")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 12
Infections as Risk Factor of Sjögren’s Syndrome
Received 17 August 2020
Accepted for publication 15 October 2020
Published 10 November 2020 Volume 2020:12 Pages 257—266
DOI https://doi.org/10.2147/OARRR.S276727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Suyud Warno Utomo,1,2 Jemima Fajarin Putri2
1Environmental Science Programme, Universitas Indonesia, Central Jakarta 10430, Indonesia; 2Department of Environmental Health Studies, Faculty of Public Health Universitas Indonesia, Depok, Indonesia
Correspondence: Suyud Warno Utomo
Environmental Science Programme, Universitas Indonesia, Building C (FKG UI), Fl. V-VI Jl. Salemba Raya No. 4, Central Jakarta 10430, Indonesia
Tel +62 812 8788 0304
Email [email protected]
Purpose: Sjögren’s syndrome (SS) is an autoimmune disease targeting exocrine glands, leading to low body fluids production, especially on the salivary and lacrimal glands. Due to the low saliva and tear production, the common symptoms of Sjögren’s syndrome are dry eyes and dry mouth, later on leading to uncomfortable sensations on the eye surface, cornea destruction, dental caries, and oral cavity infections. Several infections are known to cause similar side-effects to Sjögren’s syndrome symptoms, including low saliva flow; therefore, infection is hypothesized as one of the risk factors of Sjögren’s syndrome.
Results: Based on our literature research, there are several infectious agents which cause similar disease manifestations to Sjögren’s syndrome, including infections of hepatitis C virus, Epstein–Barr virus, cytomegalovirus, and human T-lymphotropic virus-1 (HTLV-1), and these four agents are found to cause persistent infection on the salivary gland after the first infection and later lead to organ destruction, thus causing sicca syndrome in the oral cavity. Other findings show possible Heliobacter pylori infection might lead on the increasing level of anti-Ro/SSA and anti-La/SSB of infected individuals.
Conclusion: Some research has shown persistent infection could trigger autoimmune disorders due to continuous T-cells and B-cells activation in an attempt of infected cells eradication, leading to autoimmune reaction and high autoreactive cells concentration around the healthy cells causing the immune cells to eradicate the healthy cells nearby. However, the results in this literature study found persistent infection is not the only risk factor of Sjögren’s syndrome but there are various unknown factors that trigger infection to develop into Sjögren’s syndrome.
Keywords: autoimmune diseases, Sjögren’s syndrome, infections, pathogens
Purpose
Sjögren’s syndrome (SS) is one of the autoimmune disorders generally targeting exocrine glands, leading to low body fluids production such as in salivary and lacrimal glands.1,2 Low body fluids production on salivary and lacrimal glands leads to sicca syndrome or dry eyes and mouth syndrome which might lead to further health problems such as cornea damage, blurry vision, tooth caries, and digestive problems.3,4 Worldwide, the prevalence of SS is around 0.1–0.5% of the total population,5 and t is the second highest autoimmune disorder in the US, with total population of up to 4 million people.6 Based on epidemiological studies, females have a higher risk of SS compared to males, with a ratio of 9:1, and the risk escalates at the age of 40 to the late 50s.3,7,8
There are two types of SS based on the complexity of the disease, primary SS and secondary SS. A patient is diagnosed with primary SS if they are not diagnosed with other autoimmune disorders, and diagnosed with secondary SS if they are diagnosed with another autoimmune disorder and later diagnosed with SS.5,9,10 Between the two types of SS, the main symptoms are ocular and oral cavity sicca syndrome, musculoskeletal pain, and fatigue, and among the secondary SS patients might also followed by the secondary autoimmune disorder’s manifestations.11,12 Due to the general and various symptoms, SS is known as the thousand faces disease, because of the long observation period necessary to conclusively diagnose SS.4,13 To diagnose SS among patients, the medical examiners observed and later classified the patients based on SS criteria released by the American-European Consensus.9,13 There are six criteria to define SS classification, 1) dry eyes, 2) Schirmer test results showing the patient has dry eyes, 3) dry oral cavity, 4) salivary glands test results showing low saliva production, 5) salivary glands biopsy results ≥1 FOCI per 4 mm2, and 6) immunology results showing reactivity towards anti-Ro/SSA and/or anti-La/SSB. Another SS criteria was developed by ACR-EULAR; with five criteria including 1) salivary glands biopsy shows ≥50 inflammatory mononuclear cell per 4 mm2, 2) ocular staining score ≥5 on at least one eye, 3) the presence of anti-SSA, 4) Schirmer’s test result shows low tear production by ≤5 mm/5 minutes, and 5) low unstimulated saliva production by ≤0,1 mL per minute, the ACR-EULAR criteria also mentioned the exclusion of patients with a history of head or neck radiation treatment, patients with active hepatitis C infection, patients with immunodeficiency syndrome, sarcoidosis, amyloidosis, graft versus host disease, and patients with IgG4-related diseases (Table 1).14 A patient will be diagnosed with primary SS if they fulfillat least four out of the six criteria including biopsy results and immunology results, meanwhile, a patient will be diagnosed with secondary SS if they fulfill two-to-three out of the six criteria including biopsy results and dry eyes/mouth symptoms.6,13,15
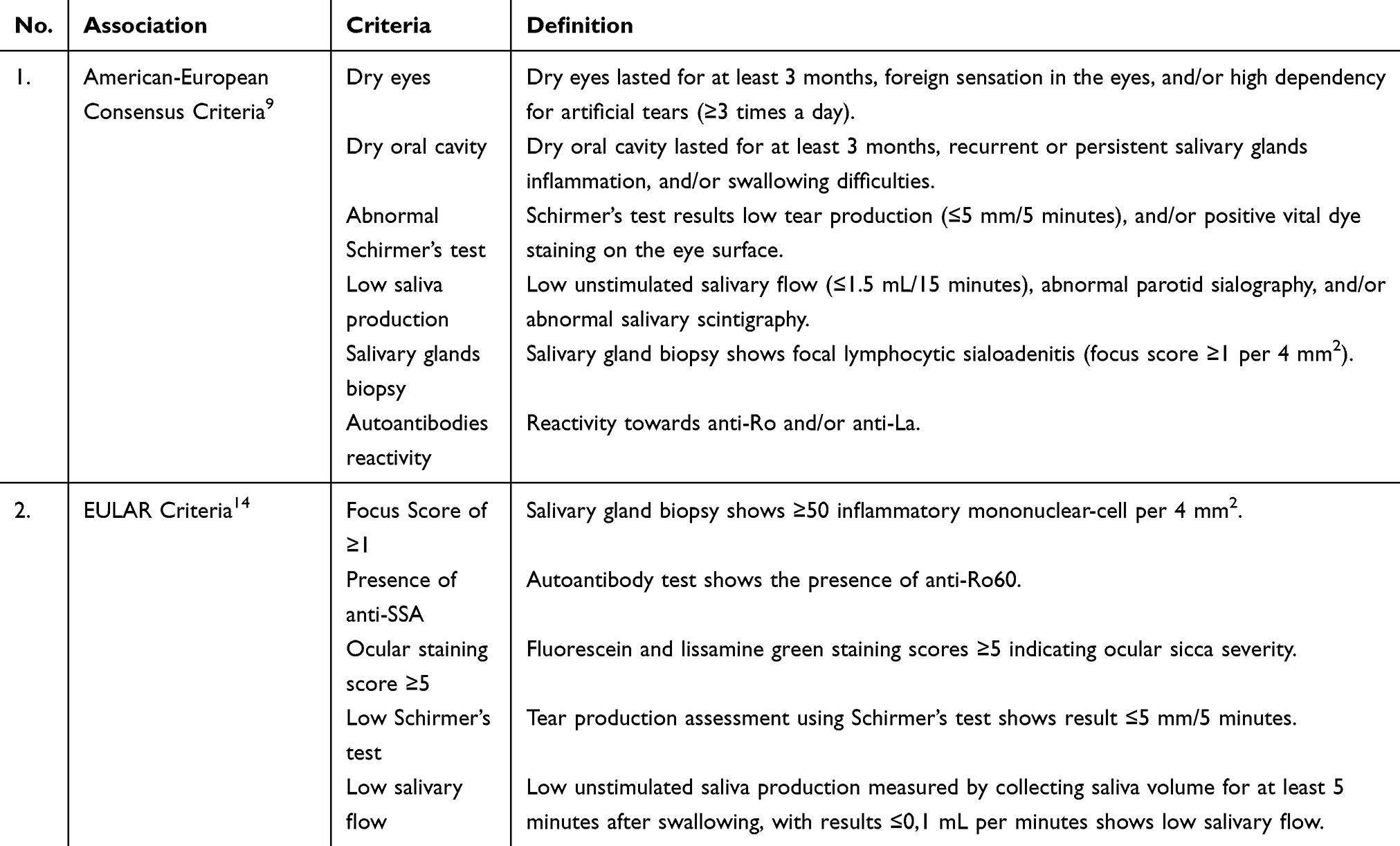 |
Table 1 Sjögren’s Syndrome Classification Criteria |
To this day, risk factors and the pathogenesis of SS remain inconclusive. But there are several suspects of SS potential risk factors, which are infection15,16 and genetic susceptibilities.17,18 Infection is believed to be one of the potential risk factors for Sjögren’s syndrome, especially persistent infections,19,20 which lead to an over-stimulated immune system and organ damage due to the high concentration of autoreactive and non-specific T-cells.21,22 Genetic susceptibility is believed to be one of the potential risks for Sjögren’s syndrome due to degeneration in DNA protein structures, leading to false protein transcription and translation, and causing the immune system to continue producing T-cell self-antigen but not followed with constant apoptosis to prevent high self-antigen cells.23,24 In this review paper, we will identify and analyze the role of infections towards the development of Sjögren’s syndrome based on various published research articles.
Method
This article is a review article towards various published research articles under the keyword “Infections and Sjögren’s syndrome”, using Google as the search engine and through journal homepages including Elsevier, Springer, ScienceDirect, NCBI, etc. The articles went through a screening process fulfilling the main criteria: 1) the article mentioned the infectious agent infecting the patient/suspect of Sjögren’s syndrome, 2) the article mentioned the patient/subject’s clinical test results including antibody test results, and 3) the article was published between 1990 and 2019. With exclusion criteria including 1) the article is a research article, not a review or systematic review article, and 2) the article is fully accessible. Articles which fulfilled all the criteria were synthesized and used to create a conclusion to answer the main purpose of this article (Figure 1).
 |
Figure 1 Article Screening and Exclusion Flow. |
Results
Under the keyword, “Infections and Sjögren’s syndrome” there were 48 published articles, consisting of research articles and review articles. After the screening and criteria fulfillment, 16 research articles were obtained to synthesize.
Infectious Agents as Potential Risk Factors of Sjögren’s Syndrome
Based on 19 research articles reviewed in this paper, there are six infectious agents which might contribute in SS development due to the similar manifestation caused by the agents and the capability of the agents to stimulate anti-Ro/SSA or anti-La/SSB production. There are several infectious agents that are suspected to play significant roles in the development of SS, such as cytomegalovirus (CMV), Epstein–Barr virus, hepatitis C virus, human T-cell lymphotropic virus-1 (HTLV-1), Staphylococcus saccharolyticus, and Heliobacter pylori (Table 2).
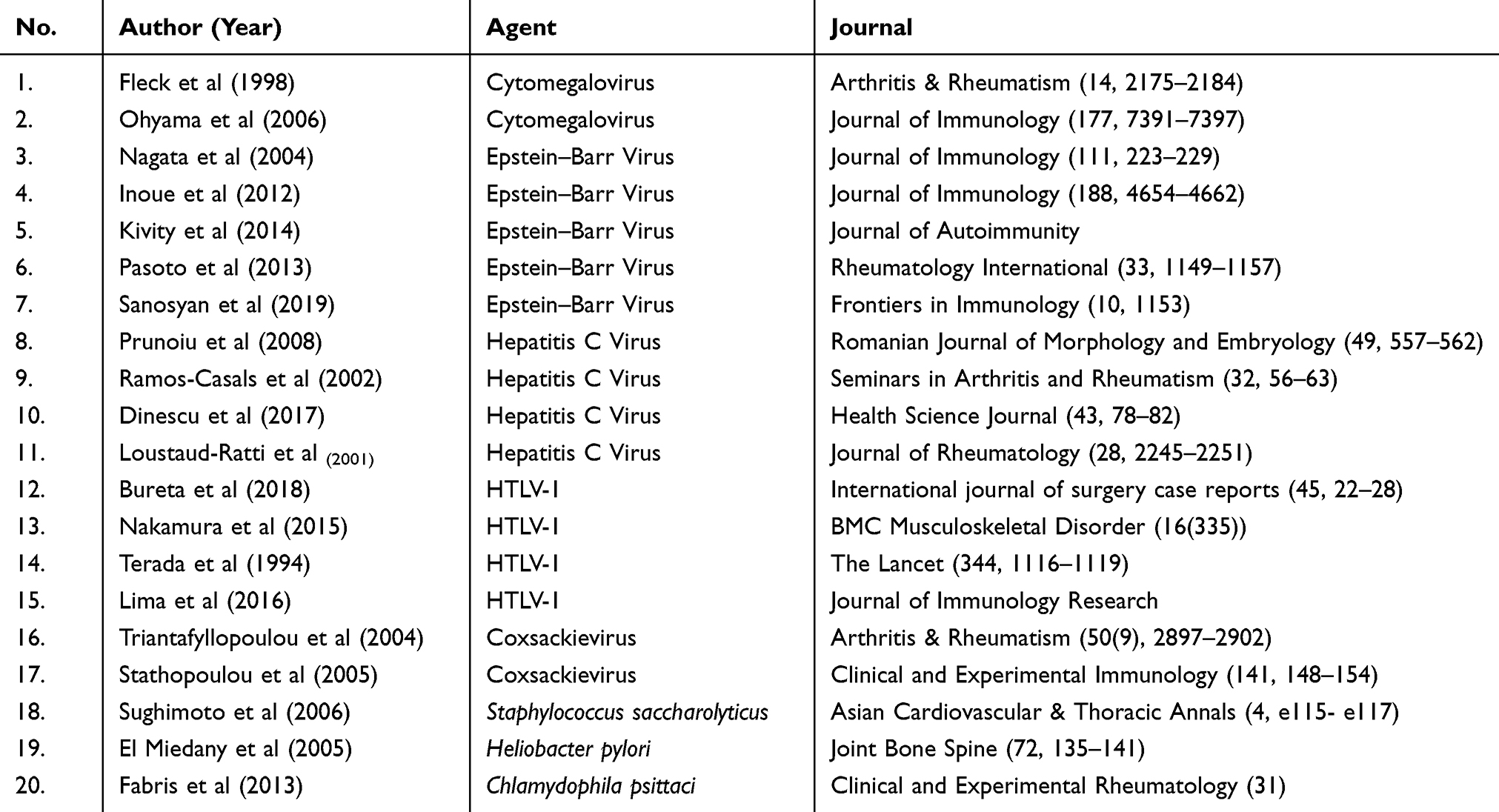 |
Table 2 Potential Infectious Agents |
Infectious Agents Role in Sjögren’s Syndrome
In this review, we describe the different roles and impacts of the six infectious agents towards the histology and physical manifestation post-infection (Table 3).
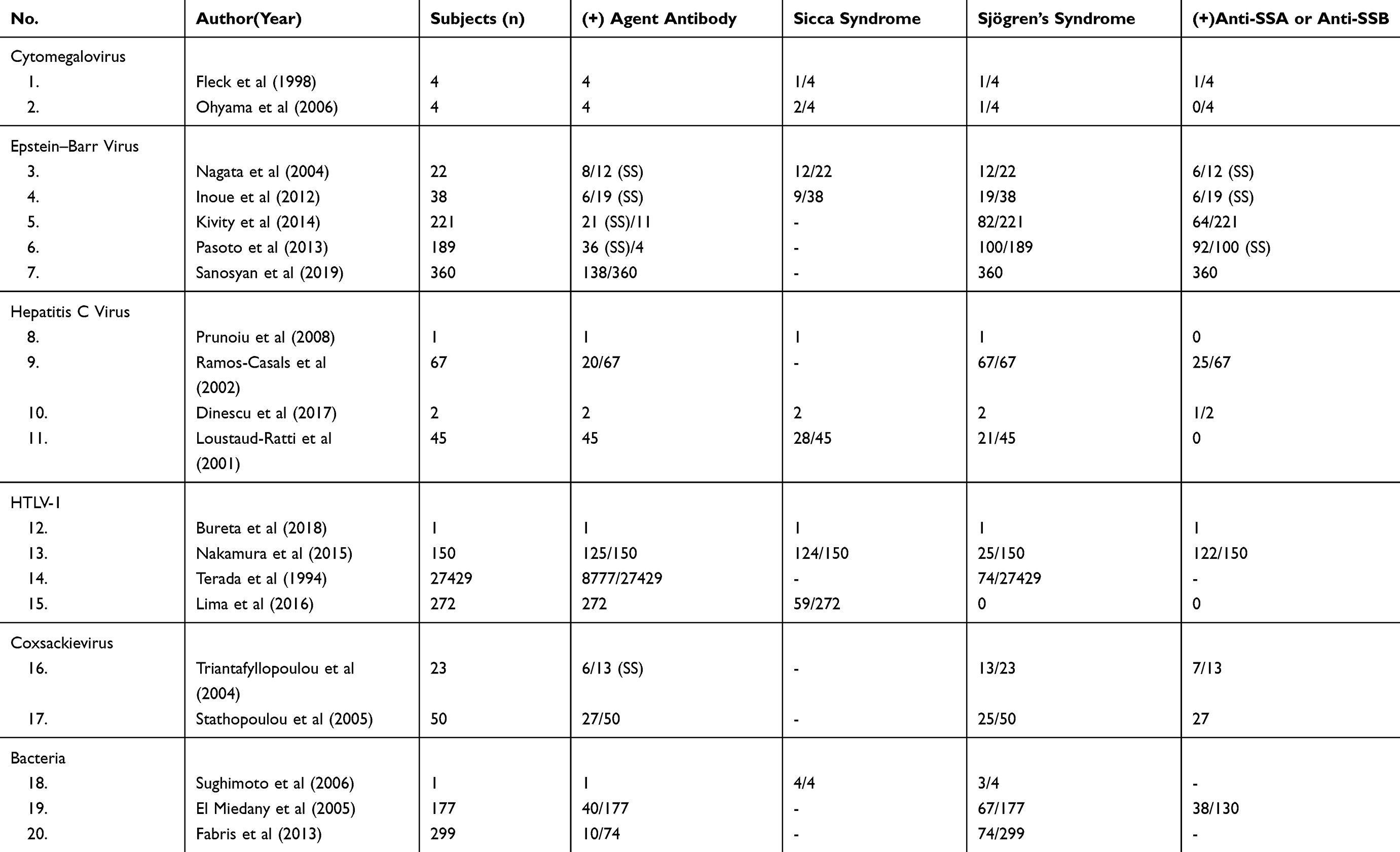 |
Table 3 Antibody and Autoantibody Prevalence, and the Manifestation of Sjögren’s Syndrome |
HTLV-1
HTLV-1 infections are known to have the capability to cause sicca syndrome, mimicking one of the SS symptoms,25,26 but the infections are not known to cause any reactivity towards anti-Ro/SSA nor anti-La/SSB.27 The prevalence of antibodies towards HTLV-1 is higher among the SS population compared to the healthy population.28 This finding shows the susceptibility towards HTLV-1 infection is rather higher among the SS population.25,28
Epstein–Barr Virus
Another potential agent is Epstein–Barr Virus (EBV), the prevalence of antibody towards EBV and viral reactivation rate among SS patients is higher compared to normal or healthy populations.29,30 It is shown in the higher EBV luciferase activity induced by saliva of SS patients, while not showing any activity when induced by healthy individual’s saliva.31,32 A higher viral reactivation rate in SS patients is hypothesized to play a role in autoantibody anti-Ro/SSA and/or anti-La/SSB production. This shows a higher concentration of anti-Ro/SSA and anti-La/SSB in SS patients with an antibody towards EBV compared to SS patients without antibody towards EBV.31,32 Other research also shows EBV infection rates are higher among SS patients with the presence of both anti-SSA and anti-SSB autoantibodies compared to SS patients with only anti-SSA or without the presence of both the autoantibodies.33 Therefore it is highly speculated that EBV reactivity increases due to the presence of both anti-SSA and anti-SSB.
Hepatitis C Virus
Hepatitis C virus infection is known to cause similar manifestations to SS which is sicca syndrome, leading to HCV patients diagnosed with SS.34–36 Despite similar sicca syndrome manifestation among HCV patients diagnosed with SS, from the histology perspective, HCV related SS patients have different immunology characteristics compared to the primary SS patients. In HCV related SS the common symptoms among the patients is un-reactivity towards anti-Ro/SSA and/or anti-La/SSB antigens,34–36 followed with different cytokines concentration with Th2 concentration is higher than Th1 concentration among HCV related SS while in primary SS patients show higher Th1 concentration than Th2.37,38 The differences between HCV related SS and primary SS indicate the HCV related Sjögren’s syndrome is classified as secondary SS.
Cytomegalovirus
Based on experiments towards several murines, which showed cytomegalovirus infection might play a role in the development of SS on specific individuals. The experiment conducted towards four different varieties of murine shows persistent infection on murine with specific genetic susceptibility towards autoimmune disorder due to Fas deficiency (B6-lpr/lpr) and on murine with zero tumor necrosis factor (TNFR).39 On murine with Fas deficiency shows persistent infection on mononuclear cells level followed by severe salivary glands damage leading to low saliva production, while on murine with zero tumor necrosis factor (TNFR) shows delayed viral eradication compared to the normal murine. High anti-Ro/SSA and anti-La/SSB production and tissue damage are shown in the salivary glands of murine with persistent infection (B6-lpr/lpr), while similar results are not shown in any other murines.39 Another experiment was conducted towards four different varieties of murines, including NZM2328, which has the tendency to develop SLE-like symptoms spontaneously by the age of 5 months, and B6-lpr/lpr murines.40 The result shows severe structural damage on the salivary glands of female NZM2328 murine and female B6-lpr/lpr murine, but this was not shown in male NZM2328 murine.40 The result contras show there is a higher risk of persistent infection followed by structural damage among female murines with genetic susceptibilities compared to male murines from similar varieties.39,40 Meanwhile, from an immunology aspect we see the elevation of anti-Ro/SSA and anti-La/SSB production on B6-lpr/lpr murine with severe salivary glands damage and persistent infection,39 while similar results are not shown in the NZM2328 murine with similar organ damage.40
Coxsackievirus
Another potential risk factor is Coxsackievirus. The results show a higher coxsackievirus infection rate among patients with primary SS compared to patients with secondary SS or healthy samples. The higher infection rates are shown in both PCR results and minor salivary glands biopsy, from the histopathology perspectives it’s hypothesized the high infection rate is due to the presence of anti-SSA autoantibody.41 Another research towards the reaction of Coxsackievirus peptide and anti-SSA/SSB autoantibodies shows out of 27 pSS patients with the presence of both anti-SSA and anti-SSB, nine of the patients recognized the Coxsackievirus peptide. The number is higher in pSS patients compared to SLE patients (7/28), while similar results are not shown among the healthy individuals. The results also show pSS patients with the presence of both anti-SSA and anti-SSB have a higher detected cross-reaction with Coxsackievirus peptide compared to pSS patients with the presence of anti-SSA.42 Both articles initiate the significant roles of anti-SSA, but it is also suggested that the presence of both autoantibodies increases the reactivity with Coxsackievirus peptide.
Bacteria Infections
Bacteria infections among SS patients show higher prevalence compared to a healthy population followed with higher infection severity.43 On Heliobacter pylori infection, there are significant infection prevalence gaps among the SS population and healthy population or SLE patients. This is shown by the higher prevalence of antibody towards Heliobacter pylori among SS patients.43,44 Another evidence of bacterial infection is the high severity of endocardium tissue damage due to Staphyloccocus saccharolyticus infection on a patient diagnosed with SS.45 Another study shows a high Chalmydophila psittaci DNA infection among secondary SS based on PCR analysis. The high Clamydophila psittaci infections are specifically detected among MALT dan MESA patients with the presence of rheumatoid factor but without the presence of anti-SSA/SSB autoantibodies.46 But from all the articles, the writers have not been able to conclude the main role of bacteria infection in the development of SS, and further investigations are needed to conclude whether bacteria infection plays a role as a risk factor or as an impact of SS itself.
Population Characteristic
From 17 research articles we can see that the population with a higher risk of developing SS or SS-like manifestation due to infections is women (88%), with the highest prevalence between the ages of 40 to the early 60s. From an ethnicity perspective, the majority of the research subjects have Asian ethnicity (41%), followed by Europeans (35%) (Table 4).
 |
Table 4 Research Subjects Characteristics |
Discussion
Persistent Infection, Bystander Activation, and Sjögren’s Syndrome
Viral infections are known to have the possibility to develop into persistent infections due to the agent’s capability to invade not only tissues but also mononuclear cells leading to undetected infection on tissue level biopsy.47 Persistent infection and inflammation due to infectious agents lead to immune system responses such as bystander activation,16,48 which is an immune response mechanism to eradicate infected cells by releasing antigen-presenting cells (APCs) and autoreactive non-specific T-cells to stimulate various immune cells to migrate towards the infected area.49 Meanwhile, at the hotspot area, CD8+ releases cytotoxin22,50 carrying the main purpose to trigger cell apoptosis to prevent further infection. To prevent further infection and inflammation, the immune system not only releases CD+ but also other various immune responses such as tumor necrosis factor (TNF)-α,50,51 lymphotoxin, and nitrite oxide. During the bystander activation, the apoptosis also affects healthy cells nearby and leads to autoimmune reactions16,52,53 due to the immune system attacking the healthy cells. In normal or healthy individuals, bystander activation could work optimally due to the body's capability to eradicate self-antigen immune cells to prevent high self-antigen antibody production.54 Meanwhile, on immunosuppressive individuals, their immune system is unable to eradicate self-antigens antibodies, leading to the high level of self-antigens antibody in the system and during persistent infection, the amount of immune response towards the infection site is high and constant, causing high self-antigens antibody release and stimulating apoptosis among the healthy cells.21,55,56
Based on the 19 research articles reviewed in this paper, Sjögren’s syndrome and persistent infection are not detected in every infected individual. The results show there is a specific factor which plays a significant role in the development of persistent infection and leads to Sjögren’s syndrome. The other potential factors, in this case, are possibly genetic susceptibilities and gender-related factors.
Genetic Susceptibility and Personal Characteristics
Genetics is believed to contribute 50% as a potential risk factor of Sjögren’s syndrome, while the other 50% is from environment stimulants.56,57 Based on cytomegalovirus infection research, persistent infection only developed in murine with specific genetic susceptibilities towards autoimmune disorder. In B6-lpr/lpr murine with Fas deficiency,20,39 persistent infection is detected on mononuclear cell level without any infection detected on tissue biopsy, while in murine with zero tumor necrosis factor shows a significant delay on viral eradication without mononuclear level infection.39 Another research on NZM2328 murines with genetic susceptibilities towards spontaneously developing SLE-like symptoms shows persistent infection only detected on female NZM2328 murine leading to severe salivary gland damage but not detected on male NZM2328.40 These results indicate there are significant roles of gender susceptibility on female murines towards the development of persistent infection.
Meanwhile, in human subjects, genetic susceptibilities and personal characteristics might contribute to the development of Sjögren’s syndrome or disease manifestation mimicking Sjögren’s syndrome.16,58 It is shown from the higher Sjögren’s syndrome prevalence among females compared to males, and genetic susceptibility is also shown from a higher prevalence of sicca syndrome due to HTLV-1 infection among the Asian population,25 meanwhile, there is a significant gap of sicca syndrome manifestation due to HTLV-1 infection among the infected population and non-infected population in South America.27 These findings show there is a role played by the different genetic structure in different races to cause susceptibility towards the development of sicca syndrome or later Sjögren’s syndrome.
Sjögren’s syndrome is known as a high complexity disease and could not be concluded from basic Mendelian law due to complex variables which may play the role of the disease development.59 The genetic role in Sjögren’s syndrome is believed not only due to single cell disruption, but more complex and multi-cells involvement,18,60 which is one of the reasons for the difficulty in diagnosing the diseases. Until these days, researchers have hypothesized the genetic susceptibility role in Sjögren’s syndrome is related to non-major histocompatibility complex (MHC) class II protein,18,61 and in humans is known as human leukocyte antigen (HLA). MHC or HLA is responsible for the immune system’s development and antibody production.18,23 Every race have different HLA structures, such as different HLA-DR and HLA-DQ due to different environment stimulants and different protein structures inherited from ancestors.23,–60–62 Different HLA structures between every race or every population with different environment stimulant lead to different susceptibility rates towards the development of Sjögren’s syndrome, this might be shown from the different anti-Ro/SSA and anti-La/SSB production level between the same races in different environment stimulants.54,–60,–63–65
Conclusion
Sjögren’s syndrome is a complex disease and has various inconclusive risk factors which may contribute to the disease development, such as environmental factors and genetic susceptibility factors. One of the suspected environmental factors of SS is persistent infections. Several infectious agents have the capability to cause post-infection manifestations mimicking SS main symptoms, such as sicca syndrome based on salivary gland biopsy and low salivary flow. In several cases the persistent infection leads to high anti-Ro/SSA and anti-La/SSB production at the infected site. But there are also various manifestations on every infectious agent in every population. In HTLV-1 and hepatitis C virus infection, both of the persistent infections lead to sicca syndrome manifestation without fulfilling the histology criteria. However, in HTLV-1 infection, the dryness symptom only occurs in a specific population while in another populations the infection did not cause any sicca syndrome. Epstein–Barr virus and cytomegalovirus infection show the persistent infection due to both of the agents leads to increasing anti-SSA/SSB production and severe salivary gland damage. It is also shown that the Epstein–Barr virus reactivity is more likely to happen among SS patients with the presence of both anti-SSA and anti-SSB, whilst the infection rate of Coxsackievirus is more likely to happen among SS patients with the presence of anti-SSA. Meanwhile, bacterial infections are still unable to be concluded and in need of further research to see whether the high prevalence and severity of the bacterial infection plays any role in SS development or the infection severity in the impact of SS, but it is shown that bacterial infection rates among SS patients are higher compared to healthy individuals. And due to the very limited research upon SS and pathogen infections, we suggest more research in epidemiological and molecular perspectives on wider populations to identify the role of personal or population characteristic in the development of Sjögren’s syndrome and to identify the role and pathogenesis of infectious agents in the development of Sjögren’s syndrome.
Acknowledgment
The authors acknowledge the division of Immunology and Allergy in RSCM (Jakarta, Indonesia), Department of Environmental Health, Faculty of Public Health Universitas Indonesia (Depok, Indonesia) and Environmental Science Programme, Universitas Indonesia (Jakarta, Indonesia).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Patel R, Shahane A. The epidemiology of Sjögren’s syndrome. Clin Epidemiol. 2014;6:247–255.
2. Foundation SsS. Sjögren’s syndrome fact sheet. Sjögren’s Syndrome Foundation; 2019.
3. Manoussakis MN. Sjögren’s syndrome. Orphanet Ency. 2004.
4. Kruszka P, O’brian R. Diagnosis and management of Sjögren syndrome. Am Fam Physician. 2009;79(6):465–470.
5. Fox PC. Autoimmune diseases and Sjögren’s syndrome: an autoimmune exocrinopathy. Ann N Y Acad Sci. 2007;1098:15–21. doi:10.1196/annals.1384.003
6. Segal B, Bowman S, Fox P, et al. Primary Sjögren’s syndrome: health experiences and predictors of health quality among patients in the United States. Health Qual Life Outcomes. 2009;7:46. doi:10.1186/1477-7525-7-46
7. Somani R, Sunil M, Khaira J, Kumar D. Sjögren’s syndrome: a review. J Indian Acad Oral Med Radiol. 2011;23(1):61–64. doi:10.5005/jp-journals-10011-1094
8. Baer A, Walitt B. Sjögren syndrome and other causes of sicca in the older adult. Clin Geriatr Med. 2017;33(1):87–103. doi:10.1016/j.cger.2016.08.007
9. Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by American-European consensus group. Ann Rheum Dis. 2002;61:554–558. doi:10.1136/ard.61.6.554
10. Tsuboi H, Asashima H, Takai C. Primary and secondary surveys on epidemiology of Sjögren’s syndrome in Japan. Mod Rheumatol. 2014;24:464–470. doi:10.3109/14397595.2013.843765
11. Vitali C, Del Papa N. Pain in primary Sjögren’s syndrome. Best Pract Res Clin Rheumatol. 2015;29:63–70. doi:10.1016/j.berh.2015.05.002
12. Macfarlane G, Croft P, Schollum J, Silman A. Widespread pain: is an improved classification possible? J Rheumatol. 1996;23:1628–1632.
13. Kassan S, Moutsopoulos H. Clinical manifestations and early diagnosis of Sjögren syndrome. Arch Intern Med. 2004;164:1275–1284. doi:10.1001/archinte.164.12.1275
14. Shiboski C, Shiboski S, Seror R, et al. 2016 ACR-EULAR classification for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheum. 2017;69(1):35–45. doi:10.1002/art.39859
15. Bayetto K, Logan R. Sjögren’s syndrome: a review of aetiology, pathogenesis, diagnosis and management. Aust Dent J. 2010;55:39–47. doi:10.1111/j.1834-7819.2010.01197.x
16. Bach J-F. Infections and autoimmune diseases. J Autoimmun. 2005;25:74–80. doi:10.1016/j.jaut.2005.09.024
17. Yamamoto K. Pathogenesis of Sjögren’s syndrome. Autoimmun Rev. 2003;2:13–18. doi:10.1016/S1568-9972(02)00121-0
18. Heward J, Gough S. Genetic susceptibility to the develoment of autoimmune disease. Clin Sci. 1997;93:479–491. doi:10.1042/cs0930479
19. Igoe A, Scofield R. Autoimmunity and infection in Sjögren’s syndrome. Curr Opin Immunol. 2013;25:480–487.
20. Burns J. Persistent cytomegalovirus infection - the etiology of Sjögren’s syndrome. Med Hypotheses. 1983;10:451–460. doi:10.1016/0306-9877(83)90011-7
21. Duke R. Self recognition by T cells. I. Bystander killing of target cells bearing syngeneic MHC antigens. J Exp Med. 1989;170:59–71. doi:10.1084/jem.170.1.59
22. Fujinami R, von Herrath M, Christen U, Whitton J. Molecular mimictry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19(1):80–94. doi:10.1128/CMR.19.1.80-94.2006
23. Tomlinson I, Bodmer W. The HLA system and the analysis of multifactorial genetic disease. Trends Genet. 1995;11:493–498. doi:10.1016/S0168-9525(00)89159-3
24. Fujinami R, Oldstone M. Amino acid homology between the ecephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi:10.1126/science.2414848
25. Nakamura H, Shimizu T, Takagi Y, et al. Reevaluation for clinical manifestations of HTLV-1-seropositive parients with Sjögren’s syndrome. BMC Musculoskelet Disord. 2015;16(335). doi:10.1186/s12891-015-0773-1
26. Bureta C, Abematsu M, Tominaga H, et al. Hypertrophic spinal pachymeningitis associated with human T-cell lymphotrophic virus-1 infection and Sjögren’s syndrome: a case report and brief literature review. Int J Surg Case Rep. 2018;45:22–28. doi:10.1016/j.ijscr.2018.03.010
27. Lima C, Santos S, Dourado A, et al. Association of sicca syndrome with proviral load and proinflammatory cytokines in HTLV-1 infection. J Immunol Res. 2016;2016. doi:10.1155/2016/8402059
28. Terada K, Katamine S, Eguchi K, et al. Prevalence of serum and salivary antibodies to HTLV-1 in Sjögren’s syndrome. Lancet. 1994;344:1116–1119. doi:10.1016/S0140-6736(94)90630-0
29. Kivity S, Arango M, Ehrenfeld M, et al. Infection and autoimmunity in Sjögren’s syndrome: a clinical study and comprehensive review. J Autoimmun. 2014. doi:10.1016/j.jaut.2014.02.008
30. Pasoto S, Natalino R, Chakkour H, et al. EBV reactivation serological profile in primary Sjögren’s syndrome: an underlying trigger of active articular involvement? Rheumatol Int. 2013;33:1149–1157. doi:10.1007/s00296-012-2504-3
31. Nagata Y, Inoue H, Yamada K, et al. Activation of Epstein-Barr virus by saliva drom Sjögren’s syndrome patients. J Immunol. 2004;111:223–229. doi:10.1111/j.0019-2805.2003.01795.x
32. Inoue H, Mishima K, Yamamoto-Yoshida S, et al. Aryl hydrocarbon receptor-mediated induction of EBV reactivation as a risk factor for Sjögren’s syndrome. J Immunol. 2012;188:4654–4662. doi:10.4049/jimmunol.1101575
33. Sanosyan A, Daien C, Nutz A, et al. Discrepancy of serological and molecular patterns of circulating Epstein-Barr virus reactivation in primary Sjögren’s syndrome. Front Immunol. 2019;10:1153. doi:10.3389/fimmu.2019.01153
34. Prunoiu C, Georgescu E, Georgescu M, Simionescu C. Sjögren’s syndrome associated with chronic hepatitis C – the benefit of the antiviral treatment. Rom J Morphol Embryol. 2008;49:557–562.
35. Dinescu S, Ciurea P, Vreju F, Sandulescu D, Musetescu A. Hepatitis C virus induced Sjögren’s syndrome - clinical and imaging features. Curr Health Sci J. 2017;43:78–82.
36. Loustaud-Ratti V, Riche A, Liozon E, et al. Prevalence and characteristics of Sjögren’s syndrome or sicca syndrome in chronic hepatitis C virus infection: a prospective study. J Rheumatol. 2001;28:2245–2251.
37. Ramos-Casals M, García-Carrasco M, Carvera R, et al. Th1/Th2 cytokine imbalance in patients with Sjögren’s syndrome secondary to hepatitis C virus infection. Semin Arthritis Rheum. 2002;32:56–63. doi:10.1053/sarh.2002.33724
38. Fan X, Liu W, Li C, Wang Z, Luo L, Tan D. Circulating Th1 and Th2 cytokines in patients with hepatitis C virus infection. Mediators Inflamm. 1998;7:295–297. doi:10.1080/09629359890992
39. Fleck M, Kern E, Zhou T, Lang B, Mountz J. Murine cytomegalovirus induces a Sjögren’s syndrome-like disease in C57B1/6-lpr/lpr mice. Arthritis Rheum. 1998;41:2175–2184.
40. Ohyama Y, Carroll V, Deshmukh U, Gaskin F, Brown M, Fu S. Severe focal sialadenitis and dacryoadenitis in NZM2328 mice induced by MCMV: a novel model for human Sjögren’s syndrome. J Immunol. 2006;177:7391. doi:10.4049/jimmunol.177.10.7391
41. Triantafyllopoulou A, Tapinos N, Moutsopoulos H. Evidence for Coxsackievir infection in primary Sjögren’s syndrome. Arthritis Rheum. 2004;50(9):2897–2902. doi:10.1002/art.20463
42. Stathopoulou E, Routsias J, Stea E, Moutsopoulos H, Tzioufas A. Cross-reaction between antibodies to the major epitope of Ro60 kD autoantigen and homologous peptide of Coxsackie virus 2B protein. Clin Exp Rheumatol. 2005;141:148–154.
43. El Miedany Y, Baddour M, Ahmed I, Fahmy H. Sjögren’s syndrome: concomitant H. Pylori infection and possible correlation with clinical parameters. Joint Bone Spine. 2005;72:135–141. doi:10.1016/j.jbspin.2004.04.005
44. De Vita S, Ferraccioli G, Avellini C, et al. WIdespread clonal B-cell disorder in Sjögren’s syndrome predisposing to Heliobacter pylori – related gastric lymphoma. Gastroenterology. 1996;110:1969–1974. doi:10.1053/gast.1996.v110.pm8964425
45. Sughimoto K, Nakano K, Gomi A, Nakatani H, Nakamura Y, Sato A. Infective endocarditis associated with Sjögren’s syndrome. Asian Cardiovasc Thorac Ann. 2006;14:115–117. doi:10.1177/021849230601400629
46. Fabris M, Dolcetti R, Pasini E, et al. High prevalence of Chlamydophila psittaci subclinical infection in Italian patients with Sjögren’s syndrome and parotid gland marginal zone B-cell lymphoma of MALT-type. Clin Exp Rheumatol. 2014;32:61–65.
47. Mariette X, Gozlan J, Clerc D, Bisson M, Morinet F. Reactivation of Epstein-Barr virus DNA by in situ hybridization and polymerase chain reaction in salivary gland biopsy specimens from patients with Sjögren’s syndrome. Am J Med. 1991;90:286–294. doi:10.1016/0002-9343(91)80007-9
48. Agmon-Levin N, Lian Z, Shoenfeld Y. Explosion of autoimmune diseases and the mosaic of old and novel factors. Cell Mol Immunol. 2011;8:189–192. doi:10.1038/cmi.2010.70
49. Hemmer B, Fleckenstein B, Vergelli M, et al. Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T-cell clone. J Exp Med. 1997;185:1651–1659. doi:10.1084/jem.185.9.1651
50. Smyth M, Sedgwick JD. Delayed kinetics of tumor decrosis factor-mediated bystander lysis by peptide-specific CD8+ cytotoxic T lymphocytes. Eur J Immunol. 1998;28:4162–4169.
51. Yasukawa M, Yakushijin Y, Hasegawa H, et al. Expression of perforin and membrane-bouncd lymphotoxin (tumor necrosis factor-beta) in virus-specific CD4+ human cytotoxic T-cell clones. Blood. 1993;81:1527–1534. doi:10.1182/blood.V81.6.1527.1527
52. Narkeviciute I, Sudzius G, Mieliauskaite D, et al. Are cytotoxic effector cells changes in peripheral blood of patients with Sjögren’s syndrome related to persistent virus infection: suggestions and conundrums. Cell Immunol. 2016;310:123–130. doi:10.1016/j.cellimm.2016.08.013
53. Pacheco Y, Acosta-Ampudia Y, Monsalve D, Chang C, Gershwin M, Anaya J. Bystander activation and autoimmunity. J Autoimmun. 2019;103:102301. doi:10.1016/j.jaut.2019.06.012
54. Manthorpe R, Morling N, Platz P, Ryder L, Svejgaard A, Thomsen M. HLA-D antigen frequencies in Sjögren’s syndrome: differences between the primary and secondary form. Scand J Rheumatol. 1981;10:124–128. doi:10.3109/03009748109095284
55. Fox R, Luppi M, Kang H, Pisa P. Reactivation of Epstein-Barr virus in Sjögren’s syndrome. Springer Semin Immunopathol. 1991;13:217–231. doi:10.1007/BF00201470
56. Cojocaru M, Cojocaru I, Silosi I. Autoimmune diseases and their environmental triggers. Maedica-J Clin Med. 2008;3(2):124–129.
57. Singh S, Wal P, Wal A, Srivastava V, Tiwari R, Sharma R. Understanding autoimmune disease: an update review. Int J Pharm Technol Biotechnol. 2016;3(3):51–65.
58. Nakamura H, Shimizu T, Kawakami A. Role of viral infections in the pathogenesis of Sjögren’s syndrome: different characteristics of Epstein-Barr virus and HTLV-1. J Clin Med. 2020;9:1459. doi:10.3390/jcm9051459
59. Bolstad A, Jonsson R. Genetic aspects of Sjögren’s syndrome. Arthritis Res. 2002;4:353–359. doi:10.1186/ar599
60. Bolstad A, Wassmuth R, Haga H, Jonsson R. HLA markers and clinical characteristics in Caucasians with primary Sjögren’s syndrome. J Rheumatol. 2001;28:1554–1562.
61. Nakken B, Jonsson R, Brokstad K, et al. Associations of MHC class II alleles in Norwegian primary Sjögren’s syndrome patients: implications for development of autoantibodies to the Ro52 autoantigen. Scand J Rheumatol. 2001;54:428–433.
62. Nepom G. MHC and autoimmune diseases. Immunol Ser. 1993;59:143–164.
63. Papasteriades C, Skopouli F, Drosos A, Andonopoulos A, Moutsopoulos H. HLA-alloantigen associations in Greek patients with Sjögren’s syndrome. J Autoimmun. 1988;1:85–90. doi:10.1016/0896-8411(88)90079-0
64. Miyagawa S, Shinohara K, Nakajima M, et al. Polymorphisms of HLA class II genes and autoimmune responses to Ro/SS-A-La/SS-B among Japanese subjects. Arthritis Rheum. 1998;41:927–934.
65. Priori R, Medda E, Conti F, et al. Risk factors for Sjögren’s syndrome: a case-control study. Clin Exp Rheumatol. 2007;25:378–384.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.