Back to Journals » Journal of Inflammation Research » Volume 14
Inflammatory Markers Related to Innate and Adaptive Immunity in Atherosclerosis: Implications for Disease Prediction and Prospective Therapeutics
Authors Hong LZ, Xue Q, Shao H ![]()
Received 3 December 2020
Accepted for publication 21 January 2021
Published 16 February 2021 Volume 2021:14 Pages 379—392
DOI https://doi.org/10.2147/JIR.S294809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Ling-Zhi Hong,1,* Qi Xue,2,* Hong Shao2
1Emergency Department, Chun’an First People’s Hospital (Zhejiang Provincial People’s Hospital Chun’an Branch), Hangzhou, 311700, Zhejiang Province, People’s Republic of China; 2Department of Cardiology, Zhejiang Provincial People’s Hospital, People’s Hospital of Hangzhou Medical College, Hangzhou, 310014, Zhejiang Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong Shao
Department of Cardiology, Zhejiang Provincial People’s Hospital (People’s Hospital of Hangzhou Medical College), No. 158 Shangtang Road, Hangzhou, 310014, Zhejiang Province, People’s Republic of China
Tel/Fax +86-571-85894283
Email [email protected]
Abstract: Several lines of evidence have linked a dysregulated inflammatory setting to the pathogenesis of atherosclerosis, which is a form of chronic vascular inflammation. Various inflammatory biomarkers have been associated with inflammation and are recognized as potential tools to monitor the progression of atherosclerosis. A well-studied inflammatory marker in the context of cardiovascular diseases is C-reactive protein (CRP) or, more accurately, highly sensitive-CRP (hs-CRP), which has been established as an inflammatory biomarker for atherosclerotic events. In addition, a growing body of investigations has attempted to disclose the potential of inflammatory cytokines, enzymes, and genetic polymorphisms related to innate and adaptive immunity as biomarkers for predicting the development of atherosclerosis. In this review article, we clarify both traditional and novel inflammatory biomarkers related to components of the innate and adaptive immune system that may mirror the progression or phases of atherosclerotic inflammation/lesions. Furthermore, the contribution of the inflammatory biomarkers in developing potential therapeutics against atherosclerotic treatment will be discussed.
Keywords: atherosclerosis, inflammation, immune response, biomarker, hs-CRP
Introduction
Since the introduction of the “response to injury” hypothesis, atherosclerosis has been considered as chronic inflammation in the vascular system.1 This hypothesis holds that endothelial injury stimulates a complex of responses, particularly inflammatory reactions, that finally manifests as atherosclerosis. The accumulation of low-density lipoprotein (LDL) in the arterial intima of vessels and its subsequent oxidization (ox-LDL) are the initial steps involved in the initiation of atherosclerosis. Then, the inflammatory state in the endothelium develops as a series of events that lead to the infiltration of monocytes and lymphocytes into the vessel wall. These events promote an inflammatory condition in the vessel wall.2 Monocytes recruited to the inflammation site differentiate into macrophages, which engulf the ox-LDL and then develop into foam cells. Consequently, these occurrences increase the size and volume of atherosclerotic plaques. The inflammatory immune cells present in the plaques release several factors, such as matrix metalloproteinases (MMPs), that contribute toward the degradation of the extracellular matrix (ECM). MMPs and other enzymes promote the loosening of the fibrous cap and plaque instability/rupture. Ultimately, atherosclerotic plaque rupture culminates in the development of a thrombus (Figure 1). Acute coronary syndrome occurs in the case of the vessel being occluded by the rupture of the plaque.3
 |
Figure 1 Schematic overview of the process of atherosclerosis. Monocytes infiltrate the inflammation site and differentiate into macrophages, which engulf the ox-LDL, and then develop into foam cells, which in turn increase the size and volume of atherosclerotic plaques. Foam cells, as well as other inflammatory immune cells present in atherosclerotic plaques, release several factors, among which extracellular matrix (ECM)-degrading enzymes such as matrix metalloproteinases (MMPs) contribute to the degradation of the ECM, resulting in loosening of the fibrous cap and plaque instability/rupture. Finally, atherosclerotic plaque rupture culminates in the development of a thrombus. |
The bulk of evidence suggests that various inflammatory factors may be increased in different phases of atherosclerosis initiation and perpetuation. These factors have been studied concerning the development of inflammatory biomarkers related to innate and adaptive immunity to identify specific and sensitive elements in the prognosis of cardiovascular risk.4 In this study, we review the research on the development of inflammatory biomarkers in the context of optimized monitoring of atherosclerosis.
Endothelial Function in Atherosclerosis
Over the past few decades, endothelial dysfunction has been suggested to be linked to cardiovascular events.5 The vessel endothelium plays a critical function in establishing homeostasis of the vessel wall. In normal physiology, the vessel endothelium modulates the relaxation of the vascular tone and establishes a lower state of oxidative stress by regulating the function of angiotensin as well as producing mediators such as endothelin, prostacyclin, and nitric oxide (NO). The vessel endothelium is also involved in controlling the permeability of the vessel wall to blood serum, adhesion and accumulation of circulating immune cells and platelets, and coagulation events such as thrombosis.6
However, various settings can cause an imbalance in the physiological state of the vessel endothelium.7 A number of stimulating factors might alter the phenotypical characteristics of the endothelium toward a condition called endothelial dysfunction, in which the mechanisms regulating homeostasis of the endothelium are abrogated. During pathological endothelial dysfunction, the vascular tone is dysregulated, the release of inflammatory and thrombosis-stimulating mediators is promoted, expression of adhesion molecules is upregulated, and generation of oxidative stress components is enhanced.8
Several lines of evidence endorse the involvement of endothelial dysfunction in the initial phases of atherosclerosis development. The formation and progression of atherosclerotic plaques have also been associated with endothelial dysfunction. People with cardiovascular risk factors, yet lacking clinical presentations of atherosclerosis, may present with endothelial dysfunction. Such individuals have an abnormal function of vessels in response to endothelial vasodilators.9 Therefore, endothelial dysfunction may be considered as a bridge between risk factors for atherosclerotic and atherosclerosis development. Moreover, in patients with risk factors for atherosclerosis and other cardiovascular disorders, endothelial dysfunction may be an independent predictor of future cardiovascular events.10
Numerous studies have testified that a prolonged inflammation related to innate and adaptive immune abnormality is involved in the development of atherosclerosis; therefore, atherosclerosis is currently regarded as an autoinflammatory disorder.11 Patients with autoimmune disorders have been shown to have an increased rate of subclinical atherosclerosis, suggesting a relationship between atherosclerosis and autoimmunity and a shared mechanism in the pathogenesis of these pathological conditions.12,13 During the autoimmune response events in atherosclerosis, several atherosclerosis-related elements, such as ox-LDL, may act as an autoantigen source to trigger inflammatory innate and adaptive cells, such as monocytes, macrophages, T and B cells, that finally promote atherosclerotic plaque formation, plaque expansion, and instability/rupture of the plaques.14,15
Research has shown that ox-LDL plays an important role in atherosclerosis development during autoimmune disorders and should be regarded as a critical triggering factor of chronic inflammation.16 Interaction of ox-LDL with the vessel wall leads to activation of the endothelial cells and overexpression of several factors, including adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), as well as the release of chemokines and cytokines, resulting in the infiltration of immune cells to the site of vessel involvement.17 Monocytes recruited to the site of endothelial injury develop into macrophages that internalize the ox-LDL, leading to the formation of foam cells that are crucially involved in the release of several inflammatory factors.18 Studies have revealed that ox-LDL interacts with C-reactive protein (CRP) to develop the ox-LDL/CRP formations, which stimulate autoimmune reactions, vascular inflammation, and atherosclerosis development.19
Several inflammatory biomarkers have been developed to assess the endothelial dysfunction during atherosclerosis development and various medications are utilized to control the inflammatory responses in atherosclerotic individuals,20,21 which will be discussed in this article.
Innate and Adaptive Immunity in Atherosclerosis
Innate Immunity in Atherosclerosis
In the early phases of the inflammatory response during atherosclerosis, studies have indicated the involvement of critical cells playing a role in the cellular arm of innate immunity, namely, monocytes/macrophages. Studies on human arterial tissues and animal models of atherosclerosis have demonstrated that the infiltration of monocytes into the intima is the initial event during atherogenesis. The infiltration of monocytes to the region involved in atherosclerosis requires the adhesion of these cells to the activated endothelial cells through adhesion molecules, named selectins and integrins. The inflammatory cytokines and chemokines modulate the infiltration of monocytes into the region involved in atherosclerosis. After migration, monocytes differentiate into macrophages, proliferate, and release a wide range of mediators. These classical events have already been reviewed in detail.22–24 Herein, we assess the hypothesis regarding the involvement of innate immunity in the process of atherogenesis.
Research has disclosed new aspects of monocyte infiltration to the intima during atherosclerosis. Surveys on the kinetics of monocyte infiltration to the involved sites in animal models of atherosclerotic indicate that monocytes migrate to the intima layer both in the early phases of lesion development and over the course of an established atherosclerotic lesion.25 This highlights that monocyte trafficking could be targeted for therapeutic purposes in atherosclerosis.
The heterogeneity in the monocyte population has been focused on as a critical issue in determining the severity and outcome of an atherosclerotic lesion.26 Observations from human samples and mouse models propose the double-morphism of monocyte phenotypes according to the disease phase.27–29 Experiments in a mouse model indicated that higher lipid levels stimulate the development of a highly pro-inflammatory phenotype of monocytes. In humans, such pro-inflammatory monocytes express high levels of P-selectin glycoprotein ligand (PSGL) on the surface.30 PSGL-expressing pro-inflammatory monocytes tend to migrate to the intima layer in atherosclerotic lesions to produce higher amounts of inflammatory mediators (particularly MMPs), promoting the inflammatory innate immune response in the intima.
Studies have also suggested the potential involvement of mast cells in the inflammatory process of atherosclerosis. Although mast cells are found less often in atherosclerotic lesions, they may play critical roles in the pathogenesis of atherosclerosis.30–32 In atherosclerotic lesions, these cells have been shown to release several mediators, including heparin, histamine, serine proteinases, and leukotrienes, which suit the conditions for inflammation.33,34 Although these data have been illustrated in preliminary experiments, further research on human samples is still required. A number of drugs has already been devised to alter the function of mast cells, and hence could be further considered in the treatment of atherosclerosis.35
The involvement of lipoproteins in the modulation of innate immune players has been established. Altered lipoproteins, such as ox-LDL, are able to bind to the scavenger receptors expressed on the macrophages and transduce pro-inflammatory signals into these cells.36,37 Some studies have focused on phospholipase A2 (PLA2), which is able to generate pro-inflammatory stimulators from the oxidized lipoproteins.38,39 PLA2 is reported to be an inflammatory biomarker of cardiovascular diseases and to be involved in the pathogenesis of atherosclerosis.40 In addition, apolipoprotein C3 has been indicated to be able to ligate to the Toll-like receptors (TLRs), particularly TLR-2, on the immune cells, promoting the inflammatory signals in these cells.41,42
A relationship between inflammation and thrombosis has been suggested.43,44 Thrombosis is proposed to be a mechanism of the innate immune system in defending against pathogenic microbes.45 For instance, the cyclooxygenase pathway generates prostaglandins that are involved in the modulation of thrombosis and inflammation.46 Hence, inhibitors of the cyclooxygenase pathway may increase the risk of thrombosis.47,48 Thrombin is a critical protein mediator that modulates coagulation and enables the production of inflammatory mediators fromvascular endothelial cells and vascular smooth muscle cells (VSMCs).49,50
Activated platelets produce and release inflammatory mediators and can upregulate the surface expression of inflammatory molecules, such as the ligand of CD40 (also known as CD154).51–53 Myeloid-related protein (MRP)-8/14, which is an inflammatory mediator, can also be generated and released by platelets.54,55 This molecule has been indicated to be a biomarker for the prognosis of adverse events of the cardiovascular system in survivors of acute coronary syndrome.56 Investigations have demonstrated that MRP-8/14 can ligate with TLR4, resulting in the activation of the innate immune system.57 MRP-8/14 is also involved in the apoptosis of endothelial cells, an event that has been associated with thrombosis in the atherosclerotic plaque.58 Taken together, the current knowledge implies a relationship between thrombosis and inflammation, proposing the involvement of these pathways in the development of atherosclerosis.
Adaptive Immunity in Atherosclerosis
Cellular Immunity
A large amount of data has stressed the critical modulatory function of adaptive immunity in atherosclerosis.59 Here, we discuss the recent findings on the implications of the adaptive immune system during atherosclerosis. A previous review has extensively described this pathway.60
T cells are activated after recognizing antigens expressed on the dendritic cells (DCs).61 DCs are frequently found in atherosclerotic plaques as well as in the nearby draining lymph nodes, and play a role in the activation of T cells through presenting antigens and co-stimulatory molecules to these cells.62 The probable antigens that can trigger T-cell activation in the lymph nodes in the vicinity of the atherosclerotic plaques are microbial components, heat-shock proteins, and a number of plasma lipoproteins.63–65 T cells recognizing these antigens proliferate and cause the amplification of the immune response.66 Then, interactions of the T cells with the same antigens culminate in the generation of the mediators and the development of inflammation. Cytotoxic T cells may also kill the cells expressing the antigens.67
After activation, CD4+ T cells may develop a distinct subclass of the effector helper T (Th) cells. The Th1 responses are generally involved in the development of the inflammatory pathways through the production of cytokines, particularly interferon (IFN)-γ. Investigations indicate that both the Th1 and the Th17 response aggravate atherosclerosis.24,68 Th2 cells may develop a reaction that modulates the inflammation; nonetheless, research on the function of Th2 in atherosclerosis is conflicting.69–73 Some studies propose that Th2 responses may stimulate the development of aortic aneurysm formation.74–76 On the other hand, regulatory T (Treg) cells are involved in the suppression of the immune responses through different mechanisms.12,77,78 Studies found that genetic manipulations of Treg cells (to interrupt their function) resulted in exacerbated atherogenesis in animal models, as presented by an increment in the size of lesions with upmodulated inflammation and the development of thrombosis.12,79 As a result, the responses by Treg and Th2 cells in relation to the inflammatory responses by Th1 and Th17 cells may contribute toward the improvement of atherosclerotic lesions.80
T cells expressing the surface marker of CD8 are involved in the recognition of antigens expressed by the major histocompatibility complex (MHC) class I. CD8+ T cells kill the target cells by triggering apoptosis in the target cells.81 Several chemokines released in the atherosclerotic lesion milieu are involved in the recruitment of the CD8+ T cells, which may then stimulate apoptosis in other immune cells and VSMCs, resulting in the development of lesions and related adverse complications.82,83 One of the functions of the natural killer (NK) T (NKT) cells is to recognize lipid antigens presented by CD1 molecules on several cells. The activated NKT cells generate diverse inflammatory mediators that augment the development of atherosclerosis.84,85
Humoral Immunity
Several lines of evidence have indicated that the humoral arm of the adaptive immune system, mediated by the antibodies produced by B cells, plays a critical role in the process of atherogenesis.86,87 Research has suggested the protective role of B cells in the development of atherosclerosis. It was shown that depletion of the B-cell colonies using splenectomy in animal models led to the exacerbation of atherosclerosis.88 In animal models with hypercholesterolemia, a humoral immune response develops against ox-LDL, resulting in decreased establishment of inflammation by atherosclerotic macrophages.89,90 In addition, immunization of animals with ox-LDL, resulting in antibody production against these molecules, has been associated with the exacerbation of atherosclerosis. In a mouse model, the antibodies produced against ox-LDL also targeted a bacterial antigen on the pneumococcus.91,92 This observation casts doubt on the notion that the host immune responses against infectious antigens may overlap with inflammatory pathways during atherogenesis. The involvement of humoral immunity in targeting ox-LDL and probable protection against atherosclerosis may open up new horizons in devising therapeutic tools to prevent or mitigate the inflammatory state in atherosclerotic patients.
Inflammatory Biomarkers in Atherosclerosis
Upon identification of the inflammatory pathways involved in the etiology and pathogenesis of atherosclerosis, it makes sense to translate the biology of these events into clinical applications. The identification of bona fide inflammatory implications has led to novel potential therapeutic strategies being designed in the context of atherosclerosis.93,94 Several currently available medications used to suppress inflammation, such as glucocorticoids,95 non-steroidal anti-inflammatory drugs (NSAIDs),96 anti-inflammatory cytokine therapy,97 and monoclonal antibodies,98,99 may be linked to the development of adverse complications that hinder their use as long-term treatments for the improvement of atherosclerosis. Several promising anti-inflammatory compounds developed for atherosclerosis therapy have not yet been approved for clinical application.100 However, the application of inflammatory biomarkers to estimate the disease risk and monitor the efficacy of therapeutics, and in personalized therapy, has indicated their significant potential for clinical utilization.101
Inflammatory Biomarkers in Prediction of Atherosclerosis Risk
An association between diverse inflammatory biomarkers and estimation of the risk of cardiovascular events has been reported. The clinical applicability of a biomarker for the prediction of a disease risk relies on several factors, including reproducibility, practicability, ease of implementation, and cost-effectiveness.102 Of the numerous inflammatory biomarkers suggested for the diagnosis of atherosclerosis, most attention has been paid to lipoprotein-associated phospholipase A2 (Lp-PLA2),103 pentraxin-3,104,105 myeloperoxidase,106,107 cytokines such as interleukin (IL)-6,108 CRP or highly sensitive CRP (hs-CRP),109 and proteases such as MMP-9.110,111
C-Reactive Protein
According to numerous pieces of evidence, CRP has been highlighted as a critical inflammatory biomarker for clinical use.112,113 In healthy subjects without inflammatory disorders or acute infections, investigations have observed stable levels of hs-CRP (compared to cholesterols) for long periods.114,115 The beneficial points of CRP as a biomarker relative to other molecules include its remarkable stability, the fact that no invasive approaches are needed for sampling, and its comparably long half-life, with no diurnal variation. Several large prospective cohort investigations have demonstrated that hs-CRP estimates the incidence of myocardial infarction, cardiovascular-related death, and stroke, even after adjusting for traditional risk factors.116 In subjects with psoriasis, the subclinical atherosclerosis risk was reported to be high and hs-CRP was found to be a beneficial biomarker for the prediction of the future risk of cardiovascular complications in these subjects.117 Unbiased computational methods have shown that hs-CRP, accompanied by parental history, augments the predictive ability of the traditional risk factors in detecting cardiovascular events.118,119 According to the Reynolds risk scores, hs-CRP plus parental history is able to perfectly recategorize subjects classified as having intermediate risk based on the traditional Framingham criteria.120 hs-CRP and fibrinogen, as inflammatory biomarkers, have been implicated in fatty liver. Non-alcoholic fatty liver disease may predict future atherosclerotic cardiovascular disease, and the predictive value increases with the addition of inflammatory biomarker levels.121 Therefore, hs-CRP has the potential to be a useful auxiliary tool to the Framingham score to predict cases at increased risk of cardiovascular complications.
Highly Sensitive C-Reactive Protein
Research has also evaluated the potential of hs-CRP as a biomarker in evaluating therapeutics in cardiovascular events.122 In the clinic, several biomarkers are being followed as an approach to monitor the efficacy of the medications and to determine the optimal dose of drugs in patients with cardiovascular diseases.123 For instance, the levels of LDL are routinely measured after the administration of lipid-lowering drugs. Considering that inflammation is involved in the different phases of atherosclerosis, it seems that hs-CRP could be used as an inflammatory biomarker to monitor the efficacy of treatments in soothing the inflammatory indices and clinical manifestations. In the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis In Myocardial Infarction 22 (PROVE IT-TIMI 22) trial, it was proposed that statins may decrease LDL levels and soothe the inflammation in the context of cardiovascular diseases, as mirrored through the decreased level of hs-CRP in patients.124,125 According to this trial, statins were suitable compounds to reduce the hs-CRP levels to below 2 mg/L.126 Other studies on the survivors of acute coronary syndromes supported the remarkable effect of statin therapy by lowering the hs-CRP levels.127,128 Despite the endorsement of this evidence on the potential of monitoring hs-CRP to measure the efficacy of statin treatment, further explorations may demonstrate more advantages of this biomarker by following the efficacy of medications in lowering inflammation in patients with cardiovascular disorders.
Neutrophil Gelatinase-Associated Lipocalin
Neutrophil gelatinase-associated lipocalin (NGAL), an acute-phase protein that regulates inflammation,129 is increased in cardiovascular disease130 and has been investigated in the context of atherosclerosis. It was reported that the NGAL level was higher in patients with high total atherosclerotic plaque volumes compared to those with low plaque volumes. In addition, a high predictive score of NGAL was seen in patients without established cardiovascular disease. Therefore, it seems that NGAL could be an encouraging inflammatory biomarker to predict cases with asymptomatic atherosclerosis.131
Endothelial Dysfunction Biomarkers
There is a need to develop efficient biomarkers to monitor the change from normal endothelium to endothelial dysfunction. The adhesion molecules expressed on the endothelium, such as ICAM-1 and VCAM-1, have been regarded as well-accepted biomarkers to survey the endothelial dysfunction.132 It was also observed that ICAM-1 and VCAM-1 were associated with the development of new post-acute myocardial infarction symptoms in patients with heart failure.133 Several inflammatory mediators, such as tumor necrosis factor (TNF)-α, are able to enhance the surface expression of adhesion molecules on the vessel endothelial cells.134 Another study suggested the potential of soluble biomarkers in monitoring endothelial dysfunction, including E selectin, von Willebrand factor, soluble endothelial cell protein C receptor, thrombomodulin, and tissue plasminogen activator.135
Studies have demonstrated that vascular homeostasis is manipulated by a balance between injury and repair of the endothelium.136–139 Reports show that the level of endothelial injury-related factors, such as endothelial microparticles (EMPs), is increased in the serum of individuals at risk for cardiovascular diseases, while the level of factors associated with endothelial repair, such as endothelial progenitor cells (EPCs), is reduced.137–139 As a consequence, enhanced EMP and decreased EPC may have potential in estimating endothelial dysfunction and the increased risk of future cardiovascular events.140,141
Despite progress in developing biomarkers of endothelial dysfunction, there are several caveats and points limiting their use in the clinic. Application of each biomarker individually may confer little efficacy; hence, a combination panel of biomarkers may offer better outcomes in distinguishing disease types, stratifying cardiovascular risk factors, revealing disease origin, and devising potential new targets for disease treatment.
Prospective and Future Directions
Biomarkers for Anti-Inflammatory Therapy in Atherosclerosis
Many investigations have endorsed the theory that statin therapy decreases the level of inflammation in patients with cardiovascular complications. Advances in determining the etiology and pathogenesis of the underlying inflammatory mechanisms in atherosclerosis have revealed potential new tools to modulate inflammation in the context of atherosclerosis. To date, no large clinical trial has demonstrated that anti-inflammatory therapy could improve the cardiovascular complications, regardless of unaltered lipid profile. Even though some systemic anti-inflammatory treatments, such as NSAIDs or corticosteroids, do not seem to be hopeful compounds for the treatment of atherosclerosis, other treatments in this category appear to be promising.
Several clinical trials are under consideration to probe the potential of inhibiting PLA2 as an anti-inflammatory treatment, even though the initial data were discouraging.142 On the other hand, several strategies to interrupt the trafficking of inflammatory cells to the atherosclerotic milieu and to inhibit the production of or neutralize the released inflammatory cytokines have gained remarkable attention for the treatment of atherosclerotic patients.143,144 Lipoprotein peptides have also been used as therapeutic vaccination in clinical trials.145 Nonetheless, many clinical considerations need to be addressed before these anti-inflammatory modalities can be introduced as inflammatory biomarkers in the practical setting.
The conventional imaging of cardiovascular complications concentrates on anatomical observations. Many functional aspects of the cardiovascular system can be investigated using magnetic resonance and nuclear imaging methods. That notwithstanding, determining the molecular players of inflammation that are involved in the process of atherogenesis has led to remarkable interest in their use as strategies for imaging. For instance, integrins, adhesion molecules (such as ICAM and VCAM), phagocytosis and glucose uptake (the function of inflammatory macrophages) evaluated using 18F-fluorodeoxyglucose, microvessels recognized via integrin-directed compounds, accumulation of modified LDL in atherosclerotic lesions, and ECM proteinases involved in plaque vulnerability have gained significant attention.146,147 Several investigations have indicated the applicability of these targeted imaging modalities and some of them are close to being applied in real clinical practice.148,149
Reactive Oxygen Species Detection as a Biomarker in Atherosclerosis
Reactive oxygen species (ROS) are unstable and highly reactive by-products of oxygen that are generally produced as metabolites of endogenous processes involving molecular oxygen.150 Studies have indicated the involvement of ROS produced by vessel wall cells in the inception and development of cardiovascular events. Among the pathophysiological complications resulting from high levels of ROS that have been implicated in the initiation and perpetuation of atherosclerosis are upregulation of adhesion molecules, apoptosis of vessel endothelial cells, activation of MMPs, and lipid oxidation.151–154 All of these events may interconnect with the elevated inflammatory state and exacerbate atherosclerosis. Therefore, a sophisticated system for the detection of ROS in the vessels seems to be critical in determining cardiovascular complications in atherosclerotic subjects.
Several approaches have been developed to assay mitochondrial superoxide (using the MitoSOX method) and superoxide (using dihydroethidium [DHE]) in mammalian cells and mouse aorta. Measurement of ROS can be accomplished using fluorescence dye-based techniques155 or flow cytometry.156 In the DHE-based detection of ROS, DHE permeates the cell membrane, leading to its oxidization via endogenous O2•−. When DHE is oxidized, two red-fluorescent compounds are generated: one is ethidium, which is produced by a non-specific redox reaction, and the other is 2-hydroxyethidium (a specific adduct of cellular O2•−). As a limiting point, these products have similar fluorescent spectra which overlap, making it difficult to apply fluorescence-based microscopic assays or confocal microscopy to detect them. As a result, a modified high-performance liquid chromatography (HPLC) method has been developed to accurately measure both ethidium and 2-hydroxyethidium. HPLC also has a limitation in its inability to discriminate the origin of the cell type that produced ROS in response to the factors triggering atherosclerosis.157 However, fluorescent microscopy is typically used in the DHE system to detect the concentration of ROS in the vessels of atherosclerotic patients.158
In the context of atherosclerosis, endothelial dysfunction (and the resulting inflammation) may underlie the high production of ROS, predominantly by mitochondria from various cells.159 The red fluorescence in the MitoSOX approach is a modified analog of DHE that is generated via the addition of a triphenylphosphonium (TPP) group. In this technique, the mitochondrial O2•− is a specific target of TPP. In analogy with the DHE method, oxidization of MitoSOX leads to the constitution of two fluorescent compounds, namely mito-ethidium and 2-hydroxy-mito-ethidium. In living cells, Mitotracker green and MitoSOX red are used for two-color staining to detect mitochondrial O2•− by means of confocal fluorescent microscopy.160 Nonetheless, owing to limitations of the MitoSOX approach, a combined approach of MitoSOX/HPLC is frequently utilized to measure mitochondrial ROS.161,162
Genetic Biomarkers of Inflammation in Atherosclerosis
Genetic association studies have also been promising in the prediction of inflammation in the context of atherosclerosis.163,164 Advances in genetics have facilitated the possibility of devising genetic biomarkers for estimatng the risk of atherosclerosis . Evaluation of diverse biomarkers, referring to the already recognized familial history of cardiovascular disorders in parents along with hs-CRP, improved the potential of the traditional risk factors in the prediction of the cardiovascular risk.118,119 As a consequence, genetic factors have a potential application in predicting the cardiovascular risk, which may not be reflected by the traditional risk factors.
The first line of investigations studied genetic variations such as single-nucleotide polymorphisms (SNPs) in the allele, genotype, haplotype, as well as inheritance models,165 which were accomplished through both small and genome-wide studies.166 CRP gene rs1205 SNP was detected to be associated with low-grade chronic inflammation.167 In multiple studies, a genetic locus on chromosome 9 was associated with susceptibility to cardiovascular disease,168–170 suggesting the potential for variations in defining novel biomarkers for cardiovascular diseases. More importantly, genome-wide association studies (GWASs) have identified the potential genetic loci in atherosclerosis with higher validity.171 A GWAS in the Han Chinese population suggested that Phosphodiesterase 4D (PDE4D) gene rs966221 polymorphism may be a valuable biomarker for predicting recurrent risks in ischemic stroke patients with large artery atherosclerosis.172 On the other hand, an individual SNP evaluation indicated the involvement of polymorphism in the receptor activator of NF-κB (RANK) as a predictor of blood pressure and, hence, cardiovascular risk.173 Identification of the functional implications of the genomic variations implicated in inflammation may disclose the corresponding biological pathways revealed by GWAS. This may increase the potential of genetic predisposing factors in identifying the inflammatory biomarkers in the context of atherosclerosis (Table 1).
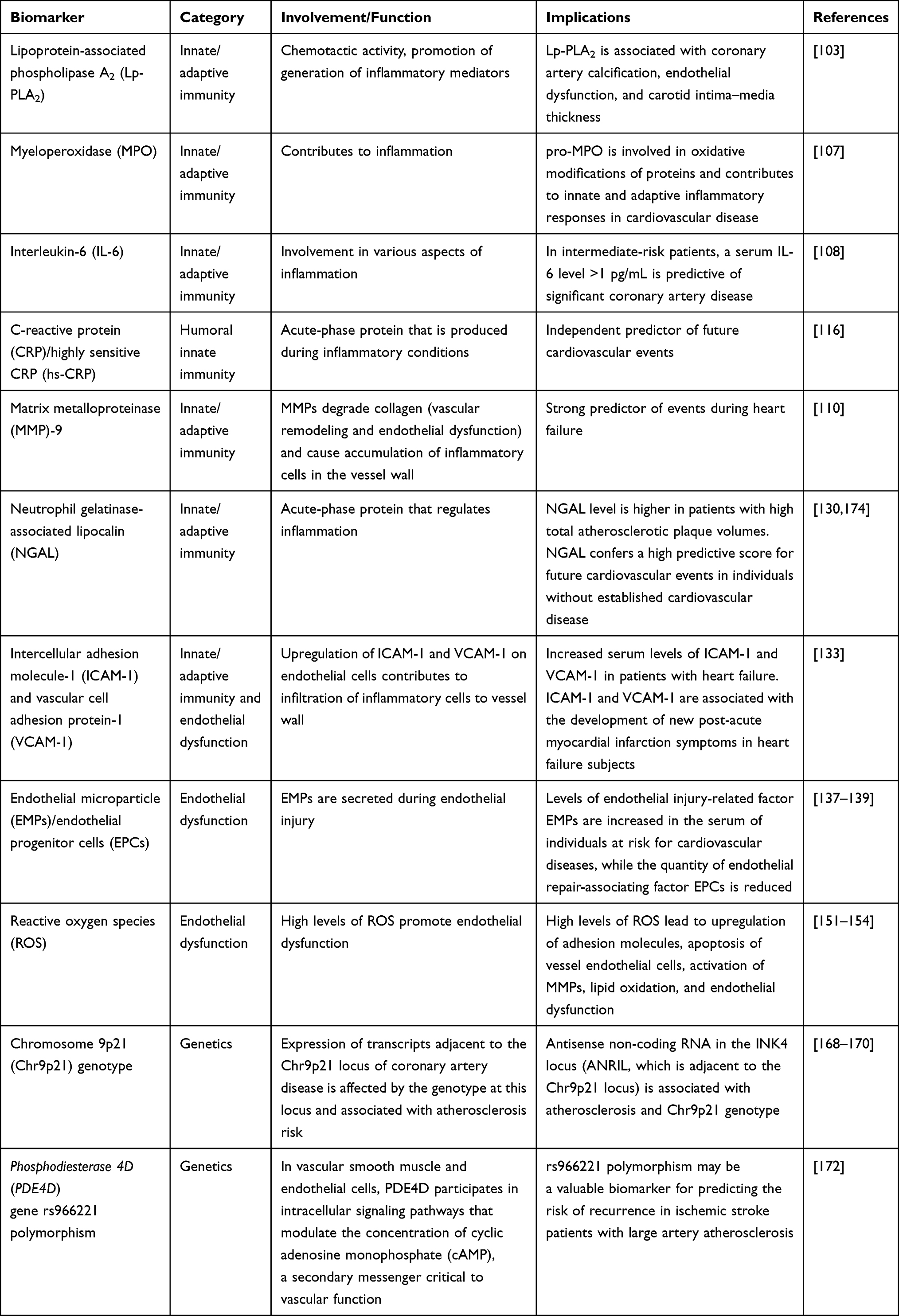 |
Table 1 Inflammation-Related Biomarkers for Prediction of Cardiovascular Diseases |
Conclusions
Over the past few years, our understanding of the implications of the innate and adaptive immune inflammatory responses in the etiology and pathogenesis of atherosclerosis has improved. We are currently on the brink of translating our knowledge on inflammation from the bench to bedside. Among the inflammatory molecules involved in the different steps of atherogenesis, hs-CRP has shown promising potential to be used as a biomarker not only in the prediction of future atherosclerosis development, but also in the recurrence of cardiovascular disorders and in the estimation of medication efficacy. The urge to translate the laboratory findings into clinical application has challenged laboratory research to revolutionize the insights to understand different aspects of the interactions between inflammation and atherosclerosis. The necessity for a fast-track utilization of the laboratory progress in clinical cardiovascular medicine is inspiing the design of sophisticated strategies for the diagnosis, prognosis, management, and monitoring of patients who are at probable riskof atherosclerosis in the years to come.
Acknowledgments
This work was supported by the Hangzhou Medical Technology Plan Project (no. OO20190052 to LZH) and Zhejiang Provincial Science and Technology Projects (no. GF20H020041 to HS).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cell. Science. 1973;180(4093):1332–1339. doi:10.1126/science.180.4093.1332
2. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551–561.
3. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325.
4. Bäck M, Hansson GK. Anti-inflammatory therapies for atherosclerosis. Nat Rev Cardiol. 2015;12(4):199.
5. Sitia S, Tomasoni L, Atzeni F, et al. From endothelial dysfunction to atherosclerosis. Autoimmun Rev. 2010;9(12):830–834.
6. Pries A, Kuebler W. Normal endothelium. In: The Vascular Endothelium I. Springer; 2006:1–40.
7. Victor VM, Rocha M, Sola E, Banuls C, Garcia-Malpartida K, Hernandez-Mijares A. Oxidative stress, endothelial dysfunction and atherosclerosis. Curr Pharm Des. 2009;15(26):2988–3002.
8. Tritto I, Ambrosio G. The Multi-Faceted Behavior of Nitric Oxide in Vascular “Inflammation”: Catchy Terminology or True Phenomenon? Elsevier Science; 2004.
9. Zardi EM, Afeltra A. Endothelial dysfunction and vascular stiffness in systemic lupus erythematosus: are they early markers of subclinical atherosclerosis? Autoimmun Rev. 2010;9(10):684–686.
10. Gokce N, Keaney JF, Hunter LM, et al. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events inpatients with peripheral vascular disease. J Am Coll Cardiol. 2003;41(10):1769–1775. doi:10.1016/S0735-1097(03)00333-4
11. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–2051.
12. Sanjadi M, Rezvanie Sichanie Z, Totonchi H, Karami J, Rezaei R, Aslani S. Atherosclerosis and autoimmunity: a growing relationship. Int J Rheum Dis. 2018;21(5):908–921.
13. Mahmoudi M, Aslani S, Fadaei R, Jamshidi AR. New insights to the mechanisms underlying atherosclerosis in rheumatoid arthritis. Int J Rheum Dis. 2017;20(3):287–297.
14. Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med. 2010;14(1‐2):70–78.
15. Ketelhuth DF, Hansson GK. Adaptive response of T and B cells in atherosclerosis. Circ Res. 2016;118(4):668–678.
16. Hulthe J, Fagerberg B. Circulating oxidized LDL is associated with subclinical atherosclerosis development and inflammatory cytokines (AIR Study). Arterioscler Thromb Vasc Biol. 2002;22(7):1162–1167.
17. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519.
18. Hahn BH, Grossman J, Chen W, McMahon M. The pathogenesis of atherosclerosis in autoimmune rheumatic diseases: roles of inflammation and dyslipidemia. J Autoimmun. 2007;28(2–3):69–75.
19. Obradovic MM, Trpkovic A, Bajic V, et al. Interrelatedness between C-reactive protein and oxidized low-density lipoprotein. Clin Chem Lab Med. 2015;53(1):29–34.
20. Dambala K, Paschou SA, Michopoulos A, et al. Biomarkers of endothelial dysfunction in women with polycystic ovary syndrome. Angiology. 2019;70(9):797–801.
21. Kershaw KN, Lane-Cordova AD, Carnethon MR, Tindle HA, Liu K. Chronic stress and endothelial dysfunction: the multi-ethnic study of atherosclerosis (MESA). Am J Hypertens. 2017;30(1):75–80.
22. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Eng J Med. 2006;354(6):610–621.
23. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124(2):315–327.
24. Gisterå A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. 2017;13(6):368.
25. Swirski FK, Pittet MJ, Kircher MF, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Nat Acad Sci. 2006;103(27):10340–10345.
26. Libby P, Nahrendorf M, Pittet MJ, Swirski FK. Diversity of denizens of the atherosclerotic plaque: not all monocytes are created equal. Circulation. 2008;117:3168–3170.
27. Swirski FK, Libby P, Aikawa E, et al. Ly-6C hi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205.
28. Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117(1):185–194.
29. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82.
30. An G, Wang H, Tang R, et al. PSGL-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117(25):3227.
31. Bot I, Biessen EA. Mast cells in atherosclerosis. Thromb Haemost. 2011;106(11):820–826.
32. Kovanen PT. Mast cells as potential accelerators of human atherosclerosis—from early to late lesions. Int J Mol Sci. 2019;20(18):4479.
33. Kritikou E, Depuydt MA, de Vries MR, et al. Flow cytometry-based characterization of mast cells in human atherosclerosis. Cells. 2019;8(4):334.
34. Hermans MA, Roeters van Lennep J, van Daele P, Bot I. Mast cells in cardiovascular disease: from bench to bedside. Int J Mol Sci. 2019;20(14):3395.
35. Kouhpeikar H, Delbari Z, Sathyapalan T, Simental-Mendía LE, Jamialahmadi T, Sahebkar A. The effect of statins through mast cells in the pathophysiology of atherosclerosis: a review. Curr Atheroscler Rep. 2020;22(5):19.
36. Khatana C, Saini NK, Chakrabarti S, et al. Mechanistic insights into the oxidized low-density lipoprotein-induced atherosclerosis. Oxid Med Cell Longev. 2020;2020.
37. Keping Y, Yunfeng S, Pengzhuo X, Liang L, Chenhong X, Jinghua M. Sestrin1 inhibits oxidized low‐density lipoprotein‐induced activation of NLRP3 inflammasome in macrophages in a murine atherosclerosis model. Eur J Immunol. 2020.
38. Patrick W, García-G HM, Pawel B, Paul E, Stefan V. Effects of the direct lipoprotein-associated phospholipase A2 inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118(1):172–182.
39. Huang F, Wang K, Shen J. Lipoprotein‐associated phospholipase A2: the story continues. Med Res Rev. 2020;40(1):79–134.
40. Santoso A, Heriansyah T, Rohman MS. Phospholipase A2 is an inflammatory predictor in cardiovascular diseases: is there any spacious room to prove the causation? Curr Cardiol Rev. 2020;16(1):3–10.
41. Krogmann AO, Lüsebrink E, Lahrmann C, Flender A, Nickenig G, Toll-like receptor 7 stimulation promotes the development of atherosclerosis in apolipoprotein E-deficient mice. Int Heart J. 2020;61(2):364–372.
42. Kawakami A, Osaka M, Aikawa M, et al. Toll-Like Receptor 2 Mediates Apolipoprotein CIII–Induced Monocyte Activation: Retracted. Am Heart Assoc; 2008:1402–1409.
43. De Caterina R, D’Ugo E, Libby P. Inflammation and thrombosis–testing the hypothesis with anti-inflammatory drug trials. Thromb Haemost. 2016.
44. Bojan IB, Badulescu O-V, Vladeanu M, Bojan A, Ciocoiu M. Correlations between inflammation and thrombosis in the pathogeny of myocardial infarction. In: Cardiac Diseases. IntechOpen; 2020.
45. Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14(1):55–61.
46. Mitchell JA, Kirkby NS, Ahmetaj-Shala B, et al. Cyclooxygenases and the cardiovascular system. Pharmacol Ther. 2020;107624.
47. Liberale L, Carbone F, Montecucco F, Sahebkar A. Statins reduce vascular inflammation in atherogenesis: a review of underlying molecular mechanisms. Int J Biochem Cell Biol. 2020;105735.
48. Lordan R, Tsoupras A, Zabetakis I. Platelet activation and prothrombotic mediators at the nexus of inflammation and atherosclerosis: potential role of antiplatelet agents. Blood Rev. 2020;100694.
49. Rahadian A, Fukuda D, Salim HM, et al. Thrombin inhibition by dabigatran attenuates endothelial dysfunction in diabetic mice. Vascul Pharmacol. 2020;124:106632.
50. Jaberi N, Soleimani A, Pashirzad M, et al. Role of thrombin in the pathogenesis of atherosclerosis. J Cell Biochem. 2019;120(4):4757–4765.
51. Schrottmaier WC, Mussbacher M, Salzmann M, Assinger A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis. 2020.
52. Michel NA, Zirlik A, Wolf D. CD40L and its receptors in atherothrombosis—an update. Front Cardiovasc Med. 2017;4:40.
53. Gerdes N, Seijkens T, Lievens D, et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler Thromb Vasc Biol. 2016;36(3):482–490.
54. Sakuma M, Tanaka A, Kotooka N, et al. Myeloid-related protein-8/14 in acute coronary syndrome. Int J Cardiol. 2017;249:25–31.
55. Wang Y, Gao H, Kessinger CW, Schmaier A, Jaffer FA, Simon DI. Myeloid-related protein-14 regulates deep vein thrombosis. JCI Insight. 2017;2(11).
56. Morrow DA, Wang Y, Croce K, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the pravastatin or atorvastatin evaluation and infection therapy: thrombolysis in myocardial infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155(1):49–55.
57. Vogl T, Tenbrock K, Ludwig S, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049.
58. Viemann D, Barczyk K, Vogl T, et al. MRP8/MRP14 impairs endothelial integrity and induces a caspase-dependent and-independent cell death program. Blood. 2007;109(6):2453–2460.
59. Miteva K, Madonna R, De Caterina R, Van Linthout S. Innate and adaptive immunity in atherosclerosis. Vascul Pharmacol. 2018;107:67–77.
60. Sima P, Vannucci L, Vetvicka V. Immunity in cancer and atherosclerosis. Ann Translat Med. 2019;7:9.
61. Wehr P, Purvis H, Law SC, Thomas R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin Exp Immunol. 2019;196(1):12–27.
62. Tai Y, Wang Q, Korner H, Zhang L, Wei W. Molecular mechanisms of T cells activation by dendritic cells in autoimmune diseases. Front Pharmacol. 2018;9:642.
63. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;1–15.
64. Cochain C, Zernecke A. Protective and pathogenic roles of CD8+ T cells in atherosclerosis. Basic Res Cardiol. 2016;111(6):71.
65. Gil-Pulido J, Zernecke A. Antigen-presenting dendritic cells in atherosclerosis. Eur J Pharmacol. 2017;816:25–31.
66. van Duijn J, Kuiper J, Slütter B. The many faces of CD8+ T cells in atherosclerosis. Curr Opin Lipidol. 2018;29(5):411–416.
67. Tse K, Tse H, Sidney J, Sette A, Ley K. T cells in atherosclerosis. Int Immunol. 2013;25(11):615–622.
68. Taleb S, Tedgui A, Mallat Z. IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb Vasc Biol. 2015;35(2):258–264.
69. Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163(3):1117–1125.
70. Binder CJ, Hartvigsen K, Chang M-K, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114(3):427–437.
71. Fredrikson GN, Andersson L, Söderberg I, et al. Atheroprotective immunization with MDA-modified apo B-100 peptide sequences is associated with activation of Th2 specific antibody expression. Autoimmunity. 2005;38(2):171–179.
72. van Wanrooij EJ, van Puijvelde GH, de Vos P, Yagita H, van Berkel TJ, Kuiper J. Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27(1):204–210.
73. Engelbertsen D, Rattik S, Knutsson A, Björkbacka H, Bengtsson E, Nilsson J. Induction of T helper 2 responses against human apolipoprotein B100 does not affect atherosclerosis in ApoE−/− mice. Cardiovasc Res. 2014;103(2):304–312.
74. Schönbeck U, Sukhova GK, Gerdes N, Libby P. TH2 predominant immune responses prevail in human abdominal aortic aneurysm. Am J Pathol. 2002;161(2):499–506.
75. Kasashima S, Kawashima A, Zen Y, et al. Upregulated interleukins (IL-6, IL-10, and IL-13) in immunoglobulin G4-related aortic aneurysm patients. J Vasc Surg. 2018;67(4):1248–1262.
76. Peshkova IO, Schaefer G, Koltsova EK. Atherosclerosis and aortic aneurysm–is inflammation a common denominator? FEBS J. 2016;283(9):1636–1652.
77. Azimi M, Aslani S, Mortezagholi S, et al. Identification, isolation, and functional assay of regulatory T cells. Immunol Invest. 2016;45(7):584–602.
78. Soltanzadeh-Yamchi M, Shahbazi M, Aslani S, Mohammadnia-Afrouzi M. MicroRNA signature of regulatory T cells in health and autoimmunity. Biomed Pharmacother. 2018;100:316–323.
79. Ou HX, Guo BB, Liu Q, et al. Regulatory T cells as a new therapeutic target for atherosclerosis. Acta Pharmacol Sin. 2018;39(8):1249–1258.
80. Pakzad B, Rajae E, Shahrabi S, et al. T-cell molecular modulation responses in atherosclerosis anergy. Lab Med. 2020.
81. Sun L, Zhang W, Zhao Y, et al. Dendritic cells and T cells, partners in atherogenesis and the translating road ahead. Front Immunol. 2020;11:1456.
82. Panigrahi S, Chen B, Fang M, et al. CX3CL1 and IL-15 Promote CD8 T cell chemoattraction in HIV and in atherosclerosis. PLoS Pathog. 2020;16(9):e1008885.
83. Kyaw T, Tipping P, Toh BH, Bobik A. Killer cells in atherosclerosis. Eur J Pharmacol. 2017;816:67–75.
84. Getz GS, Reardon CA. Natural killer T cells in atherosclerosis. Nat Rev Cardiol. 2017;14(5):304–314.
85. Li Y, Kanellakis P, Hosseini H, et al. A CD1d-dependent lipid antagonist to NKT cells ameliorates atherosclerosis in ApoE−/− mice by reducing lesion necrosis and inflammation. Cardiovasc Res. 2016;109(2):305–317.
86. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. 2019;16(3):180–196.
87. Chistiakov DA, Orekhov AN, Bobryshev YV. Immune-inflammatory responses in atherosclerosis: role of an adaptive immunity mainly driven by T and B cells. Immunobiology. 2016;221(9):1014–1033.
88. Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109(6):745–753.
89. Hartvigsen K, Chou M-Y, Hansen LF, et al. The role of innate immunity in atherogenesis. J Lipid Res. 2009;50(Supplement):S388–S393.
90. Chou MY, Hartvigsen K, Hansen LF, et al. Oxidation‐specific epitopes are important targets of innate immunity. J Intern Med. 2008;263(5):479–488.
91. Binder CJ. Natural IgM antibodies against oxidation-specific epitopes. J Clin Immunol. 2010;30(1):56–60.
92. Binder CJ, Shaw PX, Chang M-K, et al. Thematic review series: the immune system and atherogenesis. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46(7):1353–1363.
93. Gao C, Huang Q, Liu C, et al. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat Commun. 2020;11(1):1–14.
94. Kiaie N, Gorabi AM, Penson PE, et al. A new approach to the diagnosis and treatment of atherosclerosis: the era of the liposome. Drug Discov Today. 2020;25(1):58–72.
95. van der Sluis RJ, Hoekstra M. Glucocorticoids are active players and therapeutic targets in atherosclerotic cardiovascular disease. Mol Cell Endocrinol. 2020;504:110728.
96. Braun J, Baraliakos X, Westhoff T. Nonsteroidal anti-inflammatory drugs and cardiovascular risk–a matter of indication.
97. Kim M, Sahu A, Hwang Y, et al. Targeted delivery of anti-inflammatory cytokine by nanocarrier reduces atherosclerosis in Apo E−/-mice. Biomaterials. 2020;226:119550.
98. Klurfeld DM. Atherosclerotic lesions as macrophages using monoclonal antibodies. Arch Pathol Lab Med. 1985;109:445–449.
99. Bhaskar V, Yin J, Mirza AM, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. 2011;216(2):313–320.
100. Chistiakov DA, Melnichenko AA, Grechko AV, Myasoedova VA, Orekhov AN. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp Mol Pathol. 2018;104(2):114–124.
101. Soeki T, Sata M. Inflammatory biomarkers and atherosclerosis. Int Heart J. 2016;15–346.
102. Lyngbakken MN, Myhre PL, Røsjø H, Omland T. Novel biomarkers of cardiovascular disease: applications in clinical practice. Crit Rev Clin Lab Sci. 2019;56(1):33–60.
103. Younus A, Humayun C, Ahmad R, et al. Lipoprotein-associated phospholipase A2 and its relationship with markers of subclinical cardiovascular disease: a systematic review. J Clin Lipidol. 2017;11(2):328–337.
104. Ristagno G, Fumagalli F, Bottazzi B, et al. Pentraxin 3 in cardiovascular disease. Front Immunol. 2019;10:823.
105. Zlibut A, Bocsan IC, Agoston-Coldea L. Pentraxin-3 and endothelial dysfunction. Adv Clin Chem. 2019;91:163–179.
106. Correa S, Pena-Esparragoza JK, Scovner KM, Waikar SS, Mc Causland FR. Myeloperoxidase and the risk of CKD progression, cardiovascular disease, and death in the chronic renal insufficiency cohort (CRIC) study. Am J Kidney Dis. 2020;76(1):32–41.
107. Khalilova IS, Dickerhof N, Mocatta TJ, et al. A myeloperoxidase precursor, pro-myeloperoxidase, is present in human plasma and elevated in cardiovascular disease patients. PLoS One. 2018;13(3):e0192952.
108. Wainstein MV, Mossmann M, Araujo GN, et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol Metab Syndr. 2017;9(1):1–7.
109. Peikert A, Kaier K, Merz J, et al. Residual inflammatory risk in coronary heart disease: incidence of elevated high-sensitive CRP in a real-world cohort. Clin Res Cardiol. 2020;109(3):315–323.
110. Morishita T, Uzui H, Mitsuke Y, et al. Association between matrix metalloproteinase‐9 and worsening heart failure events in patients with chronic heart failure. ESC Heart Failure. 2017;4(3):321–330.
111. Lindsey ML. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat Rev Cardiol. 2018;15(8):471–479.
112. Pay JB, Shaw AM. Towards salivary C-reactive protein as a viable biomarker of systemic inflammation. Clin Biochem. 2019;68:1–8.
113. Held C, White HD, Stewart RA, et al. Inflammatory biomarkers interleukin‐6 and C‐reactive protein and outcomes in stable coronary heart disease: experiences from the STABILITY (stabilization of atherosclerotic plaque by initiation of darapladib therapy) trial. J Am Heart Assoc. 2017;6(10):e005077.
114. Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Eng J Med. 2004;350(14):1387–1397.
115. Glynn RJ, MacFadyen JG, Ridker PM. Tracking of high-sensitivity C-reactive protein after an initially elevated concentration: the JUPITER Study. Clin Chem. 2009;55(2):305–312.
116. Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49(21):2129–2138.
117. Niknezhad N, Haghighatkhah HR, Zargari O, et al. High-sensitivity C-reactive protein as a biomarker in detecting subclinical atherosclerosis in psoriasis. Dermatol Ther. 2020:e13628.
118. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611–619.
119. Ridker P, Paynter N, Rifai N, Gaziano J, Cook N. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118:2243.
120. Tattersall MC, Gangnon RE, Karmali KN, Keevil JG. Women up, men down: the clinical impact of replacing the framingham risk score with the reynolds risk score in the United States population. PLoS One. 2012;7(9):e44347.
121. Yu E, Hsu HY, Huang CY, Hwang LC. Inflammatory biomarkers and risk of atherosclerotic cardiovascular disease. Open Med. 2018;13:208–213.
122. Dadu RT, Nambi V, Ballantyne CM. Developing and assessing cardiovascular biomarkers. Translat Res. 2012;159(4):265–276.
123. Vavuranakis M, G Kariori M, I Kalogeras K, et al. Biomarkers as a guide of medical treatment in cardiovascular diseases. Curr Med Chem. 2012;19(16):2485–2496.
124. Pokharel Y, Sharma PP, Qintar M, et al. High-sensitivity C-reactive protein levels and health status outcomes after myocardial infarction. Atherosclerosis. 2017;266:16–23.
125. Ridker P. Pravastatin or atorvastatin evaluation and infection therapy-thrombolysis in myocardial infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28.
126. Ridker PM, Morrow DA, Rose LM, Rifai N, Cannon CP, Braunwald E. Relative efficacy of atorvastatin 80 mg and pravastatin 40 mg in achieving the dual goals of low-density lipoprotein cholesterol< 70 mg/dl and C-reactive protein< 2 mg/l: an analysis of the PROVE-IT TIMI-22 trial. J Am Coll Cardiol. 2005;45(10):1644–1648.
127. Morrow D, de Lemos J, Wiviott S, et al. Clinical relevance of C-reactive protein during follow-up of patients with ACS treated with statin therapy: an AtoZ substudy. Circulation. 2005.
128. Brilakis ES, de Lemos JA, Cannon CP, et al. Outcomes of patients with acute coronary syndrome and previous coronary artery bypass grafting (from the pravastatin or atorvastatin evaluation and infection therapy [PROVE IT-TIMI 22] and the Aggrastat to Zocor [A to Z] trials). Am J Cardiol. 2008;102(5):552–558.
129. Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochimica Et Biophysica Acta. 2000;1482(1–2):272–283.
130. Sivalingam Z, Larsen SB, Grove EL, Hvas A-M, Kristensen SD, Magnusson NE. Neutrophil gelatinase-associated lipocalin as a risk marker in cardiovascular disease. Clin Chem Lab Med. 2017;56(1):5–18.
131. Schreinlechner M, Noflatscher M, Lener D, et al. NGAL correlates with femoral and carotid plaque volume assessed by sonographic 3D plaque volumetry. J Clin Med. 2020;9:9.
132. Widlansky ME, Gokce N, Keaney JF, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42(7):1149–1160.
133. Lino D, Freitas I, Meneses GC, et al. Interleukin-6 and adhesion molecules VCAM-1 and ICAM-1 as biomarkers of post-acute myocardial infarction heart failure. Br J Med Biol Res. 2019;52(12).
134. Moore TC, Moore JE, Kaji Y, et al. The role of advanced glycation end products in retinal microvascular leukostasis. Invest Ophthalmol Vis Sci. 2003;44(10):4457–4464.
135. Blann AD. Assessment of endothelial dysfunction: focus on atherothrombotic disease. Pathophysiol Haemost Thromb. 2003;33(5–6):256–261.
136. Mannarino E, Pirro M. Endothelial injury and repair: a novel theory for atherosclerosis. Angiology. 2008;59(2_suppl):69S–72S.
137. Pirro M, Schillaci G, Menecali C, et al. Reduced number of circulating endothelial progenitors and HOXA9 expression in CD34+ cells of hypertensive patients. J Hypertens. 2007;25(10):2093–2099.
138. Pirro M, Schillaci G, Bagaglia F, et al. Microparticles derived from endothelial progenitor cells in patients at different cardiovascular risk. Atherosclerosis. 2008;197(2):757–767.
139. Pirro M, Schillaci G, Paltriccia R, et al. Increased ratio of CD31+/CD42− microparticles to endothelial progenitors as a novel marker of atherosclerosis in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2006;26(11):2530–2535.
140. Sinning J-M, Losch J, Walenta K, Böhm M, Nickenig G, Werner N. Circulating CD31+/Annexin V+ microparticles correlate with cardiovascular outcomes. Eur Heart J. 2011;32(16):2034–2041.
141. Schmidt-Lucke C, Rossig L, Fichtlscherer S, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111(22):2981–2987.
142. Boekholdt SM, de Winter RJ, Kastelein JJ Inhibition of lipoprotein-associated phospholipase activity by darapladib: shifting gears in cardiovascular drug development: are antiinflammatory drugs the next frontier?; 2008:1120–1122.
143. Heller EA, Liu E, Tager AM, et al. Inhibition of atherogenesis in BLT1-deficient mice reveals a role for LTB4 and BLT1 in smooth muscle cell recruitment. Circulation. 2005;112(4):578–586.
144. Erbel C, Chen L, Bea F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol. 2009;183(12):8167–8175.
145. Hansson G, Nilsson J. Introduction: atherosclerosis as inflammation: a controversial concept becomes accepted. J Intern Med. 2008;263(5):462–463.
146. Ou L-C, Zhong S, Ou J-S, Tian J-W. Application of targeted therapy strategies with nanomedicine delivery for atherosclerosis. Acta Pharmacol Sin. 2020;1–8.
147. Rudd JH, Narula J, Strauss HW, et al. Imaging atherosclerotic plaque inflammation by fluorodeoxyglucose with positron emission tomography: ready for prime time? J Am Coll Cardiol. 2010;55(23):2527–2535.
148. Tahara N, Kai H, Nakaura H, et al. The prevalence of inflammation in carotid atherosclerosis: analysis with fluorodeoxyglucose–positron emission tomography. Eur Heart J. 2007;28(18):2243–2248.
149. Kircher M, Tran-Gia J, Kemmer L, et al. Imaging inflammation in atherosclerosis with CXCR4-directed 68Ga-pentixafor PET/CT: correlation with 18F-FDG PET/CT. J Nucl Med. 2020;61(5):751–756.
150. Bayr H. Reactive oxygen species. Crit Care Med. 2005;33(12):S498–S501.
151. Förstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5(6):338–349.
152. Hsieh -C-C, Yen M-H, Yen C-H, Lau Y-T. Oxidized low density lipoprotein induces apoptosis via generation of reactive oxygen species in vascular smooth muscle cells. Cardiovasc Res. 2001;49(1):135–145.
153. Matsuoka H. Endothelial dysfunction associated with oxidative stress in human. Diabetes Res Clin Pract. 2001;54:S65–S72.
154. Siekmeier R, Grammer T, März W. Roles of oxidants, nitric oxide, and asymmetric dimethylarginine in endothelial function. J Cardiovasc Pharmacol Ther. 2008;13(4):279–297.
155. Wang Q, Zou M-H. Measurement of reactive oxygen species (ROS) and mitochondrial ROS in AMPK knockout mice blood vessels. In: AMPK. Springer; 2018:507–517.
156. Kauffman ME, Kauffman MK, Traore K, et al. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React Oxygen Species. 2016;2(5):361.
157. Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49(4):717–727.
158. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52(10):843–850.
159. Wang Q, Zhang M, Torres G, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66(1):193–205.
160. Madesh M, Zong W-X, Hawkins BJ, et al. Execution of superoxide-induced cell death by the proapoptotic Bcl-2-related proteins Bid and Bak. Mol Cell Biol. 2009;29(11):3099–3112.
161. Dikalova AE, Bikineyeva AT, Budzyn K, et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107(1):106.
162. Itani HA, Dikalova AE, McMaster WG, et al. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension. 2016;67(6):1218–1227.
163. Chen P, Chen Y, Wu W, Chen L, Yang X, Zhang S. Identification and validation of four hub genes involved in the plaque deterioration of atherosclerosis. Aging (Albany NY). 2019;11(16):6469.
164. Liu M, Gutierrez J. Genetic risk factors of intracranial atherosclerosis. Curr Atheroscler Rep. 2020;22(4):13.
165. Jung C, Evans MA, Walsh K. Genetics of age-related clonal hematopoiesis and atherosclerotic cardiovascular disease. Curr Opin Cardiol. 2020;35(3):219.
166. Reilly MP, Li M, He J, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377(9763):383–392.
167. de Santis IP, Lindenau JD, Ramos RB, et al. C-reactive protein gene rs1205 polymorphism is associated with low-grade chronic inflammation in postmenopausal women. Women’s Midlife Health. 2020;6:3.
168. Holdt LM, Beutner F, Scholz M, et al. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. 2010;30(3):620–627.
169. Liu Y, Sanoff HK, Cho H, et al. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4(4):e5027.
170. Holdt LM, Teupser D. Long noncoding RNA ANRIL: lnc-ing genetic variation at the chromosome 9p21 locus to molecular mechanisms of atherosclerosis. Front Cardiovasc Med. 2018;5:145.
171. Palmer MR, Kim DS, Crosslin DR, et al. Loci identified by a genome-wide association study of carotid artery stenosis in the eMERGE network. Genet Epidemiol. 2020.
172. Liu X, Wang Q, Zhu R. Association of GWAS-susceptibility loci with ischemic stroke recurrence in a Han Chinese population. J Gene Med. 2020:e3264.
173. Pertusa C, Tarín JJ, Cano A, García-Pérez MA. Association of a single nucleotide polymorphism of RANK gene with blood pressure in Spanish women. Medicine (Baltimore). 2020;99(40):e22436.
174. Schreinlechner M, Noflatscher M, Lener D, et al. NGAL correlates with femoral and carotid plaque volume assessed by sonographic 3D plaque volumetry. J Clin Med. 2020;9(9):2811.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.