")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Inhibition of LPA-LPAR1 and VEGF-VEGFR2 Signaling in IPF Treatment
Authors Luo YL, Li Y, Zhou W, Wang SY, Liu YQ
Received 2 April 2023
Accepted for publication 25 July 2023
Published 2 September 2023 Volume 2023:17 Pages 2679—2690
DOI https://doi.org/10.2147/DDDT.S415453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Ya-Li Luo,* Yan Li,* Wen Zhou, Si-Yu Wang, Yong-Qi Liu
Gansu University Key Laboratory for Molecular Medicine and Chinese Medicine Prevention and Treatment of Major Diseases, Gansu University of Chinese Medicine, Lanzhou, 730000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yong-Qi Liu, School of Basic Medicine, Gansu University Key Laboratory for Molecular Medicine and Chinese Medicine Prevention and Treatment of Major Diseases, Gansu University of Chinese Medicine, Lanzhou, People’s Republic of China, Email [email protected]
Abstract: Due to the complex mechanism and limited treatments available for pulmonary fibrosis, the development of targeted drugs or inhibitors based on their molecular mechanisms remains an important strategy for prevention and treatment. In this paper, the downstream signaling pathways mediated by VEGFR and LPAR1 in pulmonary cells and the role of these pathways in pulmonary fibrosis, as well as the current status of drug research on the targets of LPAR1 and VEGFR2, are described. The mechanism by which these two pathways regulate vascular leakage and collagen deposition leading to the development of pulmonary fibrosis are analyzed, and the mutual promotion of the two pathways is discussed. Here we propose the development of drugs that simultaneously target LPAR1 and VEGFR2, and discuss the important considerations in targeting and safety.
Keywords: idiopathic pulmonary fibrosis, VEGFR2, LPAR1
Background
Idiopathic pulmonary fibrosis(IPF) is a progressive, irreversible, disease with high mortality, and current treatment outcomes are limited. Molecular mechanisms of pulmonary fibrosis are complex. Symptoms of IPF include cough and dyspnoea. IPF is the most common Idiopathic interstitial pneumonia (IPF), and its incidence rate and prevalence are increasing.1 The average life expectancy after IPF diagnosis is 3–5 years.2 Although international guidelines recommend consideration of lung transplantation in certain patient populations, pirfenidone and nintedanib, multiple tyrosine kinase inhibitors, have been reported to slow disease progression and have recently been approved for use in patients with IPF. The side effects of oral pirfenidone and nintedanib lead to a great impact on the quality of life of patients.3,4 The side effect of Nintedanib is the adverse reaction of gastrointestinal tract, such as diarrhea. The research shows that 60% of patients have nausea within the first three months of Nintedanib treatment, and the intensity of these side effects is mild or moderate, which can be controlled.5 Therefore, there remains a need for effective new therapies to treat IPF. There is been development of single target drugs, multitarget drugs or similar drugs for the treatment of IPF.6
Discovering therapeutic targets related to IPF can promote further slowing down or ideally stopping the progression of IPF. Researchers have been pushing for the screening of candidate targets to accelerate drug therapy for IPF, such as Phosphoinoside 3-kinase/mammalian target of rapamycin inhibitor. However, because IPF involves multiple targets, there are still some deficiencies in targeted therapy with a single target.7,8 Lysophosphatidic acid (LPA) is a key lipid signaling molecule that regulates a series of very diverse cell events, such as motility, chemotaxis, cell cycle progression, vitality and wound healing.9 LPA participates in chronic wound healing through LPAR1, as well as idiopathic pulmonary fibrosis.10 Vascular endothelial growth factor A (VEGF-A) is essential for normal alveolar formation, rapid alveolar proliferation, and normal development of blood vessels during lung maturation.11 Its receptor, VEGFR2, is considered to be the main signal transduction receptor of VEGF biological activity, and is essential for normal development.12 Studies have shown that VEGF and LPA mediate downstream pathways by binding VEGFR2 and LPAR1, the corresponding receptors in lung tissue cells, and participate in vascular leakage, collagen deposition and other processes that underlie the continuous progression of pulmonary fibrosis.13,14 Recent studies on the development of drugs targeting VEGFR2 and LPAR1 for the treatment of pulmonary fibrosis have been reported.15,16 Current clinical medication of pulmonary fibrosis shows that there are limitations in the effect of single target drugs, and the use of multiple drugs can lead to increased adverse reactions, increased treatment costs and other drawbacks;17,18 as a result, the research and development of drugs with multi-target onsets of action has practical application prospects. At present, it is not known if LPA-LPAR1 and VEGF-VEGFR2 have an association, and whether they have an interaction in promoting the development of pulmonary fibrosis. In this study, we describe the mechanism of LPA-LPAR1 and VEGF-VEGFR2 pathways in the progression of pulmonary fibrosis, sort out and analyze the influence of the two, and provide evidence that the development of drugs that can simultaneously intervene in VEGFR2 and LPAR1 activation can open up new approaches for the treatment of pulmonary fibrosis.
Association of VEGFR2 with IPF Progression
VEGF Source
VEGF is a mitogen, which is the survival and differentiation factor of endothelial cells in the lung.19 The level of VEGF in the lung is more than 500 times that in the plasma.20 A large amount of VEGF-A is present in the normal adult lung, and the alveolar epithelium appears to be the main source,21 although smooth muscle cells, macrophages ECs and fibroblasts also express VEGF-A.22,23 In addition, bone marrow cells also secrete VEGF. Research shows that in a mouse model of pulmonary fibrosis, the loss of VEGF in bone marrow cells aggravates the damage of fibrotic tissue. The main findings include a significant decrease in the survival rate of epithelial cells, a significant increase in the invasion of myofibroblasts, the expression of hypoxia inducible factor (HIF) and Wnt/ β-catenin. Therefore, the angiogenesis process driven by VEGF derived from bone marrow cells is also very noteworthy for preventing fibrosis damage.24
VEGF-A and VEGFR2
VEGF play a very important role in physiological and pathological angiogenesis.25 VEGF is one of the most potent mediators of angiogenesis and vascular permeability to water and protein.26 VEGF has also been reported to induce fenestrations in endothelial cells (ECs) both in vivo and in vitro. In the past few years, the best-studied molecule in the VEGF family has been VEGF-A, and native VEGF is a basic, heparin-bound, 45 kDa homodimeric glycoprotein.27 VEGF-A is the most typical isoform.
The biological activity of VEGF depends on its response to specific receptors. VEGFR2 is considered the main sensor of the VEGF signal in ECs, and directly activates the PI3K/Akt signal pathway and adhesion pathway by activating adhesion kinase (FAK).28
Progress in VEGFR2 and IPF-Related Pathways
The pathogenesis of idiopathic pulmonary fibrosis (IPF) is characterized by an initial acute inflammatory response, mostly involving lung structural remodeling, extracellular matrix, and multiple cell types.29 VEGF-A acts as an important vascular endothelial growth factor, and Barratt et al30 proposed that VEGF-A may promote fibrogenesis. Evidence suggests that VEGF levels may reflect the severity of IPF and predict disease progression.31 In addition, Jaskiewicz et al32 showed that serum VEGF levels appear to positively correlate with disease severity and prognosis in IPF. However, in a small retrospective study, baseline plasma VEGF levels were significantly associated with “interstitial score” and disease progression, and 5-year survival was low.33 Studies in animal models have provided some additional insights into the role of VEGF signaling in the development of pulmonary fibrosis. In a rat adenoviral TGF-β overexpression model, co-administration of VEGF resulted in severe pulmonary fibrosis, but decreased pulmonary hypertension.34 BLM-injected animals have increased expression of VEGF in the lung, and inhibition of VEGF using an anti-VEGF antibody attenuates BLM-induced pulmonary fibrosis.35 Overexpression of VEGF in the lung of transgenic mice stimulates inflammatory remodeling and subepithelial fibrosis.36 Recent data suggests that exogenous VEGF-B can prevent the reduction of pulmonary hypertension in experimental models of fibrosis, where it increases fibrogenesis. These data indicate that serum VEGF may reflect the severity of lung disease. It is important to note that it may be important to measure the relative levels of different VEGF isoforms given the different roles of VEGF in IPF.37
Recently, many studies have shown that VEGFR2 is closely related to lung diseases, including hypoxic pulmonary arterial hypertension,38 lung cancer.39 Ou et al reported that SU5416, a VEGFR2 antagonist, attenuates BLM-induced lung fibrosis in mice. Antagonism of VEGFR2 is therefore a promising approach.40 We summarized that VEGFR2 promotes its downstream signal pathway (Figure 1).
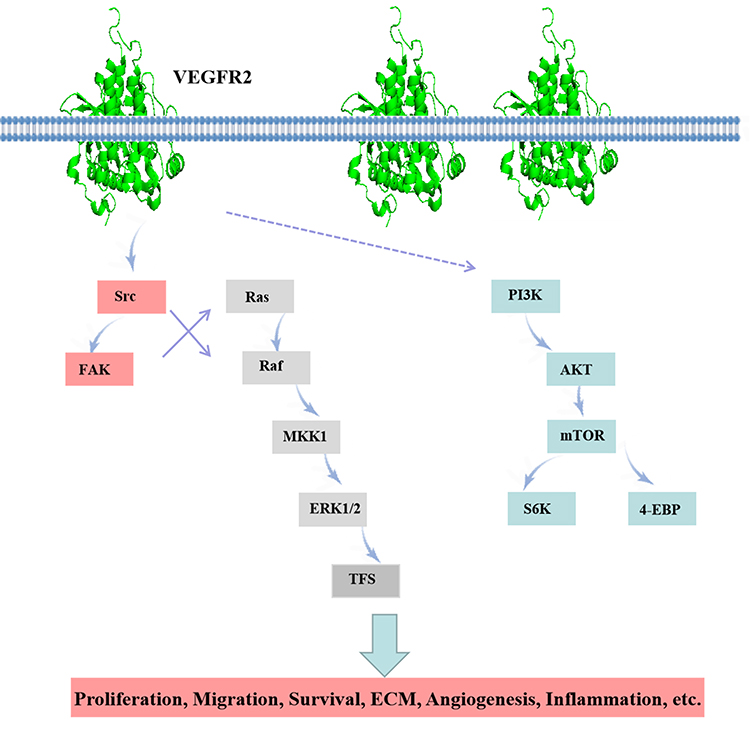 |
Figure 1 VEGFR2 related pathway diagram. |
Association of LPAR1 with IPF Progression
LPA Source
The first evidence of LPA manifested as biologically active phospholipids was obtained by Vogt in 1969.41 Lysophosphatidic acid (LPA) is a small glycerophospholipid (molecular weight: 430–480Da) present in all eukaryotic tissues and present at higher concentrations (sub-micromolar range) in plasma relative to the major phospholipid species. LPA has been shown to cause widespread cellular responses (smooth muscle contraction, platelet aggregation, calcium mobilization, chemotaxis, neurotransmitter release, cell proliferation, and cell transformation).42
LPAR1
Lysophosphatidic acid (LPA) is a lipid concentrated in serum, and in vitro, LPA is known to induce proliferation and differentiation of lung fibroblasts.43,44 LPA is considered to play an important role in the evolution of pulmonary fibrosis.45 Levels of LPA are elevated in bronchoalveolar lavage fluid samples from IPF patients46 supporting the clinical relevance of this signaling pathway in IPF development.
Many studies have shown that LPA and LPAR1 have positive roles in the pathogenesis of fibrosis. In BLM-induced lung fibrosis models, LPAR1 deletion conferred significant protection against BLM-induced lung fibrosis and mortality.45 In vivo, oral administration of LPAR1 antagonists significantly prevented BLM-induced pulmonary fibrosis in mice,47 and intraperitoneal injection of LPAR1/3 antagonists ameliorated irradiation-induced pulmonary fibrosis.48 In addition, LPA also induces fibroblast chemotaxis through an LPAR1-dependent mechanism.47 Taken together, these results suggest that LPAR1 receptor antagonism may be a useful treatment for IPF.
Progress in LPAR1 and IPF-Related Pathways
When LPA binds to LPAR1, LPA induces different downstream signaling chains through different G proteins, including Rho, PLC, MAPK, PI3K, and AC.49 LPAR signaling occurs through multiple intracellular cascades.50 We summarized that LPAR1 promotes its downstream signal pathway (Figure 2).
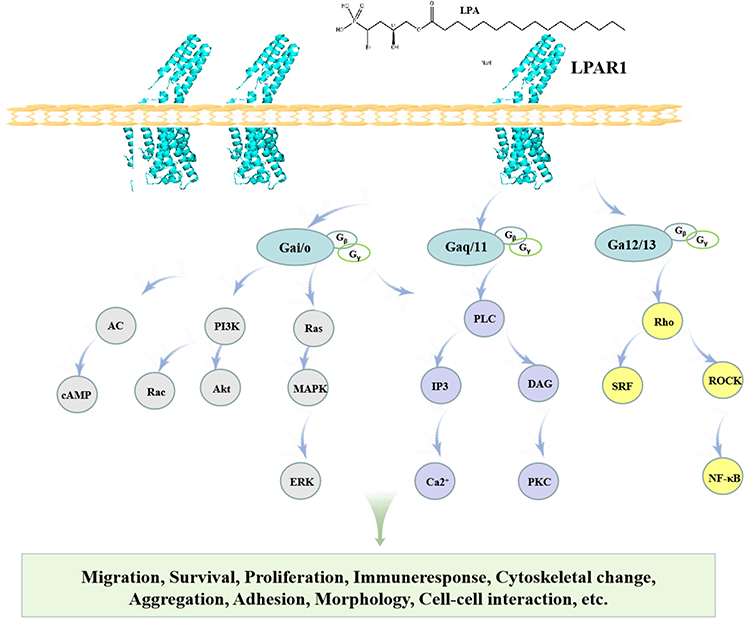 |
Figure 2 LPAR1 on the cell surface activates a downstream pathway that mediates a variety of cellular responses. |
Current Research Status of LPAR1 and VEGFR2-Targeted Drugs
Current Research Status of VEGFR2-Targeted Drugs
We summarize the FDA approved VEGFR-related drug information, as shown in Table 1. We summarize the development of drugs related to targeted inhibition of VEGFR2, as shown in Table 2. Because there are many problems such as unsatisfactory therapeutic effect, high toxicity or long R & D process, it is necessary to further optimize this R & D strategy and program.
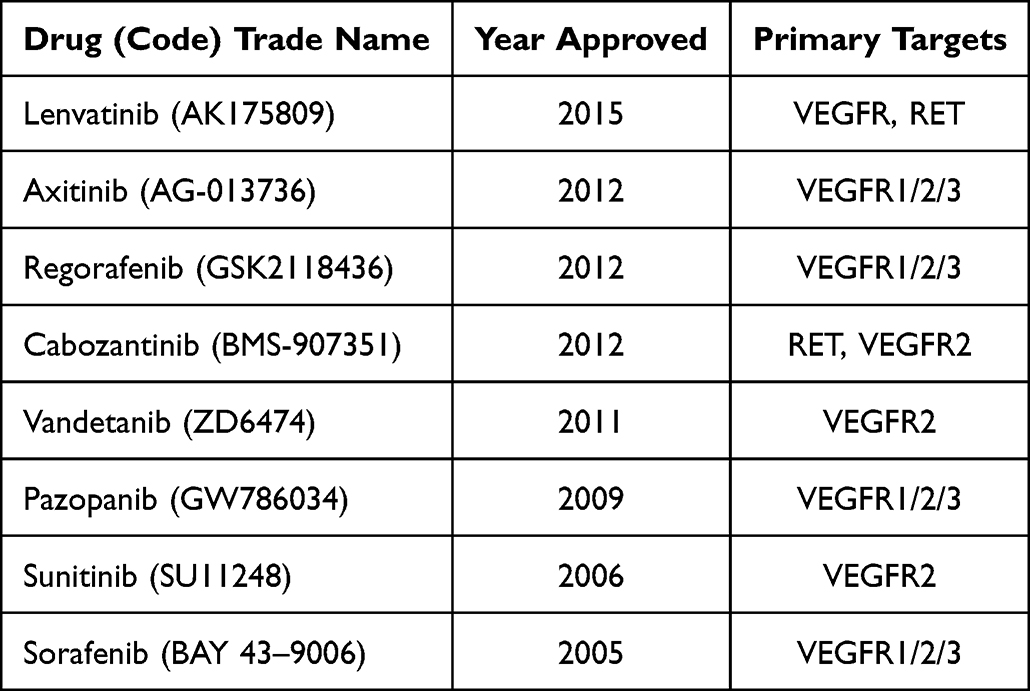 |
Table 1 FDA Approved VEGFR Related Drug Information |
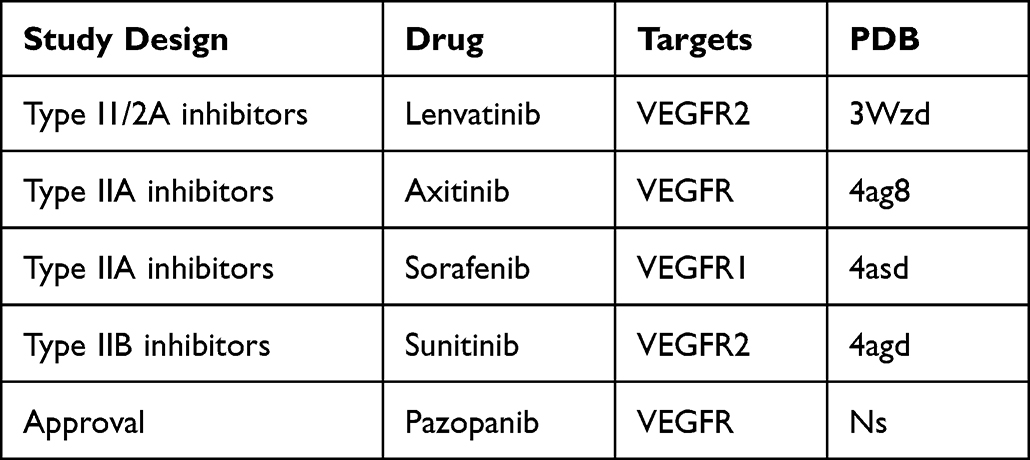 |
Table 2 Study Design, Drug Name, Target, and PDB Number Targeting VEGFR2 |
Current Status of Studies of LPAR1-Targeted Agents in IPF
It has been shown that LPAR1 is the most highly expressed LPAR on fibroblasts obtained from patients with idiopathic pulmonary fibrosis. AM095 is a selective, orally bioavailable LPAR1 antagonist with mean half-maximal inhibitory concentration values of 0.98 and 0.73 µΜ for human and mouse LPAR1, respectively.51 Swaney et al47 showed that 7 days of treatment with the selective LPAR1 antagonist AM966 or AM095 (10 mg/kg/day by gavage) conferred similar protection against BLM-induced lung fibrosis in LPAR1-KO mice. Results from a Phase 2 study showed that IPF patients treated with BMS-986020 600 mg BID showed a significantly slower rate of decline from baseline to 26 weeks compared with placebo. However, they presented in the clinic as a result of hepatobiliary toxicity characterized by treatment-related liver enzyme elevations and cholecystitis and, in addition, were discontinued prior to clinical development due to the poor nonclinical pharmacokinetic profile of BMS-986020. Preclinical studies have shown that these adverse events are off-target, drug-specific, and independent of LPAR1 antagonism. Biomarkers of fibrosis or inflammation were improved following BMS-986020 treatment, suggesting that targeting LPAR1 has potential clinical utility. Animal model experiments clearly show that LPAR1 antagonists have anti-fibrotic effects. This offers broad prospects for designing new treatments to prevent fibrosis-related diseases. However, the number of currently available effective LPAR1 antagonists remains low and none have been used in clinical trials to date.52,53 We summarize the development of drugs targeting LPAR1, as shown in Table 3. In addition, LPA, a bioactive lipid produced extracellularly by autotaxin (ATX). It is worth noting that autotaxin inhibitors also play an important role in IPF. Studies have shown that new indole-based high-efficiency ATX inhibitors play an important role in mouse pulmonary fibrosis models.54 In a 2a randomized placebo-controlled trial, GLPG1690 (Galapagos, Mechelen, Belgium) was found to be a novel, effective and selective autotaxin inhibitor with good oral availability in IPF patients.55
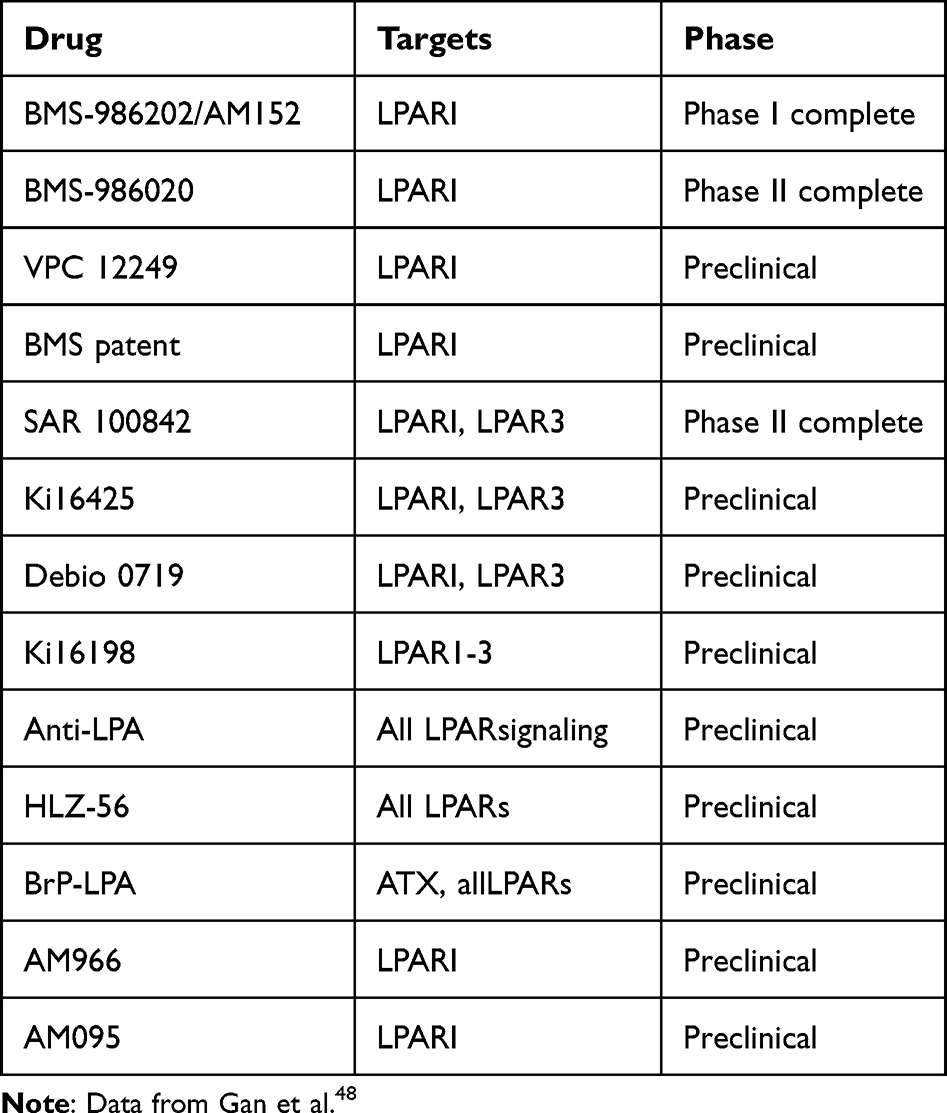 |
Table 3 Summary of Compounds That Target LPAR1 Signaling. |
Although the combination can lead to drawbacks such as increased adverse reactions and increased treatment costs,56 single target treatment regimens have limited effect, so drug research and development with multi-target effects has more practical application prospects.
Prospects for the Combination of VEGFR2 and LPAR1 Dual Targets in the Treatment of IPF
Angiogenesis, lung epithelial cells, fibroblasts, and ECM contribute to the development of pulmonary fibrosis. VEGFR2 and LPAR1 bind to their ligands and are involved in regulating vascular leakage and promoting collagen deposition in the matrix through different pathways.
Joint Involvement in Vascular Leakage
Neovascularization underlies tissue repair following injury, although the role of angiogenesis in IPF is unknown.57 Several angiogenic growth factors have been implicated in the development of pulmonary vasculature. Among them, VEGF, which is involved in angiogenesis, is the most extensively studied. VEGF is a powerful angiogenic factor and impaired expression and signaling of VEGF adversely impacts not only the development and maintenance of blood vessels, but also the structure and integrity of the entire lung.58–60 Blockade of the VEGF receptor family has been shown to result in anti-angiogenic effects.61 VEGF-A binds two tyrosine kinase receptors, VEGFR1 and VEGFR2, and regulates ECs proliferation, migration, vascular permeability, secretion, and other endothelial functions. Numerous studies have shown that VEGFR2 is considered to be a major sensor of VEGF signaling in ECs.62
The profibrotic effect of LPAR1 receptor stimulation may be explained by LPAR1 receptor-mediated vascular leakage, which is a profibrotic event. LPAR1 is involved in IPF development because of its role in regulating vascular leakage in animal models. For example, in BLM models of pulmonary fibrosis,45 LPA stimulates fibroblast migration through LPAR1, whereas LPAR1-deficient mice are protected from fibrosis by attenuating fibroblast recruitment and vascular leakage, suggesting that reducing LPA-mediated LPAR1 signaling protects the body ‘s blood vessels.
Co-Involvement in Collagen Deposition
VEGF can promote fibrosis through angiogenesis and cooperation with TGF-β, thereby enhancing ECM production by fibroblasts.63 In addition, VEGF also indirectly promotes cell migration thereby directly affecting ECM synthesis.64 In patients with IPF, fibroblasts migrate into the fibrinous rich exudate that forms in the lung space following injury; the extent of this migration generally corresponds to the severity of IPF symptoms. In BALF from IPF patients, LPA was increased, whereas inhibition of LPAR1 suppressed fibroblast chemotaxis induced by BALF from IPF patients.45 Regarding the mechanisms by which LPA and its receptors promote pulmonary fibrosis, it has been shown that after lung injury, myofibroblasts accumulate and secrete excessive ECM, and eventually form fibrotic foci.65
In summary, we summarize the relationship between VEGFR2 and LPAR1 and vascular leakage, lung epithelial cells, fibroblasts, and collagen deposition (Figure 3).
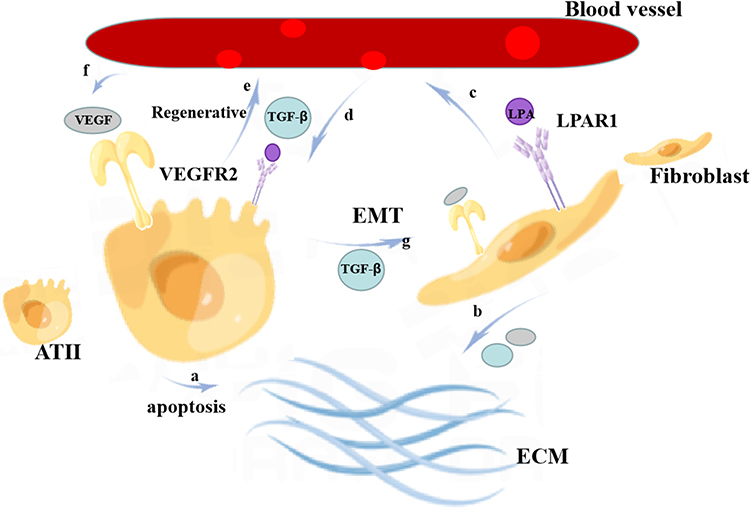 |
Figure 3 Relationship between VEGFR2 and LPAR1 and angiogenesis, lung epithelial cells, fibroblasts, and ECM. a: Type II alveolar epithelial cells (ATII) promote ECM accumulation through apoptotic pathways; b: fibroblasts promote fibrosis through angiogenesis and cooperation with TGF-β by resisting apoptosis, secreting collagen, and VEGF, thereby enhancing ECM production in fibroblasts; c: fibroblasts have the expression of LPAR1 and VEGFR2, the binding of LPA to LPAR1, VEGF to VEGFR2, and promoting fibroblast recruitment, vascular leakage, and endothelial barrier dysfunction; d, e: as a positive feedback loop, VEGF-A binds VEGFR1 and free TGF-β on ATII and regulates ATII cell injury as well as endothelial cell proliferation, migration, vascular permeability, secretion, microvessel formation, and other endothelial functions; f: VEGF from blood vessels may enhance epithelial cell proliferation and resistance to apoptosis in an autocrine manner; g: ATII transforms into fibroblasts; therefore, LPAR1 and VEGFR2 eventually lead to excessive ECM deposition and vascular leakage. |
Relationship Between LPA-LPAR and VEGF-VEGFR Pathways
There is an upstream downstream relationship between the LPA LPAR and VEGF-VEGFR signaling pathways, which is also worth further exploration. Wu PY found that LPA induced VEGFA expression in PC-3 prostate cancer cells.66 Dutta S report suggests that VEGF-VEGFR-2 signaling is involved in LPA-induced epithelial ovarian cancer invasion. In addition, the effects of LPA on VEGF-VEGFR-2 signaling and epithelial ovarian cancer invasion were found to be mediated by activation of the NF-κB pathway.67 Lin CE proposed that LPA enhanced VEGF-C expression in human ECs. In prostate cancer lymphatic metastasis experiments, LPA can enhance VEGF-C expression in a manner dependent on activation of LPAR1/3, ROS and other pathways.68 It has been shown that multiple Sp-1 sites within the VEGF promoter response region are critical for LPA-mediated transcription. By summarizing the above studies, we review the relationship between LPA-LPAR and VEGF-VEGFR pathways in disease states (Figure 4), indicating that VEGF is indeed an important mediator present downstream of LPA.
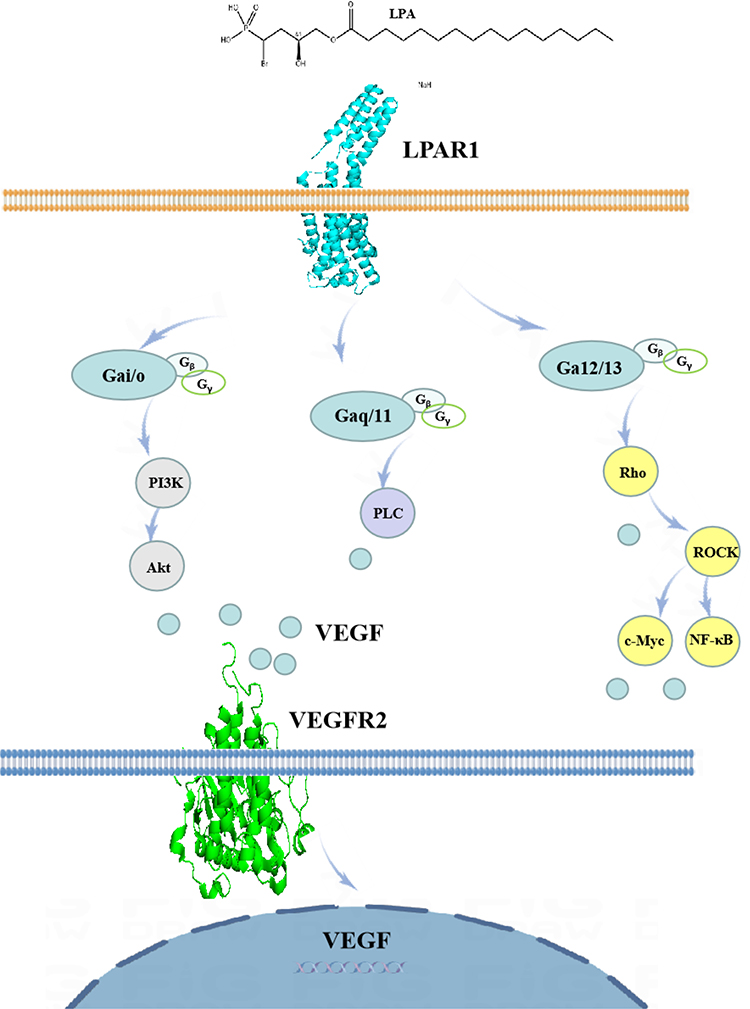 |
Figure 4 Diagram of the relationship between LPA-LPAR and VEGF-VEGFR pathways in disease states. |
Conclusion
Inhibition of VEGF helps to limit local vascular proliferation, but it may also adversely affect vascular integrity by promoting EC apoptosis.69 It was found that the physiological repair function of capillaries in nail bed was damaged due to the blocking of VEGFR. However, the linear hemorrhage under the nail can gradually disappear with the growth of the nail, without special treatment. Therefore, from the point of view of inhibiting its critical receptor, we chose to inhibit VEGFR2. In addition, inhibition of LPA acts as a key factor in the body. LPA is present in almost all cells, tissues, and body fluids,70 and once inhibited, disturbances in the internal environment of the body occur, and we consider the key receptor protein that inhibits its binding to be LPAR1. It has been shown that LPA increases in BALF from IPF patients, whereas inhibition of LPAR1 suppresses fibroblast chemotaxis induced by BALF from IPF patients.45 Interestingly, both LPAR1 and VEGFR2 are associated with angiogenesis.
Based on the current status of multi-targeted drug development, there are similar drug studies; for example, there are many reports in the literature on drugs that produce synergistic anti-angiogenic effects between VEGFR and FGFR in vivo. Similar synergy has been found between the two in lymphangiogenesis, and its inhibition by dual FGFR/VEGFR inhibitors may more readily prevent metastasis.71 In IPF drugs, multi-target synergy has long been used clinically, for example nintedanib.72 Based on the low toxicity and multi-point effect characteristics of traditional Chinese medicine, the screening and development of dual-target drugs may have obvious advantages in the treatment of IPF.73 For example, Tanshinone IIA sodium sulfonate can treat pulmonary hypertension, lung disease and other diseases by inhibiting the VEGFR receptor, and is in Phase 3 clinical trial.
However, the current study does not address the concept of dual inhibition of VEGFR2, two targets of LPAR1. We found that simultaneous inhibition of VEGFR2 and LPAR1 is very promising through the above studies. Because they mediate common signaling pathways, such as PI3K/AKT, RAS/RAF, etc.,74,75 at the same time, VEGFR2, LPAR1 are also closely related to angiogenesis and leakage, and although the relationship between angiogenesis and leakage and IPF remains unclear, reports on inhibiting the activity of the two, and thereby alleviating the progression of IPF are common. Notably, ECM, EMT is an important factor in the development of IPF. Activation of VEGFR2 and LPAR1 can promote ECM accumulation and EMT transformation. Therefore, it is very important to inhibit the expression of VEGFR2 and LPAR1 at the same time, which provides the possibility for considering one drug and two targets, or two drugs and two targets in clinical practice. Both VEGFR2 and LPAR1 targets have the effect of regulating blood vessels, and can interact with each other. They have the same characteristics in promoting disease progression and can strengthen each other. Therefore, if VEGFR2 and LPAR1 double targets are modulated at the same time, the effect of controlling angiogenesis and delaying disease progression will be more obvious than that of single target drug: VEGFR2 mainly expresses ATII while LPAR1 is mainly expressed in fibroblasts, and they act on different cells to promote extracellular matrix collagen deposition by different mechanisms. If double-target intervention is performed at the same time, the effect of hindering collagen production will be more obvious; conventional drug therapy only acts on one of the targets and its downstream pathways, and the other target and its pathway promote angiogenesis and collagen deposition are not limited, resulting in limited therapeutic effect; at the same time, inhibition of the two targets may effectively delay the progression of the disease.
However, it is worth noting that in the interaction between the two drugs and the two targets it should be considered whether the two drugs competitively bind to each other’s active site and serve as dual antagonists. In addition, the ADMET of both drugs by pH in vivo must also be considered. Because the combination of the two drugs corresponds to the combination of the two targets, the selection of its dosage should be cautious to prevent the occurrence of toxic adverse effects on the gastrointestinal tract, liver and kidney.
In conclusion, in this study, we described the mechanism of VEGF and LPA pathways in the progress of Pulmonary fibrosis, and suggested that more attention should be paid to the intervention of VEGFR2 and LPA1 activation in the development of clinical drugs in the future.
Data Sharing Statement
Relevant data and materials are available upon request to corresponding authors.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Acknowledgments
Ya-Li Luo and Yan Li are co-first authors for this study. We acknowledge Gansu University of Chinese Medicine, Provincial-level Key Laboratory for Molecular Medicine of Major Diseases and The Prevention and Treatment with Traditional Chinese Medicine Research in Gansu Colleges and University for providing support and assistance for this article. The authors would like to express their gratitude to EditSprings for the expert linguistic services provided.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from National Nature Foundation (82160846), provincial Natural Science Foundation (20JR10RA321), Gansu Provincial Higher Education Innovation Fund Project (2021A-085).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–816. doi:10.1164/rccm.200602-163OC
2. Lederer DJ, Martinez FJ, Longo DL. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811–1823. doi:10.1056/NEJMra1705751
3. Proesmans VLJ, Drent M, Elfferich MDP, Wijnen PAHM, Jessurun NT, Bast A. Self-reported gastrointestinal side effects of antifibrotic drugs in Dutch idiopathic pulmonary fibrosis patients. Lung. 2019;197(5):551–558. doi:10.1007/s00408-019-00260-1
4. Ikeda S, Sekine A, Baba T, et al. Low body surface area predicts hepatotoxicity of nintedanib in patients with idiopathic pulmonary fibrosis. Sci Rep. 2017;7(1):10811. doi:10.1038/s41598-017-11321-x
5. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–1952. doi:10.1016/S0140-6736(17)30866-8
6. Spagnolo P, Kropski JA, Jones MG, et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798. doi:10.1016/j.pharmthera.2020.107798
7. Mercer PF, Woodcock HV, Eley JD, et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax. 2016;71(8):701–711. doi:10.1136/thoraxjnl-2015-207429
8. Lukey PT, Harrison SA, Yang S, et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur Respir J. 2019;53(3):1801992. doi:10.1183/13993003.01992-2018
9. Valdés-Rives SA, González-Arenas A. Autotaxin-lysophosphatidic acid: from inflammation to cancer development. Mediators Inflamm. 2017;2017:9173090. doi:10.1155/2017/9173090
10. Gill MW, Murphy BJ, Cheng PTW, Sivaraman L, Davis M, Lehman-McKeeman L. Mechanism of hepatobiliary toxicity of the LPA1 antagonist BMS-986020 developed to treat idiopathic pulmonary fibrosis: contrasts with BMS-986234 and BMS-986278. Toxicol Appl Pharmacol. 2022;438:115885. doi:10.1016/j.taap.2022.115885
11. Mura M, Binnie M, Han B, et al. Functions of type II pneumocyte-derived vascular endothelial growth factor in alveolar structure, acute inflammation, and vascular permeability. Am J Pathol. 2010;176(4):1725–1734. doi:10.2353/ajpath.2010.090209
12. Eguchi R, Kawabe JI, Wakabayashi I. VEGF-independent angiogenic factors: beyond VEGF/VEGFR2 signaling. J Vasc Res. 2022;59(2):78–89. doi:10.1159/000521584
13. Amano H, Mastui Y, Ito Y, et al. The role of vascular endothelial growth factor receptor 1 tyrosine kinase signaling in bleomycin-induced pulmonary fibrosis. Biomed Pharmacother. 2019;117:109067. doi:10.1016/j.biopha.2019.109067
14. Shea BS, Tager AM. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc Am Thorac Soc. 2012;9(3):102–110. doi:10.1513/pats.201201-005AW
15. Huang X, Wang X, Xie X, et al. Kallistatin protects against bleomycin-induced idiopathic pulmonary fibrosis by inhibiting angiogenesis and inflammation. Am J Transl Res. 2017;9(3):999–1011.
16. Saito S, Alkhatib A, Kolls JK, Kondoh Y, Lasky JA. Pharmacotherapy and adjunctive treatment for idiopathic pulmonary fibrosis (IPF). J Thorac Dis. 2019;11(Suppl 14):S1740–S1754. doi:10.21037/jtd.2019.04.62
17. Spagnolo P, Bonella F, Ryerson CJ, Tzouvelekis A, Maher TM. Shedding light on developmental drugs for idiopathic pulmonary fibrosis. Expert Opin Investig Drugs. 2020;29(8):797–808. doi:10.1080/13543784.2020.1782885
18. Kolb M, Bonella F, Wollin L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir Med. 2017;131:49–57. doi:10.1016/j.rmed.2017.07.062
19. Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol. 2006;290:L209–L221. doi:10.1152/ajplung.00185.2005
20. Kaner RJ, Crystal RG. Compartmentalization of vascular endothelial growth factor to the epithelial surface of the human lung. Mol Med. 2001;7(4):240–246. doi:10.1007/BF03401843
21. Ke H, Masoumi KC, Ahlqvist K, Seckl MJ, Rydell-Törmänen K, Massoumi R. Nemo-like kinase regulates the expression of vascular endothelial growth factor (VEGF) in alveolar epithelial cells. Sci Rep. 2016;6(1):23987. PMID: 27035511; PMCID: PMC4817507. doi:10.1038/srep23987
22. Barratt SL, Blythe T, Jarrett C, et al. Differential expression of VEGF-Axxx isoforms is critical for development of pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(4):479–493. doi:10.1164/rccm.201603-0568OC
23. Wu WK, Llewellyn OP, Bates DO, Nicholson LB, Dick AD. IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia. Immunobiology. 2010;215(9–10):796–803. doi:10.1016/j.imbio.2010.05.025
24. Stockmann C, Kerdiles Y, Nomaksteinsky M, et al. Loss of myeloid cell-derived vascular endothelial growth factor accelerates fibrosis. Proc Natl Acad Sci U S A. 2010;107(9):4329–4334. doi:10.1073/pnas.0912766107
25. Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/ Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9:225–230. doi:10.1007/s10456-006-9055-8
26. Lin CK, Lin YH, Huang TC, Shi CS, Yang CT, Yang YL. VEGF mediates fat embolism-induced acute lung injury via VEGF receptor 2 and the MAPK cascade. Sci Rep. 2019;9(1):11713. PMID: 31406128; PMCID: PMC6690961. doi:10.1038/s41598-019-47276-4
27. Melincovici CS, Boşca AB, Şuşman S, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 2018;59(2):455–467.
28. Zachary I. VEGF Signalling: Integration and Multi-Tasking in Endothelial Cell Biology. Portland Press Limited; 2003.
29. Fehrenbach H, Kasper M, Haase M, Schuh D, Muller M. Differential immunolocalization of VEGF in rat and human adult lung, and in experimental rat lung fibrosis: light, fluorescence, and electron microscopy. Anat Rec. 1999;254(1):61–73. doi:10.1002/(SICI)1097-0185(19990101)254:1<61::AID-AR8>3.0.CO;2-D
30. Barratt SL, Blythe T, Ourradi K, et al. Effects of hypoxia and hyperoxia on the differential expression of VEGF-A isoforms and receptors in Idiopathic Pulmonary Fibrosis (IPF). Respir Res. 2018;19(1):9. doi:10.1186/s12931-017-0711-x
31. Ando M, Miyazaki E, Ito T, et al. Significance of serum vascular endothelial growth factor level in patients with idiopathic pulmonary fibrosis. Lung. 2010;188(3):247–252. doi:10.1007/s00408-009-9223-x
32. Jaskiewicz K, Mycroft K, Maskey-Warzechowska M, et al. Exhaled biomarkers in idiopathic pulmonary fibrosis-a six-month follow-up study in patients treated with pirfenidone. J Clin Med. 2020;9(8):2523. PMID: 32764328; PMCID: PMC7465603. doi:10.3390/jcm9082523
33. Simler NR. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax. 2004;59(7):581–585. doi:10.1136/thx.2003.009860
34. Farkas L, Farkas D, Ask K, et al. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest. 2009;119(5):1298–311.100. doi:10.1172/JCI36136
35. Iyer AKV, Ramesh V, Castro CA, et al. Nitric oxide mediates bleomycin‐induced angiogenesis and pulmonary fibrosis via regulation of VEGF. J Cell Biochem. 2015;116:2484–2493. doi:10.1002/jcb.25192
36. Lee CG, Link H, Baluk P, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med. 2004;10(10):1095–1103. doi:10.1038/nm1105
37. Borensztajn K, Crestani B, Kolb M. Idiopathic pulmonary fibrosis: from epithelial injury to biomarkers--insights from the bench side. Respiration. 2013;86(6):441–452. doi:10.1159/000357598
38. Zhou W, Liu K, Zeng L, et al. Targeting VEGF-A/VEGFR2 Y949 signaling-medi- ated vascular permeability alleviates hypoxic pulmonary hypertension. Circulation. 2022;146(24):1855–1881. doi:10.1161/CIRCULATIONAHA.122.061900
39. Watanabe H, Ichihara E, Kayatani H, et al. VEGFR2 blockade augments the effects of tyrosine kinase inhibitors by inhibiting angiogenesis and oncogenic signaling in oncogene-driven non-small-cell lung cancers [published correction appears in Cancer Sci.2022Jan;113(1):365]. Cancer Sci. 2021;112(5):1853–1864. doi:10.1111/cas.14801
40. Ou XM, Li WC, Liu DS, et al. VEGFR-2 antagonist SU5416 attenuates bleomycin-induced pulmonary fibrosis in mice. Int Immunopharmacol. 2009;9(1):70–79. doi:10.1016/j.intimp.2008.10.002
41. Vogt W. Pharmacologically active acidic phospholipids and glycolipids. Biochem Pharmacol. 1963;12:415–420. doi:10.1016/0006-2952(63)90074-1
42. Li ZW, Zhao YR, Zhao C, Fu R, Li ZY. [Function and biological activities of the autotaxin-LPA axis]. Sheng Li Xue Bao. 2011;63(6):601–610. Chinese
43. Yin Z, Watsky MA. Chloride channel activity in human lung fibroblasts and myofibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1110–L1116. doi:10.1152/ajplung.00344.2004
44. Shiomi T, Boudreault F, Padem N, Higashiyama S, Drazen JM, Tschumperlin DJ. Lysophosphatidic acid stimulates epidermal growth factor–family ectodomain shedding and paracrine signaling from human lung fibroblasts. Wound Repair Regen. 2011;19:229–240. doi:10.1111/j.1524-475X.2010.00655.x
45. Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. doi:10.1038/nm1685
46. Montesi SB, Mathai SK, Brenner LN, et al. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm Med. 2014;14:5. PMID: 24468008; PMCID: PMC3906883. doi:10.1186/1471-2466-14-5
47. Swaney JS, Chapman C, Correa LD, et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br J Pharmacol. 2010;160:1699–1713.
48. Gan L, Xue JX, Li X, et al. Blockade of lysophosphatidic acid receptors LPAR1/3 ameliorates lung fibrosis induced by irradiation. Biochem Biophys Res Commun. 2011;409:7–13. doi:10.1016/j.bbrc.2011.04.084
49. Cha B, Chen T, Sarker R, et al. Lysophosphatidic acid stimulation of NHE3 exocytosis in polarized epithelial cells occurs with release from NHERF2 via ERK-PLC-PKCδ signaling. Am J Physiol Cell Physiol. 2014;307(1):C55–C65. doi:10.1152/ajpcell.00045.2014
50. Plastira I, Bernhart E, Joshi L, et al. MAPK signaling determines lysophosphatidic acid (LPA)-induced inflammation in microglia. J Neuroinflammation. 2020;17(1):127. PMID: 32326963; PMCID: PMC7178949. doi:10.1186/s12974-020-01809-1
51. Swaney JS, Chapman C, Correa LD, et al. Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J Pharmacol Exp Ther. 2011;336(3):693–700. doi:10.1124/jpet.110.175901
52. Cheng PTW, Kaltenbach RF III, Hao Z, et al. Discovery of an oxycyclohexyl acid lysophosphatidic acid receptor 1 (LPA1) antagonist BMS-986278 for the treatment of pulmonary fibrotic diseases. J Med Chem. 2021;64(21):15549–15581. doi:10.1021/acs.jmedchem.1c01256
53. Palmer SM, Snyder L, Todd JL, et al. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest. 2018;154(5):1061–1069. doi:10.1016/j.chest.2018.08.1058
54. Lei H, Guo M, Li X, et al. Discovery of novel indole-based allosteric highly potent ATX inhibitors with great in vivo efficacy in a mouse lung fibrosis model. J Med Chem. 2020;63(13):7326–7346. doi:10.1021/acs.jmedchem.0c00506
55. Maher TM, van der Aar EM, Van de Steen O, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. 2018;6(8):627–635. doi:10.1016/S2213-2600(18)30181-4
56. Zhang Y, Lu P, Qin H, et al. Traditional Chinese medicine combined with pulmonary drug delivery system and idiopathic pulmonary fibrosis: rationale and therapeutic potential. Biomed Pharmacother. 2021;133:111072. PMID: 33378971; PMCID: PMC7836923. doi:10.1016/j.biopha.2020.111072
57. Garantziotis S, Zudaire E, Trempus CS, et al. Serum inter-alpha-trypsin inhibitor and matrix hyaluronan promote angiogenesis in fibrotic lung injury. Am J Respir Crit Care Med. 2008;178(9):939–947. doi:10.1164/rccm.200803-386OC
58. Ng YS, Rohan R, Sunday ME, Demello DE, D’Amore PA. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn. 2001;220:112–121. doi:10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D
59. Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1–deficient mice. Nature. 1995;376:62–66. doi:10.1038/376062a0
60. Jakkula M, Le Cras TD, Gebb S, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L600–L607. doi:10.1152/ajplung.2000.279.3.L600
61. Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang Y-S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17:1–12. doi:10.1186/s12943-018-0801-5
62. Peach CJ, Mignone VW, Arruda MA, et al. Molecular pharmacology of VEGF-A isoforms: binding and signalling at VEGFR2. Int J Mol Sci. 2018;19(4):1264. PMID: 29690653; PMCID: PMC5979509. doi:10.3390/ijms19041264
63. Elbialy ZI, Assar DH, Abdelnaby A, et al. Healing potential of Spirulina platensis for skin wounds by modulating bFGF, VEGF, TGF-ß1 and α-SMA genes expression targeting angiogenesis and scar tissue formation in the rat model. Biomed Pharmacother. 2021;137:111349. PMID: 33567349. doi:10.1016/j.biopha.2021.111349
64. Kretschmer M, Rüdiger D, Zahler S. Mechanical aspects of angiogenesis. Cancers. 2021;13(19):4987. PMID: 34638470; PMCID: PMC8508205. doi:10.3390/cancers13194987
65. Tang N, Zhao Y, Feng R, et al. Lysophosphatidic acid accelerates lung fibrosis by inducing differentiation of mesenchymal stem cells into myofibroblasts. J Cell Mol Med. 2014;18(1):156–169. doi:10.1111/jcmm.12178
66. Wu PY, Lin YC, Lan SY, Huang YL, Lee H. Aromatic hydrocarbon receptor inhibits lysophosphatidic acid-induced vascular endothelial growth factor-A expression in PC-3 prostate cancer cells. Biochem Biophys Res Commun. 2013;437(3):440–445. doi:10.1016/j.bbrc.2013.06.098
67. Dutta S, Wang FQ, Wu HS, Mukherjee TJ, Fishman DA. The NF-κB pathway mediates lysophosphatidic acid (LPA)-induced VEGF signaling and cell invasion in epithelial ovarian cancer (EOC). Gynecol Oncol. 2011;123(1):129–137. doi:10.1016/j.ygyno.2011.06.006
68. Lin CE, Chen SU, Lin CC, et al. Lysophosphatidic acid enhances vascular endothelial growth factor-C expression in human prostate cancer PC-3 cells. PLoS One. 2012;7(7):e41096. doi:10.1371/journal.pone.0041096
69. Laddha AP, Kulkarni YA. VEGF and FGF-2: promising targets for the treatment of respiratory disorders. Respir Med. 2019;156:33–46. doi:10.1016/j.rmed.2019.08.003
70. Ninou I, Magkrioti C, Aidinis V. Autotaxin in pathophysiology and pulmonary fibrosis. Front Med. 2018;5:180. PMID: 29951481; PMCID: PMC6008954. doi:10.3389/fmed.2018.00180
71. Cao R, Ji H, Feng N, et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc Natl Acad Sci U S A. 2012;109(39):15894–15899. doi:10.1073/pnas.1208324109
72. Wind S, Schmid U, Freiwald M, et al. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin Pharmacokinet. 2019;58(9):1131–1147. PMID: 31016670; PMCID: PMC6719436. doi:10.1007/s40262-019-00766-0
73. Li LC, Kan LD. Traditional Chinese medicine for pulmonary fibrosis therapy: progress and future prospects. J Ethnopharmacol. 2017;198:45–63. doi:10.1016/j.jep.2016.12.042
74. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273(2):114–127. doi:10.1111/joim.12019
75. Wang Y, Zhang F, Wang J, et al. lncRNA LOC100132354 promotes angiogenesis through VEGFA/VEGFR2 signaling pathway in lung adenocarcinoma. Cancer Manag Res. 2018;10:4257–4266. doi:10.2147/CMAR.S177327
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.