04.036
24. Ghafouri-Fard S, Shoorei H, Taheri M. Non-coding RNAs participate in the ischemia-reperfusion injury. Biomed Pharmacother. 2020;129:110419. doi:10.1016/j.biopha.2020.110419
25. Padmavathi G, Ramkumar KM. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch Biochem Biophys. 2021;698:108725. doi:10.1016/j.abb.2020.108725
26. Wang P, Liang X, Lu Y, Zhao X, Liang J. MicroRNA-93 downregulation ameliorates cerebral ischemic injury through the Nrf2/HO-1 defense pathway. Neurochem Res. 2016;41(10):2627–2635. doi:10.1007/s11064-016-1975-0
27. Guan GY, Wei N, Song T, et al. miR-448-3p alleviates diabetic vascular dysfunction by inhibiting endothelial-mesenchymal transition through DPP-4 dysregulation. J Cell Physiol. 2020;235(12):10024–10036.
28. Kyrychenko S, Kyrychenko V, Badr MA, Ikeda Y, Sadoshima J, Shirokova N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc Res. 2015;108(3):324–334. doi:10.1093/cvr/cvv238
29. Hu Y, Xu R, He Y, et al. Downregulation of microRNA‑106a‑5p alleviates ox‑LDL‑mediated endothelial cell injury by targeting STAT3. Mol Med Rep. 2020;22(2):783–791. doi:10.3892/mmr.2020.11147
30. Ma J, Wang W, Azhati B, Wang Y. miR-106a-5p Functions as a tumor suppressor by targeting VEGFA in renal cell carcinoma. Dis Markers. 2020;2020:8837941.
31. He QY, Wang GC, Zhang H, et al. miR-106a-5p Suppresses the proliferation, migration, and invasion of osteosarcoma cells by targeting HMGA2. DNA Cell Biol. 2016;35(9):506–520. doi:10.1089/dna.2015.3121
32. Wei P, Yang J, Zhang D, Cui M, Li L. lncRNA HAND2-AS1 regulates prostate cancer cell growth through targeting the miR-106a-5p/RBM24 axis. Onco Targets Ther. 2020;13:4523–4531. doi:10.2147/OTT.S246274
33. Chao H, Zhang M, Hou H, Zhang Z, Li N. HOTAIRM1 suppresses cell proliferation and invasion in ovarian cancer through facilitating ARHGAP24 expression by sponging miR-106a-5p. Life Sci. 2020;243:117296. doi:10.1016/j.lfs.2020.117296
34. Zhao X, Jin Y, Li L, et al. MicroRNA-128-3p aggravates doxorubicin-induced liver injury by promoting oxidative stress via targeting Sirtuin-1. Pharmacol Res. 2019;146:104276. doi:10.1016/j.phrs.2019.104276
35. Qu C, Yan C, Cao W, et al. miR-128-3p contributes to mitochondrial dysfunction and induces apoptosis in glioma cells via targeting pyruvate dehydrogenase kinase 1. IUBMB Life. 2020;72(3):465–475.
36. Shao D, Wu Z, Bai S, Fu G, Zou Z. The function of miRNA‑153 against isoflurane‑induced neurotoxicity via Nrf2/ARE cytoprotection. Mol Med Rep. 2019;19(5):4001–4010.
37. Sun W, Yi Y, Xia G, et al. Nrf2-miR-129-3p-mTOR axis controls an miRNA regulatory network involved in HDACi-induced autophagy. Mol Ther. 2019;27(5):1039–1050. doi:10.1016/j.ymthe.2019.02.010
38. Chu SF, Zhang Z, Zhou X, et al. Ginsenoside Rg1 protects against ischemic/reperfusion-induced neuronal injury through miR-144/Nrf2/ARE pathway. Acta Pharmacol Sin. 2019;40(1):13–25. doi:10.1038/s41401-018-0154-z
39. Zhao L, Qi Y, Xu L, et al. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018;15:284–296. doi:10.1016/j.redox.2017.12.013
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.