")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Inhibition of cardiac Kv1.5 potassium current by the anesthetic midazolam: mode of action
Authors Vonderlin N, Fischer F, Zitron E, Seyler C, Scherer D, Thomas D , Katus HA, Scholz E
Received 1 July 2014
Accepted for publication 31 July 2014
Published 7 November 2014 Volume 2014:8 Pages 2263—2271
DOI https://doi.org/10.2147/DDDT.S70461
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Nadine Vonderlin,1 Fathima Fischer,1 Edgar Zitron,1,2 Claudia Seyler,1 Daniel Scherer,1 Dierk Thomas,1,2 Hugo A Katus,1,2 Eberhard P Scholz1
1Department of Internal Medicine III, University Hospital Heidelberg, Heidelberg, Germany; 2German Centre for Cardiovascular Research (DZHK), Partner Site Heidelberg/Mannheim, Heidelberg, Germany
Abstract: Midazolam is a short-acting benzodiazepine that is widely used in anesthesia. Despite its widespread clinical use, detailed information about cardiac side effects of midazolam is largely lacking. Using the double-electrode voltage clamp technique, we studied pharmacological effects of midazolam on heterologously expressed Kv1.5 channels underlying atrial repolarizing current IKur. Midazolam dose-dependently inhibited Kv1.5 current, yielding an IC50 of 17 µM in an HEK cell line and an IC50 of 104 µM in Xenopus oocytes. We further showed that midazolam did not affect the half-maximal activation voltage of Kv1.5 channels. However, a small negative shift of the inactivation curve could be observed. Midazolam acted as a typical open-channel inhibitor with rapid onset of block and without frequency dependence of block. Taken together, midazolam is an open channel inhibitor of cardiac Kv1.5 channels. These data add to the current understanding of the pharmacological profile of midazolam.
Keywords: anesthetics, potassium channels, pharmacology
Introduction
Midazolam is a short-acting imidazobenzodiazepine that is widely used in anesthesia.1,2 Similar to classic benzodiazepines, midazolam potentiates the effect of GABA by increasing the frequency of chloride channel opening in the brain.3,4 From a clinical point of view, midazolam bears a number of beneficial characteristics, such as a rapid onset of action as well as the possibility of pharmacological antagonism.1 Considering its favorable pharmacological profile, midazolam is increasingly used by cardiologists in patients undergoing electrophysiological procedures such as electrical cardioversion, catheter ablation of atrial fibrillation, or catheter ablation of ventricular tachycardia.5–7 Within this context, detailed information on the pharmacological side effects of midazolam on cardiac electrophysiology seems necessary. Interestingly, a variety of other anesthetics have been shown to interfere with cardiac ion channels, thereby exerting potential pro- or antiarrhythmic side effects (for review, see Hüneke et al8 and Staikou et al9). For example, Miró et al published a small case series of patients exhibiting a conversion from atrial fibrillation to sinus rhythm after intravenous administration of propofol.10 From the literature available so far, midazolam has not been associated with severe pro- or antiarrhythmic side effects. However, it is well recognized that midazolam application might influence cardiac repolarization, thereby prolonging the QT interval.9,11 In experimental studies, midazolam has been shown to exert inhibitory effects on the calcium-independent transient outward current Ito and the inward rectifier current IK1, as well as on a neural delayed rectifier current IK(DR).12,13 However, effects of midazolam on the main repolarizing atrial delayed rectifier current IKur or its molecular correlate, the Kv1.5 ion channel, have not yet been reported.
The family of voltage-gated K+ channels (Kv) is characterized by a pore-forming α-subunit containing six membrane-spanning domains, a highly charged voltage-sensing S4 domain, and a K+ selectivity filter.1,14 Kv channels play an essential role in the regulation of the cardiac action potential. As a consequence, pharmacological inhibition of Kv channels represents a cornerstone of antiarrhythmic drug therapy. Among the family of Kv channels, Kv1.5 belongs to the first cloned Kv subfamily, termed “Shaker”. When heterologously expressed, Kv1.5 channels give rise to ultra-rapidly activating and merely inactivating potassium current. Kv1.5 has been identified as the molecular basis of the repolarizing ultra-rapidly activating delayed rectifier current IKur in human atrial myocytes.15,16 Hence, inhibition of IKur results in action potential prolongation in isolated trabeculae from human atria.17 Nearly absent in the ventricles and highly expressed in human atria, Kv1.5 represents a promising pharmacological target for atrial selective antiarrhythmic therapy.18 As a consequence, much effort is being devoted to identifying small molecule-inhibitors of Kv1.5.
Pharmacological inhibition of cardiac ion channels is a well-recognized side effect that has been reported for a number of clinically established anesthetics.19,20 However, despite its widespread clinical use, the effects of midazolam on cardiac ion channels have not yet been systematically studied. So far, inhibitory effects of midazolam on the calcium-independent transient outward current Ito, the inward rectifier potassium current IK1, and a neural delayed rectifier current (IK(DR)) have been described.12,13 The aim of the present study was to characterize pharmacological effects of midazolam on cardiac Kv1.5 channels underlying the important atrial repolarizing potassium current IKur. These data might, on the one hand, provide better insights into the pharmacological profile of midazolam and, on the other hand, help identify new lead compounds for the development of Kv1.5 inhibitors.
Methods
Drugs, chemicals, and drug administration
Midazolam (Hoffman-La Roche Ltd., Basel, Switzerland) was stored and handled according to the manufacturer’s specifications. On the day of the experiment, the stock solution was further diluted to the desired concentration with external solution. External and internal solutions were stored at 4°C. Double-electrode voltage-clamp experiments were carried out using a standard external solution containing 5 mM KCl, 100 mM NaCl, 1.5 mM CaCl2, 2 mM MgCl2, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH adjusted to 7.4 with NaOH). Electrodes were back-filled with 3 M KCl solution. As soon as a control measurement had been performed, electrodes were removed from the oocytes and cells were placed in small (10 mL) dishes for drug incubation (30 minutes). After incubation, cells were again placed in the recording chamber and the experiment was repeated. For the analysis of frequency dependence, cells were kept in the recording chamber after the control measurement without pulsing (at the holding potential). Midazolam was washed-in over a period of 15 minutes, and the measurements were repeated. For whole-cell patch-clamp experiments, the bath solution contained 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM HEPES, 1.8 mM CaCl2, and 10 mM glucose (pH adjusted to 7.4 with NaOH). The pipette solution contained 100 mM K-aspartate, 10 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 10 mM ethylene glycol tetraacetic acid (EGTA), 10 mM HEPES, and 40 mM glucose (pH adjusted to 7.2 with KOH). All electrophysiological measurements were carried out at room temperature (22°C).
Heterologous expression of Kv1.5 channel subunits in Xenopus oocytes
The Kv1.5 (GenBank accession number NM_002234) potassium channel clone was a generous gift of Dr B Wible (Cleveland, OH, USA). Out of the pSP64 plasmid, complementary Kv1.5-RNA was prepared using the mMESSAGE mMACHINE® in vitro transcription kit (Ambion, Huntingdon, UK) by use of SP6 polymerase after linearization with EcoRI (Hoffman-La Roche Ltd.). RNA injection into stage V and VI defolliculated oocytes was performed using a Nanoject automatic injector (Drummond, Broomall, USA). Xenopus frogs were kept under species-appropriate conditions at the Interfacultary Biomedical Faculty (IBF) of the University of Heidelberg, Heidelberg, Germany. Frogs were anesthetized with a solution containing 0.3% ethyl-3-aminobenzoate-methanesulfonic acid (15°C) for oocyte extraction. As soon as 600 to 800 oocytes had been carefully removed, the muscle layer and skin were sutured and the animal was revitalized. The volume of injected complementary RNA (cRNA) solution was 46 nL per oocyte, and measurements were carried out 1 to 3 days after injection. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No 85-23, revised 1996) and was approved by the regional administrative council (Regierungspräsidium Karlsruhe, Karlsruhe, Germany) (application number G-211/12).
Heterologous expression of Kv1.5 channels and immunostaining
Human embryonic kidney (HEK) 293 cells stably expressing Kv1.5 channels were obtained from CLS Cell Line Service (Eppelheim, Germany). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, and 100 μg/mL gentamicin in an atmosphere of 95% humidified air and 5% CO2 at 37°C. Cells were passaged regularly with trypsin/ethylenediaminetetraacetic acid (EDTA) and subcultured prior to treatment in 3 mL dishes.
Electrophysiological experiments and data analysis
Kv1.5 currents in Xenopus oocytes were obtained using the double-electrode voltage-clamp technique as reported previously.21 Microelectrodes had tip resistances ranging from 1 to 5 MΩ. Data were low-pass-filtered at 1 to 2 kHz (-3 dB, four-pole Bessel filter) before digitalization at 5 to 10 kHz. Recordings were performed using a commercially available amplifier (Warner OC-725A; Warner Instruments, Hamden, CT, USA) and the pCLAMP software package (Molecular Devices LLC, Sunnyvale, CA, USA) for data acquisition and analysis. For patch-clamp experiments, cells were transferred from the incubator into a recording chamber which was continuously rinsed with bath solution. For measurements, single cells were selected. Pipettes had resistances of 3–4 MΩ and whole-cell currents were measured with an RK-400 amplifier (Bio-Logic SAS, Claix, France) and analyzed with the pCLAMP software package. Dose-response curves were fitted to the Hill function:
Y = A1 + {[A2 − A1]/[1+10^log(X0 − X)*nH]}, | (1) |
with A1 being the bottom asymptote, A2 the top asymptote, logX0 being the center, and nH being the Hill slope. Activation and inactivation curves were fitted to a Boltzmann function:
Y = {{(A1 − A2)/[1 + (e^(X − X0)/k)]} + A2}, | (2) |
with A1 being the initial value, A2 being the final value, X0 representing the half-maximal activation potential, Y the degree of activation, and k the slope factor. Statistical data are expressed as mean ± standard error, with “n” representing the number of experiments performed. Statistical significance was evaluated using the independent Student’s t-test or the analysis of variance (ANOVA), followed by the Bonferroni post hoc analysis where necessary. Differences were considered significant when the P-value was <0.05.
Results
Midazolam inhibits heterologously expressed Kv1.5 channels
Inhibitory effects of midazolam on cloned Kv1.5 channels were first analyzed using the double-electrode voltage-clamp technique in Xenopus oocytes. From a holding potential of -80 mV, cells were depolarized by a variable voltage step (1,250 ms) ranging from -90 to +50 mV (10 mV increments). When activated with the depolarizing voltage step, Kv1.5 channels gave rise to a rapidly activating and slowly inactivating outward potassium current, reminiscent of cardiac ultra-rapidly delayed rectifier current IKur. Figure 1A exemplarily displays a single Kv1.5 current trace elicited by a voltage step to +50 mV. Incubation with midazolam (100 μM) resulted in a reduction of Kv1.5 current (representative experiment of n=8). Under control conditions, Kv1.5 currents exhibited a small run-up to 131.6%±25.1% (n=9). Incubation with increasing midazolam concentrations (10, 30, 100, 200, 300, and 1,000 μM) resulted in a dose-dependent change of Kv1.5 current amplitude by +4.8%±12.4%, -12.3%±12.3%, -39.1%±14.3%, -72.0%±8.9%, -86.2%±10.7%, and -94.9%±3.4%, respectively (n=6–9). The IC50 in the Xenopus oocyte expression system was 104.1±27.7 μM (n=6–9, nH=1.66±0.66) (Figure 1B). To underline the relevance of the observed effects, inhibitory effects of midazolam were next analyzed in a mammalian cell line (HEK 293) stably expressing Kv1.5 channels using the patch-clamp technique. Kv1.5 current was elicited using a single-step voltage protocol (600 ms) ranging from +60 to -80 mV (20 mV decrements). Figure 1C displays a typical current trace elicited by a step to +60 mV before and after application of 30 μM midazolam. As expected, compared to the Xenopus expression system, applications of 1, 3, 10, 30, and 100 μM midazolam resulted in more pronounced reductions, of 16.2%±5.7%, 25.7%±6.4%, 34.9%±2.8%, 54.6%±2.7%, and 65.6%±11.6%, respectively. The IC50 of inhibition was 17.0±4.2 μM (n=5–10, nH=1.50±0.4) (Figure 1D).
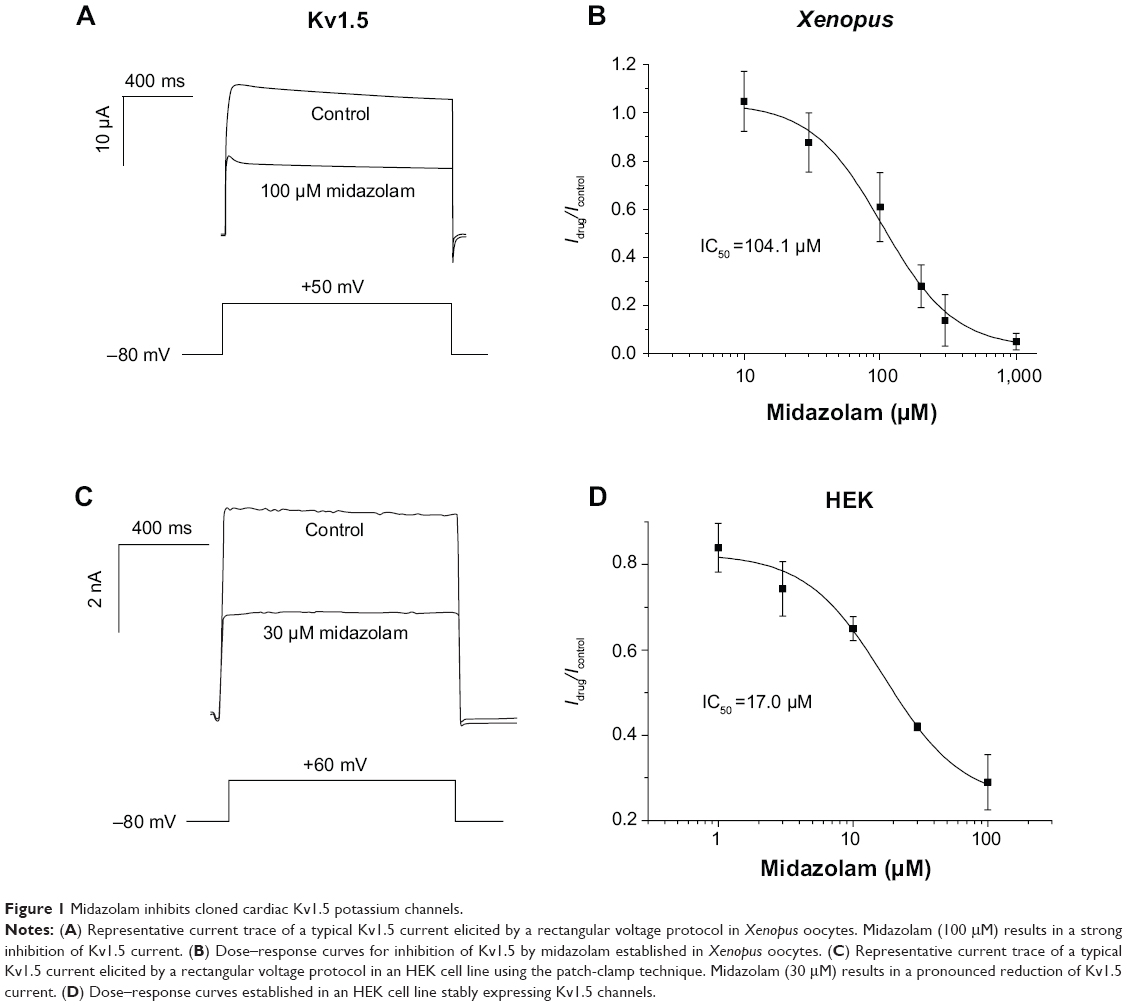 | Figure 1 Midazolam inhibits cloned cardiac Kv1.5 potassium channels. |
Pharmacological properties of Kv1.5 channel inhibition
Effects of midazolam on biophysical properties of Kv1.5 channels were further analyzed using a double-step voltage protocol. From a holding potential of -80 mV, cells were depolarized by a variable voltage step (1,250 ms) ranging from -90 to +50 mV (10 mV increments). This variable step was followed by a constant return pulse to +50 mV (50 ms) eliciting outward tail currents. Figure 2A and B display typical families of Kv1.5 current traces before and after application of midazolam (100 μM) (see inset for voltage protocol). The corresponding current–voltage relationship measured at peak current amplitude (measured at the first dashed line in Figure 2A and B) is displayed in Figure 2C. Kv1.5 channel activation curves were established, as reported previously, by dividing current amplitude by the corresponding electrochemical driving force (Figure 2D).21 The I-V relationship of each cell before and after midazolam application was fitted with a Boltzmann function in order to obtain half-maximal activation voltages (V1/2). Under control conditions, Kv1.5 channels revealed a mean V1/2 of 1.93±0.85 mV (n=6) (Figure 2D). Inhibition by midazolam did not significantly influence the V1/2 (V1/2 =3.21±1.47 mV, n=6, P>0.05) (Figure 2D).
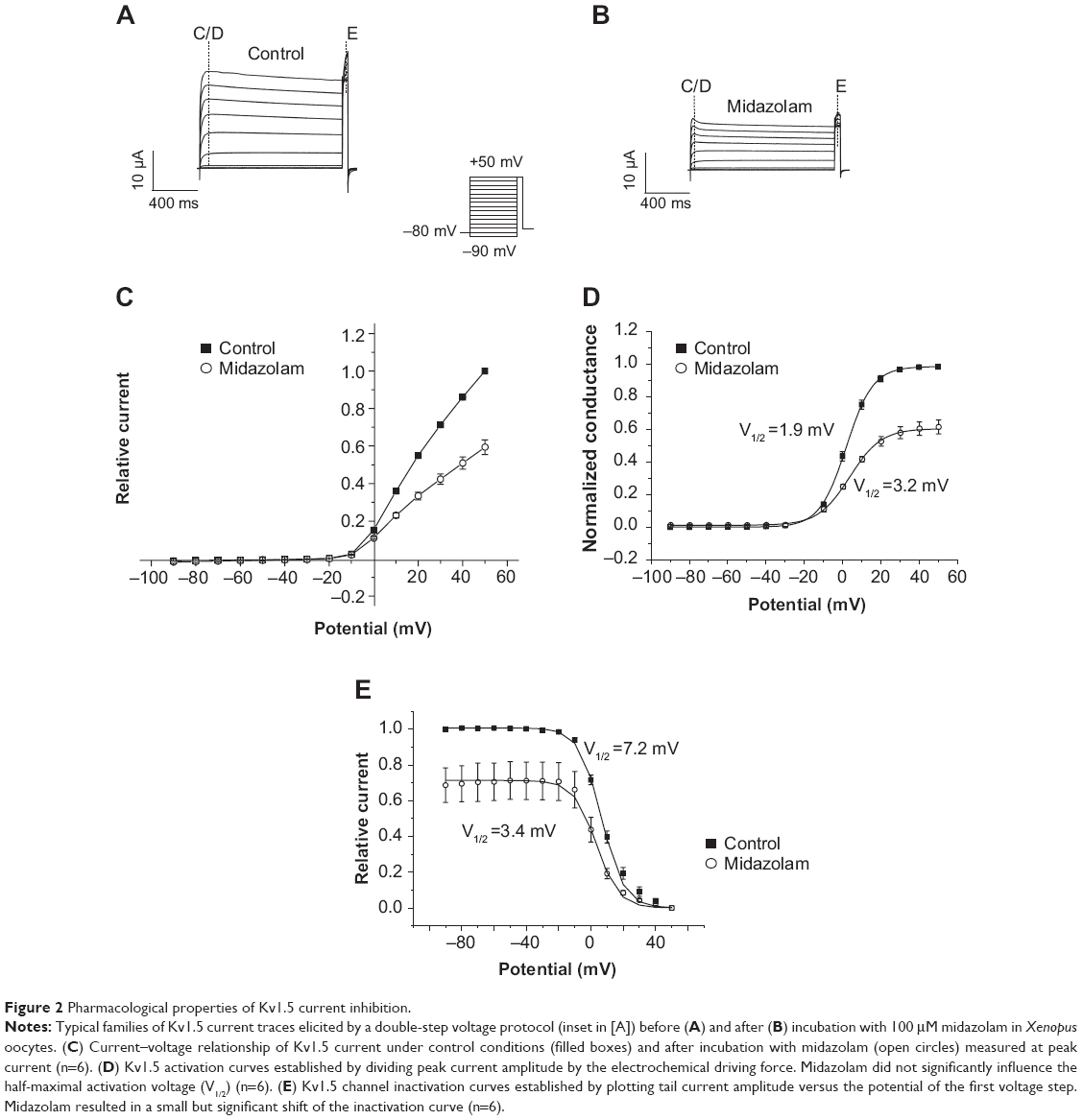 | Figure 2 Pharmacological properties of Kv1.5 current inhibition. |
Channel inactivation curves were established by plotting tail current amplitude during the constant return pulse (measured at the second dashed line in Figure 2A and B) versus the test pulse potential of the preceding variable voltage step (Figure 2E). Half-maximal inactivation voltages before and after midazolam application were obtained by fitting the data with a Boltzmann function. Under control conditions, Kv1.5 channels revealed a mean half-maximal inactivation voltage of 7.15±1.12 mV (n=6). Application of midazolam (100 μM) resulted in a small but significant leftward shift of the mean half-maximal inactivation voltage (V1/2 =3.41±0.74 mV, n=6, P<0.05) (Figure 2E).
Time course of block development
Block development was next analyzed using a single depolarizing voltage step to +50 mV. Kv1.5 current was measured under control conditions and after incubation with either 30 μM or 1,000 μM (Figure 3A and B). Incubation with 30 μM and 1,000 μM midazolam resulted in a current reduction of 12.25%±12% and 94.91%±3%, respectively (n=7) (Figure 3A and B). Time course of block development could be obtained by dividing the current trace after midazolam incubation by the corresponding current trace under control conditions (Figure 3C and D). In both cases, block development was fast, yielding a mean time constant of 9.0±2.1 ms for 30 μM midazolam (Figure 3C) and 4.1±0.8 ms for 1,000 μM midazolam (Figure 3D) (representative experiment of n=6).
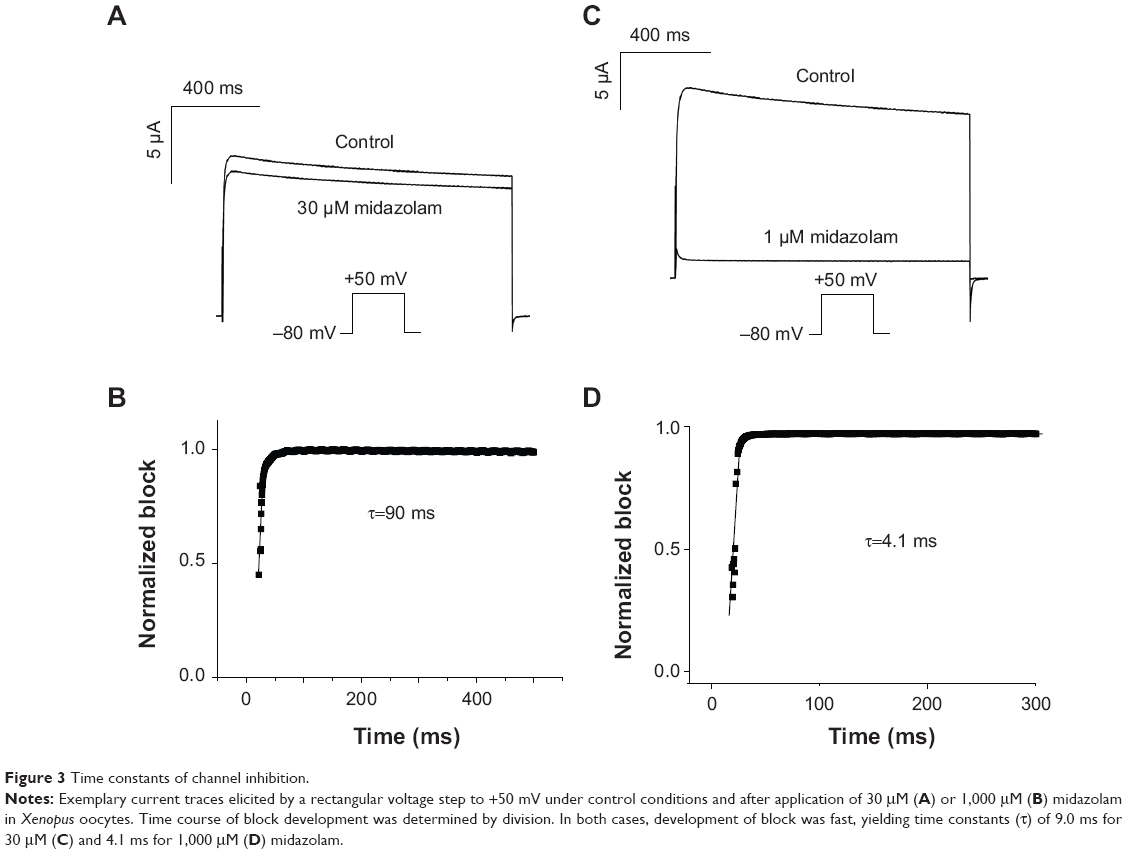 | Figure 3 Time constants of channel inhibition. |
Inhibition of Kv1.5 channels by midazolam exhibits no frequency dependence
Frequency dependence of block is a common finding among ion channel inhibitors. In order to analyze if Kv1.5 channels exhibit more pronounced inhibition at higher pacing rates, a set of three different voltage protocols was established. Cells were depolarized by a test pulse to +50 mV (200 ms) that was repeated with a frequency of 1, 2, or 4 Hz for duration of 16 seconds (Figure 4A, inset). Having obtained a control measurement, 200 μM midazolam was washed-in over a period of 15 minutes and the measurement was repeated. Peak current amplitude of each pulse before and after drug incubation was measured to determine the degree of block. Figure 4A–C displays the first (at 0 second) and the last (at 16 seconds) recordings after midazolam incubation for 1 Hz, 3 Hz, and 4 Hz (representative experiment of n=6 each). Figure 4D summarizes the inhibitory effects of midazolam on current amplitude. After 16 seconds, midazolam (200 μM) resulted in relative reductions of Kv1.5 current of 0.64%±0.033%, 0.66%±0.034%, and 0.67%±0.033% for 1 Hz, 2 Hz, and 4 Hz, respectively (n=6). However, at this point in time, there was no significant frequency dependence of block (P<0.05, ANOVA). In order to achieve a clear presentation, data are only presented for every full second (Figure 4D).
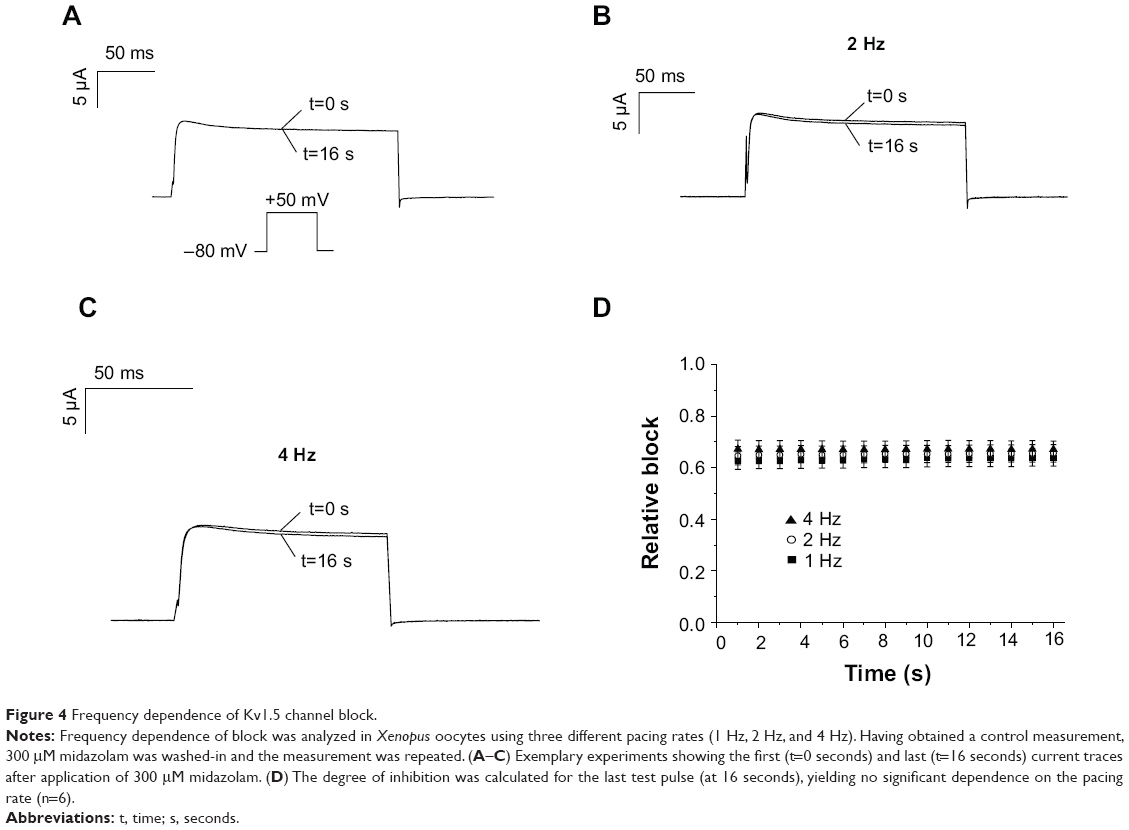 | Figure 4 Frequency dependence of Kv1.5 channel block. |
Discussion
We have shown that the anesthetic midazolam is a typical open-channel blocker of cardiac Kv1.5 potassium channels. So far, inhibitory effects of midazolam on Kv1.5 channels have not been described.
Pharmacological effects of midazolam
Pharmacological effects of midazolam on Kv1.5 channels were analyzed using two different heterologous expression systems (Xenopus and HEK 293). Compared to native cardiomyocytes, the main advantage of these simplified cell models mainly relates to the absence of interfering ion currents. Strategies aimed at blocking these currents, either by using elaborated voltage protocols or small molecule inhibitors, might negatively influence the current under analysis. Within several pharmacological studies, we were able to prove that the human kidney cell line HEK 293 represents a suitable cell model for the expression and analysis of cardiac ion channels.22–25 Midazolam inhibited cloned Kv1.5 channels in a dose-dependent manner, yielding an IC50 within the low micromolar range when analyzed in a mammalian cell line (HEK 293). As expected, when analyzed in Xenopus oocytes, we found that considerably higher midazolam concentrations were necessary to achieve similar effects. This phenomenon is well recognized and has been attributed to diffusion barriers within Xenopus oocytes caused by the vitelline membrane and yolk sac.26 As a consequence, results obtained from the mammalian HEK cell line might be better suited to questioning the physiological relevance of our findings. The dose–response relationship obtained in these cells is in line with previously published data on pharmacological inhibition of ion channels by the anesthetic midazolam. For example, an IC50 of 10 μM has been observed for calcium channels.27 Similarly, the whole-cell potassium outward current IK, measured in tracheal smooth muscle cells, as well as the calcium-independent transient outward current Ito, were reduced by approximately 20% after application of 100 or 60 μM midazolam, respectively.12,27
For the induction of anesthesia, the recommended intravenous midazolam dose is approximately 0.15 to 0.4 mg/kg.28 Depending on individual metabolic variations, reported peak plasma concentrations during induction of anesthesia can vary widely, and may reach values of up to 2 mg/L (~5 μmol/L).29,30 Interestingly, some healthy subjects may exhibit prolonged elimination half-time of midazolam.31 This might be partly explained by individual differences in the activity of hepatic enzymes, namely the members of the cytochrome P450 enzyme family, 3A4 and 3A5.32 Furthermore, in patients with renal or hepatic dysfunction, clearance from blood may be reduced, resulting in high blood levels over long time periods.33 When analyzed in a mammalian cell line, we found that application of 3 μM midazolam resulted in a reduction of Kv1.5 current of 25.7%. Considering the reported peak plasma levels of ~5 μmol/L, these effects seem small. However, it has to be taken into account that midazolam acts as a multichannel inhibitor. Only recently, So et al reported inhibitory effects of midazolam on a neural delayed rectifier potassium current IK(DR).13 We recently observed that midazolam acts as a low-affinity inhibitor of cardiac iou channel gene (hERG) channels (Vouderlin et al, unpublished data, 2014). Considering these additional effects, it is tempting to speculate that inhibitory effects on cardiac potassium channels might act in an additive manner. On the other hand, midazolam has also been shown to interfere with depolarizing ion channels.13,27 Simultaneous inhibition of depolarizing and repolarizing ion channels might cancel each other out, thereby accounting for the obvious gap between positive in vitro data and the paucity of in vivo evidence. Action potential recordings obtained from human atrial myocytes would help with analysis of the net effect of midazolam on atrial electrophysiology. Taking all evidence, due to the complex electrophysiological mechanisms, definite conclusions of midazolam-induced effects on the shape and duration of cardiac action potential can only be drawn with great caution. From the in vitro and in vivo data available, the use of midazolam seems safe in clinical practice and the clinical relevance of its side effects on cardiac electrophysiology seem negligible under therapeutic dosages. However, the data presented herein add to the current understanding of the pharmacological profile of midazolam. Furthermore, only a few small molecule inhibitors of Kv1.5 have been reported so far. Within this context, midazolam might help in the identification of new lead compounds for the development of structurally new Kv1.5 inhibitors.
Decrease of Kv1.5 current could be due either to a decrease in open probability or a decrease of the number of available channels. However, when analyzing properties of Kv1.5 channel inhibition in detail, we found that midazolam exhibits a fast onset of block. From the kinetics of block development, it can be concluded that inhibition of the Kv1.5 channel is state dependent, and does not occur in the closed state. A similar behavior was recently reported for the inhibition of Kv1.5 channels by the antiarrhythmic ajmaline.21 These effects strongly favor an inhibition via a decrease in open probability due to channel blockade. We further showed that midazolam caused a shift of the Kv1.5 channel inactivation curve. However, this shift was small, and the physiological relevance of this observation remains to be confirmed.
Midazolam did not alter channel activation kinetics and induced no frequency dependence of block. Analyzing binding sites of structurally unrelated Kv1.5 channel inhibitors, Decher et al were able to identify a number of critical amino acid residues located within the pore helix as well as in the S6 domain.34,35 Interestingly, they further showed that the exact binding site is dependent on the size and shape of the inhibitor with larger molecules (eg, AVE0118) interfering with biophysical properties of the channel (“trapping mechanism”).34 Following this hypothesis, it is tempting to speculate that midazolam, similarly to the Kv1.5 inhibitor S0100176, might bind to amino acids located within the central cavity of Kv1.5 channels.34 Accordingly, no pronounced effects on channel gating were observed. However, this hypothesis needs to be further validated by alanine-scanning mutagenesis. Taking all evidence midazolam represents a typical open channel inhibitor of cardiac Kv1.5 currents that does not have marked effects on channel gating kinetics.
Study limitations
The transferability of the present results might be limited by technical shortcomings. For example, all experiments were conducted at room temperature. However, it is well recognized that ion channel function is temperature dependent. Therefore, it cannot be excluded that pharmacological effects might differ slightly when analyzed at physiological temperature. Furthermore, it is well recognized that midazolam acts as a multichannel inhibitor with reported inhibitory effects on both depolarizing and repolarizing ion currents. Thus, the net effect of midazolam on cardiac electrophysiology is expected to be complex and cannot simply be derived from observed effects on isolated ion channels. Action potential recordings obtained in isolated human cardiomyocytes would aid in further understanding of the sum effect.
Conclusion
We have shown that the anesthetic midazolam is a typical open-channel blocker of cardiac Kv1.5 channels. Our findings add to the current understanding of the pharmacological profile of the compound midazolam. These data may assist with identification of new lead compounds for the development of Kv1.5 inhibitors.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) Scho1350/2-1 Zi1177/1-1 to EPS and EZ. FF was supported by the Hartmut Hoffmann-Berling International Graduate School of Molecular and Cellular Biology. The skillful assistance of Christine Jeckel is gratefully acknowledged. DS was supported by a grant from the University of Heidelberg: Postdoc Fellowship Program of the Faculty of Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Thummel KE, O’Shea D, Paine MF, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59(5):491–502. | ||
Nordt SP, Clark RF. Midazolam: a review of therapeutic uses and toxicity. J Emerg Med. 1997;15(3):357–365. | ||
Nonaka A, Kashimoto S, Imamura M, Furuya A, Kumazawa T. Mechanism of the negative inotropic effect of midazolam and diazepam in cultured foetal mouse cardiac myocytes. Eur J Anaesthesiol. 1997;14(5):481–487. | ||
Study RE, Barker JL. Diazepam and (–)-pentobarbital: fluctuation analysis reveals different mechanisms for potentiation of gamma-aminobutyric acid responses in cultured central neurons. Proc Natl Acad Sci U S A. 1981;78(11):7180–7184. | ||
Morani G, Bergamini C, Angheben C, et al. General anaesthesia for external electrical cardioversion of atrial fibrillation: experience of an exclusively cardiological procedural management. Europace. 2010;12(11):1558–1563. | ||
Wutzler A, Loehr L, Huemer M, et al. Deep sedation during catheter ablation for atrial fibrillation in elderly patients. J Interv Card Electrophysiol. 2013;38(2):115–121. | ||
Wutzler A, Mueller A, Loehr L, et al. Minimal and deep sedation during ablation of ventricular tachycardia. Int J Cardiol. 2014;172(1):161–164. | ||
Hüneke R, Fassl J, Rossaint R, Lückhoff A. Effects of volatile anesthetics on cardiac ion channels. Acta Anaesthesiol Scand. 2004;48(5):547–561. | ||
Staikou C, Stamelos M, Stavroulakis E. Impact of anaesthetic drugs and adjuvants on ECG markers of torsadogenicity. Br J Anaesth. 2014;112(2):217–230. | ||
Miró O, de la Red G, Fontanals J. Cessation of paroxysmal atrial fibrillation during acute intravenous propofol administration. Anesthesiology. 2000;92(3):910. | ||
Michaloudis DG, Kanakoudis FS, Petrou AM, Konstantinidou AS, Pollard BJ. The effects of midazolam or propofol followed by suxamethonium on the QT interval in humans. Eur J Anaesthesiol. 1996;13(4):364–368. | ||
Buljubasic N, Marijic J, Berczi V, Supan DF, Kampine JP, Bosnjak ZJ. Differential effects of etomidate, propofol, and midazolam on calcium and potassium channel currents in canine myocardial cells. Anesthesiology. 1996;85(5):1092–1099. | ||
So EC, Wu KC, Kao FC, Wu SN. Effects of midazolam on ion currents and membrane potential in differentiated motor neuron-like NSC-34 and NG108-15 cells. Eur J Pharmacol. 2014;724:152–160. | ||
Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525 Pt 2:285–298. | ||
Kanto JH. Midazolam: the first water-soluble benzodiazepine. Pharmacology, pharmacokinetics and efficacy in insomnia and anesthesia. Pharmacotherapy. 1985;5(3):138–155. | ||
Feng J, Wible B, Li GR, Wang Z, Nattel S. Antisense oligodeoxynucleotides directed against Kv1.5 mRNA specifically inhibit ultrarapid delayed rectifier K+ current in cultured adult human atrial myocytes. Circ Res. 1997;80(4):572–579. | ||
Wettwer E, Hála O, Christ T, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110(16):2299–2306. | ||
Li GR, Feng J, Yue L, Carrier M, Nattel S. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res. 1996;78(4):689–696. | ||
Tibbs GR, Rowley TJ, Sanford RL, et al. HCN1 channels as targets for anesthetic and nonanesthetic propofol analogs in the amelioration of mechanical and thermal hyperalgesia in a mouse model of neuropathic pain. J Pharmacol Exp Ther. 2013;345(3):363–373. | ||
López-Izquierdo A, Ponce-Balbuena D, Ferrer T, Rodríguez-Menchaca AA, Sánchez-Chapula JA. Thiopental inhibits function of different inward rectifying potassium channel isoforms by a similar mechanism. Eur J Pharmacol. 2010;638(1–3):33–41. | ||
Fischer F, Vonderlin N, Zitron E, et al. Inhibition of cardiac Kv1.5 and Kv4.3 potassium channels by the class Ia anti-arrhythmic ajmaline: mode of action. Naunyn Schmiedebergs Arch Pharmacol. 2013;386(11):991–999. | ||
Fischer F, Vonderlin N, Seyler C, et al. Acute and subacute effects of the selective serotonin-noradrenaline reuptake inhibitor duloxetine on cardiac hERG channels. Naunyn Schmiedebergs Arch Pharmacol. 2013;386(9):795–804. | ||
Scholz EP, Konrad FM, Weiss DL, et al. Anticholinergic antiparkinson drug orphenadrine inhibits HERG channels: block attenuation by mutations of the pore residues Y652 or F656. Naunyn Schmiedebergs Arch Pharmacol. 2007;376(4):275–284. | ||
Scholz EP, Alter M, Zitron E, et al. In vitro modulation of HERG channels by organochlorine solvent trichlormethane as potential explanation for proarrhythmic effects of chloroform. Toxicol Lett. 2006;165(2):156–166. | ||
Zitron E, Scholz E, Owen RW, et al. QTc prolongation by grapefruit juice and its potential pharmacological basis: HERG channel blockade by flavonoids. Circulation. 2005;111(7):835–838. | ||
Madeja M, Musshoff U, Speckmann EJ. Follicular tissues reduce drug effects on ion channels in oocytes of Xenopus laevis. Eur J Neurosci. 1997;9(3):599–604. | ||
Yamakage M, Matsuzaki T, Tsujiguchi N, Honma Y, Namiki A. Inhibitory effects of diazepam and midazolam on Ca2+ and K+ channels in canine tracheal smooth muscle cells. Anesthesiology. 1999;90(1):197–207. | ||
Reves JG, Fragen RJ, Vinik HR, Greenblatt DJ. Midazolam: pharmacology and uses. Anesthesiology. 1985;62(3):310–324. | ||
Crevat-Pisano P, Dragna S, Granthil C, Coassolo P, Cano JP, Francois G. Plasma concentrations and pharmacokinetics of midazolam during anaesthesia. J Pharm Pharmacol. 1986;38(8):578–582. | ||
Persson P, Nilsson A, Hartvig P, Tamsen A. Pharmacokinetics of midazolam in total i.v. anaesthesia. Br J Anaesth. 1987;59(5):548–556. | ||
Dundee JW, Halliday NJ, Harper KW, Brogden RN. Midazolam. A review of its pharmacological properties and therapeutic use. Drugs. 1984;28(6):519–543. | ||
Wandel C, Böcker R, Böhrer H, Browne A, Rügheimer E, Martin E. Midazolam is metabolized by at least three different cytochrome P450 enzymes. Br J Anaesth. 1994;73(5):658–661. | ||
Kulka PJ, Lauven PM, Schüttler J, Apffelstaedt C. Methohexital vs midazolam/flumazenil anaesthesia during laryngoscopy under jet ventilation. Acta Anaesthesiol Scand Suppl. 1990;92:90–95; discussion 107. | ||
Decher N, Kumar P, Gonzalez T, Pirard B, Sanguinetti MC. Binding site of a novel Kv1.5 blocker: a “foot in the door” against atrial fibrillation. Mol Pharmacol. 2006;70(4):1204–1211. | ||
Decher N, Pirard B, Bundis F, et al. Molecular basis for Kv1.5 channel block: conservation of drug binding sites among voltage-gated K+ channels. J Biol Chem. 2004;279(1):394–400. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.