")
Back to Journals » Biologics: Targets and Therapy » Volume 12
Infliximab dose adjustment can improve the clinical and radiographic outcomes of rheumatoid arthritis patients: REVIVE study results
Authors Nozaki Y , Nagare Y, Ashida C , Tomita D, Okada A, Inoue A, Kinoshita K, Funauchi M, Matsumura I
Received 18 September 2018
Accepted for publication 27 October 2018
Published 27 November 2018 Volume 2018:12 Pages 171—182
DOI https://doi.org/10.2147/BTT.S187998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Doris Benbrook
Yuji Nozaki, Yasuaki Nagare, Chisato Ashida, Daisuke Tomita, Akinori Okada, Asuka Inoue, Koji Kinoshita, Masanori Funauchi, Itaru Matsumura
Department of Hematology and Rheumatology, Kindai University School of Medicine, Osaka, Japan
Purpose: We evaluated the clinical responses and radiographic outcomes of 90 patients with rheumatoid arthritis (RA) undergoing continuous or dose-adjusted infliximab treatment over 104 weeks.
Patients and methods: Patients received 3 mg/kg infliximab continuously (the contin group; n=50), or the dose escalation and de-escalation of infliximab (3, 6, and 10 mg/kg) from week 14 (the adjusted group; n=40) based on the patient’s Disease Activity Score in 28 joints (DAS28). The retention rate, clinical response, and radiographic assessment were determined at week 104.
Results: The contin and adjusted groups’ retention rates at week 104 were 56.8 and 66.7%, and the groups’ low disease activity in the DAS28 was 39.1 and 66.7%, respectively. Remission based on the DAS28 and the American College of Rheumatology (ACR)/European League against Rheumatism (EULAR) Boolean-based criteria was significantly increased in the adjusted group. In the radiographic assessment, there was also a significant reduction in the mean changes in total Sharp score. The cumulative rates of any adverse effects showed no significant difference between the groups.
Conclusion: In an assessment of adequate DAS28 results, the RA patients who did not respond to the initial dose of infliximab showed improved clinical responses and radiographic assessment after a dose adjustment of infliximab, without an increased risk of serious adverse events.
Keywords: rheumatoid arthritis, DAS28, infliximab, mTSS, dose adjustment, REVIVE
Introduction
The development of antitumor necrosis factor α (anti-TNFα) therapy provided a major advance in the treatment of patients with rheumatoid arthritis (RA).1 Infliximab is a monoclonal antibody specific for TNFα that, when administered in combination with methotrexate (MTX), has been shown to be effective in treating patients with active RA. The pivotal multinational clinical study, ie, the Anti-TNF Trial in Rheumatoid Arthritis with Concomitant Therapy (ATTRACT), showed that repeated treatment with 3 or 10 mg/kg infliximab was more effective than MTX alone in reducing the disease activity of RA, inhibiting the subjects’ joint damage, and improving their physical function.2,3
It is well known that some RA patients need a dose escalation from their initial dose of infliximab in order to maintain their level of physical activity. The ATTRACT trial and the Safety Trial for Rheumatoid Arthritis with Remicade Therapy (START) study also demonstrated a significant association between RA patients’ clinical responses and their trough serum infliximab levels.4,5 A dose-escalating study (RISING) that examined the impact of infliximab with MTX therapy on RA patients’ radiographic and clinical responses based on their trough serum levels demonstrated a significant correlation between the trough serum level and the European League against Rheumatism (EULAR) response or Disease Activity Score in 28 joints (DAS28) remission.6 However, there was no significant difference in the incidence of adverse events (AEs) regardless of the infliximab dose.6 The RISING study also demonstrated that infliximab inhibited the progression of joint damage in most of the patients.
In the present retrospective study (the REVIVE study; Remicade in validation and effectiveness of the clinical response and radiographic outcomes in the dose escalation and de-escalation strategy), we compared the clinical and radiographic outcomes in the following two groups: patients who were administered the combination therapy of MTX with 3 mg/kg infliximab continuously (the contin group) as the controls and patients who were administered the combination therapy of MTX with 3, 6, or 10 mg/kg infliximab based on the assessment of their DAS28 values as the dose escalation and de-escalation strategy (the adjusted group). The results of our analyses indicate that based on the assessment of the DAS28 at week 104 in this series of RA patients, compared to the patients who took the minimum infliximab dose (3 mg/kg) continuously, the patients whose infliximab dose was adjusted showed improved disease activity and an inhibition of the progression of joint damage.
Patients and methods
Patients
The REVIVE study enrolled 21–81-year-old Japanese patients who had suffered from RA for ≥6 months and showed active disease based on the American College of Rheumatology (ACR) criteria7 despite previous treatment with a conventional synthetic disease-modifying antirheumatic drug (csDMARD) and who had received a stable MTX dose (up to 16 mg/week). The patients’ RA was defined as ≥6 swollen joints and ≥6 tender joints and two or more of the following: C-reactive protein (CRP) ≥2.0 mg/dL; erythrocyte sedimentation rate (ESR) ≥28 mm/h; a global health score ≥20 mm on a 0–100 mm scale, ie, the patient’s global assessment of disease activity (PtGA), where 0= best and 100= worst; investigator-documented evidence of bone erosion on radiographs; and a positive finding of anticyclic citrullinated peptide antibodies (ACPA) or rheumatoid factor (RF). The patients were screened for latent and active tuberculosis. Concomitant oral corticosteroids (stable dose ≤10 mg of prednisolone/day or equivalent) were permitted.
Patients were excluded if they had any other connective tissue disease with joint symptoms or therapy with other biological agents within 4 months before the initial infliximab infusion. Other exclusion criteria were as follows: a history of serious or opportunistic infection within 6 months before registration; active tuberculosis; hepatitis B virus, hepatitis C virus, or HIV carriers; and those with chronic infectious diseases.
Study design
This study was conducted from 2009 to 2017 at a single center, Kindai University Hospital (Osaka, Japan). Two patient groups were studied from week 0 (initiation of infliximab) to week 104. All of the patients received 3 mg/kg infliximab at weeks 0, 2, and 6. In one group, the patients received the combination therapy of MTX with 3 mg/kg infliximab continuously for the 104 weeks in accordance with their physician’s judgment (the contin group). In the other group, at week 14, the decision for infliximab dose escalation/ de-escalation was made (with the patient’s agreement) based on the patient’s clinical response (the adjusted group).
In the adjusted group, the decision of whether to adjust the infliximab dose was then made every 8 weeks based on the patient’s DAS28–ESR,8 which was calculated by a physician who treated the allocated treatment group during the entire study period. If the patient did not reach a DAS28 of <3.2 for 4 months, the treating physician immediately adjusted the therapy by increasing the dose of infliximab. If the patient’s clinical response was consistently adequate (a DAS28 of <3.2 for ≥6 months), the dose of infliximab was decreased until it reached a stable dose. Following the starting dose of infliximab at 3 mg/kg, the dose of 3, 6, or 10 mg/kg infliximab was administered every 8 weeks from weeks 14 to 104 and the efficacy of the treatment was evaluated at weeks 26, 52, and 104.
The DAS28 calculations for dose adjustments were performed every 8 weeks, within 4 weeks before the next infusion of infliximab. If the patient’s DAS28 was ≥3.2, the dose of the next infusion was increased to 6 mg/kg every 8 weeks and finally to 10 mg/kg every 8 weeks. If a patient still had a DAS28 of ≥3.2 while receiving MTX along with 10 mg/kg infliximab, the infliximab was switched to another biological agent. In the case of a persistent good response (a DAS28 of <3.2 for ≥6 months), the dose of infliximab was reduced (from 10 to 6 and then to 3 mg/kg) at each next infusion.
Over the entire 2-year study period, csDMARDs (other than tacrolimus, iguratimod, and salazosulfapyridine), nonsteroidal anti-inflammatory drugs (NSAIDs), oral glucocorticoids (prednisolone 10 mg/day), and folic acid preparations were permitted at the stable dose from ≥4 weeks before the initial infliximab infusion. All patients received concomitant MTX throughout the study. The patient’s dose of MTX had to be stable (ie, ≥6 mg/week; the approved maximum dose of MTX for RA in Japan is 16 mg/week) for >4 weeks just before the initial infliximab infusion and over the entire study period. Prednisolone could be tapered and discontinued. If a patient’s disease activity flared (ie, a DAS28 of ≥3.2 was observed) after the prednisone was tapered, the last effective dose was reintroduced. Prednisolone could be reintroduced only once: if, after a second discontinuation, the DAS28 increased again to ≥3.2, then, the next step in the protocol was taken. If side effects occurred, the responsible drug was reduced to the lowest tolerated dose. If a medication was not tolerated at all or was contraindicated, it was discontinued.
This study was conducted according to the principles expressed in the Declaration of Helsinki of 1983, and it was approved by the Research Ethics Committee of Kindai University of Medicine. In the guidelines for postmarketing observational studies in Japan, it is not necessary to obtain consent in all patients.9 However, we guaranteed the opportunity for refusal in an agreement with the patients and physicians because a written consent from all patients was not necessary.
AEs were evaluated until week 104. In the patients in whom treatment was discontinued, AEs were assessed until 12 weeks after the final medication administration.
Radiographic assessment
Plain radiographs of each patient’s hands and feet were taken at baseline and at week 104 and evaluated and scored using the Steinbrocker class and the modified Sharp/van der Heijde scoring system.10,11 The baseline and week 104 radiographs were evaluated by two independent readers in accordance with reported methods.10,11 We calculated the mean changes from baseline in the modified total Sharp score (mTSS), the erosion score (ES), and the joint space narrowing score (JSN), with a range of 0–390. Radiographic nonprogression was defined as an mTSS score of ≤0.5. The smallest detectable change (SDC) is an estimate of the measurement error between readers of the films.12
Assessment of clinical assessment
The RA patients’ demographic characteristics recorded at baseline included age, sex, disease duration, and current therapy. The endpoint for clinical response was the DAS28 at weeks 0, 26, 52, and 104. The EULAR response13 was evaluated at weeks 26, 52, and 104. At each visit, the following laboratory tests were performed. Efficacy endpoints included DAS28 values and the proportion of patients with DAS28 in EULAR responses, and the tender joint count (TJC), the swelling joint count (SJC) in 28 joints (both measured by the treating physician), and the PtGA. The laboratory parameters of CRP (mg/dL) and ESR (mm/h) were similarly assessed every 4 weeks from weeks 0 to 104. The RF (U/mL), ACPA (U/mL), and matrix metalloproteinase-3 (MMP-3; ng/mL) values were measured at baseline. The patient’s physical function at baseline was evaluated as the Health Assessment Questionnaire-Disability Index (HAQ-DI).14
Clinical remission was defined as achieving a DAS28 of <2.6 and one or none of the following criteria (ACR/EULAR Boolean-based criteria)15: the TJC, the SJC, the CRP, and the PtGA (a 100 mm VAS data converted to centimeter).
Safety
The treating physician recorded all AEs and serious AEs and, if necessary, made treatment adjustments in accordance with the protocol described earlier. Serious AEs were defined as any adverse reaction resulting in any of the following outcomes: a life-threatening condition or death, a significant or permanent disability, a malignancy, hospitalization, or prolongation of hospitalization, a congenital abnormality, and a birth defect. Laboratory test results (hematological, blood chemistry, and urinalysis), chest radiographs, and electrocardiograms were also evaluated.
Statistical analyses
We used the GraphPad Prism software (GraphPad Software, San Diego, CA, USA) and JMP Statistical Software (SAS Institute Inc., Cary, NC, USA) for the statistical analyses. Summary statistics of mean and SD or median and IQR, when appropriate, is presented for continuous variables. Categorical variables are presented as percentages. Comparisons between independent means were conducted with the Mann–Whitney U-test. Relationships between categorical variables were evaluated by the Chi-squared test. We assessed the survival rate for infliximab treatment using the Kaplan–Meier method, and the difference in retention curves was examined by means of a log-rank test. P-values <0.05 were considered statistically significant.
Results
Baseline characteristics and patient disposition
We retrospectively analyzed the cases of 90 patients with RA who received the combination therapy of MTX and infliximab. Of them, 50 patients were treated with infliximab 3 mg/kg continuously throughout the study (the contin group) and the other 40 patients underwent the dose escalation and de-escalation of infliximab (3, 6, and 10 mg/kg) on the basis of their DAS28 at week 14 (the adjusted group). Table 1 summarizes the baseline characteristics, laboratory findings, and treatment in the two groups.
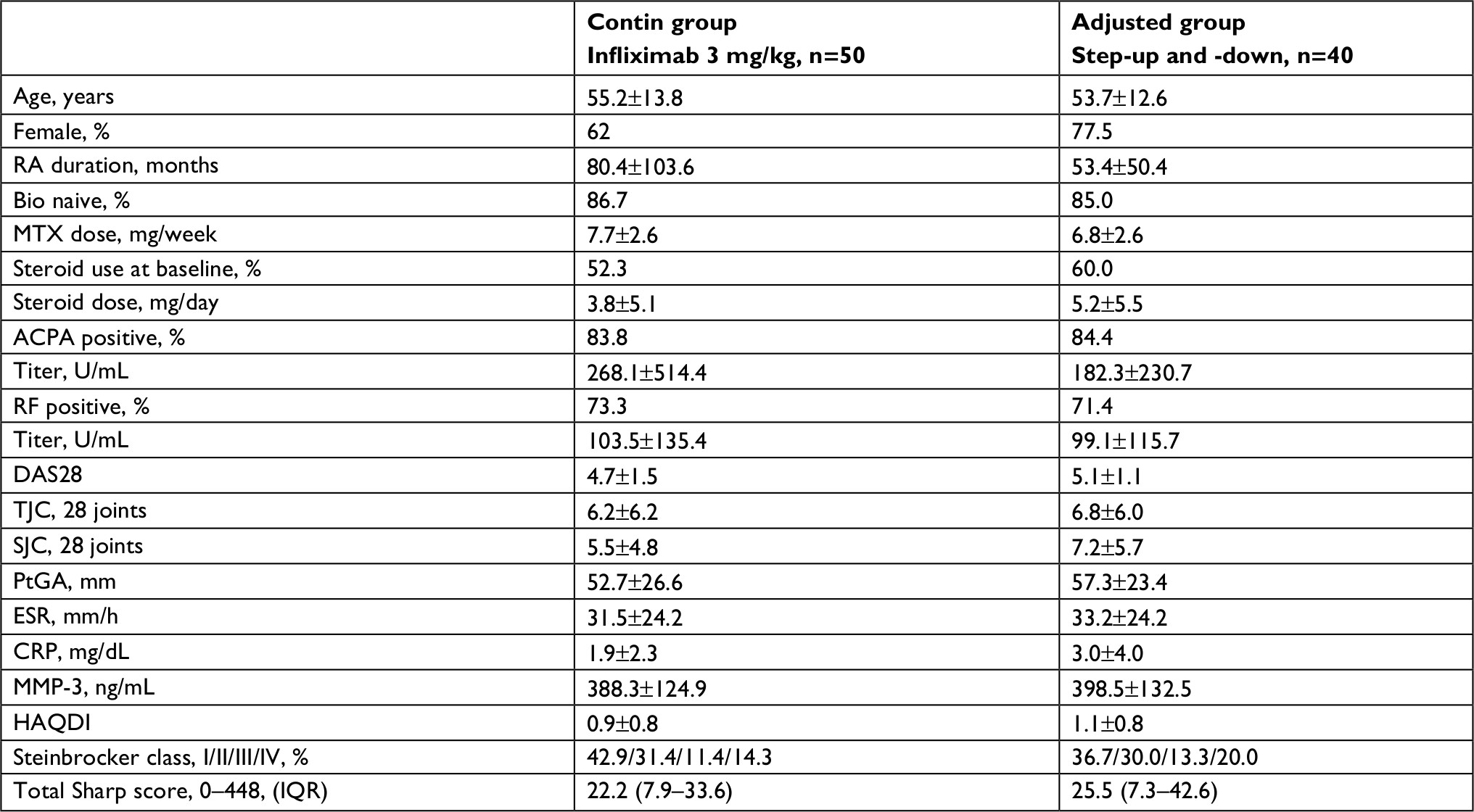 | Table 1 Baseline demographic and disease characteristics Note: Values are median (25th to 75th percentiles) or mean±SD, unless otherwise indicated. Abbreviations: ACPA, anticitrullinated peptide antibody; CRP, C-reactive protein; DAS, Disease Activity Score; ESR, erythrocyte sedimentation rate; HAQDI, Health Assessment Questionnaire Disability Index; MMP-3, matrix metalloproteinase-3; MTX, methotrexate; PtGA, patient’s global assessment of disease activity; RA, rheumatoid arthritis; RF, rheumatoid factor; SJC, swollen joint count; TJC, tender joint count. |
There were no significant differences in the background between the two groups in age, sex, the disease duration, the percentage of bio-naive patients, the proportion of Steinbrocker class, the steroid and MTX dose, the positive percentage and titers of ACPA, RF, and MMP-3, clinical disease activity, HAQ-DI, or total Sharp score at baseline. Twenty-seven of the 50 patients (54.0%) in the contin group and 29 of the 40 patients (72.5%) in the adjusted group completed the study (Figure 1). In the contin group, the main reason for discontinuation was lack of efficacy (n=17) and there were AEs (n=6). In the adjusted group, the main reason for discontinuation was the lack of efficacy (n=6), economic problems (n=2) and the moving away (n=2) as the patient’s request, and maintaining remission (n=1). There were no AEs in the adjusted group.
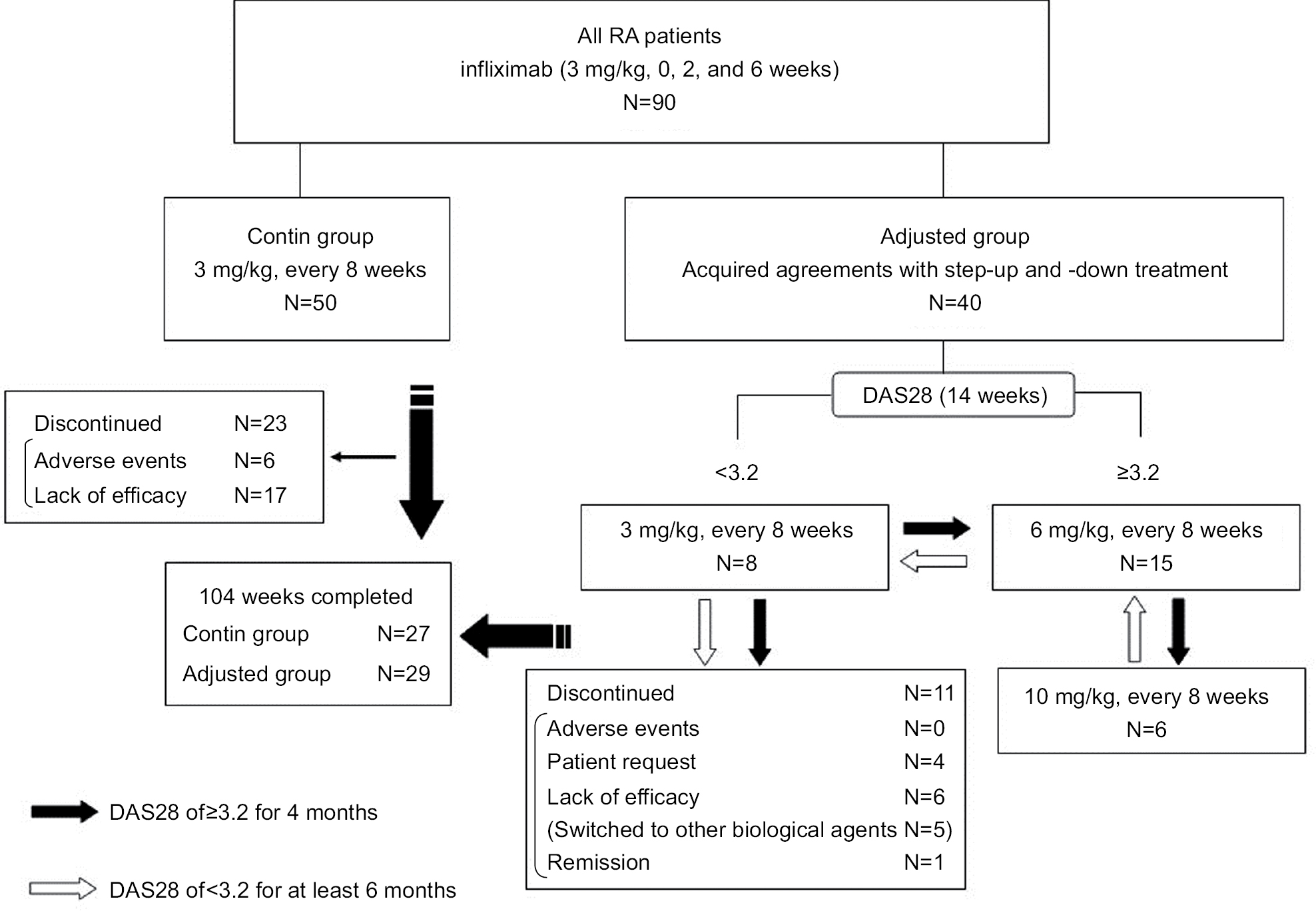 | Figure 1 Patient flow chart. Notes: As controls, the contin group received 3 mg/kg infliximab continuously without DAS28 assessments from baseline (week 0) to week 104. The patients in adjusted group received 3, 6, or 10 mg/kg infliximab with DAS28 assessments as the strategy of dose escalation and de-escalation. If the patient did not reach a DAS28 of ≥3.2 for 4 months, the treating physician immediately adjusted the regimen by proceeding to the dose-escalation protocol. If the clinical response was consistently adequate (DAS28<3.2 for ≥6 months), the infliximab was gradually de-escalated until it remained at a maintenance dose. Starting from infliximab 3 mg/kg, infliximab at doses 3, 6, or 10 mg/kg was administered every 8 weeks from weeks 6 to 104, and the efficacy was evaluated at weeks 26, 52, and 104. Abbreviations: DAS28, Disease Activity Score in 28 joints; RA, rheumatoid arthritis. |
Infliximab treatment
At week 14, the decision to use the dose escalation and de-escalation strategy with infliximab was performed based on the individual patient’s DAS28. Eighteen (45.0%) and nine (22.5%) of the 40 patients in the adjusted group received 3 mg/kg infliximab continuously at weeks 26 and 52, respectively (Figure S1). Eight (20.0%) patients were taking 3 mg/kg infliximab at week 104: two patients who underwent dose reduction from 6 mg/kg infliximab and nine patients whose treatment was discontinued (four patients due to the lack of efficacy and four patients at the patient’s request). After week 14, 17 (42.5%) and 18 (45.0%) patients underwent a dose escalation from 3 to 6 mg/kg at weeks 26 and 52, respectively. Fifteen (40.0%) patients were taking 6 mg/kg infliximab at week 104: two patients who underwent a dose reduction to 3 mg/kg and one patient who underwent a dose escalation to 10 mg/kg. Similarly, after week 14, three (7.5%) and five (12.5%) patients underwent a dose escalation from 6 to 10 mg/kg at weeks 26 and 52, respectively. Overall, six (15.0%) patients were taking 10 mg/kg at week 104 (including one patient who underwent a dose escalation from 6 mg/kg). The details of the dose escalation and de-escalation procedure are shown in Figure S1.
Retention rate
The overall retention rate over the 104 weeks in the RA patients whose infliximab was withdrawn due to the lack of efficacy, the patient’s request, or sustained remission is illustrated in Figure 2A. In the contin and adjusted groups, the retention rates (95% CI) were 58.3% (52.3–66.1) and 80.0% (71.1–87.1) at week 52 and 54.0% (47.1–61.3) and 72.5% (65.3–78.0) at week 104, respectively. The overall retention rate in the adjusted group was thus significantly higher than that in the contin group at weeks 52 and 104. The retention rate among the patients who experienced a lack of efficacy is shown in Figure 2B. The retention rates (95% CI) were 61.2% (55.1–67.3) and 88.8% (79.2–91.1) at week 52 and 61.4% (52.3–68.2) and 83.3% (75.3–88.0) at week 104 in the contin and adjusted groups, respectively. The retention rate in the adjusted group was significantly and notably higher than that in the contin group at weeks 52 and 104.
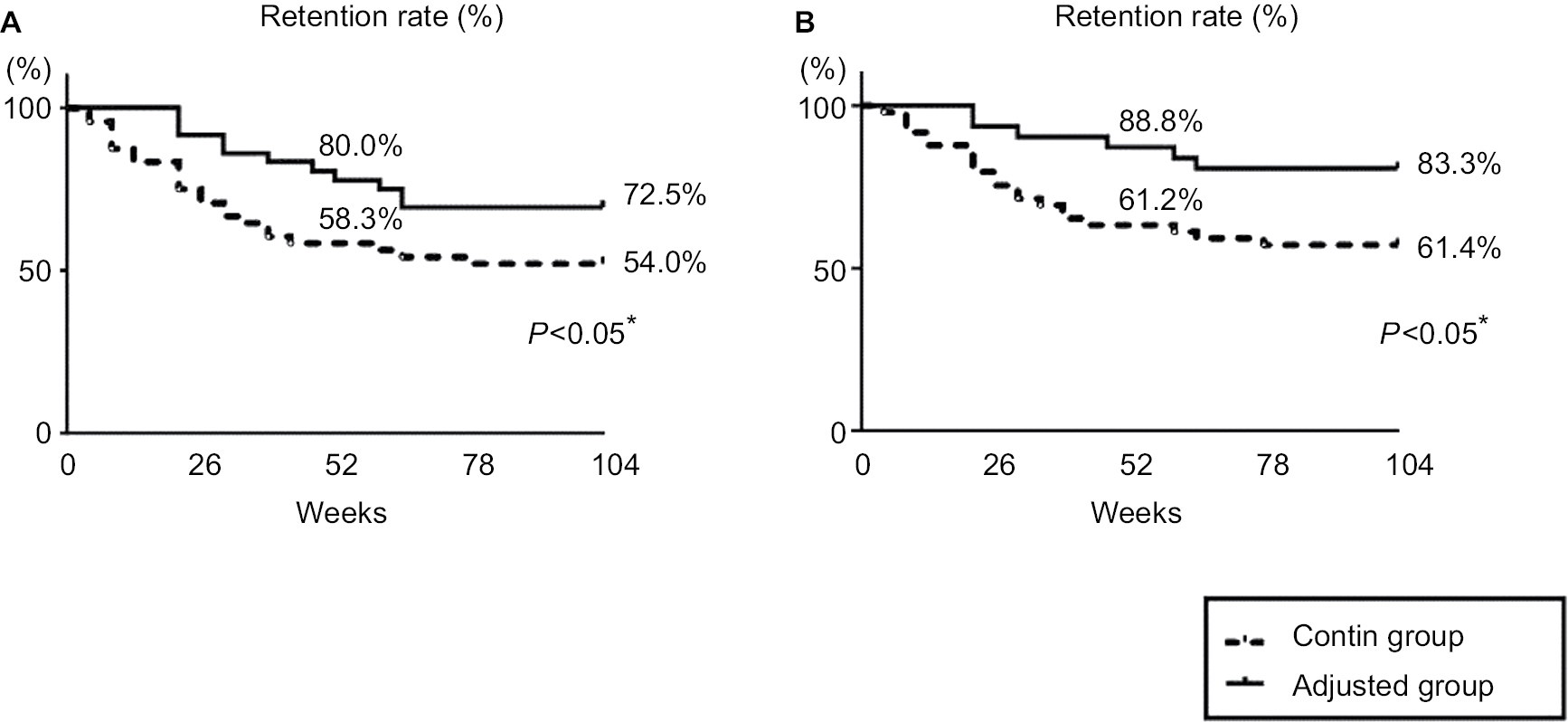 | Figure 2 Retention rate. Note: Kaplan–Meier curves for the contin and adjusted groups regarding the time to withdrawal for any reason due to the lack of efficacy, adverse effects, or the patient’s request (A) and due to the lack of efficacy (B) from the start of infliximab to week 104. *P<0.05, contin group vs adjusted group. |
Clinical efficacy
Figure 3 shows the patients’ DAS28 scores and EULAR responses at weeks 0, 26, 52, and 104. At weeks 26 and 52, there were no significant differences between the two groups. At week 104, the DAS28 scores were significantly higher in the contin group compared to the adjusted group (4.0±1.9 and 2.5±1.3; Figure 3A). Figure 3B shows the changes from the baseline in DAS28 score at weeks 26, 52, and 104. In the adjusted group, the changes from the baseline in DAS28 were significant compared to the contin group (−2.5±−2.1 vs −1.1±−1.9, respectively).
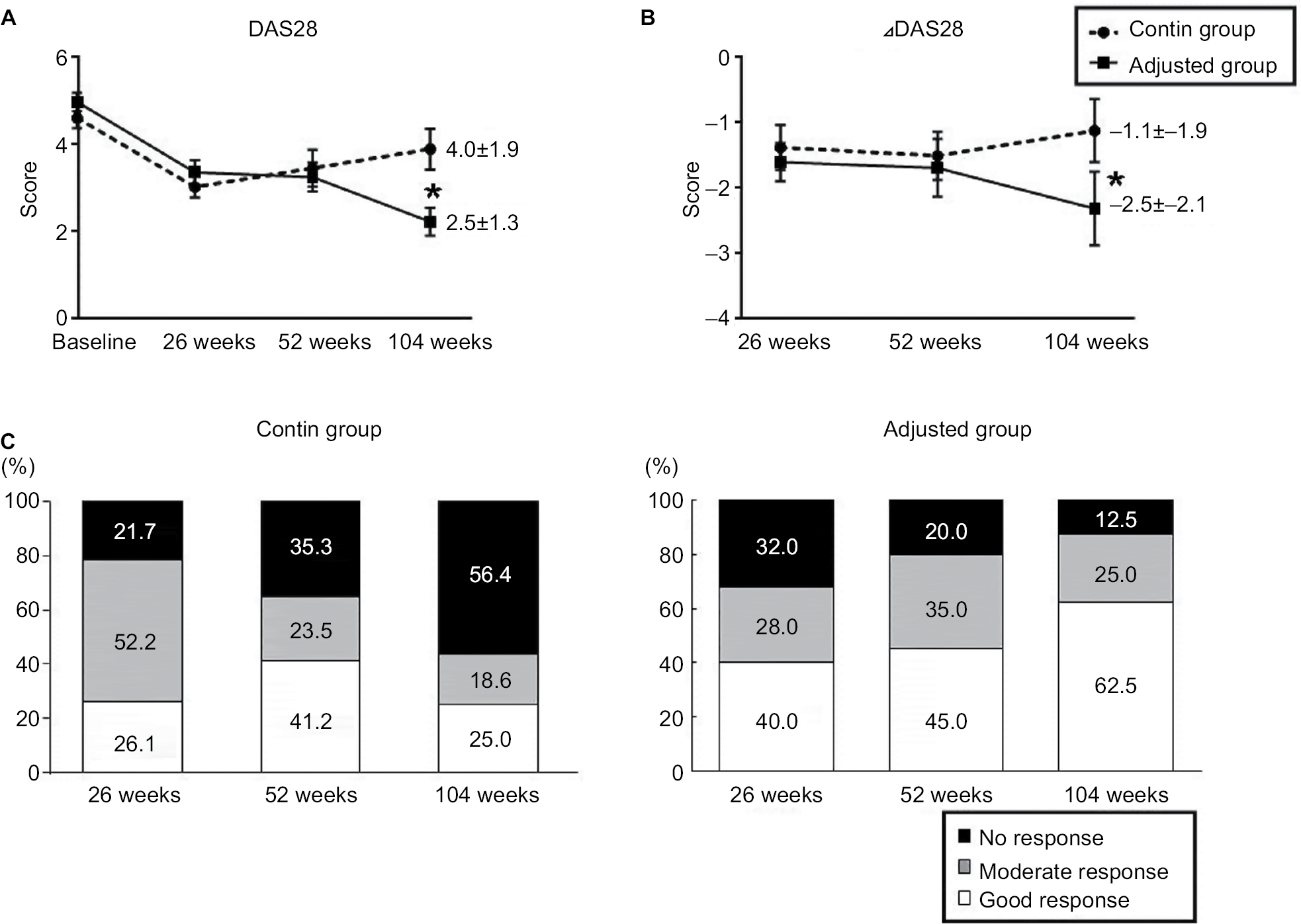 | Figure 3 DAS28 and the EULAR response. Notes: (A) Time course of the DAS28 through week 104 following the initiation of infliximab treatment in the contin and adjusted groups (*P<0.05). (B) Time course of the changes in the DAS28 from the baseline at weeks 26, 52, and 104 as the clinical response to infliximab treatment (*P<0.05). (C) By comparing the DAS28 scores of a patient at different time points of baseline and the estimation at weeks 26, 52, and 104, it is possible to define an improved response. The rate of responders (ie, those with a good or moderate response) at week 104 was 43.6% in the contin group and 87.5% in the adjusted group (*P<0.05). The EULAR responses were categorized as follows. No response: DAS28 improvement ≤0.6 in present DAS28≤3.2, >3.2, ≤5.1, and >5.1 and DAS28 improvement >0.6 and ≤1.2 in present DAS28>5.1. Moderate response: DAS28 improvement >0.6 and ≤1.2 in present DAS28≤3.2, >3.2, and ≤5.1 and DAS28 improvement >1.2 in present DAS28>5.1, >3.2, and ≤5.1. Good response: DAS28 improvement >1.2 in present DAS28≤3.2. *P<0.05, contin group vs adjusted group. Abbreviations: DAS28, Disease Activity Score in 28 joints; EULAR, European League against Rheumatism. |
Figure 3C illustrates the EULAR responses at weeks 26, 52, and 104 in the two groups. The rate of responders (ie, those with a good or moderate response) at week 104 was 43.6% in the contin group and 87.5% in the adjusted group (*P<0.05). Conversely, the proportions of nonresponder patients at week 104 were 56.4% in the contin group and 12.5% in the adjusted group.
The proportions of disease activity based on DAS28 and ACR/EULAR Boolean-based criteria were evaluated at baseline and weeks 26, 52, and 104 (Figure 4A). In addition, the proportions of patients achieving low disease activity (LDA) at weeks 26, 52, and 104 – when LDA and remission based on the DAS28 (<3.2 and <2.6) and ACR/EULAR Boolean-based remission were evaluated – were significantly different between the contin and adjusted groups (Figure 4B). At week 104, LDA based on the DAS28 (<3.2) was achieved in 35% of the contin patients and in 68.5% of the adjusted patients. Remission based on DAS28 (<2.6) was also achieved in 25% of the contin patients and in 47.5% of the adjusted patients. The rates of ACR/EULAR Boolean-based remission were 2.2 vs 22.7, 15.4 vs 26.5, and 16.7 vs 39% at weeks 26, 52, and 104 in the contin and adjusted groups, respectively.
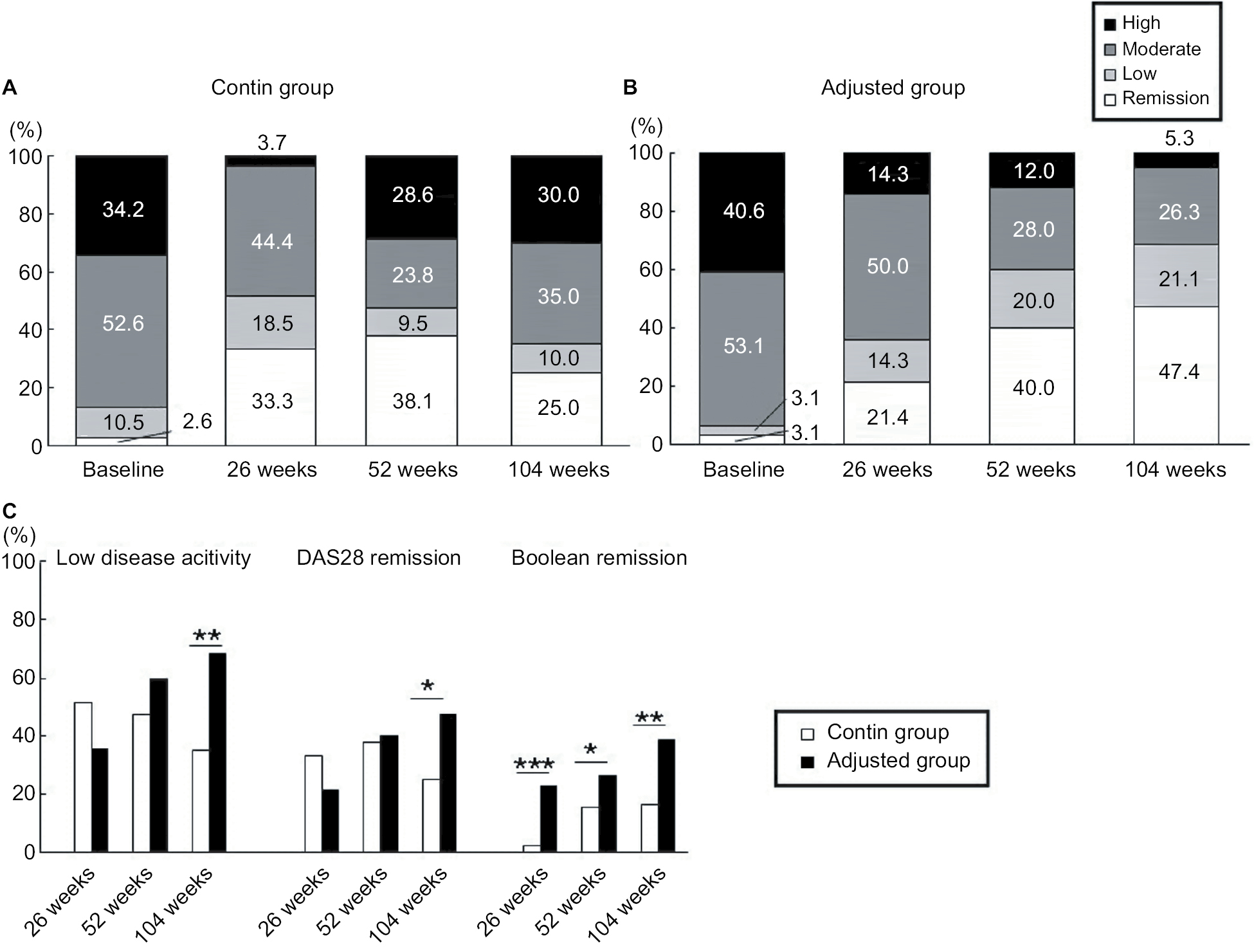 | Figure 4 Remission rate in DAS28 and ACR/EULAR Boolean-based criteria. Notes: (A) Clinical response at baseline and weeks 26, 52, and 104 in the contin and adjusted groups according to DAS28 criteria. (B) DAS28-based low disease activity and remission and ACR/EULAR Boolean-based remission rates at weeks 26, 52, and 104 by the Chi-squared test (*P<0.05, **P<0.01, and ***P<0.001). The disease activity and criteria in the DAS28 and ACR/EULAR Boolean-based remission were categorized as follows. The DAS28 criteria: high: 5.1< DAS28, moderate: 3.2≤ DAS28≤5.1. Low: 2.6≤ DAS28<3.2. Remission: DAS28<2.6. ACR/EULAR Boolean-based remission: TJC, SJC, PtGA, and CRP all ≤1. Abbreviations: ACR, American College of Rheumatology; CRP, C-reactive protein; DAS28, Disease Activity Score in 28 joints; EULAR, European League against Rheumatism; PtGA, patient’s global assessment of disease activity; SJC, swelling joint count; TJC, tender joint count. |
We also estimated the effect of steroid tapering in the contin and adjusted groups (Table 2). At baseline, the percentage of steroid users (52.3 vs 60.0%) and the steroid dose (3.8±5.1 vs 5.2±5.5 mg/day) were similar in the contin and adjusted groups. At week 104, the percentage of steroid users (36.4 vs 39.1%) and the steroid dose (1.7±2.5 vs 2.0±2.8 mg/day) in the contin and adjusted groups were decreased and the steroid dose had been tapered significantly compared to the values at baseline (*P<0.05,). In contrast, the cumulative steroid dose at week 104 was not significantly different between the contin and adjusted groups at 1,866.1±2,285.7 vs 2,118.5±2,629.9 mg, respectively. However, the reduction of steroid dose in the adjusted group tended to be smaller compared to that in the contin group (−3.2±2.7 vs −2.1±2.6 mg) at week 104.
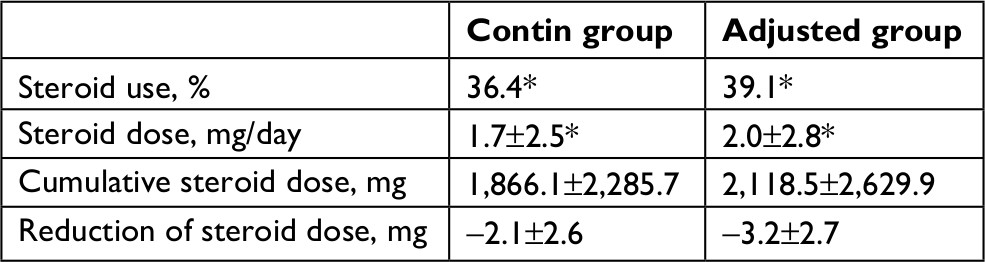 | Table 2 Steroid treatment at week 104 Notes: Values are mean±SD, unless otherwise indicated. *P<0.05, the percentage and dose at baseline vs those at week 104. |
Radiographic progression
At baseline and at week 104, 57 radiographs were assessed (32 in the contin group and 25 in the adjusted group). The two groups were similar at baseline with respect to the number of erosions, joint space narrowing, and TSS (Tables 1 and 3). The comparison of the contin and adjusted groups’ baseline and week 104 radiographs showed a higher TSS with more erosions and joint narrowing spaces, but the difference was not significant at week 104 (Table 3).
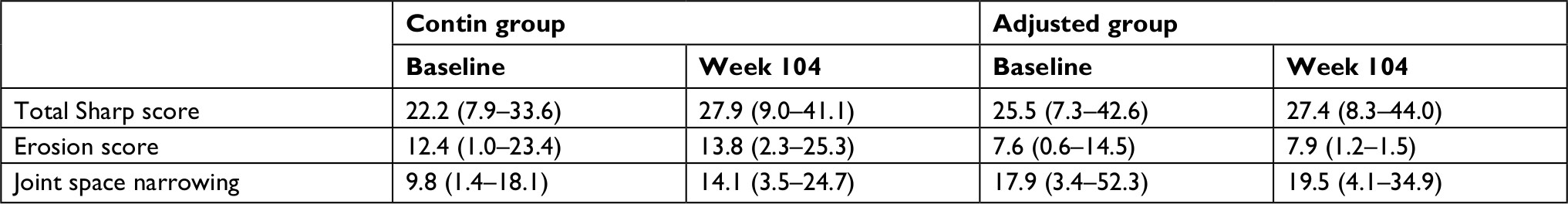 | Table 3 Radiographic progression Note: Data are median and (IQR). |
At week 104, there were significant differences between the contin and adjusted groups in the mean changes in TSS at 5.7 (5.1–6.3) vs 1.9 (1.5–2.3) and in erosions at 1.6 (1.2–2.0) vs 0.3 (0.1–0.5), respectively. There was no significant difference in joint space narrowing: 4.1 (3.7–4.5) vs 1.6 (1.1–2.1) in the contin and adjusted groups, respectively (Figure 5A). The percentages of patients with no progression of joint damage (improved or no change; ΔmTSS ≤0.5) were 40.0 in the contin group and 64.3 in the adjusted group (a nonsignificant difference; Figure 5B).
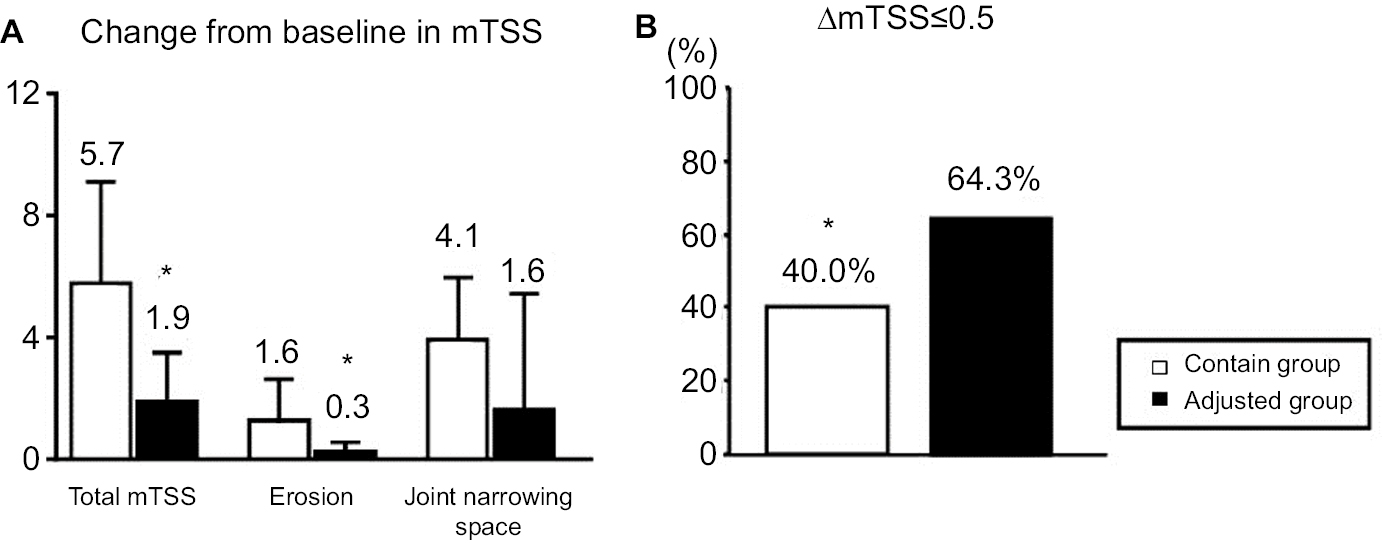 | Figure 5 Joint damage in radiographic assessment. Notes: The progression of joint damage in the contin and adjusted groups according to the mTSS at week 104. (A) The change in mTSS from baseline. (B) The rate of patients with progression, no change, or improvement in the mTSS (ΔmTSS ≤0.5), *P<0.05. Abbreviation: mTSS, modified total Sharp score. |
Safety assessments
At week 104, the cumulative rates of any AEs in the contin and adjusted groups were 34 and 30% and the cumulative rates of serious AEs were 18 and 12.5%, respectively (Table 4). Serious AEs occurred in 18% of the contin group: five patients developed a serious infection (one patient with herpes zoster, one patient with cellulitis, and three patients with pneumonia), one patient incurred interstitial pneumonia, and three patients experienced an injection site reaction. Serious AEs occurred in 15% of the adjusted group: five patients developed a serious infection (three patients with herpes zoster and two patients with pneumonia) and one patient developed a malignancy. Discontinuations due to AEs occurred in 10% of the patients in the contin group: one patient developed interstitial pneumonia, one patient showed a drug rash, and three patients had injection site reactions. No opportunistic infections, tuberculosis, and lymphomas were observed. Overall, 20% of the contin and adjusted groups had an infection through week 104, with nasopharyngitis and upper respiratory tract infections the most frequently reported. None of the serious infections in either group led to treatment discontinuation, although almost all of the serious infections led to hospitalization. Throughout the 104-week study period, there were no significant differences between the contin and adjusted groups in the number of cases of interstitial pneumonia (2 vs 0%), malignancies (breast cancer; 0 vs 2.5%), or injection site reaction (12 vs 7.5%), respectively.
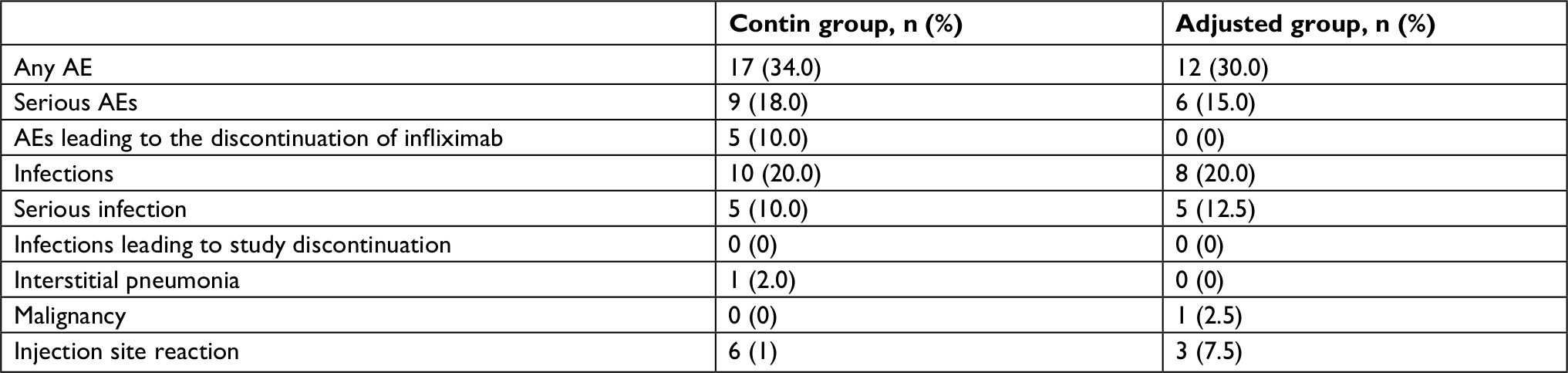 | Table 4 Safety assessment of the infliximab + methotrexate regimen Notes: Serious AEs as judged by the patients’ treating physicians. The patients who exhibited one or more AEs leading to the discontinuation of infliximab were only in the contin group. Discontinuations due to AEs occurred in five patients (one patient with interstitial pneumonia, one patient with cellulitis, and three patients with injection site reactions). Abbreviation: AEs, adverse events. |
Discussion
In a previous study, infliximab at 3 and 10 mg/kg given every 4–8 weeks for up to 2 years produced significant clinical, radiographic, and functional benefits in RA patients when added to background MTX therapy.2,3 However, some RA patients needed to be switched from infliximab treatment to other biological DMARDs due to an inadequate response. In general, switching these agents is a reasonable treatment strategy. In the SWITCH study, etanercept or adalimumab with an adequate or lower response in which RA patients were switched to infliximab.16 At week 10, approximately one-half of that study’s patients achieved a significantly improved DAS28–ESR, but approximately one-half of the responding patients required an infliximab dose escalation. The START trial17 assessed infliximab dose escalation in RA patients who showed an inadequate response to 3 mg/kg infliximab or whose disease flared following an initial response. Approximately 30% of the infliximab-treated patients in the START trial underwent dose escalation, and 80% of the patients who received up to three 1.5 mg/kg dose escalations achieved a 20% improvement in the total tender and swollen joint count after their last infliximab dose.
In contrast, an analysis of data from the Stockholm Biologics Registry revealed that the improvement in efficacy following infliximab dose escalation was small.18 In the ATTRACT study, the ACR response in patients treated with 3 mg/kg every 8 weeks tended to be lower than the responses obtained with a higher dose or a shorter perfusion interval.4 The pharmacokinetic analysis of serum samples from the ATTRACT patients showed that the serum level of infliximab was significantly more frequently undetectable in the patients treated with 3 mg/kg every 8 weeks compared to the patients treated with a higher dose (10 mg/kg) or a shorter treatment interval (every 4 weeks).4 More patients with detectable levels (>0.1 g/mL) of serum infliximab preinfusion showed ACR responses compared to the patients without detectable infliximab levels. The ATTRACT authors suggested that shortening of the dose interval would be preferable to increasing the dose at a constant interval.4
The RISING study investigated the efficacy and safety of RA treatment with infliximab, comparing 10 and 6 mg/kg with 3 mg/kg.6 The clinical response of mean percentage ACR improvement, the primary endpoint of that study, was 58.3% in the 10 mg/kg group and 51.3% in the 3 mg/kg group, presenting a significant difference. In addition, regarding the changes in the DAS28 and the EULAR response criteria, significantly higher responses were observed in the 10 mg/kg group compared to the 3 mg/kg group. In the radiographic assessment, the progression of joint damage was inhibited in most of the patients. The percentages of patients with no progression of joint damage (improved or no change) in the 3, 6, and 10 mg/kg groups were 93.0, 87.0, and 94.7%, respectively. However, these results were not significantly different.6 The START and RISING studies were also limited by their 1-year study duration. Some of the patients in these studies might have responded if the study had continued for longer than 1 year.
As an extended investigation, the REVIVE study was conducted to evaluate the clinical responses and radiographic outcomes of RA patients who were eligible to undergo an adjustment of infliximab in a dose escalation strategy and a de-escalation strategy based on their DAS28 scores for 2 years. About 20% of the patients in the adjusted group did not require any dose escalation or de-escalation and continued to receive 3 mg/kg infliximab throughout the 104 weeks. At weeks 26 and 52, there were no significant differences in DAS28 scores between the adjusted and contin groups. At week 104, remarkable changes in the DAS28 and the EULAR response criteria were observed; a significantly higher percentage of good responses was observed in the adjusted group compared to the contin group.
In addition, at week 104, nearly 50% of the patients achieved a significantly higher remission rate in the DAS28 (25 and 47.4% of the contin and adjusted groups, respectively) and in the ACR/EULAR Boolean-based criteria (16.7 and 39% of the contin and adjusted groups, respectively). Regarding the joint damage progression shown by the radiographic assessment, the changes in mTSS from baseline were significantly inhibited and radiographic nonprogression at week 104 was achieved at a higher rate in the adjusted group (64.3 and 40% in the adjusted and contin groups, respectively). These results indicate the importance of RA patients’ clinical responses and radiographic assessment, as highlighted by the difference between the two groups after week 52.
Infliximab treatment has shown overall lower retention rates compared to adalimumab and etanercept.19 The retention rate for infliximab is in accordance with that in the Danish DANBIO registry, with a half-life of −2 years, and the authors of that study19 reported that the difference in the retention rate was most pronounced for withdrawal due to AEs but was also significant for infliximab compared to etanercept in the patients who withdrew due to the lack of efficacy. In contrast, a German registry showed that the short-term drug survival rates were similar for etanercept and infliximab.20 In the REVIVE study, the overall retention rate was higher than that in the DANBIO report at week 104.19 In addition, the retention rate in our adjusted group was increased compared to that in the contin group, but there was no significant difference in the retention rate between the two groups through week 104.
In contrast, the rate of adverse effects (including serious infections) in our patients who underwent the dose escalation and de-escalation strategies was not increased compared to the rate among the patients who underwent the continuous 3 mg/kg infliximab treatment. As observed in other investigations,5,6 patients with and without dose escalations did not show increased rates of AEs, serious AEs, infections, or serious infections.
As a retrospective study, the REVIVE study has some limitations. First, the lack of randomization and blinding may have resulted in bias by indication, channeling bias, and performance bias. Second, the disease duration in the adjusted group was shorter than that in the contin group, although no significant between-group differences in disease activity or radiographic findings were seen at baseline. Third, the study was also limited by the 2-year study duration. We cannot discuss the retention rate, clinical response, radiographic assessment, or safety profile at time points beyond 2 years.
The availability of biological agents has changed the treatment goals in patients with RA. Since treatments with biological agents are expensive, the increasing use of these agents represents a significant economic burden to society.21 In Japan, the mean total drug costs for 1 year of 3, 6, and 10 mg/kg infliximab therapies are 965,000 yen ($8,600 US dollars), 1,930,000 ($17,000), and 2,900,000 ($26,000), respectively – calculated using 60 kg (~132 lbs) as the average adult Japanese body weight. In Japan’s universal health care insurance system, the expenditures for the treatment of nonelderly and elderly patients differ. In general, the costs paid by patients <75 and ≥75 years are 30 and 10% of the total drug costs, respectively. In the present study, the infliximab therapy of two adjusted group patients who were <75 years had to be suspended due to the patients’ financial issues. The initial intensive therapy approach that used in the REVIVE study may be favorably economical for patients. Intensive therapy for the rapid achievement of stable LDA or remission has been associated with the maintenance of response upon tapering/withdrawal.22 The possibility of the tapering and the discontinuation of biological agents after LDA and remission are achieved must be considered, because of both the potential long-term safety issues and the economic burden associated with their expense.
Conclusion
RA patients who did not respond to the initial dose of 3 mg/kg infliximab and those who initially responded but subsequently flared showed improved clinical responses and radiographic assessments after a dose escalation of infliximab treatment based on the assessment of the DAS28, without an increased risk of serious AEs, including serious infections.
Acknowledgment
We thank all of the investigators who participated in the REVIVE study: K Kishimoto, T Shiga, S Hino, K Sakai, and J Ri.
Disclosure
The authors report no conflicts of interest in this work.
References
Furst DE, Breedveld FC, Kalden JR, et al. Updated consensus statement on biological agents for the treatment of rheumatoid arthritis and other immune mediated inflammatory diseases (May 2003). Ann Rheum Dis. 2003;62 Suppl 2:ii2–ii9. | ||
Lipsky PE, van der Heijde DM, St Clair EW, et al; Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. Infliximab and methotrexate in the treatment of rheumatoid arthritis. N Engl J Med. 2000;343(22):1594–1602. | ||
Maini RN, Breedveld FC, Kalden JR, et al; Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50(4):1051–1065. | ||
St Clair EW, Wagner CL, Fasanmade AA, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(6):1451–1459. | ||
Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66(9):1233–1238. | ||
Takeuchi T, Miyasaka N, Inoue K, Abe T, Koike T; RISING study. Impact of trough serum level on radiographic and clinical response to infliximab plus methotrexate in patients with rheumatoid arthritis: results from the RISING study. Mod Rheumatol. 2009;19(5):478–487. | ||
Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–324. | ||
Prevoo ML, van ’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38(1):44–48. | ||
Takeuchi T, Nakajima R, Komatsu S, et al. Impact of adalimumab on work productivity and activity impairment in Japanese patients with rheumatoid arthritis: Large-scale, prospective, single-cohort ANOUVEAU study. Adv Ther. 2017;34(3):686–702. | ||
Steinbrocker O, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. J Am Med Assoc. 1949;140(8):659–662. | ||
van der Heijde DM, van Riel PL, Nuver-Zwart IH, Gribnau FW, vad de Putte LB. Effects of hydroxychloroquine and sulphasalazine on progression of joint damage in rheumatoid arthritis. Lancet. 1989;1(8646):1036–1038. | ||
Bruynesteyn K, Boers M, Kostense P, van der Linden S, van der Heijde D. Deciding on progression of joint damage in paired films of individual patients: smallest detectable difference or change. Ann Rheum Dis. 2005;64(2):179–182. | ||
Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010;69(6):964–975. | ||
Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2):137–145. | ||
Felson DT, Smolen JS, Wells G, et al. American college of rheumatology/European league against rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011;70(3):404–413. | ||
Fleischmann R, Goldman JA, Leirisalo-Repo M, et al. Infliximab efficacy in rheumatoid arthritis after an inadequate response to etanercept or adalimumab: results of a target-driven active switch study. Curr Med Res Opin. 2014;30(11):2139–2149. | ||
Westhovens R, Yocum D, Han J, et al; START Study Group. The safety of infliximab, combined with background treatments, among patients with rheumatoid arthritis and various comorbidities: a large, randomized, placebo-controlled trial. Arthritis Rheum. 2006;54(4):1075–1086. | ||
van Vollenhoven RF, Klareskog L. Infliximab dosage and infusion frequency in clinical practice: experiences in the Stockholm biologics registry STURE. Scand J Rheumatol. 2007;36(6):418–423. | ||
Hetland ML, Christensen IJ, Tarp U, et al; All Departments of Rheumatology in Denmark. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum. 2010;62(1):22–32. | ||
Zink A, Listing J, Kary S, et al. Treatment continuation in patients receiving biological agents or conventional DMARD therapy. Ann Rheum Dis. 2005;64(9):1274–1279. | ||
Kobelt G. Health economic issues in rheumatoid arthritis. Scand J Rheumatol. 2006;35(6):415–425. | ||
Tanaka Y, Hirata S. Is it possible to withdraw biologics from therapy in rheumatoid arthritis? Clin Ther. 2013;35(12):2028–2035. |
Supplementary material
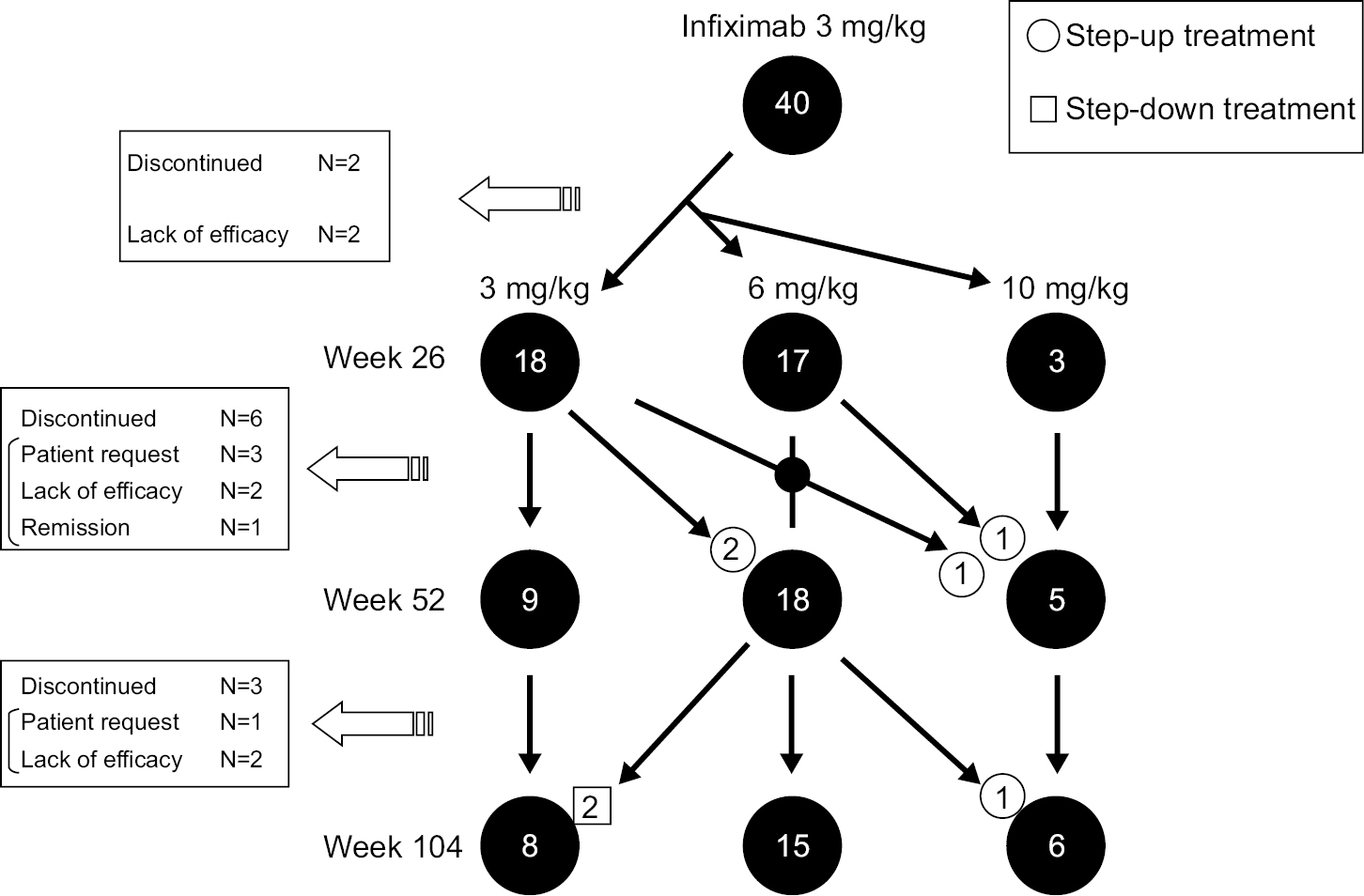 | Figure S1 Patients’ disposition. Notes: The primary reasons for discontinuation are listed. In the adjusted group, the escalation and de-escalation of the infliximab dose were selected based on the individual patient’s DAS28 with the clinical response at week 14. The decision was reconsidered every 8 weeks. If the patient did not reach a DAS28 of ≥3.2 for 4 months, the infliximab dose was increased (6 or 10 mg/kg). If the clinical response was consistently adequate (DAS28<3.2 for ≥6 months), the infliximab dose was gradually de-escalated until the dose remained at a maintenance dose or the infliximab was discontinued (for patients in remission). The starting infliximab dose of 3 mg/kg (n=40) at week 14 was adjusted as 3, 6, or 10 mg/kg every 8 weeks. At week 26, the numbers of patients who were treated with infliximab at 3, 6, and 10 mg/kg were 18, 17, and 3, respectively. The treatment of two patients was discontinued at week 26 (lack of efficacy). At week 52, the numbers of patients treated with infliximab at 3, 6, and 10 mg/kg were 9, 18, and 5, respectively. Some patients were escalated from 3 to 6 mg/kg (n=2) and 10 mg/kg (n=1) via 6 mg/kg and from 6 to 10 mg/kg (n=1). As of week 52, the treatment of six patients was discontinued due to the lack of efficacy (n=2), the patient’s request (n=3), or remission (n=1). At week 104 as the endpoint, the numbers of patients treated at 3, 6, and 10 mg/kg infliximab were 6, 10, and 6, respectively. Some patients were de-escalated from infliximab 6 to 3 mg/kg (n=2) and escalated from infliximab 6 to 10 mg/kg (n=1). As of week 104, the treatment of four patients was discontinued due to the lack of efficacy (n=2) and the patient’s request (n=1). Abbreviation: DAS28, Disease Activity Score in 28 joints. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.