Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Increased von Willebrand Factor Processing in COPD, Reflecting Lung Epithelium Damage, Is Associated with Emphysema, Exacerbations and Elevated Mortality Risk
Authors Langholm LL ![]() , Rønnow SR
, Rønnow SR ![]() , Sand JMB, Leeming DJ, Tal-Singer R
, Sand JMB, Leeming DJ, Tal-Singer R ![]() , Miller BE
, Miller BE ![]() , Vestbo J
, Vestbo J ![]() , Karsdal MA
, Karsdal MA ![]() , Manon-Jensen T
, Manon-Jensen T
Received 22 October 2019
Accepted for publication 21 February 2020
Published 9 March 2020 Volume 2020:15 Pages 543—552
DOI https://doi.org/10.2147/COPD.S235673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Lasse L Langholm,1,2 Sarah Rank Rønnow,1,3 Jannie MB Sand,1 Diana Julie Leeming,1 Ruth Tal-Singer,4 Bruce E Miller,4 Jørgen Vestbo,5 Morten A Karsdal,1 Tina Manon-Jensen1
1Nordic Bioscience A/S, Herlev, Denmark; 2University of Copenhagen, Faculty of Health and Medical Sciences, Department of Biomedical Sciences, Copenhagen, Denmark; 3University of Southern Denmark, The Faculty of Health Science, Odense, Denmark; 4Respiratory Medical Innovation, Value Evidence & Outcomes, GSK R&D, Collegeville, PA, USA; 5Division of Infection, Immunity and Respiratory Medicine, University of Manchester, and Manchester University NHS Foundation Trust, Manchester, UK
Correspondence: Lasse L Langholm
Nordic Bioscience A/S, Herlev Hovedgade 205-207, Herlev 2730, Denmark
Tel +45 4452 5252
Fax +45 4452 5251
Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is characterized by chronic inflammation and lung tissue deterioration. Given the high vascularity of the lung, von Willebrand factor (VWF), a central component of wound healing initiation, has previously been assessed in COPD. VWF processing, which is crucial for regulating the primary response of wound healing, has not been assessed directly. Therefore, this study aimed to characterize wound healing initiation in COPD using dynamic VWF-processing biomarkers and to evaluate how these relate to disease severity and mortality.
Methods: A cross-sectional analysis of plasma samples from the ECLIPSE study collected at year 1 from moderate to very severe COPD subjects (GOLD 2– 4, n=984) was performed. We applied competitive neo-epitope ELISAs specifically targeting the formation of and ADAMTS13-processed form of VWF, VWF-N and VWF-A, respectively.
Results: VWF-A and VWF-N were significantly increased (VWF-N, p=0.01; VWF-A, p=0.0001) in plasma of symptomatic (mMRC score ≥ 2) compared to asymptomatic/mild symptomatic COPD subjects. Increased VWF-N and VWF-A levels were specifically associated with emphysema (VWF-N, p< 0.0001) or prior exacerbations (VWF-A, p=0.01). When dichotomized, high levels of both biomarkers were associated with increased risk of all-cause mortality (VWF-N, HR 3.5; VWF-A, HR 2.64).
Conclusion: We demonstrate that changes in VWF processing were related to different pathophysiological aspects of COPD. VWF-N relates to the chronic condition of emphysema, while VWF-A was associated with the more acute events of exacerbations. This study indicates that VWF-A and VWF-N may be relevant markers for characterization of disease phenotype and are associated with mortality in COPD.
Study Identifier: NCT00292552; GSK study code SCO104960.
Keywords: von Willebrand factor processing, COPD, emphysema, exacerbations, increased mortality risk
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by chronic airway obstruction and includes individuals with chronic bronchitis and/or emphysema who may experience acute worsenings or exacerbations, which accelerate the degeneration of lung tissue14,37 and increase mortality risk.39 An abnormal inflammatory response, including disruption of normal tissue repair processes and activation of wound healing, is believed to be among the underlying drivers of COPD, leading to small airway fibrosis.4,25,31 Subjects with COPD present with very heterogeneous disease characteristics,1 which complicates identification of mortality predictors and lung function decline. Identifying subjects with progressive phenotypes such as emphysema and exacerbations using biomarkers reflecting inflammation and tissue damage could be a valuable tool in assessing mortality risk in COPD.
Protein components of the wound healing cascade and inflammatory signaling are affected in COPD and could be potential biomarkers to help characterize COPD phenotypes and severity.2,11 Platelets are essential for blood clotting during wound healing and have shown highly increased activation in COPD.24 Furthermore, the lungs are a major contributor to platelet biogenesis and a reservoir for platelet-producing megakaryocytes.23 von Willebrand factor (VWF) is an important player in platelet activation and a biomarker of endothelial dysfunction and inflammation in COPD.35 VWF is a large multimeric glycoprotein that circulates in the bloodstream or is stored in either platelet α-granules or the Weibel-Palade bodies of endothelial cells.30,43 In response to endothelial damage, VWF is activated by shear-stress, unfolding the multimers and thereby facilitating platelet-tethering to damaged sub-endothelial extracellular matrix (ECM) leading to bleeding cessation.3,38 During VWF unfolding, a cleavage-site for the metalloproteinase ADAMTS13 at Tyr1605-Met1606 is exposed, resulting in cleavage and reduction of VWF multimer size, which also regulates VWF activity.32 Notably, only in the active unfolded conformation can ADAMTS13 cleave VWF.13,32 Both VWF and platelets have been shown to have altered levels and activities in inflammatory disorders,22,29 indicating a function beyond just facilitating hemostasis. Increased VWF levels could therefore potentially reflect the persistence of chronic inflammation in COPD.
VWF levels and relative activity have been found to be increased in COPD,8 but VWF-processing products reflecting endothelial release and activation, which is the central axis of initiation of the primary wound healing response have, to our knowledge, not been assessed directly in COPD subjects.
To emphasize the role that the vascular endothelium plays in lung tissue destruction and inflammation in COPD, we applied two biomarkers of VWF-processing measuring VWF formation by targeting the released pro-peptide (VWF-N) and ADAMTS13-cleaved activated VWF (VWF-A) using neo-epitope specific monoclonal antibodies. We hypothesized that ADAMTS13-cleaved VWF generates a dynamic biomarker of tissue damage, inflammation and platelet activation, while VWF-N reflect newly released VWF into the circulation. The aim of this study was to characterize tissue damage and hemostatic processes in COPD, to describe how these dynamic VWF-processing biomarkers relate to disease severity and mortality.
Materials and Methods
The study population consisted of a cross-sectional subset of plasma samples from the Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) study collected at year 1 from moderate to very severe COPD patients (GOLD stage 2–4, n=957) and non-COPD controls including both smoker- (n=203) and non-smoker-controls (n=96). The study design of ECLIPSE (clinicaltrials.gov identifier NCT00292552; GSK study code SCO104960) has been fully described previously.40 In short, ECLIPSE is a non-interventional, observational, multicentre study in patients with COPD (n=2164) and control subjects (n=582). Participants were monitored at specific time points during the study, which ran for three years in total. Inclusion criteria were baseline post-bronchodilator forced expiratory volume in one second (FEV1) values of <80% of predicted, an FEV1/FVC (forced vital capacity) ratio of ≤0.7 and ≥10 pack-years of previous smoking history. The controls were divided into a non-smoker and smoker control group defined by less than or more than 10 pack-years of smoking history, respectively, in addition to normal lung function.40 In our analyses, we used biomarker and other clinically relevant data collected at baseline and year 1, as well as follow-up data on mortality.
Heparin plasma samples were prepared from whole blood collected by venipuncture in vacutainer tubes from fasting participants. Plasma was obtained by centrifuging the samples for 10–15 min at 2000 x g, before storage at −80°C until analyzed.
Competitive enzyme-linked immunosorbent assay (ELISA) format was used to assess protein fragments of VWF release (VWF-N) and ADAMTS13-processing (VWF-A) using neo-epitope specific mouse monoclonal antibodies28 (Nordic Bioscience A/S, Herlev, Denmark). The term neo-epitope defines a specific amino acid sequence generated by specific proteolytic cleavage. The antibodies are specific for only the proteolytically processed protein form. Briefly, the assays were performed on a 96-well streptavidin plate coated with 100µL of dissolved specific biotinylated synthetic peptide in an optimized coating buffer, incubated for 30 min at 20°C. 20 µL of sample or standard peptide diluted in incubation buffer were applied to the plate followed by 100 µL of antibody solution, incubated overnight (20 hrs) at 4°C with shaking. Subsequently, 100 µL of horseradish peroxidase (HRP) labeled secondary anti-mouse antibody was added and incubated for 1 hr at 20°C. Finally, the plates were incubated with 100 µL of tetramethylbenzidine (TMB) for 15 min at 20°C in the dark. The HRP reaction was stopped by adding 100 µL of stopping solution (1% H2SO4). All steps, aside from the TMB to stop-solution, were followed by a plate wash cycle to remove unbound and excess reagents. Plates were read in a SpectraMax M5 (Molecular Devices, CA, USA) at 450nm with 650nm as a reference.
We included analyses of CRP, previously measured in the ECLIPSE study.41 Prospective analyses were based on follow-up on all-cause mortality (n=30) two years post blood-sampling (to day 1080 from study start).
Statistics
D’Agostino-Pearson testing found biomarker data not to be normally distributed; therefore, the statistical analyses were performed by non-parametric methods. One-way ANOVA and chi-squared test were used to evaluate patient demographics and clinical biomarkers. Mann–Whitney t-test and Kruskal–Wallis testing were applied to compare biomarker levels between groups. ROC curve analysis was utilized to define biomarker cut-offs from Youden Index criterion based on mortality data and survival curves were displayed using Kaplan-Meier plotting. Cox proportional hazard regression analysis was used to estimate hazard ratios (HR) per 1 SD change in biomarker for predicting mortality. HR was adjusted for the following confounders of mortality identified from univariate analysis: age, smoking history, high-sensitivity C-reactive protein (hsCRP), 6 min walking distance (6MWD), dyspnea scale (mMRC) and prior hospitalizations. The software used was GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla California USA) and MedCalc Statistical Software version 14.8.1 (MedCalc Software bvba, Ostend, Belgium). A p-value <0.05 was considered statistically significant. Data presented as median + 95% Confidence interval (CI), unless stated otherwise.
Results
Mean age, BMI and percent of predicted FEV1 (FEV1% predicted) for the COPD subjects were 63.1, 26.8 and 50.4, respectively, thereby being significantly older than the controls and having a lower BMI than the non-smoker controls. There were also significantly higher percentage of men in the COPD group. The FEV1% predicted confirmed their highly decreased lung capacity (Table 1).
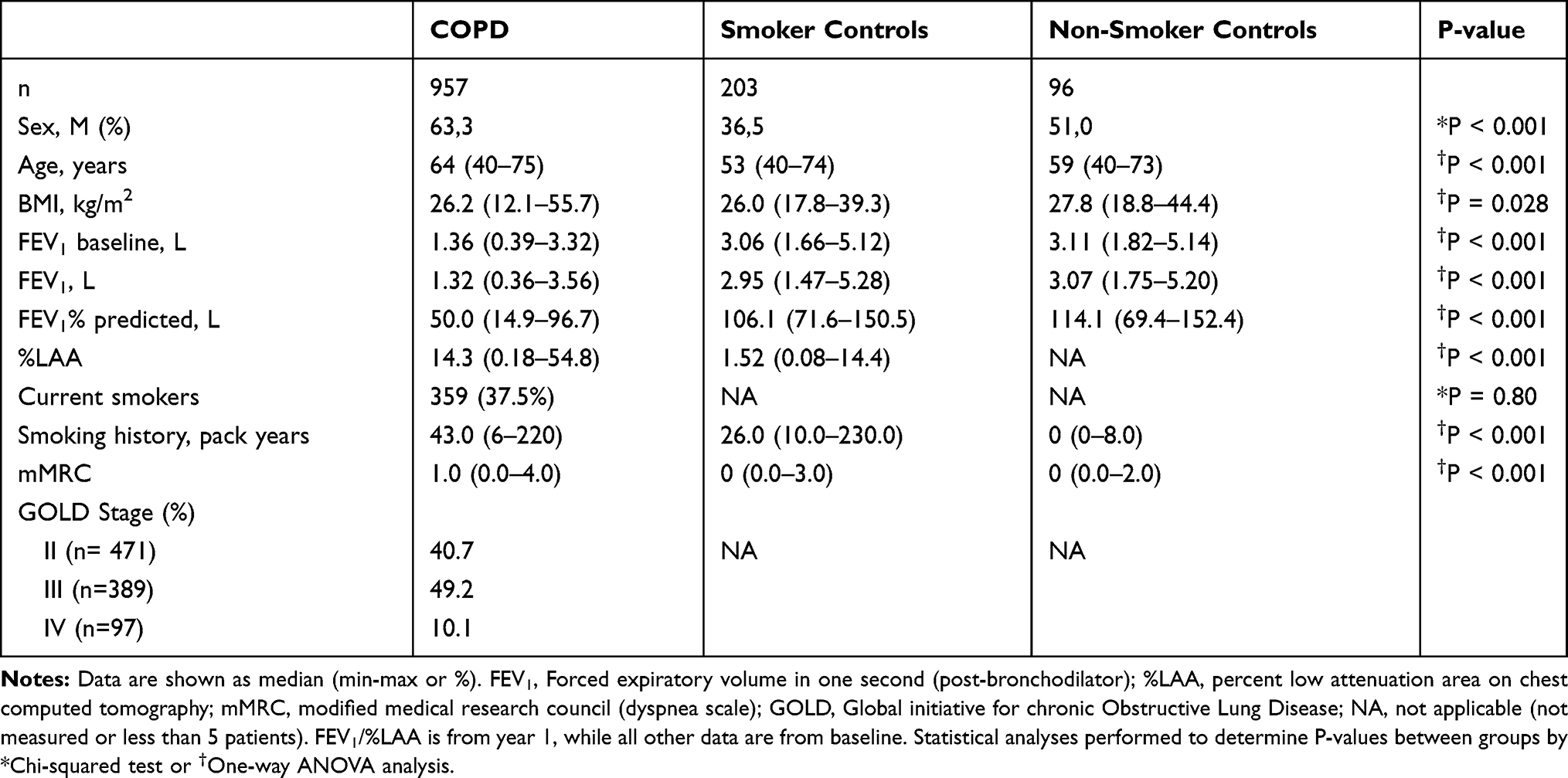 |
Table 1 Participant Demographics |
Plasma VWF-N and VWF-A were significantly elevated in COPD subjects compared to smoker controls (p<0.05) but not compared to non-smoker controls (Figure 1A). Both biomarkers proved to be significantly elevated in COPD subjects compared to the combined control groups (VWF-N, VWF-A, p=0.01), data not shown.
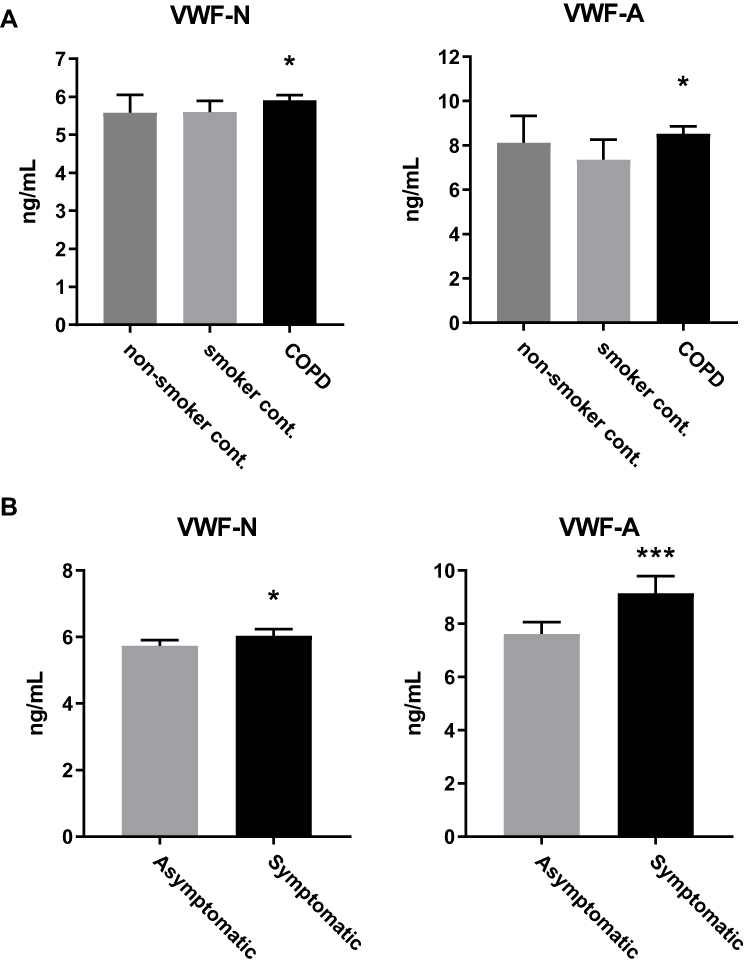 |
Figure 1 VWF processing was increased in COPD and symptomatic disease. (A) VWF-N and VWF-A were significantly increased in COPD subjects (n=957) compared to smoker controls (n=203), but not non-smoker controls (n=96). (B) Both VWF-N and VWF-A were significantly increased in symptomatic (mMRC ≥2) COPD subjects (n=458) compared to non-symptomatic/mild COPD (n=462). Data presented as median + 95% CI. *p<0.05, ***p<0.001. |
We wanted to investigate disease activity of COPD and how this relates to VWF processing, firstly by addressing symptomatic COPD. We divided the COPD subjects into symptomatic and asymptomatic/mild subjects, based on a modified Medical Research Council (mMRC) dyspnea scale cut off of two as previously described in the GOLD guidelines.42 Both VWF biomarkers were significantly increased in symptomatic (mMRC ≥2) compared to asymptomatic/mild (mMRC ≥2) COPD subjects, with VWF-A being the more statistically significant of the two (VWF-N, p=0.0002; VWF-A, p<0.0001; Figure 1B).
Next, we sought to investigate if there was a specific VWF-processing profile between different underlying symptoms or subtypes of COPD, such as emphysema and exacerbations. Emphysema was defined as low attenuation area of −950 Hounsfield units of more than 10% (%LAA) on chest computed tomography,41 while exacerbations were self-reported events within the year prior to the blood sampling used for our biomarker measurements. Twenty-four percent of patients with GOLD stage 2, 32% with stage 3 and 44% in stage 4 were considered frequent exacerbators, having had two or more exacerbations within the year prior to sampling.20 We observed increased VWF-A and not VWF-N levels in subjects who had experienced exacerbations (VWF-A, p=0.009; VWF-N, p=0.87; Figure 2A), while for emphysematous subjects, only VWF-N levels were significantly elevated (p<0.0001) while VWF-A levels were unchanged (p=0.09) (Figure 2B).
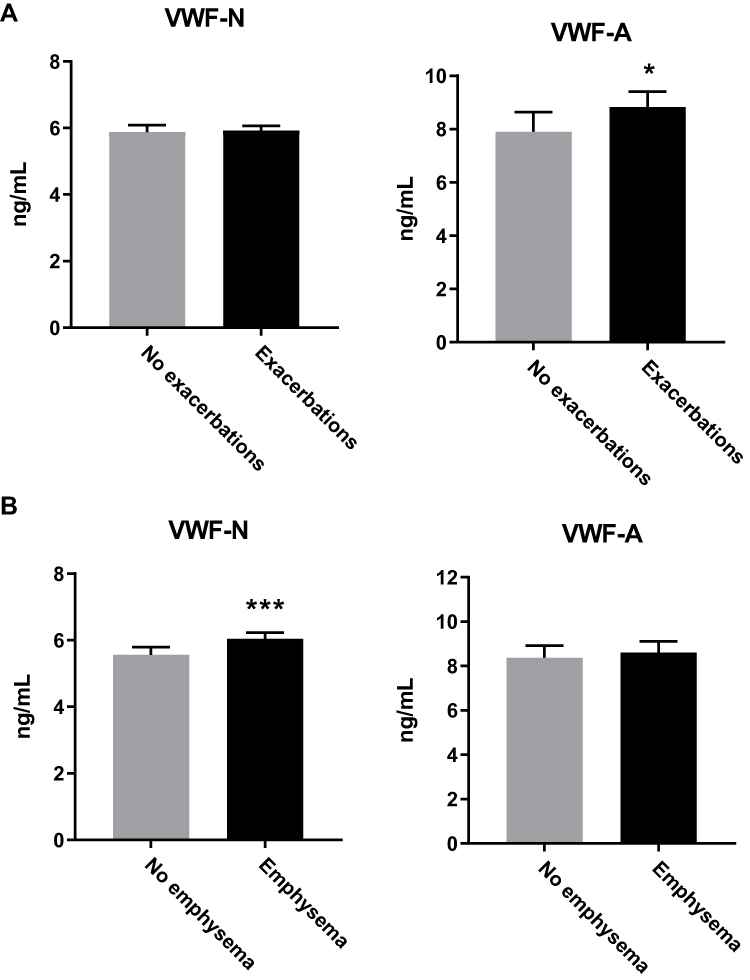 |
Figure 2 VWF processing was different between subjects with emphysema and exacerbations. (A) VWF-A but not VWF-N was increased in COPD subjects who suffered from one or more exacerbations within the previous year (n=522), compared to the no exacerbation group (n=418). (B) VWF-N but not VWF-A is increased in COPD subjects with emphysema (n=584) compared to non-emphysematous subjects (n=310). Data presented as median + 95% CI. *p<0.05, ***p<0.001. |
Knowing that emphysema and exacerbations are related to decreasing survival rates, we wanted to investigate if VWF processing was associated with risk of mortality. Both VWF biomarkers were significantly increased in subjects that died (n=30) within the follow-up period (VWF-N, p=0.0001; VWF-A, p=0.0056; Figure 3A). We then wanted to define a cut off for high VWF levels, associated with increased mortality. We dichotomized the VWF biomarkers using ROC curve analysis of mortality data, resulting in a Youden index criterion of 6.7 ng/mL and 9.7 ng/mL for VWF-N and VWF-A, respectively. Using these cutoffs to define high versus low levels of VWF-N and VWF-A, we analyzed survival times using Kaplan-Meier survival curves (Figure 3B).
 |
Figure 3 Increased VWF processing was associated with increased risk of mortality. (A) VWF-N and VWF-A levels were increased in subjects that died within a two-year follow-up period (n=30) compared to survivors (n=910). (B) VWF biomarkers were dichotomized using AUROC analysis. Kaplan-Meier survival curves showed high biomarker levels associated with decreased survival time. Data presented as median + 95% CI. **p<0.01, ***p<0.0001. |
To address this further we investigated if the biomarkers were independently associated with mortality, considering possible confounding factors of mortality using cox proportional hazards regression. We identified age, 6 min walking distance (6 MWD), hsCRP, mMRC, prior hospitalizations and current smoking status as confounders to all-cause mortality (Table 2). Adjusting for these covariates we could define an adjusted hazard ratio of 1.214 per 1 SD increase in biomarker levels (95% CI 1.089; 1.352) for VWF-N (p<0.001) and 1.26 per 1 SD for VWF-A (95% CI 0.963; 1.652) (p=0.018) (Table 3).
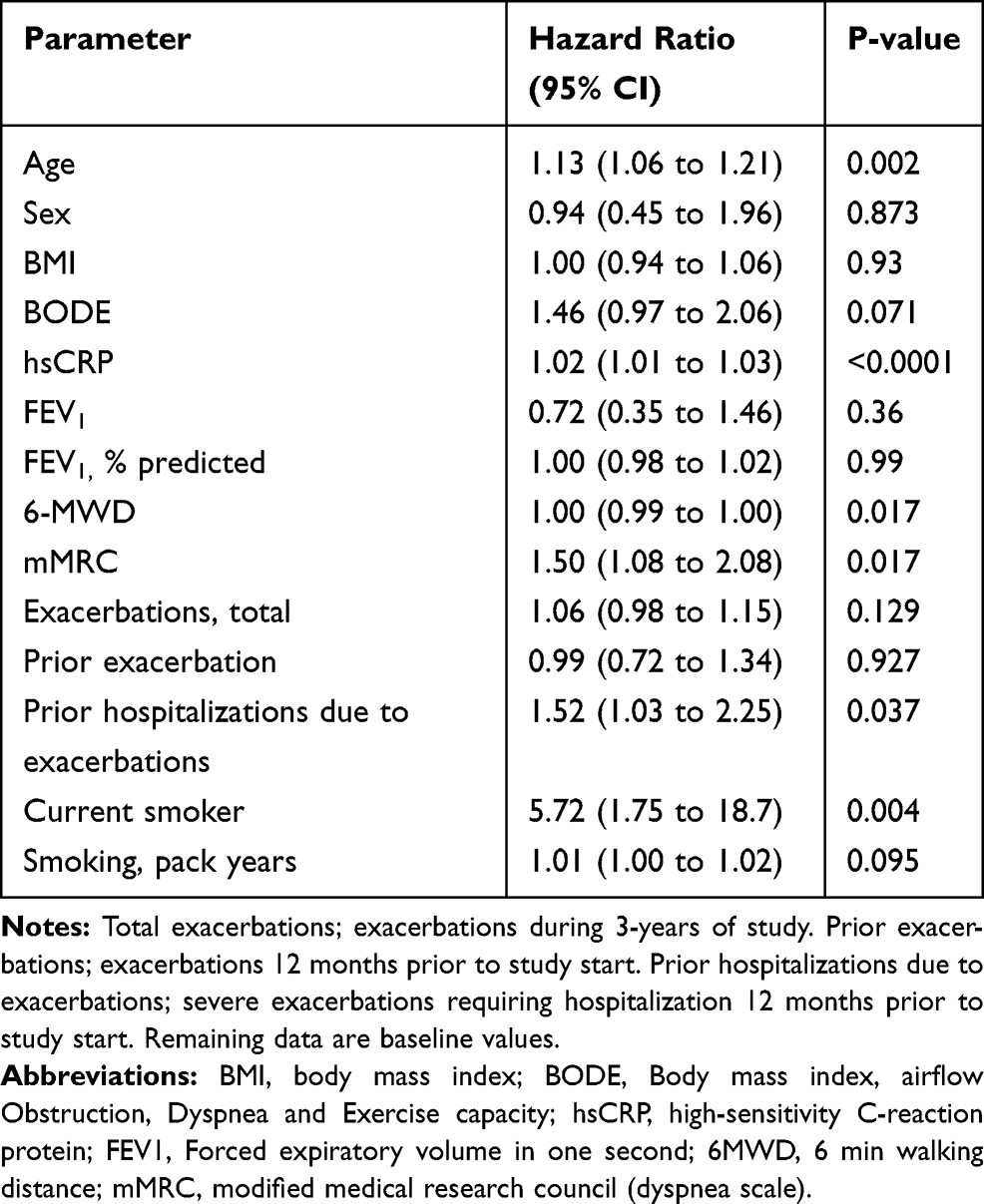 |
Table 2 Univariate Regression Analysis of Confounding Factors of Mortality in COPD Subjects |
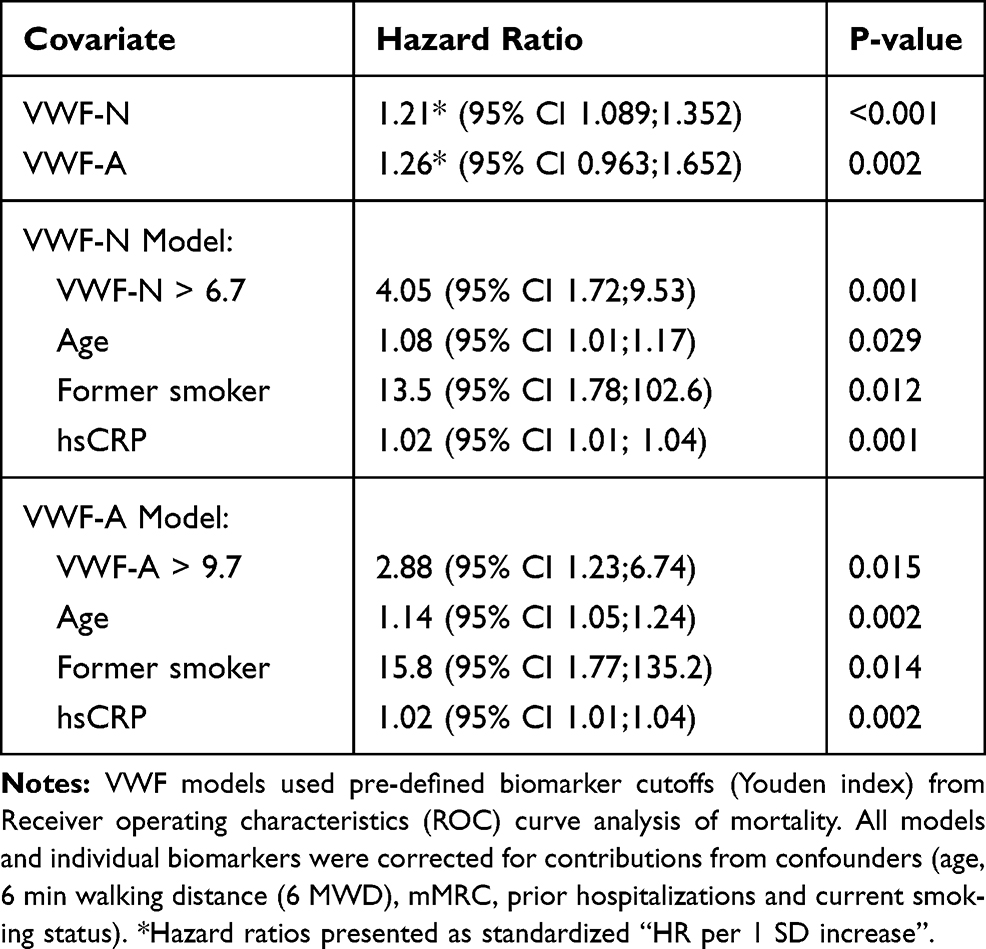 |
Table 3 Multivariate Analyses of VWF Biomarkers as Predicters of Mortality |
Evaluating the dichotomized biomarker data we could devise the best cox regression models including all confounders. The resulting models (adjusted for significant confounders) indicate that high levels of VWF-N (>6.7 ng/mL) and VWF-A (>9.7 ng/mL) were independently associated with mortality with an HR of 3.5 (95% CI 1.57; 7.79) and 2.64 (95% CI 1.18; 5.91), respectively (Table 3).
Discussion
In this study, we addressed the role of wound healing initiation in COPD pathology, and found that VWF processing is associated with emphysema and exacerbations and has prognostic value for assessing mortality risk. The pathophysiological changes that occur in COPD are not limited to the lungs, and the disease etiology is characterized by an increasing degree of systemic inflammation and endothelial damage,6,15,17 also evident from previous studies finding VWF to be increased in COPD.8 Since VWF processing is central to the primary response of wound healing, biomarkers measuring the dynamic processing of VWF could potentially aid in characterizing the pathophysiological changes in COPD. The activity of VWF can be investigated by several methods, for example by the use of ristocetin-dependent platelet aggregation,36 albeit this method measures only VWF-processing indirectly and has been reported to be less sensitive at low VWF levels.19 Here we apply the neo-epitope technology, specifically targeting ADAMTS13-cleavage fragments of activated VWF (VWF-A), together with newly endothelial released VWF (VWF-N).
Biomarker levels of VWF-N and VWF-A were increased in subjects with COPD compared to smoker controls. Cigarette smokers with normal lung function also experience pulmonary inflammation, but COPD subjects are even more affected by smoking which gives them an amplified inflammatory response7 that persists beyond smoking cessation.17 Surprisingly we did not observe any significant difference between COPD and non-smoker controls, although when combining the two control groups we did observe significantly lower levels of both markers. This could partially be due to the limited amount of very severe COPD participants (Table 1), although these results could also be influenced by the difference in age, gender and BMI between the groups, as hemostasis potential in general is changed with aging,16 including VWF.12 hsCRP data showed a significant difference between COPD and both control groups, indicating an overall increase in inflammatory state in COPD compared to controls. This could mask any potential disease-related change in VWF and therefore, these results indicate that the VWF-processing biomarkers are not to be considered stand-alone markers for disease diagnosis. Further studies should seek to elucidate this potential bias.
We speculated that wound healing would be initiated to a higher degree in symptomatic subjects which are known to have a higher risk of exacerbations and mortality.42 Both biomarkers could discriminate symptomatic from asymptomatic/mild COPD, underlining the increased damage response in symptomatic disease as seen from VWF-N levels, and suggesting ongoing active wound healing reflected in the increased VWF-A levels. This is in line with a previous study showing increased VWF activity in symptomatic subjects, but interestingly no change in total VWF was found in that study.8 We speculate that this is due to the endothelial release of VWF being a more dynamic process, while total VWF reflect a ratio between synthesis and degradation of VWF, which might not be significantly changed in symptomatic disease. Notably, no change was observed of either VWF biomarker when dividing the cohort into GOLD severity groups. Results similar to these have previously been reported,8 but could also be due to our cohort consisting of no mild COPD (GOLD stage I) and only a limited number of patients with very severe COPD (GOLD stage IV) (Table 1). Further studies should address this in a study representing the full range of COPD severities.
Interestingly, we found that only VWF-N reflected the more chronic progressive condition of emphysema, indicating a sustained increase of VWF release into the circulation. Emphysema is a result of chronic inflammatory responses that induce slow progressive epithelial tissue damage9 which is in agreement with VWF as a proposed marker of endothelial dysfunction.18 To our knowledge, no previous studies have shown VWF to be associated with emphysema. The VWF-N biomarker specifically reflects newly released VWF since it targets the C-terminal of the VWF pro-peptide, which is only accessible when released from the endothelia and thereby does not measure total circulating VWF, a characteristic of (total) VWF-antigen assays. The dynamic nature of this biomarker is also underlined by studies showing that half-life of the pro-peptide is significantly shorter than mature VWF (2 vs 12 hrs, respectively),10 thereby reflecting only recent or ongoing endothelial damage.
VWF-A showed to be associated with the more acute phenotype of exacerbations. This was also highlighted by the fact that VWF-A had a better association with symptomatic disease than VWF-N. Previous studies have found VWF-antigen but not VWF activity,8 to be increased in exacerbating patients.34,35 The VWF-A assay targets a neo-epitope within the A2-domain of VWF, which arises from ADAMTS13-cleavage which is specific for active VWF.28 This cleavage does not completely render VWF inactive, but rather decreases VWF multimeric size to regulate activity and dampens the coagulation response to limit the risk of thrombosis.13 As previous exacerbations are the best predictor for future exacerbations20 the ability to identify patients at risk using biomarkers like VWF-A might help prevent future events. It has been suggested that endothelial cell response is modulated when inflammation changes from acute to chronic,33 which might be reflected by VWF processing. We propose that chronic inflammation and endothelial damage in emphysema subjects induce a state of continuous VWF synthesis and release from endothelial cells, but with a lower rate of activation, while the acute damage which arises during exacerbations results in an increased activation of VWF.
We suspected that the degenerative nature of endothelial dysfunction and damage in COPD would be reflected in the increased airflow limitation (FEV1). Although we saw significantly decreased FEV1 levels in COPD (Table 1) this was not associated with poorer outcome in this sub-cohort (Table 2), which could indicate that airflow limitations might not be a universal tool for clinical assessment.1 The limited number of GOLD stage I and IV patients in our cohort (Table 1) could possibly limit the association between FEV1 and outcome. Interestingly, increased levels of VWF-N and VWF-A biomarkers were both associated with all-cause mortality and decreased survival time. We have recently compared biomarkers of fibrinogen turnover to fibrinogen measured in the ECLIPSE study.27 Plasma fibrinogen is currently the only FDA approved prognostic marker for all-cause mortality in COPD.26 Indeed, fibrinogen was found to be an independent predictor of mortality, comparable to the results obtained for the VWF biomarkers in this paper. We chose to compare the VWF biomarkers to hsCRP as both they all reflect the state of inflammation. Fibrinogen was excluded here although Fibrinogen and VWF are associated with similar processes of wound healing activation and platelet aggregation, since VWF can also independently lead to platelet aggregation and subsequent wound healing activation during high shear stress conditions known in COPD.5 We have discussed fibrinogen in relation to wound healing in this cohort more thoroughly elsewhere.27
To address the difference in age between the disease and control groups we included this parameter in our analyses. Interestingly, although age was found to be associated with mortality, both VWF-N and VWF-A were found to be independently associated with increased mortality risk, when adjusting for confounders, including age. This has previously been investigated in systemic inflammatory response syndrome where active VWF was found to correlate with mortality, while total VWF was not.21
Collectively, these data suggest that lung endothelial damage and initiation of the wound healing cascade in COPD are associated with an increased mortality risk. As COPD pathology manifests with tissue destruction and small airway fibrosis it is not believed that anti–inflammatory treatments will cure the disease, but early intervention might slow down or halt progression entirely.7 Dynamic biomarkers of VWF processing reflecting emphysema and exacerbation phenotypes might be a supplement to existing biomarkers to help identify patients with high risk of progression that could benefit from anti–inflammatory treatments.
A limitation of our study is the relatively low mortality rate (3%), hence a larger validation cohort is needed in order to properly evaluate the prognostic value and help define potential biomarker cut offs. A longitudinal study setup would also be favoured over a cross-sectional study to correlate progression and outcome with longitudinal biomarker measurements. The effect of age and BMI on VWF levels should also be addressed in future studies when addressing diagnostic potential of the biomarkers, although adjusting for age in our study supports the prognostic potential of the VWF biomarkers.
Conclusion
We demonstrate that differential processing of VWF is associated with different pathophysiological aspects of COPD, as we found that VWF-N reflected the chronic progressive condition of emphysema, while VWF-A was associated with acute exacerbations. These results generally support other studies investigating total VWF, but also propose a novel application for VWF biomarkers measuring dynamic changes in VWF processing as accurate and precise biochemical markers to characterize the heterogeneity of COPD.
Data Sharing Statement
The datasets of used and/or analysed patient and biomarker data are available upon reasonable request, and can be acquired from the corresponding author.
Ethics Approval and Participant Consent
The ECLIPSE study was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice Guidelines, and has been approved by the relevant ethics committees (see Supplementary Table 1). All participants provided written informed consent prior to the study.
Acknowledgments
The authors acknowledge all participants, medical, nursing, and technical staff involved in the ECLIPSE study. The study was sponsored by GlaxoSmithKline; the Danish Agency for Science, Technology and Innovation; and the Danish Research Foundation. The study sponsors did not place any restrictions with regard to statements made in the final paper.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
RTS, BEM, and JV are part of the Evaluation of COPD Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigators. LLL, SRR, JMBS, DJL, MAK and TMJ are fulltime employees of Nordic Bioscience A/S. BEM is a fulltime employee and shareholder of GlaxoSmithKline (GSK). RTS is a former employee and current shareholder of GSK. JV is supported by the National Institute of Health Research Manchester Biomedical Research Centre (NIHR Manchester BRC). JV reports personal fees from AstraZeneca, grants from Boehringer-Ingelheim, personal fees from Boehringer-Ingelheim, personal fees from Chiesi, personal fees from GSK, and personal fees from Novartis, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Agusti A, Calverley PMA, Celli B, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) investigators. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res. 2010;11:122. doi:10.1186/1465-9921-11-122
2. Alkady O, Fattouh M. Role of inflammatory biomarkers in COPD. In: 5.2 Monitoring Airway Disease. European Respiratory Society:PA1010. 2016. Available from: https://erj.ersjournals.com/content/48/suppl_60/PA1010 . Accessed March 04, 2020.
3. André P, Denis CV, Ware J, et al. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood. 2000;96:3322–3328. doi:10.1182/blood.V96.10.3322
4. Angelis N, Porpodis K, Zarogoulidis P, et al. Airway inflammation in chronic obstructive pulmonary disease. J Thorac Dis. 2014;6(Suppl 1):S167–72. doi:10.3978/j.issn.2072-1439.2014.03.07
5. Barak OF, Mladinov S, Hoiland RL, et al. Disturbed blood flow worsens endothelial dysfunction in moderate-severe chronic obstructive pulmonary disease. Sci Rep. 2017;7. doi:10.1038/s41598-017-17249-6.
6. Barnes PJ. Chronic obstructive pulmonary disease: effects beyond the lungs. PLoS Med. 2010;7:e1000220. doi:10.1371/journal.pmed.1000220
7. Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov. 2013;12:543–559. doi:10.1038/nrd4025
8. Bártholo TP, da Costa CH, Rufino R. Evaluation of von Willebrand factor in COPD patients. J Bras Pneumol. 2014;40:373–379. doi:10.1590/S1806-37132014000400004
9. Bihlet AR, Karsdal MA, Sand JMB, et al. Biomarkers of extracellular matrix turnover are associated with emphysema and eosinophilic-bronchitis in COPD. Respir Res. 2017;18:22. doi:10.1186/s12931-017-0509-x
10. Borchiellini A, Fijnvandraat K, Ten Cate JW, et al. Quantitative analysis of von Willebrand factor propeptide release in vivo: effect of experimental endotoxemia and administration of 1-deamino-8-D-arginine vasopressin in humans. Blood. 1996;88:2951–2958. doi:10.1182/blood.V88.8.2951.bloodjournal8882951
11. Cockayne DA, Cheng DT, Waschki B, et al. Systemic biomarkers of neutrophilic inflammation, tissue injury and repair in COPD patients with differing levels of disease severity. PLoS One. 2012;7:e38629. doi:10.1371/journal.pone.0038629
12. Coppola R, Mari D, Lattuada A, Franceschi C. Von Willebrand factor in Italian centenarians. Haematologica. 2003;88:39–43.
13. Crawley JTB, de Groot R, Xiang Y, Luken BM, Lane DA, De Groot R. Unravelling the scissile bond: how ADAMTS13 recognises and cleaves von Willebrand factor. Blood. 2011;118:3212–3221. doi:10.1182/blood-2011-02-306597
14. Donaldson G, Seemungal T, Bhowmik A, Wedzicha J. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. doi:10.1136/thorax.57.10.847
15. Fabbri LM, Rabe KF. From COPD to chronic systemic inflammatory syndrome? Lancet. 2007;370:797–799. doi:10.1016/S0140-6736(07)61383-X
16. Franchini M. Hemostasis and aging. Crit Rev Oncol Hematol. 2006;60:144–151. doi:10.1016/j.critrevonc.2006.06.004
17. Gan WQ, Man SFP, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59:574–580. doi:10.1136/thx.2003.019588
18. Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet (London, England). 2004;364:709–721. doi:10.1016/S0140-6736(04)16900-6
19. Hulstein JJJ, de Groot PG, Silence K, Veyradier A, Fijnheer R, Lenting PJ. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood. 2005;106:3035–3042. doi:10.1182/blood-2005-03-1153
20. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138. doi:10.1056/NEJMoa0909883
21. Hyseni A, Kemperman H, de Lange DW, Kesecioglu J, de Groot PG, Roest M. Active von Willebrand factor predicts 28-day mortality in patients with systemic inflammatory response syndrome. Blood. 2014;123:2153–2156. doi:10.1182/blood-2013-08-508093
22. Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15:1285–1294. doi:10.1111/jth.2017.15.issue-7
23. Lefrançais E, Ortiz-Muñoz G, Caudrillier A, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105–109. doi:10.1038/nature21706
24. Maclay JD, McAllister DA, Johnston S, et al. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax. 2011;66:769–774. doi:10.1136/thx.2010.157529
25. MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:
26. Mannino D, Tal-Singer R, Lomas D, et al. Plasma fibrinogen as a biomarker for mortality and hospitalized exacerbations in people with COPD. Chronic Obstr Pulm Dis J COPD Found. 2014;2:23–34. doi:10.15326/jcopdf.2.1.2014.0138
27. Manon-Jensen T, Langholm LL, Rønnow SR, et al. End-product of fibrinogen is elevated in emphysematous chronic obstructive pulmonary disease and is predictive of mortality in the ECLIPSE cohort. Respir Med. 2019;160:105814. doi:10.1016/j.rmed.2019.105814
28. Manon-Jensen T, Langholm LL, Sun S, et al. Initiation of the wound healing cascade in inflammatory bowel disease: assessment of Von Willebrand factor ADAMTS-13 processing and formation in crohn’s disease. EC Gastroenterol Dig Syst. 2019;6:143–154.
29. van Mourik JA, Romani de Wit T. Von Willebrand factor propeptide in vascular disorders. Thromb Haemost. 2001;86:164–171. doi:10.1055/s-0037-1616214
30. Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J Thromb Haemost. 2013;11:192–201. doi:10.1111/jth.12225
31. Perotin J-M, Adam D, Vella-Boucaud J, et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir Res. 2014;15:151. doi:10.1186/s12931-014-0151-9
32. Pimanda J, Hogg P. Control of von Willebrand factor multimer size and implications for disease. Blood Rev. 2002;16:185–192. doi:10.1016/S0268-960X(02)00017-6
33. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi:10.1038/nri2171
34. Polatli M, Çakir A, Cildag O, Bolaman AZ, Yenisey C, Yenicerioglu Y. Microalbuminuria, von Willebrand factor and fibrinogen levels as markers of the severity in COPD exacerbation. J Thromb Thrombolysis. 2008;26:97–102. doi:10.1007/s11239-007-0073-1
35. Polosa R, Malerba M, Cacciola RR, et al. Effect of acute exacerbations on circulating endothelial, clotting and fibrinolytic markers in COPD patients. Intern Emerg Med. 2013;8:567–574. doi:10.1007/s11739-011-0636-1
36. Sagheer S, Rodgers S, Yacoub O, Dauer R, Mcrae S, Duncan E. Comparison of von Willebrand factor (VWF) activity levels determined by HemosIL AcuStar assay and HemosIL LIA assay with ristocetin cofactor assay by aggregometry. Haemophilia. 2016;22:e200–e207. doi:10.1111/hae.2016.22.issue-3
37. Sand JMB, Knox AJ, Lange P, et al. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir Res. 2015;16:69. doi:10.1186/s12931-015-0225-3
38. Schneider SW, Nuschele S, Wixforth A, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci U S A. 2007;104:7899–7903. doi:10.1073/pnas.0608422104
39. Soler-Cataluña JJ, Martínez-García MÁ, Sánchez PR, Salcedo E, Navarro M, Ochando R. Disease patients with chronic obstructive pulmonary severe acute exacerbations and mortality in topic collections severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:925–931. doi:10.1136/thx.2005.040527
40. Vestbo J, Anderson W, Coxson HO, et al. Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE). Eur Respir J. 2008;31:869–873. doi:10.1183/09031936.00111707
41. Vestbo J, Edwards LD, Scanlon PD, et al.; ECLIPSE Investigators. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365:1184–1192. doi:10.1056/NEJMoa1105482
42. Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi:10.1164/rccm.201204-0596PP
43. Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–246. doi:10.1146/annurev.cb.06.110190.001245
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.