Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management?
Authors Stockley R ![]() , Edgar R
, Edgar R ![]() , Pillai A, Turner A
, Pillai A, Turner A ![]()
Received 27 April 2016
Accepted for publication 17 June 2016
Published 1 August 2016 Volume 2016:11(1) Pages 1745—1756
DOI https://doi.org/10.2147/COPD.S111508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Robert A Stockley,1 Ross G Edgar,1 Anilkumar Pillai,1 Alice M Turner2
1Department of Lung Function and Sleep, University Hospitals Birmingham NHS Foundation Trust, 2Department of Inflammation and Ageing, University of Birmingham, Edgbaston, Birmingham, UK
Background: Chronic obstructive pulmonary disease (COPD) is characterized by fixed airflow obstruction and accelerated decline of forced expired volume in 1 second (FEV1). Alpha-1-antitrypsin deficiency is a genetic cause of COPD and associated with more rapid decline in lung function, even in some never smokers (NS) but the potential for individualized assessment to reveal differences when compared to group analyses has rarely been considered.
Methods: We analyzed decline in post-bronchodilator FEV1 and gas transfer (% predicted) over at least 3 years (mean= 6.11, 95% CI 5.80–6.41) in our unique data set of 482 patients with alpha-1-antitrypsin deficiency (PiZ) to determine individual rates of decline, implications for prognosis, and potential clinical management.
Findings: There was a marked variation in individual rates of FEV1 decline from levels consistent with normal aging (observed in 23.5% of patients with established COPD, 57.5% of those without) to those of rapidly declining COPD. Gas transfer did not decline in 12.8% of NS and 20.7% of ex-smokers with established COPD (33.3% and 25.0%, respectively, for those without COPD). There was no correlation between decline in gas transfer and FEV1 for those with COPD, although a weak relationship existed for those without (r=0.218; P<0.025).
Conclusion: These data confirm differing individual rates of lung function decline in alpha-1-antitrypsin deficiency, indicating the importance of comprehensive physiological assessment and a personalized approach to patient management.
Keywords: alpha-1-antitrypsin deficiency, COPD, emphysema, lung function
Introduction
Chronic obstructive pulmonary disease (COPD) is a slowly progressive and destructive condition diagnosed by an impaired forced expired volume in 1 second (FEV1) expressed as a ratio of the total forced expired volume (forced vital capacity, FVC). The FEV1 has long been regarded as the most important physiological parameter because it relates to health status and respiratory as well as all-cause mortality.1–3 Thus, short-term improvement and/or stabilization of COPD progression (as determined by the FEV1) remain a long-term aim of current and future therapeutic development. In health, FEV1 is influenced by age, sex, height, and ethnicity, and all factors are taken into account to provide a normal range and, by inference, identification of “abnormality” throughout life. At diagnosis, patients’ results are placed in context of this normal range, and FEV1 values are expressed as “% predicted”.4 At diagnosis, % predicted reflects historical progression and not future progression because the starting baseline is unknown, and smoking cessation, for instance, may lead to subsequent disease stabilization. The progressive decline in FEV1 has conventionally been assessed as mL/year and, in general, patients with COPD have a more rapid decline than “usual.” Decline is a feature of natural aging and the reduced FEV1 of COPD has led to the concept of COPD reflecting an “accelerated ageing” process.5 However, recent cohort studies have highlighted that the decline in FEV1 in COPD differs between subjects, ranging from none to substantial annual loss.6–8 This has led to categorization of some patients as “rapid decliners” although this characterization is determined by arbitrary cutoff points.
The concept of accelerated decline is particularly true in alpha-1-antitrypsin deficiency (AATD), which is a recognized genetic predisposition to COPD characterized by emphysema and more rapid disease progression than in nondeficient COPD due to the poorly controlled release of tissue damaging proteases.9 Observational studies have suggested that in AATD decline in FEV1 can be partly modified by regular augmentation of circulating AAT.10–12 However, it is not clear whether this is indicated to prevent physiological progression in all individuals, as smoking cessation alone can stabilize FEV1 decline13 and subsequent individual differences are unknown. Nevertheless in many countries, AAT augmentation is the standard therapy to retard emphysema progression and stabilize lung function.14
Because of this uncertainty and the lifelong nature of augmentation therapy members of the Alpha-1-antitrypsin International Registry proposed a pragmatic approach based on physiological decline expressed as change in % predicted.15 The reasoning for this is described in detail elsewhere;15 a key point is that using % predicted accounts for age-related deterioration, such that if the % declines then the individual is deteriorating faster than normal aging, whereas a value in mL/year for FEV1 is unable to separate people declining normally from those with an excessive decline. This approach might be critical for health systems to adopt in order to maximize the benefit from augmentation, by restricting to those with greatest potential to benefit. The present study explores the implications of this approach using unique data collected prospectively as part of the ADAPT (Antitrypsin Deficiency Assessment and Program for Treatment) registry of AATD patients never treated with augmentation therapy.
Methods
All patients referred to the ADAPT program who had the PiZ phenotype (n=482) were studied providing at least four annual lung function data points were available for analysis (average 5.92: 95% confidence interval [CI] 5.67–6.17 data points collected over an average of 6.11 years [95% CI 5.80–6.41]). Augmentation therapy is not available in the UK, but any patient who had received augmentation therapy or any other potential disease modifying therapy as part of a previous clinical trial was excluded from the analysis. The study was approved by the South Birmingham Local Research Ethics Committee (LREC 3359), and all patients gave written informed consent.
All the patients were seen and assessed at the Birmingham center, for post bronchodilator lung function testing, including measurement of lung volumes and gas transfer (DLco and Kco), all of which were performed according to accepted guidelines.16 In addition, we documented demographics including smoking history and current status, quality of life, exacerbation history and drug history as described previously.17 Index cases were defined as those tested because of presentation with respiratory symptoms. Nonindex patients were those identified through family screening. For the purposes of the current paper, the presence of COPD was defined as subjects with a post-bronchodilator FEV1/FVC ratio <70%.
Decline in lung function was determined by the change in % predicted for age, sex, height, and ethnicity using published equations18 and linear regression determined for all annual data points (provided ≥4) for each patient and expressed as change in % predicted/year. Results were divided into those whose lung function decline (either FEV1 or gas transfer adjusted for effective alveolar volume (Kco) was consistent with normal aging (change <−0.1% predicted per year), slow (−0.1% to <−0.5%), moderate (−0.5% to <−1.0%), and rapid decline (>−1.0%) as described previously.15
Statistical analysis
Statistical analyses were performed using IBM SPSS statistics version 21.0.0.0 for windows (IBM Corporation, Armonk, NY), stratifying for the presence of COPD. Chi-squared analysis was used to determine any differences between and across groups for categorical data and the Mann–Whitney U-test was used for all comparisons of differences between groups for continuous data as it was nonparametric in distribution. The Kruskal–Wallis test was used to determine differences across groups for continuous data (statistical significance taken as P<0.05). Multivariate analysis was performed using (backwards) stepwise linear regression with body mass index, age, sex, index status, smoking status (ex/never smokers), and inhaled therapy (combination, LAMA, ICS) taken into account to determine any factor that related to the % predicted decline.
Results
Demographic differences
The characteristics of the cohort are shown in Tables 1 and 2. Overall the PiZ non-COPD patients consisted of more never smokers, less index patients, and more females compared to PiZ patients with COPD (P<0.0001 for all comparisons). The non-COPD group consisted of more patients with no FEV1 decline and conversely consisted of less patients with rapid decline than the group with established COPD (P<0.0001 for all comparisons). These data confirmed many prior associations of FEV1 decline in AATD,17 including a difference in the prevalence of bronchodilator reversibility between slow (22.4% of patients) and rapidly declining individuals (33.0% of patients; P<0.024). However, the decline rates for gas transfer were similar between the non-COPD group and the group with COPD (P>0.25 for all rates) and were not associated with factors previously shown to relate to FEV1 decline (eg, bronchodilator reversibility, P>0.40). For the PiZ non-COPD group, no baseline demographic feature predicted the rate of decline of gas transfer, although rapid decline in FEV1 was a feature of those with lower baseline FEV1/FVC ratio and lower gas transfer compared with those with no decline (P=0.003 and 0.004, respectively, Table 2).
 | Table 1 Characteristics of the patients with and without COPD |
 | Table 2 Characteristics of patients without COPD substratified by FEV1 decline |
Average demographic data for the two patient cohorts are shown together with the standard deviation and subject number analyzed for each feature. The proportion of subjects in each of the subgroups is shown in percentage. Statistical differences between patients with and without COPD (P) are shown.
Demographics are shown for the AATD subjects without COPD in each of the four FEV1 decline groups. Any differences across groups and between the nondeclining group and those with rapid decline are shown (P) and significant values are highlighted. N/A indicates no statistics were applied since by definition (“Methods” section) the four groups did not overlap.
Demographic data are shown for the AATD subjects with established COPD in each of the FEV1 decline groups. Statistical differences across groups and between the non- and rapid-decliner group are shown (P).
In the group with COPD, all were taking short-acting bronchodilators, and 68% were taking at least one other established long-acting bronchodilator (eg, LAMA, LABA/ICS). For the PiZ group with established COPD, rapid decline was more likely in those with a higher baseline FEV1 and FEV1/FVC ratio than those with no decline (P<0.0001 and P=0.017, respectively) as summarized in Table 3. Again, no demographic features identified those with differing rates of gas transfer decline.
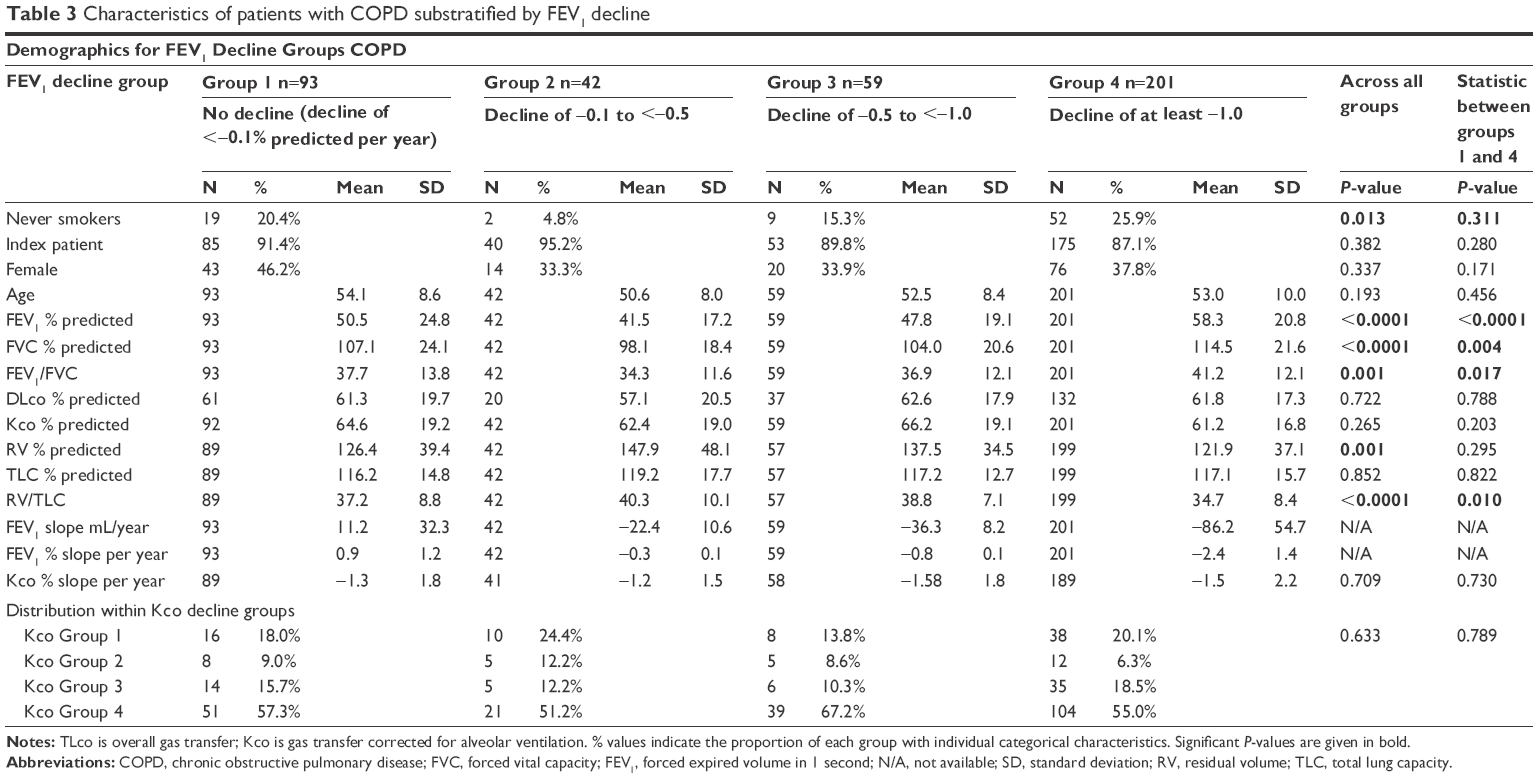 | Table 3 Characteristics of patients with COPD substratified by FEV1 decline |
Of the 482 patients with sufficient data for analysis, 27 (5.5%) were current smokers at baseline assessment and most ceased smoking during data collection; hence, analysis of the impact of current smoking on decline could not provide meaningful data, and such patients were excluded from analysis.
Decline in lung function
Decline is summarized in Tables 2 (non-COPD subjects) and 3 (COPD patients). For the 87 patients without established COPD, 50 (57.5%) showed no excessive decline in FEV1, whereas five (5.7%), ten (11.5%), and 22 (25.3%) showed slow, moderate, and rapid decline, respectively. The decline rate was similar for the slow moderate and rapid decline groups of never and ex-smokers. The average rapid decline for the ex-smokers (n=6) was greater (−4.17% predicted/year, standard deviation ±3.36) than the 16 never smokers (−2.64±1.00) although not statistically significant. The rapidly declining FEV1 group of the six ex-smokers however had a more rapid decline in gas transfer than the 16 never smokers (−2.94±1.81% predicted/year vs −0.35±2.16% predicted/year; P=0.022). For the COPD patients (ex- and never smokers) 93/395 (23.5%) showed no decline in FEV1% predicted. Forty-two (10.6%) showed a slow decline (−0.1 to <−0.5% predicted/year), 59 (14.9%) showed a moderate decline (−0.5 to <−1.0% predicted/year), and 201 (50.9%) showed a rapid decline (>−1.0% predicted/year). There was no significant difference in decline rate between never and ex-smokers. However, the proportion of ex-smokers and rapid and non-decliners differed between those with and without COPD as well as at different stages of COPD severity defined by the FEV1.4 These data are summarized in Figures 1 and 2, respectively.
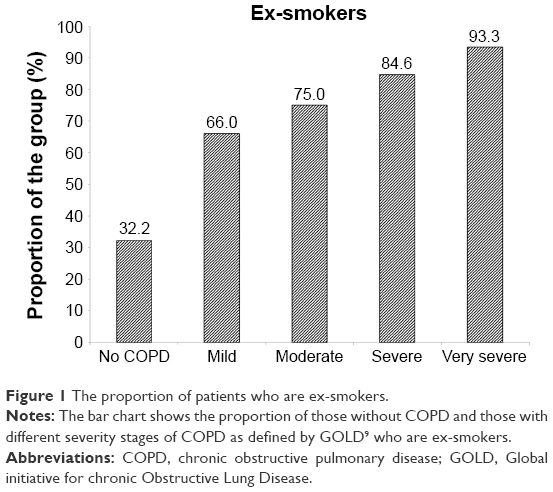 | Figure 1 The proportion of patients who are ex-smokers. |
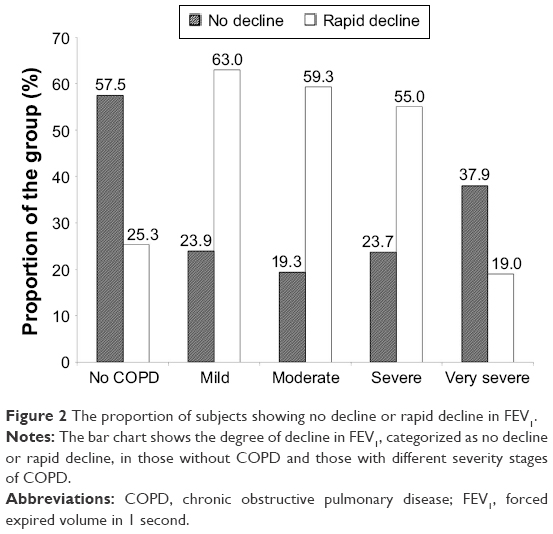 | Figure 2 The proportion of subjects showing no decline or rapid decline in FEV1. |
There was a significant but weak correlation between the declines in gas transfer and FEV1 in the patients without COPD (r=0.218; P<0.025). However, there was no correlation in AATD patients with established COPD (r=0.008; P=not significant) indicating that progression in these two physiological parameters is generally independent (Figure 3).
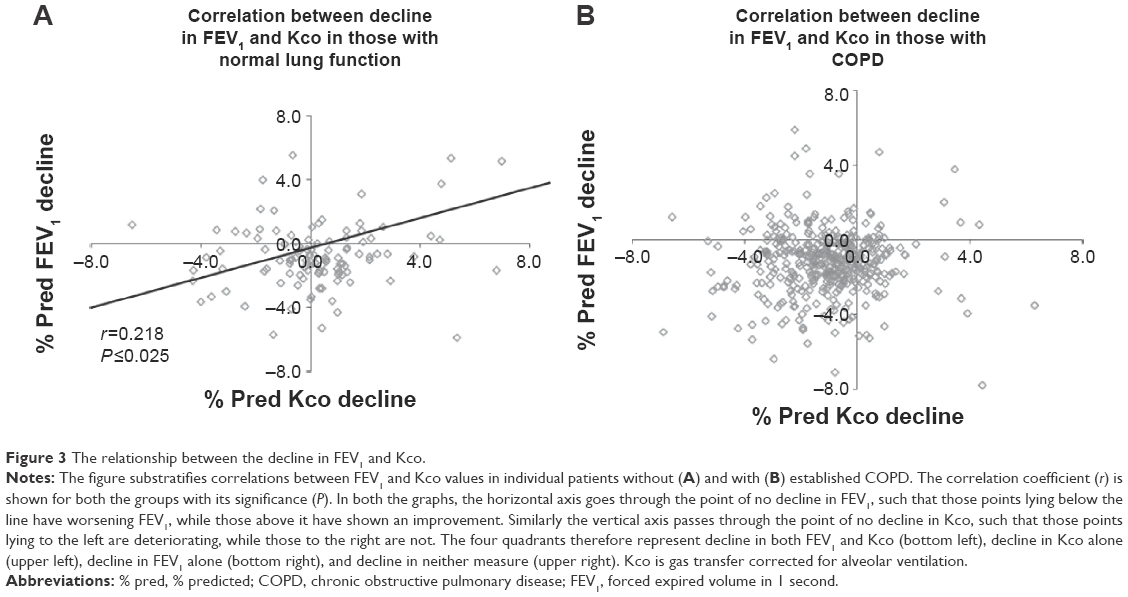 | Figure 3 The relationship between the decline in FEV1 and Kco. |
The decline rates for gas transfer in the PiZ COPD and non-COPD patients are summarized in Tables 2 and 3, respectively. In general 12.8% of COPD never smokers (n=81 where gas transfer was obtained for three or more consecutive years) had no decline in gas transfer with 7.7%, 20.5%, and 59.0% showing slow, moderate, or rapid decline, respectively (equivalent, on average, to −0.30%±0.12% predicted, −0.77%±0.13% predicted, and −2.56%±1.69% predicted, respectively). Data for the ex-smokers (n=302) were similar with 20.7% showing no decline and 8.0%, 14.7%, and 56.5% showing slow, moderate, and rapid decline, respectively.
For those without COPD, 33.3% of the never smokers had no excessive decline in gas transfer with 3.5%, 18.6%, and 47.5% showing slow, moderate, and rapid decline, respectively. These data were again similar for the ex-smokers (25.0%, 14.3%, 7.1%, and 53.6% showing no, slow, moderate, or rapid decline, respectively).
Predicting progression through COPD severity stages
COPD is usually staged by the degree of impairment of FEV1 expressed as a % predicted.4 Cross-sectional measurement does not provide historical evidence of decline since baseline starting point is unknown and neither is any influence of previous smoking. The rate of decline quantified here (for an individual) has to relate to the point of patient acquisition but would permit a general observation and prediction of the time needed to develop moderate (FEV1 50%–80% predicted), severe (FEV1 30%–50% predicted), and very severe (FEV1 <30% predicted) COPD, assuming decline is and remains linear for given individuals (as demonstrated in Figures 4 and 5).
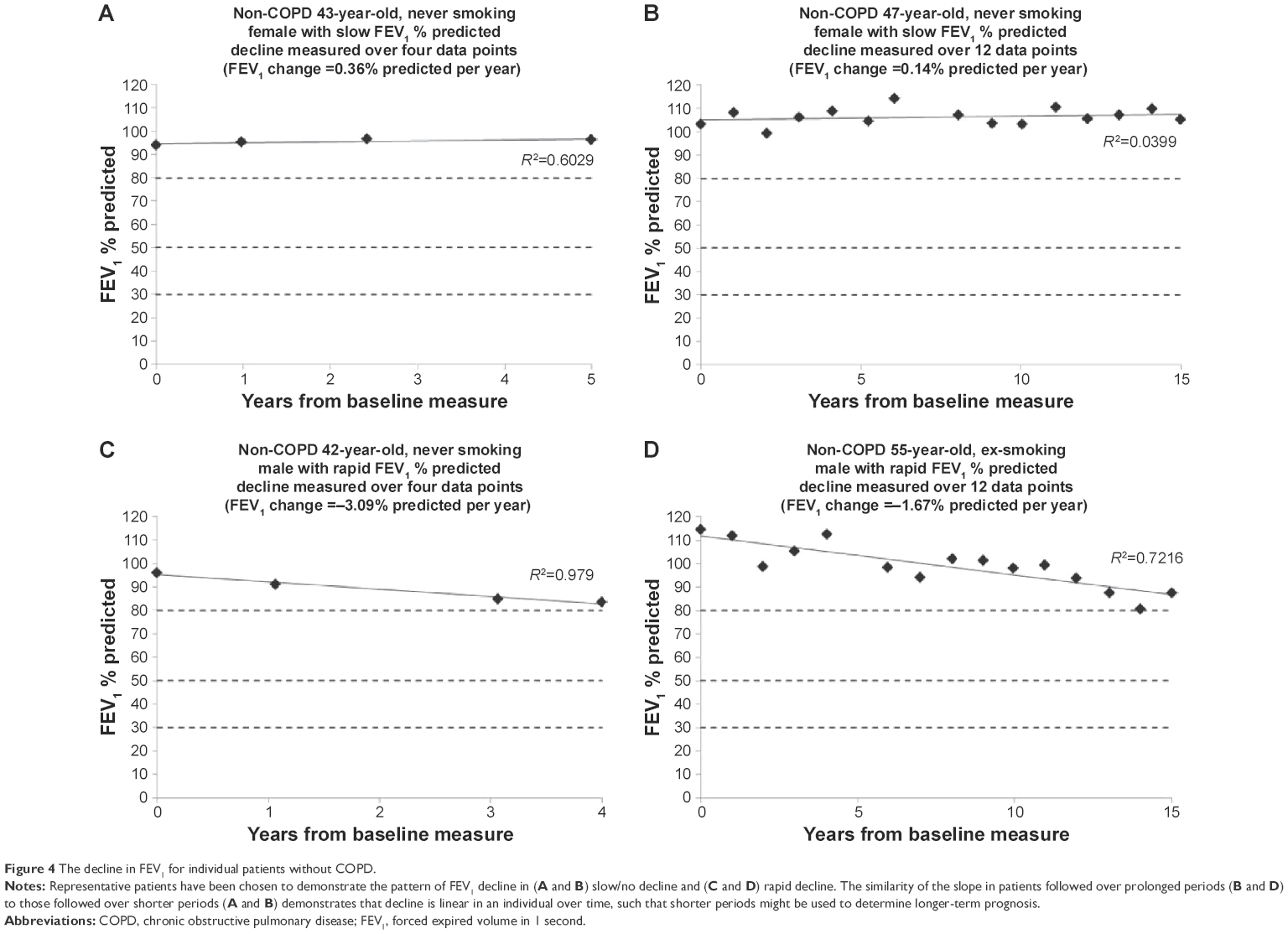 | Figure 4 The decline in FEV1 for individual patients without COPD. |
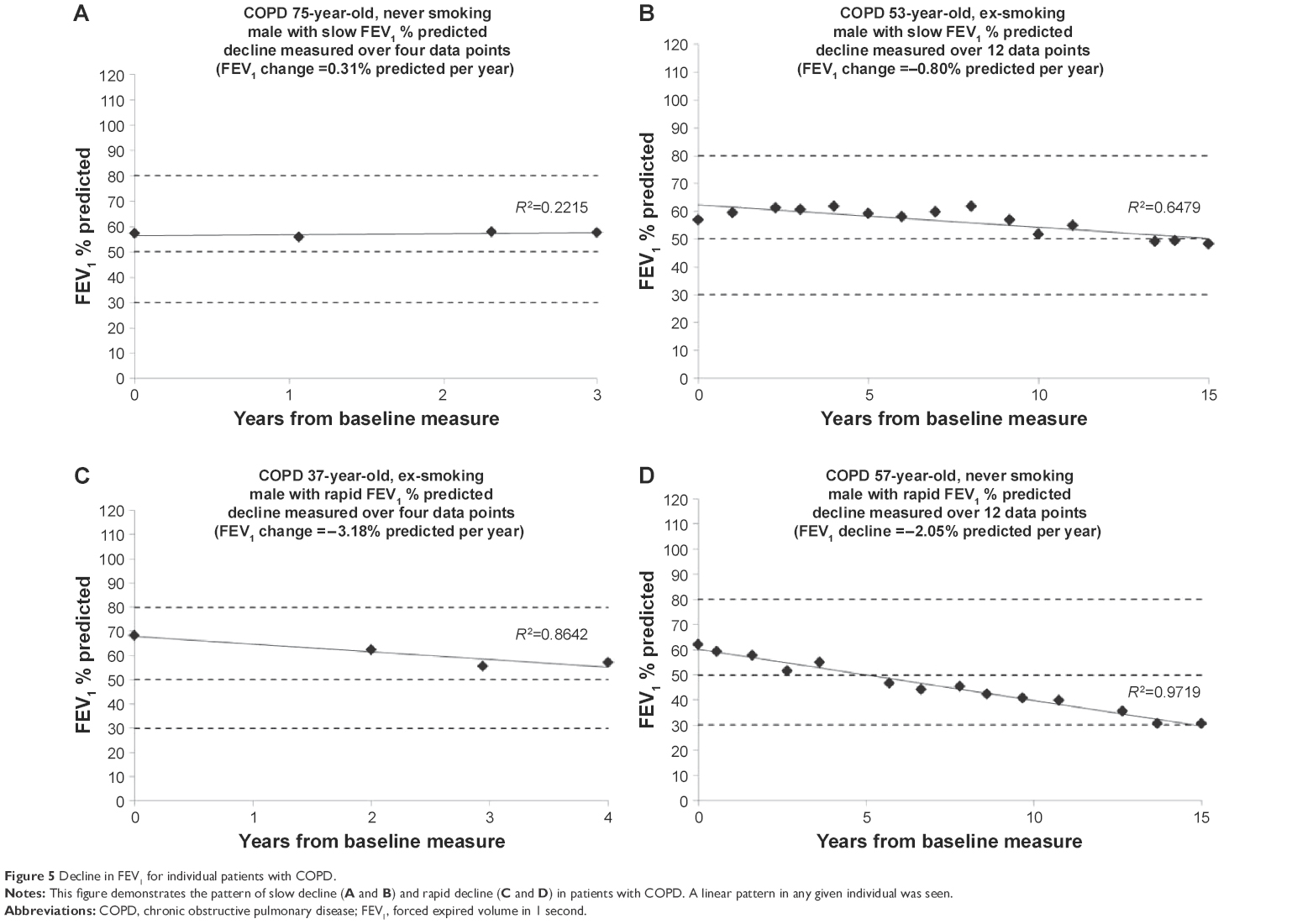 | Figure 5 Decline in FEV1 for individual patients with COPD. |
To determine the relationship between progression determined over 3 years with four full data points and that seen over longer periods of time, we compared the decline determined by linear regression for 123 patients where both four and eleven annual data points were available. These data are summarized in Figure 6 showing a good correlation between the short- and long-term data. Using our criteria, the positive predicted value of a slow decliner determined over 3 years was 84.5% when compared to 11 years decline and that for rapid decliners was 58.8%.
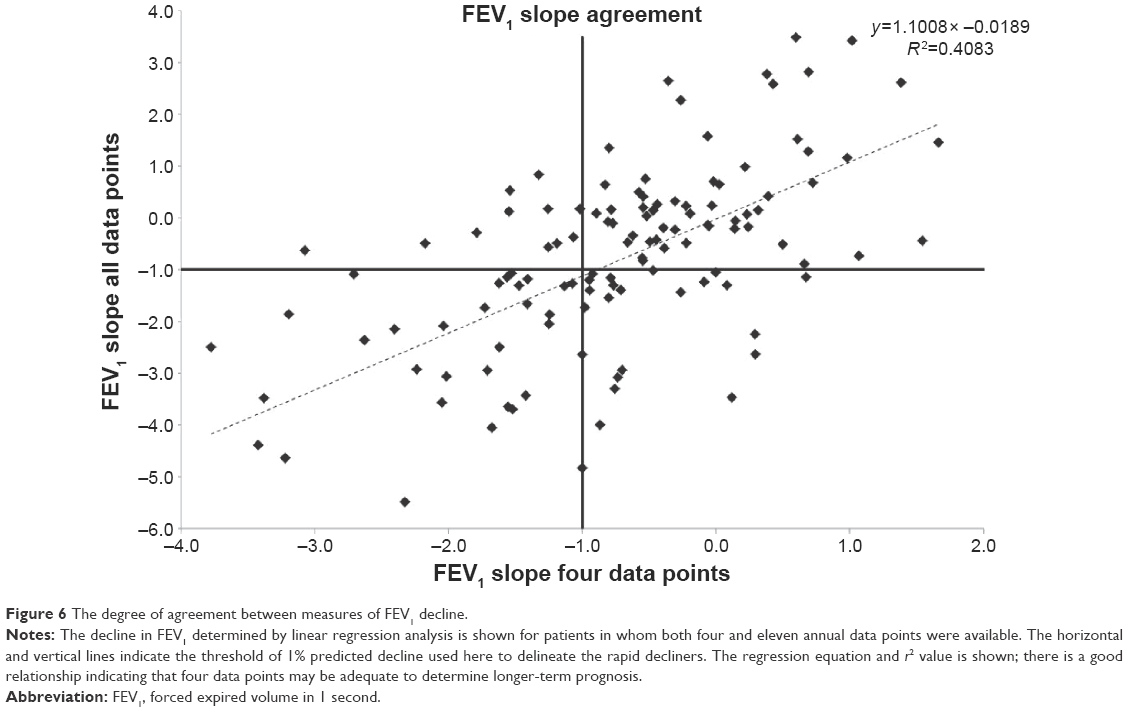 | Figure 6 The degree of agreement between measures of FEV1 decline. |
Discussion
The data presented here provide unique information on the physiological natural history of an untreated population of AATD patients assessed and monitored in a single center with high quality control for all measurements. The data indicate that physiological decline differs between subjects at all stages of lung function impairment for both index and nonindex patients as well as never smokers and ex-smokers.
The data have several important implications for patient management and particularly the consideration of augmentation therapy which is both expensive and requires weekly infusions of AAT for life. The aim of such therapy is to slow down or prevent progression of lung disease and is prescribed for AATD patients who have never smoked or who have stopped smoking. At present, the indications are generally restricted to those patients with an FEV1 between 35% and 60% predicted (the “severe” COPD stage). This is based on the observation that a statistically significant slower decline in FEV1 was observed in this physiological range of subjects who had received augmentation therapy but not at higher or lower levels in the National Heart Lung and Blood Institute observational study.14 Whereas the observation is partly informative about the average effect on FEV1 decline, overall it would not be expected to be effective/required if the FEV1 were stable. Our data show that at all stages of COPD severity, defined by the FEV1, there are patients who do not decline during the observational period as well as those who decline rapidly.
Our data, divided into patients within the widely recognized Global initiative for chronic Obstructive Lung Disease stages,4 show that the proportion of rapid decliners (as defined here) differs with severity. However, there remains a significant proportion of individuals who decline less rapidly or not at all, at all stages of disease severity. In addition, the proportion of ex-smokers within each FEV1 stage increases with severity suggesting their decline while smoking was far more rapid than that after smoking cessation. Since never and ex-smokers generally decline at the same rate, the data explain (at least in part) why Stage 4 COPD which consists of virtually all (94%) ex-smokers has a slower decline compared with the less severe stages. Nevertheless, rapid and nondecliners are present at all stages of the disease process. In general, the lower the FEV1 the greater the likelihood of death19 and the need for transplantation. Thus, knowledge of FEV1 decline will play a key role in decision making about augmentation therapy aimed to slow or stop decline in an individual patient. Furthermore, knowledge of decline may allow enrichment of patient cohorts for future studies by selecting rapid decliners and ensuring adequate power where physiological decline is the outcome.
Use of a patients’ longitudinal data to decide about augmentation would have a number of impacts on clinical practice. In some patients, rapid decline might be apparent early in follow-up, in which case documenting this before treatment would provide a baseline to monitor the effects of augmentation.20 In most there would be a delay of up to 3 years to make a decision about treatment, especially ex-smokers where precessation and subsequent decline may differ. The positive predictive value for identifying slow/nondecliners with this method would be 84.5% (Figure 6), indicating that with four annual data points, those in whom augmentation therapy would have less impact on FEV1 decline can be identified with reasonable confidence. Conversely, the positive predictive value of identifying long-term rapid decliners with four annual data points is less (58.8%), which indicates little change over the initial 3-year period in a proportion who have a significant decline confirmed subsequently. Longer follow-up might ensure that such individuals obtained augmentation therapy at an appropriate later time point. Alternatively monitoring a more specific physiological measure of emphysema such as gas transfer may be as or more appropriate for decision making. Gas transfer relates to lung density, especially in the upper zones,21 and this anatomical effect explains why the gas transfer decline is greatest late in the disease17,19 as the classical basal emphysema becomes more apparent in the upper zones. This region of the lung, however, may be less responsive to augmentation therapy.22 Furthermore, it does not relate to the FEV1 decline except in early disease (Figure 1). Whether monitoring gas transfer decline can be an indicator for the need for augmentation therapy in the absence of an FEV1 decline requires further study perhaps before and after the start of augmentation therapy.
It is possible that the eventual pool of patients eligible for augmentation could be extended. The identification of rapid decliners with initial lung function in the normal range, or with mild COPD, suggests that such individuals should also be considered for augmentation therapy, where current guidelines would not recommend this, as their FEV1 is too well maintained. Interestingly the idea of earlier treatment in COPD to prevent decline is one that has also been raised in patients with COPD unrelated to AATD.23
In such patients, knowledge of the decline rate allows informed outcome prediction and earlier decision making about augmentation therapy even outside the current prescribing limits. This could have great potential societal and economic benefits, such as patients remaining in work.
Prevailing FEV1 alone does not provide this information on an individual patient basis even in lifetime never smokers. Mild impairment in middle age may be perceived to reflect slow decline. However, since the initial FEV1 at maturity is unknown, this could reflect both a slow decline for those whose FEV1 started at the bottom of the normal range and a rapid decline for those who started at the top of the normal range. In addition ex-smokers, even with severe impairment, may stabilize their FEV1. For these reasons, a period of observation of FEV1 decline would appear mandatory before implementing augmentation therapy.
Knowledge of FEV1 decline therefore allows both a projection of likely future progression24 and prognosis and more informed confidence of the need and subsequent expected effects of augmentation therapy. Furthermore, the presence of rapid or nondecline can indicate patients who most (or least) require augmentation therapy to influence prognosis.
Clinical trials to demonstrate the efficacy of augmentation therapy have proven impractical with FEV1 as a primary outcome. Indeed its sensitivity to change is low,24 suggesting large numbers of patients are required over prolonged periods to confirm or refute efficacy. FEV1 is however only a surrogate of the emphysema process and hence the acceptance of CT densitometry as an alternative outcome. This is far more sensitive to change,25 is related cross-sectionally to lung physiology and health status26 and mortality27 and the decline in FEV1 in longitudinal studies.28,29 Longitudinal follow-up of density in our untreated patients also suggests that density decline relates to mortality.30 Clinical trials using density decline as an outcome have been consistent in demonstrating benefit31–33 and the recent adequately powered study alone has confirmed a treatment effect.31 However, not all AATD patients show a decline in densitometry,29,31 and our published data show that neither FEV1 nor gas transfer measurement is capable of identifying all patients whose CT density is deteriorating. This implies that monitoring density decline might also be necessary for decision making, though benefit from serial scans identifying emphysema progression would need to be balanced against the requirement for specialist interpretation and repeated radiation exposure.
Conclusion
The data show that lung function decline in AATD differs between patients but that rapid decline is present in both patients with and patients without COPD. The decline determined for an individual, especially with respect to FEV1, appears linear, permitting longer-term projection to be estimated particularly in ex-smokers (where the decline rate may have changed). This would allow identification of those more likely to need/benefit from augmentation therapy and also provide a means of assessing response to this by close subsequent monitoring.
Acknowledgments
Funding for the ADAPT program was provided by noncommercial grant funding from Grifols and CSL Behring. Project grant funding was supplied by The West Midlands Chest Fund, National Institute of Health Research, and the Alpha1 Foundation. The Birmingham and Black Country comprehensive local research network funded the fellowship of AP.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
RAS and AMT have attended advisory board meetings for Grifols and CSL Behring. AMT has received noncommercial grant funding from CSL Behring and RAS has received noncommercial grant funding from Grifols and CSL Behring. The authors report no other conflicts of interest in this work.
References
Ketelaars CA, Schlösser MA, Mostert R, et al. Determinants of health-related quality of life in patients with chronic obstructive pulmonary disease. Thorax. 1996;51:39–43. | ||
Ebi-Kryston KL. Respiratory symptoms and pulmonary function as predictors of 10-year mortality from respiratory disease, cardiovascular disease, and all causes in the Whitehall study. J Clin Epidemiol. 1988;4:251–260. | ||
Tockman MS, Comstock GW. Respiratory risk factors and mortality: longitudinal studies in Washington County, Maryland. Am Rev Respir Dis. 1989;140(3 Pt 2):S56–S63. | ||
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease, GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. | ||
Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174:886–893. | ||
Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359:1543–1554. | ||
Jenkins CR, Jones PW, Calverley PM, et al. Efficacy of salmeterol/fluticasone propionate by GOLD stage of chronic obstructive pulmonary disease: analysis from the randomised, placebo-controlled TORCH study. Respir Res. 2009;10:59. | ||
Vestbo J, Agusti A, Wouters EF, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints Study Investigators. Should we view chronic obstructive pulmonary disease differently after ECLIPSE? A clinical perspective from the study team. Am J Respir Crit Care Med. 2014;189:1022–1030. | ||
Stockley RA. Proteases and Antiproteases in Chronic Obstructive Pulmonary Disease: Pathogenesis to Treatment. Chadwick D, Goode JA, editors. Wiley: Chichester, UK; 2001:189–204. | ||
The Alpha-l-Antitrypsin Deficiency Registry Study Group. Survival and FEVl decline in individuals with severe deficiency of alpha-l antitrypsin. Am J Respir Crit Care Med. 1998;158:49–59. | ||
Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J. 1997;10:2260–2263. | ||
Chapman KR, Stockley RA, Dawkins C, et al. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6:177–184. | ||
Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA. 1994;272:1497–1505. | ||
American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society Statement: standards for the diagnosis and management of individuals with alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818–900. | ||
Stockley RA, Miravitlles M, Vogelmeier C. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149. | ||
BTS/ARTP guidelines. Guidelines for the measurement of respiratory function. Resp Med. 1994;88:165–194. | ||
Dawkins PA, Dawkins CL, Wood AM, et al. Rate of progression of lung function impairment in alpha-1 antitrypsin deficiency. Eur Respir J. 2009;33:1338–1344. | ||
Quanjer PH, Tammeling GJ, Cotes JE, et al. Lung volumes and forced ventilatory flows. Report Working Party Standardization of Lung Function Tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur Respir J. 1993;16:5–40. | ||
Pillai AP, Turner AM, Stockley RA. Relationship of the 2011 Global Initiative for Chronic Obstructive Lung Disease strategy to clinically relevant outcomes in individuals with α1-antitrypsin deficiency. Ann Am Thoracic Soc. 2014;11:859–864. | ||
Wencker M, Fuhrmann B, Banik N, et al; Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest. 2001;119:737–744. | ||
Parr DG, Stoel BC, Stolk J, et al. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med. 2004;170:1172–1178. | ||
Parr DG, Sevenoaks M, Deng C, et al. Detection of emphysema progression in alpha 1-antitrypsin deficiency using CT densitometry; methodological advances. Respir Res. 2008;13;(9):21. | ||
Tantucci C, Modina D. Lung function decline in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:95–99. | ||
Wang ML, Avahia BH, Petsonk EL. Interpreting periodic lung function tests in individuals: the relationship between 1- to 5-year and long-term FEV1 changes. Chest. 2006;130:493–499. | ||
Stolk J, Putter H, Bakker EM, et al. Progression parameters for emphysema: a clinical investigation. Respir Med. 2007;101:1924–1930. | ||
Dowson LJ, Guest PJ, Hill SL, et al. High-resolution computed tomography scanning in a1-antitrypsin deficiency: relationship to lung function and health status. Eur Respir J. 2001;17:1097–1104. | ||
Dawkins PA, Dowson LJ, Guest PJ, et al. Predictors of mortality in – antitrypsin deficiency. Thorax. 2003;58:1020–1026. | ||
Parr DG, Stoel BC, Stolk J, et al. Validation of computed tomographic lung densitometry for monitoring emphysema in a-1-antitrypsin deficiency. Thorax. 2006;61;485–490. | ||
Stolk J, Stockley RA, Piitulainen E, et al. Relationship between change in lung density and long-term progression of lung function. Am J Respir Crit Care Med. 2015;192:114–116. | ||
Green CE, Parr DG, Edgar RG, Stockley RA, Turner AM. Lung density associates with survival in alpha 1 antitrypsin deficient patients. Respir Med. 2016;112:81–87. | ||
Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha-1-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160:1468–1472. | ||
Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha-1-antitrypsin deficiency. Eur Respir J. 2009;33:1345–1353. | ||
Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe a1 antitrypsin deficiency (RAPID): a randomised, double blind, placebo controlled trial. Lancet. 2015;386:360–368. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.