")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Increased expression of retinoic acid-induced gene 1 in the dorsolateral prefrontal cortex in schizophrenia, bipolar disorder, and major depression
Authors Haybaeck J, Postruznik M, Miller CL, Dulay JR, Llenos IC, Weis S
Received 10 August 2014
Accepted for publication 15 October 2014
Published 4 February 2015 Volume 2015:11 Pages 279—289
DOI https://doi.org/10.2147/NDT.S72536
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
Johannes Haybaeck,1 Magdalena Postruznik,1 Christine L Miller,2 Jeannette R Dulay,3 Ida C Llenos,3,4 Serge Weis3,4
1Department of Neuropathology, Institute of Pathology, Medical University Graz, Graz, Austria; 2Department of Pediatrics, Johns Hopkins University, Baltimore, 3Laboratory of Brain Research and Neuropathology, Departments of Psychiatry and Pathology, Uniformed Services University of the Health Sciences, and Stanley Medical Research Institute, Bethesda, MD, USA; 4Laboratory of Neuropathology, Department of Pathology and Neuropathology, State Neuropsychiatric Hospital Wagner-Jauregg, Medical School, Johannes Kepler University, Linz, Austria
Background: Retinoids regulate gene expression in different cells and tissues at the transcriptional level. Retinoic acid transcriptionally regulates downstream regulatory molecules, including enzymes, transcription factors, cytokines, and cytokine receptors. Animal models indicate an involvement of retinoid signaling pathways in the regulation of synaptic plasticity and learning, especially in the hippocampus. Retinoic acid-inducible or induced gene 1 (RAI-1) is induced during neuronal differentiation, and was associated with the severity of the phenotype and response to medication in schizophrenic patients.
Methods: In the present study, we used immunohistochemistry to investigate the expression of RAI-1 in 60 brains from the Stanley Neuropathology Consortium (15 cases each from controls and from patients with schizophrenia, bipolar disorder, and major depression). Rating scores for density and intensity were determined in the dorsolateral prefrontal cortex.
Results: All four groups showed high interindividual variation. RAI-1-positive cells were identified as neurons and astrocytes. Significantly increased intensities in cortical neurons were noted in all three major psychiatric groups compared with controls. The density of RAI-1-positive neurons was increased (P=0.06) in schizophrenia and bipolar disorder. In bipolar disorder, RAI-1-positive astrocytes in gray matter showed a significantly increased intensity and compound value. Thus, a significant increase in the parameters measured was found in schizophrenia, bipolar disorder, and major depression.
Conclusion: Our study shows a significant increase in expression of RAI-1 in the brains from patients with schizophrenia, bipolar disorder, or major depression. The increased expression might reflect altered signaling pathways, like that for retinoic acid. The underlying mechanisms leading to the increased expression and its functional consequences are so far unknown, and remain to be investigated in future studies.
Keywords: retinoic acid-inducible gene 1, dorsolateral prefrontal cortex, schizophrenia, bipolar disorder, major depression
Introduction
Schizophrenia, bipolar disorder, and major depression are complex, multigenetic, multifactorial diseases. They lead to disruptive psychopathologies involving thought, perception, behavior, emotion, cognition, movement, and mood.1,2 The possible underlying pathogenic mechanisms are not fully understood. The morphological changes seen in the brains of these patients constitute the platform upon which various pathogenic factors may act at different time points.3 The morphological changes include reduced volume in different brain regions, focally disturbed cytoarchitecture in various cortical areas, changed synaptic numbers and proteins, loss of myelin proteins, and a not yet fully understood reaction pattern of astrocytes and microglia (for review, see Soares and Gershon1). Large-scale microarray studies showed that a multitude of genes representing various pathways are upregulated or downregulated in these disorders.4,5 The etiology of these multifaceted changes remains obscure.
Retinoic acid-inducible gene 1 (RAI-1) is a protein, expression of which is modulated by retinoic acid (RA). Very little is known about its role in the human body; however, it occurs intracellularly and functions as a pattern recognition receptor by sensing viral double-stranded RNA, like in measles,6 influenza,7 or hepatitis B or C infection.8,9
Besides its roles as a virus sensor, RAI-1 plays an important role in tumor cell proliferation, where it is able to inhibit the proliferation of leukemia cells. It can indirectly lead to apoptosis in hepatoma and melanoma cells through pathways that are not yet fully understood. One mechanism could be the induction of interferon-β which has proapoptotic properties. Further, RAI-1 plays a crucial role in stress-related pathways, such as DNA damage.10
Joober et al demonstrated that the number of CAG repeats in STS GCT10D04 (Accession number G09710) was associated with the severity of the phenotype and the response to medication in schizophrenic patients.11 Database searches revealed this STS to be homologous to a part of the mouse retinoic acid inducible-1 gene (Rai1, Accession number D29801), which is induced by RA during neuronal differentiation of P19 embryonal carcinoma cells.12
A congenital disorder in which RAI-1 plays a crucial role is the Smith-Magenis syndrome (SMS). Patients show craniofacial and behavioral abnormalities such as brachydactyly, mental retardation, self-injurious behavior, and behavioral stereotypes. The underlying etiology in 70% of cases is a de novo deletion on chromosome 17 in 17p11.2, which encompasses about 3.7 Mb.13 Another factor that leads to an SMS-like phenotype is a haploinsufficiency of the RAI-1 gene. Vilboux et al found that patients with SMS-like features who show either the deletion on chromosome 17 or a de novo RAI-1 mutation have a lower mRNA level of RAI-1.14 In their study, decreased expression of RAI-1 led to ocular and behavioral abnormalities like polyembolokoilomania (insertion of foreign objects, usually into the ears). These findings match with other studies that showed RAI-1 mutations occurring in patients with SMS-features.15,16 Rai-1 knockout mouse models showed deficits in locomotor and learning abilities if the animals survived.17 Most of the knockout mice died in utero, indicating an important role of RAI-1 during fetal development.18
Carmona-Mora et al found that a mutation on RAI-1 can impair its function by prohibiting its translocation into the nucleus and therefore impairing its action as a transcription factor.19 As RAI-1 under normal circumstances induces expression of brain-derived neurotrophic factor,20 which plays a role in neurogenesis and neuroprotective actions, it could counteract the mental retardation in SMS.
RA is a metabolic product of vitamin A which is taken up through food and is stored in the liver as retinyl esters.21 RA leaves the cell nucleus towards the cytoplasm where it is catabolized by cytochrome P450 enzymes.21 The data suggest different physiological roles of RA during the life cycle in an individual as well as in various brain regions, particularly in areas with a high turnover of cell connections.22
RA signaling levels are highest in areas with high synaptic plasticity. Examples are the olfactory system and the limbic area, in particular the hippocampus. The receptors RARα, RARβ, and RARγ were reported to be present at different subcellular localizations in the hippocampus.23 Here, RA influences neurogenesis in the granular layer of the dentate gyrus, and can enhance synaptic strength by increasing postsynaptic glutamate receptor 1 expression. The RA-triggered increase in synaptic strength was reported to be inactivated by activity blockade-driven synaptic scaling.24 Interestingly, in this situation, RA acts at the translational but not at the transcriptional level.24
Moreover, RA is involved in the maintenance of the differentiated state of adult neurons. Destruction of RA signaling pathways in the adult leads to degeneration of motor neurons, development of Alzheimer’s disease,25–29 and perhaps to a certain extent, development of Parkinson’s disease.30,31
The data published so far regarding the possible functions of RA and consequently RAI-1 make the RA pathway a potential pathogenic candidate for severe mental disorders. In this work, we aimed to elucidate the immunohistochemical expression level and pattern of RAI-1 in the brains of patients with schizophrenia, bipolar disorder, or major depression.
Materials and methods
Materials
In the present study, the dorsolateral prefrontal cortex was investigated in 15 brains each from a control group and from patients with schizophrenia, bipolar disorder, or major depression. The brains are part of the Stanley Neuropathology Consortium Collection, details of which were reported elsewhere2 (Table 1). A public statement about the Stanley Brain Collection from January 2007 (updated August 2009) is available online (http://www.stanleyresearch.org).
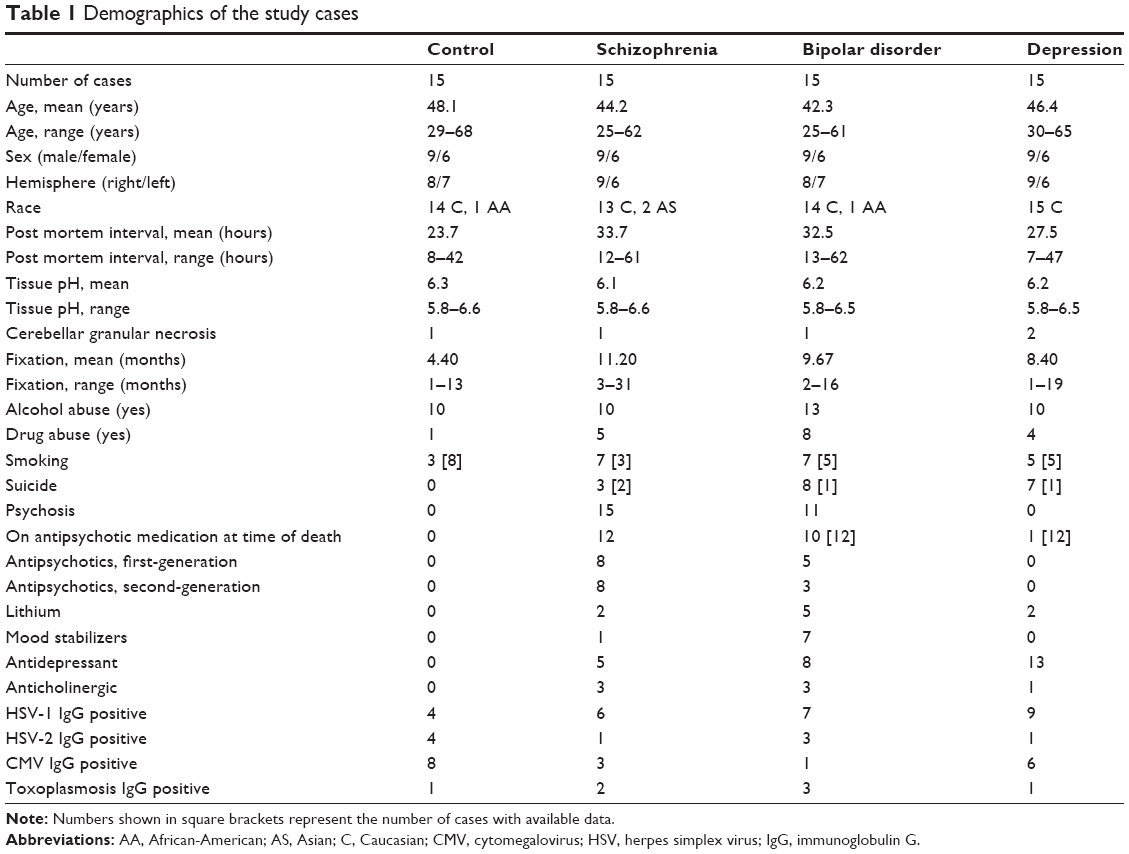 | Table 1 Demographics of the study cases |
Immunohistochemistry
Immunohistochemistry was performed on 10 μm thick frozen sections on Superfrost Plus slides (M6146-Plus, Allegiance, McGraw Park, IL, USA). Rehydrated sections underwent antigen retrieval using 2 mmol/L HCl for 20 minutes in a water bath at 95°–100°. All subsequent steps were carried out using the S3400 autostainer immunostaining system and EnVison™ + kit (code K4007, DakoCytomation, Carpinteria, CA, USA). Sections were treated with 3% H2O2 for 5 minutes to block endogenous peroxidase followed by a protein block for 5 minutes.3,32
Antibody generation
Polyclonal antibodies were generated to regions of the RAI-1. The program Protean (DNASTAR) was used to select optimal peptide epitopes 15–20 residues in size. Freeware NetPhos was employed to check for probability of phosphorylation and freeware COILS was employed to avoid coiled-coil regions. Three different antibodies were generated as follows.
A RAI-1 antibody (AB)-1 was generated by injecting rabbits with a peptide composed of a sequence found near epitope A near the N-terminus of RAI-1, ie, KQQNYQQTSQETSRLEC, with a C residue added at the carboxy terminus for adjuvant purposes. The region selected was predicted to be without phosphorylation and with a high probability of surface exposure.
A RAI-1 AB-2 was generated by injecting rabbits with a peptide composed of a sequence found in a central region with a low probability of phosphorylation, ie, TRAQKQPGHTNYSSYSK, with a C residue added at the carboxy terminus to increase antigenicity.
A RAI-1 AB-3 was generated by injecting rabbits with a peptide composed of a sequence found in a central region of RAI-1 with a low probability of phosphorylation, ie, GKEERPEGSPTLFKRM, with a C residue added at the carboxy terminus to increase antigenicity.
Polyclonal antibody generation and enzyme-linked immunosorbent assay measurements were carried out by GeneMed Synthesis (San Francisco, CA, USA). The enzyme-linked immunosorbent assay results showed significant peptide-specific reactions at serum dilutions of 1:1,000 and 1:10,000.
None of the antibodies worked on formalin-fixed, paraffin-embedded tissue; AB3 gave reliable results when used on frozen tissue. The AB-3 was used at a concentration of 1:25 for 60 minutes. Sections were incubated with the secondary anti-rabbit antibody for 30 minutes. The reaction product was visualized using diaminobenzidine chromogen (liquid DAB+, K3468, DakoCytomation) for 5 minutes. The sections were then counterstained with Gill 2 hematoxylin (Richard-Allan Scientific, Kalamazoo, MI, USA). As a negative control, the primary antibody was omitted and replaced by normal rabbit serum (code X0903, DakoCytomation).
Evaluation of immunohistochemical stains
On each immunohistochemically stained section, neurons and glial cells were analyzed separately for gray and white matter.3,32 For each cell type, the staining intensity was score-rated as follows: (0) no staining, (1) weak staining, (2) moderate staining, and (3) strong staining. The density (% of positive cells relative to the density of the total population of that specific cell type) of stained cells was rated as follows: (0) no cell stained, (1) low density (<25%), (2) moderate density (25%–50%), and (3) high density (>50%). The histological analysis was performed by an experienced neuropathologist blinded to the diagnosis. For each single analyzed cell type (ie, neurons and glial cells), compound values were calculated by multiplying the staining intensity score by the density score. Finally, sums were calculated for gray matter by adding the compound values of neurons and glial cells, and for the total brain by adding the compound values of neurons and glial cells in gray matter and of glial cells in white matter.3,32
Statistical analysis
Differences between the control and diagnostic groups were calculated using the non-parametric Kolmogorov-Smirnov test for overall differences and the Mann–Whitney U-test for post hoc testing between the various groups.
Confounding variables, grouped according to their scaling properties, were used to assess their influence on the dependent variables. These included age, sex, side of the hemisphere examined, suicide status, smoking at time of death, post mortem interval, brain pH, cerebellar granular cell layer necrosis, rapidity of death, lifetime antipsychotic intake (in fluphenazine mg equivalents), data on intake of first-generation and second-generation generic antipsychotic drugs, generic mood stabilizer drugs, lithium, generic antidepressant drugs, and generic anticholinergic drugs.
At first, we assessed if there was a significant difference for the confounding variables between the various diagnostic groups either by analysis of variance (ANOVA) or logistic regression. As a second step, to determine if the effect of a confounding variable could account for the differences we obtained in our immunohistochemistry, significant differences between disease groups and measured dependent variables as well as confounders and measured dependent variables were compared. In the event that a confounding variable was significant in step one, it was checked if this variable also showed a significant change in step two. Only confounding variables that significantly influenced all measured variables were added to the ANOVA/regression model to remove the confounding effect on the disease/outcome relationship. For example, if fixation time was different between the studied groups, fixation time would only be added to the ANOVA/regression model if we observed a significant effect on staining density and intensity in neurons and glial cells. In other words, we did not include confounding variables in our ANOVA/regression model if they did not contribute to the observed differences. A critical value of α=0.05 was used for all analyses.
Results
Immunohistochemistry
RAI-1 immunoreactivities were seen in neurons and astroglial cells (Figures 1 and 2). Astroglial cells were identified based on their histological characteristics, ie, a large, pale nucleus and scant cytoplasm. Double-labeling immunohistochemistry was not done as only a minor percentage of astrocytes stain with glial fibrillary acidic protein.33 Neurons cannot be reliably stained with antibodies against NeuN in the autopsy setting.
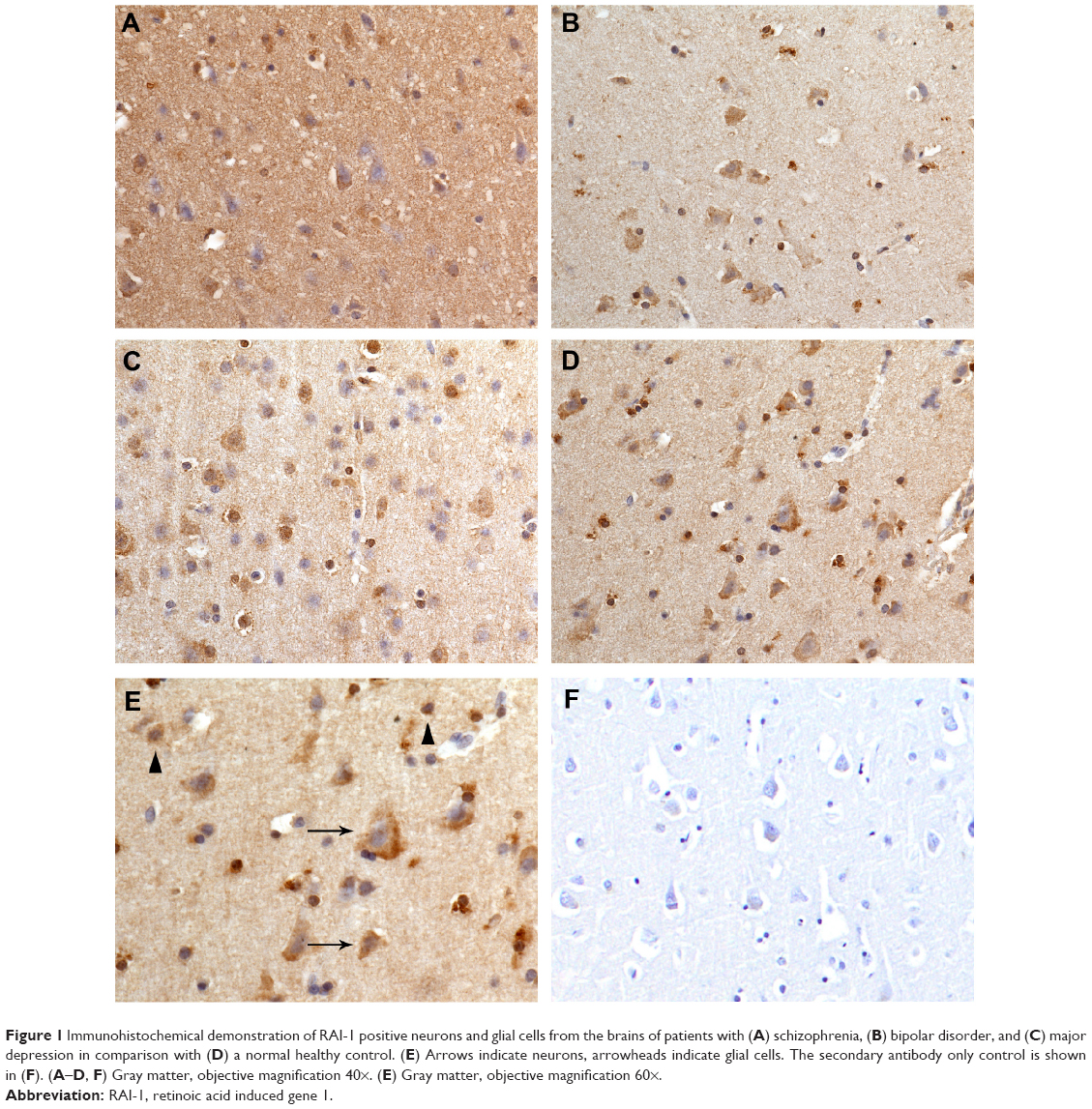 | Figure 1 Immunohistochemical demonstration of RAI-1 positive neurons and glial cells from the brains of patients with (A) schizophrenia, (B) bipolar disorder, and (C) major depression in comparison with (D) a normal healthy control. (E) Arrows indicate neurons, arrowheads indicate glial cells. The secondary antibody only control is shown in (F). (A–D, F) Gray matter, objective magnification 40×. (E) Gray matter, objective magnification 60×. |
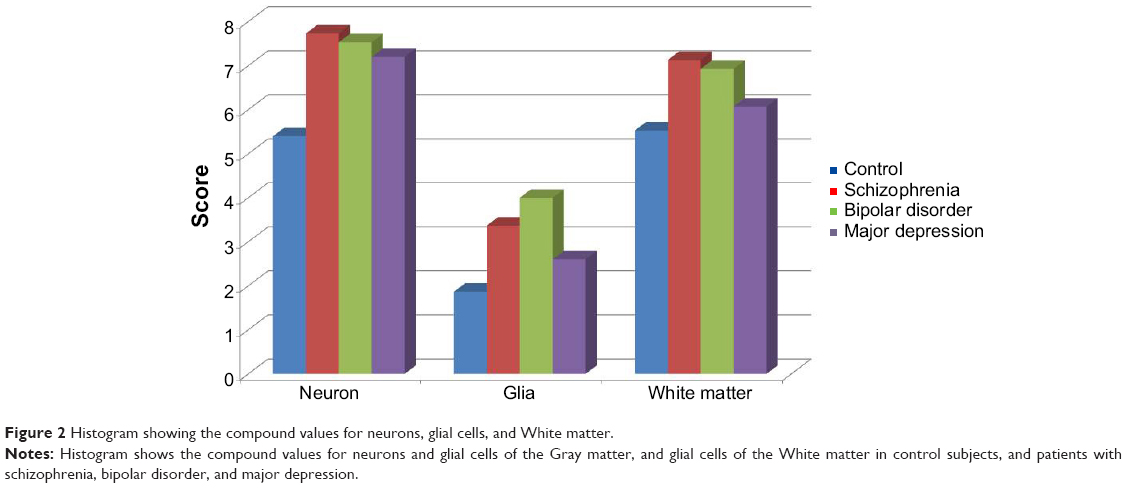 | Figure 2 Histogram showing the compound values for neurons, glial cells, and White matter. |
Confounding variables
The following confounding variables differed significantly compared with controls: post mortem time in schizophrenia; time between death and body refrigeration, suicide, and heavy drug abuse in schizophrenia, bipolar disorder and depression; rapidity of death, being on drugs of abuse, cocaine abuse, and heavy alcohol abuse in bipolar disorder.
Rating scales
Using rating scores and compound values, the following results were obtained. All four groups showed a high interindividual variation (Table 2). Significantly increased intensities of RAI-1 protein expression were noted in cortical neurons from all three major psychiatric groups compared with controls. The density of RAI-1-positive neurons was increased (P=0.06) in schizophrenia and bipolar disorder (P=0.06). RAI-1-positive glial cells in gray matter showed a significantly increased intensity in bipolar disorder. The compound values for the cortex were significantly increased in schizophrenia, bipolar disorder, and major depression (P=0.01, P=0.00, and P=0.04 respectively). No significant changes were noted for the white matter.
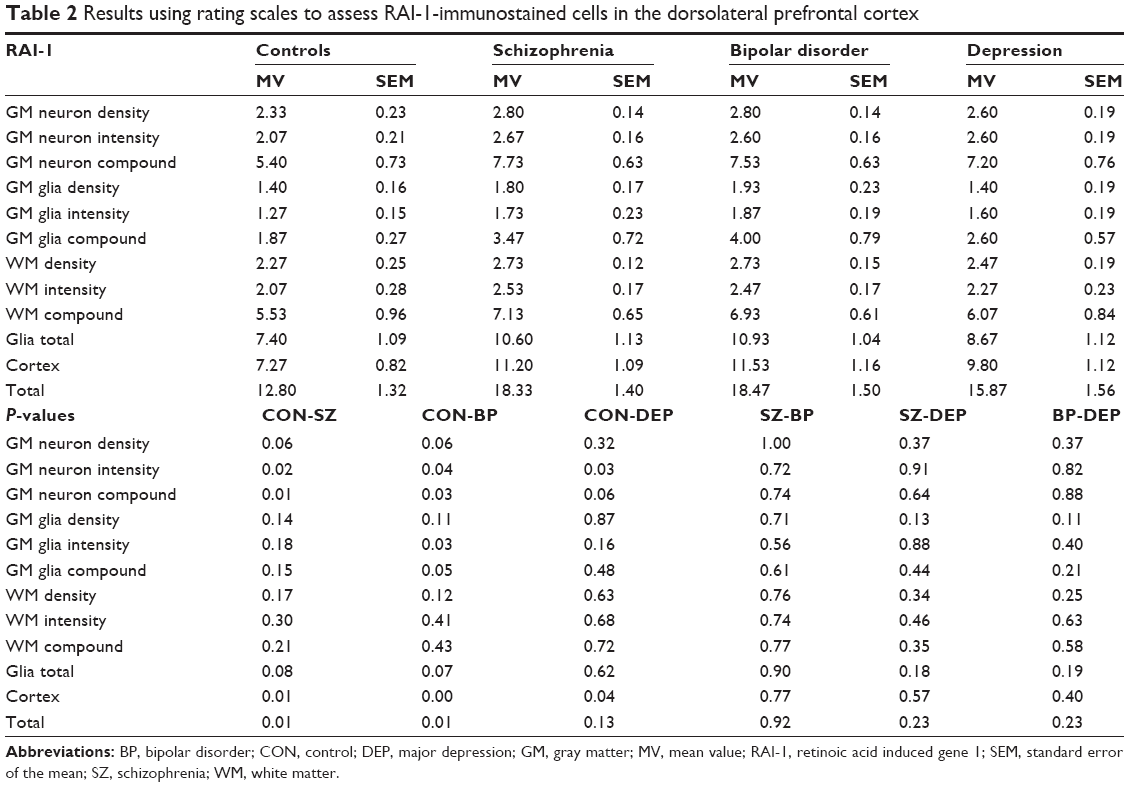 | Table 2 Results using rating scales to assess RAI-1-immunostained cells in the dorsolateral prefrontal cortex |
Effect of confounding variables
Post mortem time as well as time between death and body refrigeration had no effect on any variable assessed in RAI-positive cells from each diagnostic group. The effect of other confounding variables is shown in Table 3. For the whole group, granular cell layer necrosis of the cerebellum (assessed on the formalin-fixed, paraffin-embedded cerebellar tissue of the contralateral side) was associated with a significant increase in gray matter glial density (P=0.03), gray matter glial compound (P=0.05), and glia total (P=0.02); in brains of persons with alcohol intake, white matter glial density was significantly increased (P=0.02). However, when analyzing these variables for each diagnostic group separately, there were no significant changes.
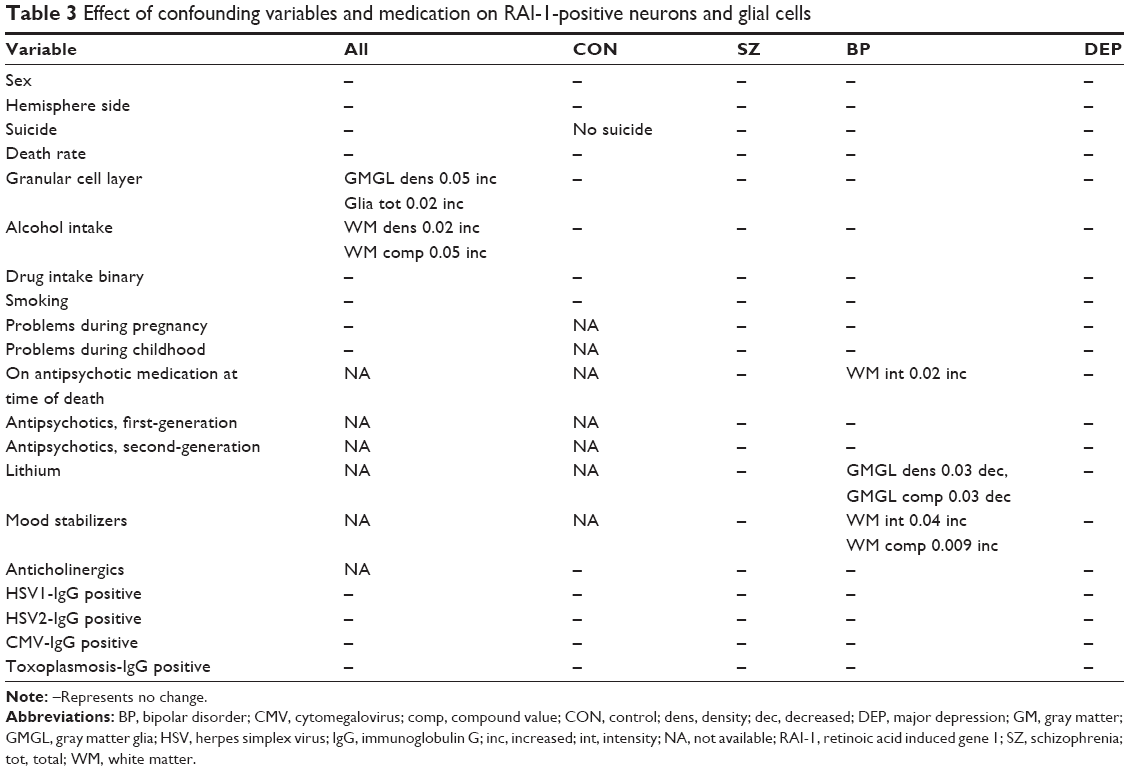 | Table 3 Effect of confounding variables and medication on RAI-1-positive neurons and glial cells |
Effect of medication
The effect of medication is shown in Table 3. The intake of lithium in the bipolar group led to a significant decrease in gray matter glial RAI-1 density and gray matter glial compound value. Within the same diagnostic group of bipolar patients, intake of mood stabilizers induced an increased expression of RAI-1, reflected as an increased intensity in white matter glial cells and white matter compound value. In bipolar patients, being on antipsychotic medication at the time of death resulted in an increased intensity of RAI-1 in white matter glial cells.
Discussion
In the present study, immunoreactivity for RAI-1 was seen in neurons and astroglial cells from brains of patients with schizophrenia, bipolar disorder, or major depression. The major changes noted were a significantly increased intensity, ie, increased expression, of RAI-1 in cortical neurons in the three disorders examined and in cortical astroglial cells in bipolar disorder, as well as an increased density of RAI-1-positive neurons in schizophrenia and bipolar disorder. A high interindividual variability within each group and a high intergroup variability of RAI-1-immunoreactive neurons and astroglial cells were noted. Although part of this variability can be explained by confounding variables (ie, medication), the factors regulating low and high expression of RAI-1 in brain cells have still to be elucidated.
RAI-1 in schizophrenia and bipolar disorder
The increased expression of RAI-1 protein reported here is likely a reflection of an increased tone of retinoic acid in schizophrenia and bipolar disorder and, therefore, is likely associated with concomitant effects on the development of dopaminergic neurons. Although expression in the adult brain is not necessarily illustrative of the retinoid status during development, adult RAI-1 expression does have relevance to any ongoing commitment of stem cells to a dopaminergic path and is certainly of relevance to dopaminergic cell maintenance potentially leading to a build-up of cytotoxic compounds.
The human RAI-1 gene is very similar to its mouse ortholog, both in DNA and protein sequences and in expression patterns. If we take into consideration that a polyglutamine polymorphism of a homolog of RAI-1 is associated with schizophrenia and that it is highly expressed in neuronal brain regions, RAI-1 protein might be an important modulator of susceptibility to schizophrenia by influencing some aspects of neuron differentiation or function. RAI-1 is likely to function as a transcription factor.15 It is about 50% homologous to TCF20 (another transcription cofactor), and like many other proteins that can induce gene expression, it has a polyglutamine stretch that modulates activation of transcription.34 In 2005, a study showed that mouse RAI-1 protein has moderate transcription activity in HeLa cells.18 In 2010, a study assessed the activity of RAI-1 in Neuro-2a cells and found that it has much stronger transactivation activity than that in HeLa cells.35
Another study reported that particular genes are dysregulated in haploinsufficient HEK293T (human embryonic kidney) cells. The genes that were most significantly upregulated included SCN12A, PSEN2, ZIC1, RXRβ, and CLN8. These genes are involved in sensory transmission, nociceptive behavior, Alzheimer’s disease, neurogenesis, neurological function, epilepsy, and mental retardation. The genes downregulated were NF1, MLL, and NRID2, which are involved in neurofibromatosis type I, schizophrenia, circadian activity, and transcription.36 Haploinsufficiency of Rai-1 in mice (Rai-1) and in humans (RAI-1) leads to downregulation of brain-derived neurotrophic factor,20 which is involved in neuronal maintenance and regulation of cell survival, differentiation, and growth during development.16 Since haploinsufficiency may trigger schizophrenia-like conditions and RAI-1 represents a growth-promoting factor through induction of transcription of brain-derived neurotrophic factor, a clinical potential for RAI-1 might be foreseen.20 Taking into consideration the reported increase in RAI-1 expression, one might assume that RAI is involved in the pathogenesis of major psychiatric disorders and could also possibly serve as potential therapeutic target.
RA induces the differentiation of various types of neurons and glia by activating the transcription of different families of genes.37–39 A role for all-trans retinoic acids (ATRA) in the physiological function of the hippocampus is suggested by the presence of components of retinoid signaling pathways.40 RARα and RXR-α, RXR-β, and RXR-γ mRNA transcripts were seen in the hippocampus.23,41 As the hippocampus is a center for memory and learning, retinoid signaling might show an effect on these coordinated processes. Obviously, retinoid signaling has a physiological role in synaptic plasticity, learning, and memory. Vitamin A deprivation in adult mice and rats highlights the importance of adequate vitamin A status for such cognitive functions; however, the precise targets of the retinoid signaling pathways underlying these altered behaviors remain to be identified.
RA pathway in schizophrenia and bipolar disorder
Schizophrenia and bipolar disorder are complex diseases, with multiple neurotransmitter systems being involved. There is evidence that altered dopaminergic function underlies the psychotic aspects of these disorders, as drugs that increase dopamine at the synapse exacerbate positive symptoms (reviewed by Seeman42 and Davis et al43). Although retinoid analogs have been proposed as possible therapeutic drugs for schizophrenia,44,45 it is not yet clear that the multifaceted effects of retinoic acid agonists or antagonists on the dopaminergic system could reliably be expected to be therapeutic. Thus, retinoic acid is involved in directing stem cells to form dopaminergic neurons46–48 and increases the expression of dopaminergic D2R receptors;49 however, it can also exert negative effects on the integrity of dopaminergic neurons in the substantia nigra and ventral tegmental areas.
Although the mechanism of action of antipsychotics is not yet clearly understood, both the typical and atypical antipsychotic drugs antagonize dopamine D2 receptors (reviewed by Davis et al43), an effect which at a minimum contributes to side effects such as blunting of affect. Retinoid therapy that would increase expression of D2 receptors might therefore be expected to help minimize the D2-related effects of antipsychotics. Of note, chronic treatment of adult rats with haloperidol, but not clozapine, resulted in a small but significant increase in abundance of mRNA for RARβ and RXRγ, the predominant isoforms expressed in the striatum; however, increased mRNA abundance does not always translate into increased protein expression.28,29 The fact that the effect was limited in degree and restricted to haloperidol is consistent with antipsychotics not being a major confounder of RAI-1 expression data.
RA in depression
An increase in depressive episodes through hypervitaminosis A is well documented.50,51 Potential targets for retinoid signaling in depression include dopaminergic, serotonergic, and noradrenergic pathways or a complex interaction between these neurotransmitter systems. ATRA have been reported to regulate the differentiation of serotonergic neurons in mouse neural crest cells, to promote the proliferation of these neurons,52 and to induce the expression of 5-HT1A receptors in neuronal cells.53 Interestingly, whereas ATRA have been shown to promote adult neurogenesis in vitro,54,55 chronic administration of 13-cis-RA in vivo results in a significant decrease in hippocampal neurogenesis in adults.56
Maret et al57 showed that retinoids play a role in synchronizing cortical activity, whereby RARβ determines the contribution of delta oscillations to the sleep electroencephalogram. Desynchronization of cortical activity is a conspicuous feature in schizophrenia58 and autism.59 In Spencer et al, patients described the following changes: absence of the posterior component of the early visual gamma band response to Gestalt stimuli; abnormalities in the topography, latency, and frequency of the anterior component of this response; delayed onset of phase coherence changes; and decreased pattern of interhemispheric coherence.58 Thus, a failure of gamma band synchronization, especially in the 40 Hz range, is present in schizophrenia. RA signaling, which is involved in the patterning of the brain and dopaminergic pathways, regulates cortical synchrony in the adult.
Given the large number of neuronal genes that could potentially be transcriptionally regulated by retinoids in the adult brain, surprisingly little is known about the impact of ATRA signaling on other brain functions. The use of transgenic mouse models has indicated a role for retinoids in the regulation of striatal dopaminergic function and the control of locomotor activity. However, such models do not clearly delineate between the well described developmental effects of retinoids and any novel adult-specific effects.
Relevance of RA to other CNS disorders
RA pathway in Alzheimer’s disease
Retinoid signaling has been implicated in the pathology of Alzheimer’s disease.60 Application of ATRA to cells upregulates amyloid precursor protein (APP) mRNA61,62 which is likely mediated by a direct effect on a RARE in the APP promoter. Amyloid is the central component of senile plaques, and these plaques may be generated as a result of overproduction or disturbed metabolism of APP,63 or due to differential expression of APP mRNA transcripts.64 Ono et al65 showed that vitamin A had antiamyloidogenic and destabilizing effects on fibril formation in vitro.65 This effect was independent of any changes in gene expression and most likely resulted from the antioxidant properties of retinoids.65 Divergent results by Rinaldi et al66 reported that plasma vitamin A levels are reduced in patients suffering from Alzheimer’s disease, whereas Connor and Sidell67 have shown that hippocampal retinoid content is similar in Alzheimer’s disease and control groups. Corocan et al68 reported a decreased abundance of RARα and RALDH2 in brains with Alzheimer’s disease, suggesting that RA signaling is likely to be compromised in patients.
RA pathway in Parkinson’s disease
Parkinson’s disease is characterized by loss of dopaminergic neurons in the substantia nigra and intracytoplasmic accumulation of Lewy bodies, with alpha synuclein being the most abundant component of Lewy bodies. The RA pathway might play a role in the pathogenesis of Parkinson’s disease, but also provides a platform for possible treatment approaches.
RA is required for the establishment of proper mesodiencephalic dopaminergic neuronal identity.48 Early and specific exposure to retinoic acid improved the regional identity of neural progenitor cells derived from human embryonic stem cells, Parkinson’s disease, or healthy subject-specific induced pluripotent stem cells.69 ATRA induce differentiation of cord blood-derived multipotent stem cells into dopaminergic neurons, thus opening up new ways for alternative therapy for Parkinson’s disease.70
Embryonic stem cells treated by retinoic acid and transplanted into Parkinsonian rats relieved apomorphine-induced asymmetric motor behavior; the transplanted cells showed tyrosine hydroxylase immunoreactivity and displayed neuronal and glial features.71 Ulusoy et al72 showed that pioglitazone and retinoic acid have some beneficial effects in a rotenone-induced model of Parkinson’s disease in rats whereby pioglitazone seemed to be more effective than retinoic acid. Pioglitazone, but not retinoic acid, significantly reversed the reduced striatal dopamine level. Data from Yin et al73 suggested that early post-treatment with 9-cis-retinoic acid has a protective effect against neurodegeneration in nigrostriatal dopaminergic neurons in an animal model of Parkinson’s disease.
RA promotes differentiation and α-synuclein oligomer formation in neuroblastoma cells.74 Addition of retinoic acid to SHSY-5Y cells75 and 3D5-transfected cells76 leads to formation of alpha-synuclein oligomers and promotes assembly of alpha-synuclein aggregates, respectively.
Conclusion
Retinoids, retinoid agonists and antagonists offer some potential as therapeutics in schizophrenia, depression, Alzheimer’s disease, and possibly even Parkinson’s disease. In the present study, we observed by immunohistochemistry a significant increase in expression of RAI-1 in the brains of patients with schizophrenia, bipolar disorder, or major depression. One might assume that RAI-1 is increased due to its modulatory function in neuronal maintenance, altered signaling pathways, and cell survival. The factors leading to increased RAI-1 expression are still not known.
Acknowledgments
The authors thank Theodore and Vada Stanley, as well as E Fuller Torrey, for their generous support.
Disclosure
The authors report no conflicts of interest in this work.
References
Soares JC, Gershon S. The diagnostic boundaries of bipolar disorder. Bipolar Disord. 2000;2:1–2. | ||
Torrey EF, Webster M, Knable M, Johnston N, Yolken RH. The Stanley Foundation Brain Collection and Neuropathology Consortium. Schizophr Res. 2000;44:151–155. | ||
Weis S, Llenos IC, Dulay JR, Verma N, Sabunciyan S, Yolken RH. Changes in region- and cell type-specific expression patterns of neutral amino acid transporter 1 (ASCT-1) in the anterior cingulate cortex and hippocampus in schizophrenia, bipolar disorder and major depression. J Neural Transm. 2007;114:261–271. | ||
Tang B, Thornton-Wells T, Askland KD. Comparative linkage meta-analysis reveals regionally-distinct, disparate genetic architectures: application to bipolar disorder and schizophrenia. PLoS One. 2011;6:e19073. | ||
Wang KS, Liu XF, Aragam N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr Res. 2010;124:192–199. | ||
Clifford HD, Yerkovich ST, Khoo SK, et al. TLR3 and RIG-I gene variants: associations with functional effects on receptor expression and responses to measles virus and vaccine in vaccinated infants. Hum Immunol. 2012;73:677–685. | ||
Mi Z, Ma Y, Tong Y. Avian influenza virus H5N1 induces rapid interferon-beta production but shows more potent inhibition to retinoic acid-inducible gene I expression than H1N1 in vitro. Virol J. 2012;9:145. | ||
Wang Y, Ye L, Wang X, Li J, Song L, Ho W. Retinoic acid inducible gene-I (RIG-I) signaling of hepatic stellate cells inhibits hepatitis C virus replication in hepatocytes. Innate Immun. 2013;19:193–202. | ||
Zhao G, An B, Zhou H, et al. Impairment of the retinoic acid-inducible gene-I-IFN-beta signaling pathway in chronic hepatitis B virus infection. Int J Mol Med. 2012;30:1498–1504. | ||
Liu F, Gu J. Retinoic acid inducible gene-I, more than a virus sensor. Protein Cell. 2011;2:351–357. | ||
Joober R, Benkelfat C, Toulouse A, et al. Analysis of 14 CAG repeat-containing genes in schizophrenia. Am J Med Genet. 1999;88: 694–699. | ||
Toulouse A, Rochefort D, Roussel J, Joober R, Rouleau GA. Molecular cloning and characterization of human RAI1, a gene associated with schizophrenia. Genomics. 2003;82:162–171. | ||
Juyal RC, Figuera LE, Hauge X, et al. Molecular analyses of 17p11.2 deletions in 62 Smith-Magenis syndrome patients. Am J Hum Genet. 1996;58:998–1007. | ||
Vilboux T, Ciccone C, Blancato JK, et al. Molecular analysis of the retinoic acid induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS One. 2011;6: e22861. | ||
Bi W, Saifi GM, Shaw CJ, et al. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet. 2004;115:515–524. | ||
Carmona-Mora P, Walz K. Retinoic acid induced 1, RAI1: a dosage sensitive gene related to neurobehavioral alterations including autistic behavior. Curr Genomics. 2010;11:607–617. | ||
Bi W, Yan J, Shi X, et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum Mol Genet. 2007;16:1802–1813. | ||
Bi W, Ohyama T, Nakamura H, et al. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith-Magenis syndrome. Hum Mol Genet. 2005;14:983–995. | ||
Carmona-Mora P, Canales CP, Cao L, et al. RAI1 transcription factor activity is impaired in mutants associated with Smith-Magenis syndrome. PLoS One. 2012;7:e45155. | ||
Burns B, Schmidt K, Williams SR, Kim S, Girirajan S, Elsea SH. Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum Mol Genet. 2010;19: 4026–4042. | ||
Maden M. Retinoid signalling in the development of the central nervous system. Nat Rev Neurosci. 2002;3:843–853. | ||
Nomoto M, Takeda Y, Uchida S, et al. Dysfunction of the RAR/RXR signaling pathway in the forebrain impairs hippocampal memory and synaptic plasticity. Mol Brain. 2012;5:8. | ||
Fragoso YD, Shearer KD, Sementilli A, de Carvalho LV, McCaffery PJ. High expression of retinoic acid receptors and synthetic enzymes in the human hippocampus. Brain Struct Funct. 2012;217:473–483. | ||
Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60: 308–320. | ||
Miller CL, Murakami P, Ruczinski I, et al. Two complex genotypes relevant to the kynurenine pathway and melanotropin function show association with schizophrenia and bipolar disorder. Schizophr Res. S2009;113:259–267. | ||
Severinsen JE, Als TD, Binderup H, et al. Association analyses suggest GPR24 as a shared susceptibility gene for bipolar affective disorder and schizophrenia. “http://www.ncbi.nlm.nih.gov/pubmed?term=Association+analyses+suggest+GPR24+as+a+shared+susceptibility+gene+for+bipolar+affective+disorder+and+schizophrenia&TransSchema=title&cmd=detailssearch”\o “American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics.” Am J Med Genet B Neuropsychiatr Genet. 2006;141B:524–533. | ||
Dempster EL, Pidsley R, Schalkwyk LC, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. | ||
Miller CL, Dulay JR. The high-affinity niacin receptor HM74A is decreased in the anterior cingulate cortex of individuals with schizophrenia. Brain Res Bull. 2008;77:33–41. | ||
Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry. 2006;11: 737–747. | ||
de Oliveira MR, Silvestrin RB, Mello e Souza T, Moreira JC. Therapeutic vitamin A doses increase the levels of markers of oxidative insult in substantia nigra and decrease locomotory and exploratory activity in rats after acute and chronic supplementation. Neurochem Res. 2008;33: 378–383. | ||
Bremner JD, Shearer KD, McCaffery PJ. Retinoic acid and affective disorders: the evidence for an association. J Clin Psychiatry. 2012;73: 37–50. | ||
Weis S, Llenos IC, Sabunciyan S, et al. Reduced expression of human endogenous retrovirus (HERV)-W GAG protein in the cingulate gyrus and hippocampus in schizophrenia, bipolar disorder, and depression. J Neural Transm. 2007;114:645–655. | ||
Weis S, Haug H, Budka H. Astroglial changes in the cerebral cortex of AIDS brains: a morphometric and immunohistochemical investigation. Neuropathol Appl Neurobiol. 1993;19:329–335. | ||
Gerber HP, Seipel K, Georgiev O, et al. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science. 1994;263:808–811. | ||
Carmona-Mora P, Encina CA, Canales CP, et al. Functional and cellular characterization of human retinoic acid induced 1 (RAI1) mutations associated with Smith-Magenis syndrome. BMC Mol Biol. 2010; 11:63. | ||
Girirajan S, Truong HT, Blanchard CL, Elsea SH. A functional network module for Smith-Magenis syndrome. Clin Genet. 2009;75:364–374. | ||
Andrews PW. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol. 1984;103: 285–293. | ||
Kuff EL, Fewell JW. Induction of neural-like cells and acetylcholinesterase activity in cultures of F9 teratocarcinoma treated with retinoic acid and dibutyryl cyclic adenosine monophosphate. Dev Biol. 1980;77: 103–115. | ||
Maden M. Role and distribution of retinoic acid during CNS development. Int Rev Cytol. 2001;209:1–77. | ||
Wagner E, Luo T, Drager UC. Retinoic acid synthesis in the postnatal mouse brain marks distinct developmental stages and functional systems. Cereb Cortex. 2002;12:1244–1253. | ||
Zetterstrom RH, Lindqvist E, Mata de Urquiza A, et al. Role of retinoids in the CNS: differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. Eur J Neurosci. 1999;11:407–416. | ||
Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1:133–152. | ||
Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:474–1486. | ||
Goodman AB. Three independent lines of evidence suggest retinoids as causal to schizophrenia. Proc Natl Acad Sci U S A. 1998;95:7240–7244. | ||
Citver AS, Shields AM, Ciaccia LM, Schulingkamp RJ, Raffa RB. Indirect modulation of dopamine D2 receptors as potential pharmacotherapy for schizophrenia: III. Retinoids. J Clin Pharm Ther. 2002;27:161–168. | ||
Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci. 1999;19:10–20. | ||
Castro DS, Hermanson E, Joseph B, et al. Induction of cell cycle arrest and morphological differentiation by Nurr1 and retinoids in dopamine MN9D cells. J Biol Chem. 2001;276:43277–43284. | ||
Jacobs FM, Smits SM, Noorlander CW, et al. Retinoic acid counteracts developmental defects in the substantia nigra caused by Pitx3 deficiency. Development. 2007;134:2673–2684. | ||
Samad TA, Krezel W, Chambon P, Borrelli E. Regulation of dopaminergic pathways by retinoids: activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family. Proc Natl Acad Sci U S A. 1997;94:14349–14354. | ||
Restak RM. Pseudotumor cerebri, psychosis, and hypervitaminosis A. J Nerv Ment Dis. 1972;155:72–75. | ||
Wieland RG, Hendricks FH, Amat y Leon F, Gutierrez L, Jones JC. Hypervitaminosis A with hypercalcaemia. Lancet. 1971;1:698. | ||
Ito K, Morita T. Role of retinoic acid in mouse neural crest cell development in vitro. Dev Dyn. 1995;204:211–218. | ||
Charest A, Wainer BH, Albert PR. Cloning and differentiation-induced expression of a murine serotonin1A receptor in a septal cell line. J Neurosci. 1993;13:5164–5171. | ||
Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. 1997;8:389–404. | ||
Takahashi J, Palmer TD, Gage FH. Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol. 1999;38:65–81. | ||
Crandall J, Sakai Y, Zhang J, et al. 13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice. Proc Natl Acad Sci U S A. 2004;101:5111–5116. | ||
Maret S, Franken P, Dauvilliers Y, Ghyselinck NB, Chambon P, Tafti M. Retinoic acid signaling affects cortical synchrony during sleep. Scienc. 2005;310(57450):111–113. | ||
Spencer KM, Nestor PG, Niznikiewicz MA, Salisbury DF, Shenton ME, McCarley RW. Abnormal neural synchrony in schizophrenia. J Neurosci. 2003;23:7407–7411. | ||
Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2: 255–267. | ||
Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2003;100:2901–2905. | ||
Konig G, Masters CL, Beyreuther K. Retinoic acid induced differentiated neuroblastoma cells show increased expression of the beta A4 amyloid gene of Alzheimer’s disease and an altered splicing pattern. FEBS Lett. 1990;269:305–310. | ||
Fukuchi K, Deeb SS, Kamino K, et al. Increased expression of beta-amyloid protein precursor and microtubule-associated protein tau during the differentiation of murine embryonal carcinoma cells. J Neurochem. 1992;58:1863–1873. | ||
Robakis NK, Anderson JP, Refolo LM, Wallace W. Expression of the Alzheimer amyloid precursor in brain tissue and effects of NGF and EGF on its metabolism. Clin Neuropharmacol. 1991;14 Suppl 1: S15–S23. | ||
Demontis D, Nyegaard M, Christensen JH, et al. The gene encoding the melanin-concentrating hormone receptor 1 is associated with schizophrenia in a Danish case-control sample. Psychiatr Genet. 2012; 22:62–69. | ||
Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Vitamin A exhibits potent antiamyloidogenic and fibril-destabilizing effects in vitro. Exp Neurol. 2004;189:380–392. | ||
Rinaldi P, Polidori MC, Metastasio A, et al. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol Aging. 2003;24:915–919. | ||
Connor MJ, Sidell N. Retinoic acid synthesis in normal and Alzheimer diseased brain and human neural cells. Mol Chem Neuropathol. 1997;30:239–252. | ||
Corcoran JP, So PL, Maden M. Disruption of the retinoid signalling pathway causes a deposition of amyloid beta in the adult rat brain. Eur J Neurosci. 2004;20:896–902. | ||
Cooper O, Hargus G, Deleidi M, et al. Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci. 2010;45:258–266. | ||
Li X, Li H, Bi J, Chen Y, Jain S, Zhao Y. Human cord blood-derived multipotent stem cells (CB-SCs) treated with all-trans-retinoic acid (ATRA) give rise to dopamine neurons. Biochem Biophys Res Commun. 2012;419:110–116. | ||
Fathi F, Altiraihi T, Mowla SJ, Movahedin M. Transplantation of retinoic acid treated murine embryonic stem cells and behavioural deficit in Parkinsonian rats. Indian J Med Res. 2010;131:536–544. | ||
Ulusoy GK, Celik T, Kayir H, Gursoy M, Isik AT, Uzbay TI. Effects of pioglitazone and retinoic acid in a rotenone model of Parkinson’s disease. Brain Res Bull. 2011;85:380–384. | ||
Yin LH, Shen H, Diaz-Ruiz O, et al. Early post-treatment with 9-cis retinoic acid reduces neurodegeneration of dopaminergic neurons in a rat model of Parkinson’s disease. BMC Neurosci. 2012;13:120. | ||
Vamvaca K, Lansbury PT Jr, Stefanis L. N-terminal deletion does not affect alpha-synuclein membrane binding, self-association and toxicity in human neuroblastoma cells, unlike yeast. J Neurochem. 2011;119: 389–397. | ||
Vekrellis K, Xilouri M, Emmanouilidou E, Stefanis L. Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J Neurochem. 2009;109: 1348–1362. | ||
Ko LW, Ko HH, Lin WL, Kulathingal JG, Yen SH. Aggregates assembled from overexpression of wild-type alpha-synuclein are not toxic to human neuronal cells. J Neuropathol Exp Neurol. 2008;67: 1084–1096. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.