Back to Journals » International Journal of Nanomedicine » Volume 15
In vivo Glioblastoma Therapy Using Targeted Liposomal Cisplatin
Authors Ashrafzadeh MS, Akbarzadeh A, Heydarinasab A ![]() , Ardjmand M
, Ardjmand M ![]()
Received 7 April 2020
Accepted for publication 27 August 2020
Published 23 September 2020 Volume 2020:15 Pages 7035—7049
DOI https://doi.org/10.2147/IJN.S255902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Maryam Sadat Ashrafzadeh,1 Azim Akbarzadeh,2 Amir Heydarinasab,1 Mehdi Ardjmand3
1Department of Chemical Engineering, Science and Research Branch, Islamic Azad University, Tehran, Iran; 2Department of Pilot Nanobiotechnology, Pasteur Institute of Iran, Tehran, Iran; 3Department of Chemical Engineering, South Tehran Branch, Islamic Azad University, Tehran, Iran
Correspondence: Azim Akbarzadeh
Department of Pilot Nanobiotechnology, Pasteur Institute of Iran, Tehran, Iran
Tel +989128387017
Email [email protected]
Background: Drug delivery systems have demonstrated promising results to cross blood–brain barrier (BBB) and deliver the loaded therapeutics to the brain tumor. This study aims to utilize the transferrin receptor (TR)-targeted liposomal cisplatin (Cispt) for transporting Cispt across the BBB and deliver Cispt to the brain tumor.
Methods: Targeted pegylated liposomal cisplatin (TPL-Cispt) was synthesized using reverse phase evaporation method and thiolated OX26 monoclonal antibody. The formulation was characterized in terms of size, size distribution, zeta potential, drug encapsulation and loading efficiencies, bioactivity, drug release profile, stability and cellular uptake using dynamic light scattering, flame atomic absorption spectroscopy (AAS), ELISA, dialysis membrane, and fluorescence assay. Next, the potency of the formulation to increase the therapeutic effects of Cispt and decrease its toxicity effects was evaluated in the brain tumor-bearing rats through measuring the mean survival time (MST), blood factors and histopathological studies.
Results: The results showed that TPL-Cispt with a size of 157± 8 nm and drug encapsulation efficiency of 24%± 1.22 was synthesized, that was biologically active and released Cispt in a slow-controlled manner. The formulation compared to Cispt-loaded PEGylated liposome nanoparticles (PL-Cispt) caused an increase in the cellular uptake by 1.43-fold, as well as an increase in the MST of the brain tumor-bearing rats by 1.7-fold compared to the PL-Cispt (P< 0.001). TPL-Cispt was potent enough to cause a significant decrease in Cispt toxicity effects (P< 0.001).
Conclusion: Overall, the results suggest that targeting the Cispt-loaded PEGylated liposome is a promising approach to develop formulation with enhanced efficacy and reduced toxicity for the treatment of brain tumor.
Keywords: liposome, targeted drug delivery, brain tumor, blood brain barrier, cisplatin
Introduction
Glioblastoma multiforme (GBM) is known as the most aggressive brain tumor,1 in which GBM patients live on average 9.9 months after surgical resection, and 14.6 months after radiation and adjuvant temozolomide therapy.2 Clinical application of chemotherapeutic agents for the treatment of GBM is limited due to the presence of the blood–brain barrier (BBB).3 Some approaches are available for brain drug delivery such as disrupting the BBB integrity or preparing lipid-soluble derivatives of the active agents. These approaches, however, have specific issues such as toxin entrance into the brain or change in pharmacokinetic properties of the original drug.4 In this regard, drug delivery systems such as liposomes seem more beneficial as they preserve both the drug and barrier properties.4
Liposomes are bilayer vesicular structures that are constituted of phospholipid and cholesterol, surrounding an aqueous core. They can be unilamellar or multilamellar, and due to their unique properties, they are able to encapsulate both hydrophilic and hydrophobic therapeutics. They are known as biocompatible and biodegradable carriers with less toxicity and target specificity, and they can release the loaded drug in a controlled manner. Moreover, their surface can be modified by addition of various macromolecules such as polymers and antibodies to improve their blood circulation time and targeted brain delivery.5 If their surfaces are modified with antibodies, then immunoliposomes will be produced, allowing for an active tissue targeting (eg, brain) through binding to specific receptors (eg, transferrin receptor; TR), available in BBB.6–8
TR is an interesting and unique target for brain drug delivery, since endothelial cells of the brain capillaries are one of the main cells that express TR.9 The density of cellular TR is in direct correlation with the degree of cell growth and division in which neoplastic cells such as glioma cells express more TR due to their faster cell division compared to the surrounding cells with normal cell division. The extent and diffuseness of TR are directly correlated with the glioma severity. Thus, TR can be exploited as a proper target for brain drug delivery.10 OX26—a mouse monoclonal antibody—is able to target the rat TR.11 Immunoliposomes grafted with OX26 can recognize TR at the BBB and transport the receptor across a rat BBB model via transcytosis.12,13 Researchers in various studies have used Cispt as a chemotherapeutic agent for GBM treatment.3,14
Cisp is an antitumor agent and functions by binding to DNA molecules and induction of apoptosis. Despite proper anticancer activity, it has some severe side effects such as kidney toxicity, audiotoxicity and neurotoxicity that limit its clinical application.15,16 Encapsulation of the drug into liposome nanoparticles can lead to a reduction in these side effects and an enhancement of its antitumor activity.17,18
In this study, cisplatin- (Cispt) loaded PEGylated liposomes, targeted with OX26 monoclonal antibody (targeted PEGylated liposomal Cispt; TPL-Cispt) were synthesized and after characterization, their therapeutic and toxicity effects were evaluated and compared with Cispt-loaded PEGylated liposome nanoparticles (PL-Cispt) and Cispt in an in-vivo experimental model of a brain tumor. For this purpose, the mean survival time (MST) and the blood concentrations of blood urea nitrogen (BUN), creatinine, alanine transaminase (ALT), aspartate transaminase (AST), and alkaline phosphatase (ALP) as the kidney and liver biochemical markers19 were measured. Also, histopathological studies were performed to confirm the results of toxicity measurement.
Experimental
Materials
Cispt was kindly supplied by Sobhan Oncology Company (Iran). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000](DSPE-PEG 2000) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG(2000) Maleimide) were purchased from Biochempeg Scientific Inc. (Watertown, MA, USA). Egg lecithin, cholesterol, PBS tablet, EDTA, FBS, DMEM (high glucose), penicillin/streptomycin antibiotics, 2-imionothiolan hydrochloride, maltose, ketamine, xylazine, diethyl ether, endothelial cell growth supplement (ECGS), basic fibroblast growth factor (bFGF), Hanks’ Balanced Salt Solution (HBSS), gelatin, dialysis bag cellulose membrane (cutoff 6 KDa), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), chloroform, coumarin-6, BSA, citric acid, 2.2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), H2O2, Triton X™-100 and formaldehyde were purchased from Sigma-Aldrich (USA). The secondary antibody goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, horseradish peroxidase (HRP) conjugate was purchased from Thermo Scientific-Pierce Antibodies. Twelve-well cell culture inserts (polyethylene terephtalate membrane, 3 μm pore size) were purchased from Millipore (Billerica, USA). OX26 monoclonal antibody was purchased from AbD Serotec (Kidlington, Oxfordshire, UK). Centriprep-30 concentrator (molecular weight cutoff: 30,000) was purchased from Millipore Inc. (Billerica, USA). Rat glioma C6 cell line and Wistar rats were purchased from Pasteur Institute of Iran. Distilled water was used throughout the study. All the chemicals used in this study were of analytical grade and were consumed as received.
Preparation of PL-Cispt
These nanoparticles were synthesized using reverse phase evaporation technique.20 Firstly, DSPE-PEG 2000 (7 mg), DSPE-PEG(2000) maleimide (5.8 mg), lecithin (16.48 mg), Cispt (14.4 mg) and cholesterol (13.12 mg) were dissolved in chloroform (10 mL). The solvent was then evaporated using a rotary evaporator (37°C, 50 rpm) and a thin lipid film was formed on the flask wall. Next, 7.2 mL PBS (10 mM, pH 7.4) was added to the lipid film and stirred (180 rpm, 10 min). The prepared vesicles were sonicated in a bath sonicator (Bandelin Sonorex Digitec, 60 Hz) to obtain unilamellar vesicles (ULVs) and decrease their size. Then, PL-Cispt were stored in 2 mL vials in a 4°C refrigerator for subsequent characterization. Empty liposomes were synthesized with the same method except that Cispt was not added to the medium.
Preparation of Thiolated OX26 Antibody
Thiolated antibody was synthesized based on the method described by Huwyler et al7 using 2-imionothiolan hydrochloride. For this purpose, OX26 antibody solution (1 mL, 0.5 mg/mL) was firstly concentrated using centriprep-30 concentrator (4000 rpm, 20 min) and secondly it was dissolved in sodium-borate buffer (0.5 mL, 0.15 M, pH 8.5) containing 0.1 mM EDTA. Then, 202 µg of 2-immunothiolane hydrochloride was added and incubated at room temperature for one hour. The solution was next concentrated again and the buffer was exchanged with 0.1 M Na-phosphate buffer (pH 8.0). The thiolated antibody instantaneously used for coupling to PL-Cispt. OX26/2-imionothiolan hydrochloride was used at the molar ratio of 1:40.
Coupling of Cispt-Loaded Pegylated Liposomes to OX26
To couple thiolated OX26 antibody to PL-Cispt, the antibody was incubated with the particles (PL-Cispt) for 18 h at a 1:20 weight ratio of peptide/phospholipid. The TPL-Cispt were separated by the use of a Sepharose CL-4B column and elution with PBS buffer (0.001 M, PH 7.4) from the reaction mixture. The amount of uncoupled OX26 was measured using NanoOrange protein quantitation kit (Invitrogen, Carlsbad, CA, USA) and the coupling efficiency (CE) was calculated using the formula below.
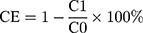
Where, C0 and C1, are the total and uncoupled amount of OX26, respectively.
Nanoparticle Characterization
The prepared formulations were characterized in vitro environment in terms of size, size distribution, zeta potential, drug encapsulation and loading efficiencies using dynamic light scattering (DLS) and flame atomic absorption spectroscopy (AAS) methods. In addition, morphology of the nanoparticles was evaluated using scanning electron microscopy (SEM). For this purpose, maltose was added to the suspensions of PL-Cispt and TPL-Cispt (weight ratio of 2:1) and the formulations were lyophilized. Next, the powder form of each formulation (1 mg) was individually coated with gold, and then evaluated using SEM (XL30, Philips, The Netherlands) for visual observation.
Drug Release Study
The kinetic of drug release from both PL-Cispt and TPL-Cispt formulations was evaluated using dialysis membrane technique.21 For this purpose, the suspension of both formulations was centrifuged (13,000 rpm, 30 min, 4°C) and the pellets were obtained. Next, the pellets containing 5 mg Cispt were resuspended infresh PBS (5 mL, pH 7.4) and individually transferred into dialysis bags. A solution of the standard Cispt (5 mL; 1 mg/mL in PBS, pH 7.4) was prepared and poured to a separate dialysis bag. Both ends of the bags were tightly closed and they were immersed in a beaker containing 100 mL PBS (pH 7.4) as the acceptor medium, and stirred (150 rpm, room temperature). The water solubility of Cispt is 1.0 mg/mL,22 ensures maintaining the sink conditions during the study.15 At the predetermined time intervals, 2 mL of PBS were taken and immediately 2 mL of fresh PBS were added. The platinum concentration in the samples was calculated using AAS method, and the cumulative percentage of drug release was measured by the formula below:
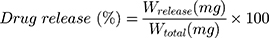
Where Wtotal is the quantity of the loaded drug into nanoparticles, and Wrelease is the amount of released drug from nanoparticles into the acceptor medium at the different time intervals.
ELISA Studies
The binding affinity of OX26 modified liposomal Cispt to TR was evaluated using ELISA. For this purpose, the surface of the wells of a 96-well plate were treated with TR and incubated overnight at 4°C. To block the plate, BSA was used and incubated for two hours at room temperature. The PL-Cispt and TPL-Cispt were then added, and incubated for one hour. After that, the wells were washed with PBS and the peroxidase-conjugated secondary antibody (goat anti-mouse) was added and incubated for 45 min at room temperature. To display the samples, a solution of citric acid, ABTS and H2O2 was used and the absorbance was read at 405 nm using a fluorescence microplate reader (H1M, BioTek Co., USA). Nonmodified nanoparticles as negative control were used.23
Nanoparticles Stability
The nanoparticles stability was studied by simulating the in vivo environment using the DLS method. For this purpose, a suspension of both PL-Cispt and TPL-Cispt formulations was provided in FBS (200 µL in 1 mL FBS) and stirred (37°C, 200 rpm). The study was performed under sterile condition to prevent bacterial contamination which could influence samples readings. At the predetermined time intervals, 10 µL of each formulation were taken, diluted with PBS (1:50) and its absorbance was read at 630 nm. Then, the changes in size, size distribution, and zeta potential were measured using Zetasizer instrument.
In vitro Cellular Uptake
The cellular uptake of PL-Cispt and TPL-Cispt was measured using a fluorescence assay and coumarin-C6. Briefly, PL-Cispt and TPL-Cispt containing coumarin-C6 were synthesized using reverse phase evaporation method as mentioned above, while 0.2 mg C6-coumarin was added to the formulations. C6 cells at the density of 1×105/well were cultured in 96-well plates. The culture medium was DMEM supplemented with 10% FBS and 1% penicillin/streptomycin antibiotics. After 24 h, the culture medium was replaced with the culture medium containing coumarin-C6-loaded PL-Cispt and coumarin-C6-loaded TPL-Cispt at the drug concentration of 128 µM and incubated for two hours. After that, to eliminate the noninternalized nanoparticles, the cells were washed with cold PBS and then were lysed with 0.1% Triton X-100 in 0.1 N NaOH solution. Next, the fluorescence intensity resulted from coumarin-C6-loaded nanoparticles was calculated by means of fluorescence microplate reader (H1M, BioTek Co., USA) with excitation and emission wavelengths set at 430 and 485 nm, respectively.
In vitro BBB Model
The BBB model was established using brain capillary endothelial cells (BCECs) according to the method described previously.24 For this purpose, BCECs were isolated based on the method described by Lu et al.25 All animal experiments were approved by the ethics committee of Pasteur Institute of Iran, and all procedures were performed in accordance with the National Institute of Health Guidelines for Care and Use of Laboratory Animals. Next, 12-well cell culture inserts were precoated with 2% gelatin for 30 min. Then, 7.5×104 BCECs/insert were cultured in inserts on day one. The culture media were changed every 48 h. On the fifth day, permeation assay (four hours) was carried out and transendothelial electrical resistance (TEER) values of the BBB were calculated by means of TEER instrument (Word Precision Instruments, Sarasota, FL, USA). Only the BBB models with TEER value over 250 Ωcm2 and no medium permeation in four hours were included in the study.
Evaluation of the Effects of Formulations on the BBB Integrity
Formulations including standard cisplatin, PL-Cispt and TPL-Cispt at the drug concentration of 256 µM were added to the inserts of BBB models. After four hours the effects of all formulations on BBB integrity were evaluated by measuring TEER values during the experiment.
BBB Targeting Effects of the Formulations
To measure the ability of the formulations to cross BBB, a BCECs/C6 cells co-culture model was established.26 Briefly, the selected BBB models provided in the inserts were transferred to another 12-well plate where C6 cells had been cultured for one day. Next, the standard Cispt, PL-Cispt and TPL-Cispt at the drug concentration of 254 μM were added into these inserts at the drug concentration of 254 μM for four hours, and then the inserts were removed. The cell viability of C6 glioma cells in the basolateral compartment was calculated after 48 h incubation by means of MTT assay.24
Evaluating the in vivo Biological Effects of the Formulations
Establishment of Glioblastoma Tumor in Animal
First, the GBM tumor was developed in male Wistar rats (10 weeks old, 240±10 g) using C6 cells according to the previous study.27 Depending on the type of formulations, the overall duration of the study ranged from 24 to 52 days. Briefly, animals were kept in animal house at room temperature, with 50–60% humidity and 12 h dark/light cycle. They had free access to food and water. After a week, the animals were anesthetized using intraperitoneal injection of a mixture of Ketamine (40 mg/kg) and xylazine (8 mg/kg). Next, a median incision (1 cm) was developed on the skull skin and a cranial cavity was developed at the right frontal trepanation, 2 mm in front of the coronal suture and 2 mm to the side of the sagittal using a mini electric drill. Next, 5×104 cells/10 μL of PBS were injected at the depth of 3 mm using a 10 µL Hamilton syringe and the stereotaxic technique. Inoculation was slowly carried out over a five-minute period and the needle was held in position for five minutes to prevent refluxing along the needle trajectory. The needle was then removed and the skin was sutured using two separate 5–0 mononylon stitches (Ethicon). After the surgical procedure, the rat was returned to the cage with free access to water and food.
The Treatment Protocol and Measurement of the Survival Time
Two days after tumor cell inoculation, the animals were randomly divided into four groups (n=8) and received standard Cispt, PL-Cispt, TPL-Cispt, and PBS as the control group. This number of animals per each group was selected according to previous study.3 The Cispt concentration in the formulations was 1 mg/kg, and the formulations were administered intraperitoneally with 72-h time intervals and two, five, eight, and 11 days after tumor cell inoculation. The MST of animals as the main factor for evaluating the therapeutic potential of the formulations was measured. For this purpose, the animals were monitored daily in terms of body weight, alertness, gait disturbance, and responses to contact and were overdosed with anesthetic when they showed the following symptoms together; a 20% body weight loss, uncoordination as well as ophthalmic hemorrhage. Animals which showed these symptoms were considered terminated and unlikely to recover. The MST was determined and analyzed using a Kaplan–Meier analysis.
Measurement of the Cisplatin Concentration in Brain Tumor Tissue
At indicated time points, animals were anesthetized by inhalation of diethyl ether and were sacrificed by cervical dislocation and decapitated. The blood samples were collected from the heart of the animals. Before removing the brain from the skull, the scalp tissue was investigated for extracranial tumor mass and associated blood vessels. Brain tissue containing tumor and extracranial tumor masses if present were weighed and fixed in 10% formalin. The brains were then investigated grossly and microscopically, and to evaluate histologically, they were stained with H&E. A section of brain tumor was removed and digested in a microwave digester (Sineo, MDS-10 model). The platinum concentration in the tumor tissue was measured after dilution in distilled water, using AAS method.28
Toxicity Studies
The toxicity effects of the formulations were evaluated by measuring the serum concentration of BUN, creatinine, ALT, AST and ALP as the kidney and liver biochemical markers.19 The toxicity effects were evaluated by histopathological studies. For this purpose, kidneys and liver from all the animals in all groups were harvested. The organs were fixed in 10% formalin, paraffinized, sectioned (5 µm) and stained with H&E. Next, the toxicity effects were evaluated using a semiquantitative scoring system by a pathologist in blind analysis, in which organ toxicity was considered 0 when no toxicity symptoms were observed, 1 when any slight change was perceived, and 2 when medium organ changes were observed.
Statistical Analysis
GraphPad Prism software version 8.00 was used for all statistical analyses. Statistical differences were analyzed by one-way ANOVA test. All data was expressed as mean ±standard deviation (SD, n=3).
Results
Nanoparticles Characterization
The results of drug encapsulation and loading efficiencies as well as size, size distribution, and zeta potential of blank, PL-Cispt and TPL-Cispt were demonstrated in Table 1. As the results show, nanoscale particles were obtained. The results of morphology evaluation by SEM showed that nanoparticles were formed monodispersed with a smooth surface and without surface fractures or pitting (Figure 1A and B). In addition, the coupling efficiency of OX26 for TPL-Cispt was calculated to be 38±4.7%.
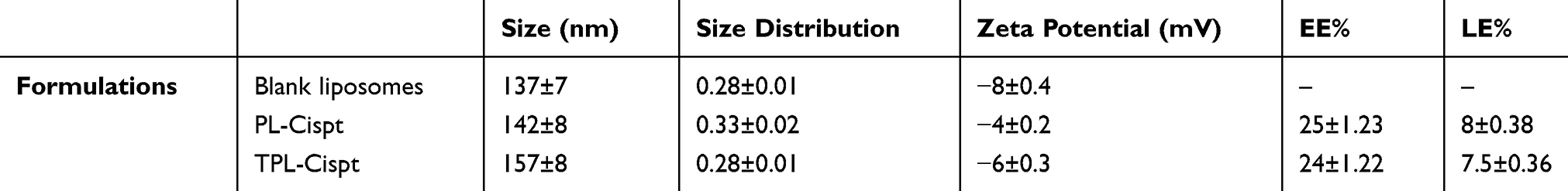 |
Table 1 Size, Size Distribution, Zeta Potential, Drug Encapsulation and Loading Efficiencies in Different Nanoformulations |
 |
Figure 1 SEM results of (A) PL-Cispt (scale bar: 1 µM, Mag: 15,000×) and (B) TPL-Cispt (scale bar: 2 µM, Mag. 10,000×). As the Figure shows, spherical monodispersed nanoparticles were formed with a smooth surface and without surface fractures or pitting. |
ELISA Studies
The ability of OX26-modified PEGylated liposomal Cispt to bind to TR was studied using ELISA assay. The results demonstrated that TPL-Cispt had a significant higher absorbance at 405 nm (0.77±0.04) compared to PL-Cispt (0.25±0.01) (P<0.05). These results indicated that TPL-Cispt preserved its bioactivity after conjugation of OX26 to the nanoparticles.
Drug Release Study
As the results of the drug release study show in Figure 2, in the first two hours of the study, 80% of Cispt was released from the Cispt solution, while both nanoformulations showed a pattern of slow-controlled drug release. In both nanoformulations, a burst release occurred in the first 30 min of the study in which 10% of the loaded drug was released. It was continued with a steady trend until the end of the study, when 60 and 64% of the loaded Cispt were released from PL-Cispt and TPL-Cist, respectively, after 72 h. Overall, the formulation was found as a controlled drug delivery system that released Cispt in a slow-controlled manner.
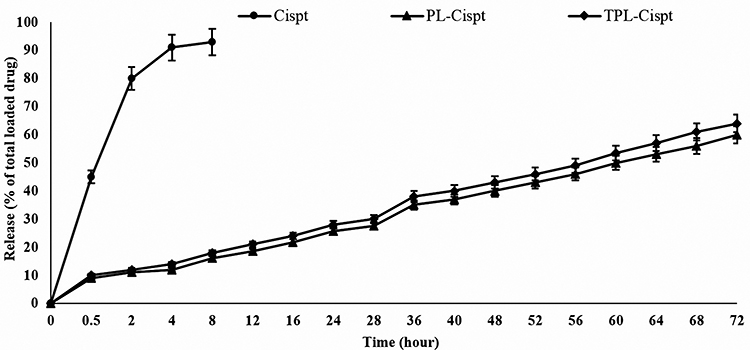 |
Figure 2 The profile of Cispt release from PL-Cispt and TPL-Cispt compared to the standard drug. While both formulations released Cispt in a slow-controlled manner (60 and 64% of the loaded Cispt were released from PL-Cispt and TPL-Cist, respectively after 72 h), Cispt was released from its solution rapidly, in which approximately all of it was released in the first eight hours of the study. Statistical analyses were performed using one-way ANOVA, Tukey’s post hoc test. The results were expressed as mean ±SD (n=3). |
Nanoparticle Stability
The stability results of PL-Cispt and TPL-Cispt in human serum showed that the size of both formulations was decreased by 9% (Figure 3; 142±7.1 to 129±6.4 nm and 157±8.1 to 143±7.1 nm for PL-Cispt and TPL-Cispt, respectively) in 48 h. Both formulations demonstrated approximately 17% increase in size distribution (0.33±0.02 to 0.40±0.02 and 0.28±0.01 to 0.34±0.02 in LP-Cispt and TLP-Cispt, respectively). In addition, the values of zeta potential in both nanoformulations were decreased at the end of the study by approximately 26% (−4±0.2 to −5.4±0.3 mV and −6±0.3 to −7.5±0.4 mV, for PL-Cispt and TPL-Cispt, respectively).
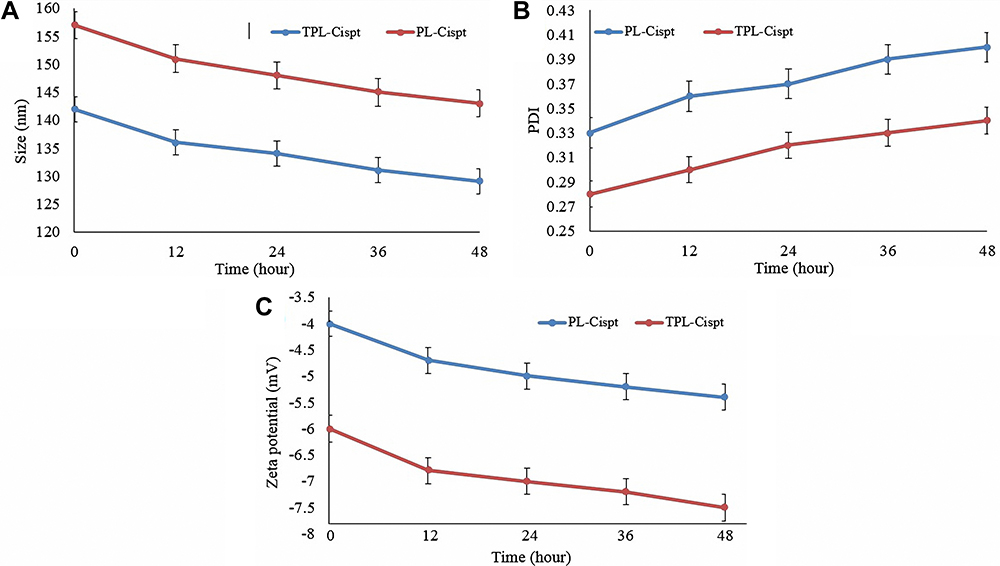 |
Figure 3 The results of changes in (A) the size, (B) size distribution and (C) zeta potential of PL-Cispt and TPL-Cispt, obtained from stability study in human serum. As the results demonstrated, while size and zeta potential of the nanoformulations (PL-Cispt and TPL-Cispt) were decreased by incubation in human serum, The PDI values were increased. The results were expressed as mean ±SD (n=3). |
In vitro Cellular Uptake
The cellular uptake of PL-Cispt and TPL-Cispt was calculated after two hours incubation using fluorescent dye, coumarin-C6. The results demonstrated that TPL-Cispt compared to PL-Cispt had a significantly higher cellular uptake, in which the cellular uptake of TPL-Cispt was 1.43-fold higher than the PL-Cispt (Figure 4).
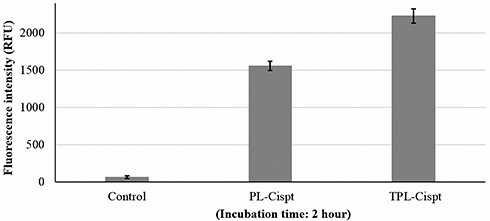 |
Figure 4 The cellular uptake results for PL-Cispt and TPL-Cispt which was measured by fluorescence assay after 2 hours incubation. As the results demonstrated the targeted nanoformulation (TPL-Cispt) compared to that non-targeted formulation (PL-Cispt) had a 1.43-fold higher cellular uptake. |
BBB Targeting Effects of the Formulations
An in vitro BBB model was created based on the method described by Yue et al24 by means of BCECs. The integrity of the BBB was evaluated and proved by calculating the TEER. The integrity was maintained during four hours incubation with the Cispt formulations at Cispt concentration of 256 µM. The cell viability results demonstrated that Cispt caused 75% cell viability, while this value for PL-Cispt and TPL-Cispt was 68 and 28%, respectively (Figure 5). In other words, TPL-Cispt compared to PL-Cispt and the standard drug, caused an increase in the cytotoxicity effects of Cispt by 2.4-fold (P<0.01) and 2.7-fold (P<0.01), respectively (cell viability of 75±3.7, 68±3.3 and 28±1.3 for Cispt, PL-Cispt and TPL-Cispt, respectively). Liposome as a Cispt carrier increased the cytotoxicity effects of the drug by 1.1-fold.
 |
Figure 5 The effects of Cispt, PL-Cispt, and TPL-Cispt on the C6 cell viability which was evaluated in the BBB targeting experiment after 48 h incubation. As the Figure shows, TPL-Cispt compared to PL-Cispt and Cispt, caused a significant lower cell viability (cell viability of 75±3.7, 68±3.3 and 28±1.3 for Cispt, PL-Cispt and TPL-Cispt, respectively). Statistical analyses were carried out using one-way ANOVA, Tukey’s post hoc test. The results were expressed as mean ±SD (n=3). **P<0.01. |
Evaluation of the in vivo Biological Effects of the Formulations
Establishment of Glioblastoma Tumor in Animal
Glioblastoma tumors were successfully established as the animals exhibit its symptoms including decreased agility and movement, ophthalmic and nasal hemorrhage which were intensified by tumor progression. These findings were more prominent in PBS group. Compared to healthy brain with the smooth surface (Figure 6A), the tumor was perceived as a swollen mass in frontal lobe of tumor-bearing brain (Figure 6B). However, the tumor did not develop in one animal in each group.
 |
Figure 6 Brain tissue in (A) healthy and (B) brain tumor-bearing rat. While normal brain has a smooth surface without protrusions, the tumor mass can be seen in the frontal lobe, indicated with an arrow. |
Measurement of the Survival Time
Regarding the therapeutic effects of the formulations, the MST was 17 days in the control group. The value of MST in TPL-Cispt, PL-Cispt and Cispt receiver rats was equal to 45, 27, and 25 days, respectively (Figure 7). In other words, TPL-Cispt caused an increase in the MST of the brain tumor-bearing rats by 1.7- and 1.8-fold compared to the PL-Cispt and Cispt in tumor-bearing rats, respectively (P<0.001). Moreover, the brain hemorrhage was observed in one tumor-bearing rat, treated with Cispt, while it was absent in the groups treated with PL-Cispt and TPL-Cispt (Figure 8).
 |
Figure 7 Kaplan–Meier survival curve comparing survival of brain tumor-bearing rats, treated with various formulations with P-value 0.001 by log-rank statistics test. As the Figure shows, the MST value in TPL-Cispt receiver group compared to that of PL-Cispt and Cispt groups was increased by 1.7- and 1.8-fold, respectively (MST: 45, 27, and 25 days for TPL-Cispt, PL-Cispt and Cispt groups, respectively, P<0.001). |
 |
Figure 8 Histological characterization of brain tissue using H&E staining in (A) the brain tumor bearing rat, treated with TLP-Cispt, and (B) the brain tumor-bearing rat, treated with the standard Cispt. As the results show, brain hemorrhage occurs in the Cispt receiver rat, while brain hemorrhage is not observed in TLP-Cispt group (magnification size ×4, scale bar: 500 µm). |
Measurement of Cisplatin Concentration in the Brain Tumor Tissue
The platinum concentration in the brain tumor of rats treated with Cispt, PL-Cispt and TPL-Cispt was 0.65±0.03, 0.75±0.04 and 1.4±0.06 μg/g, respectively. In other words, TLP-Cispt increased Cispt concentration in brain tumor by 1.9- and 2.1-fold compared to that of PL-Cispt and Cispt, respectively.
Toxicity Studies
Toxicity was evaluated by measuring the serum concentration of ALT, AST, ALP, BUN and creatinine and histopathological studies. The results showed that the serum concentrations of these factors were significantly increased in brain tumor-bearing rats that received Cispt compared to PBS group (P<0.001), while Cispt encapsulation in liposome caused a significant decrease in the serum concentrations of these factors compared to that of brain tumor-bearing rats who received Cispt (P<0.001, Table 2). Also, the results of histopathological studies showed a significant reduction in pathological lesions in tumor-bearing rats who received PL-Cispt and TPL-Cispt compared to the Cispt receiver group, in which a higher rate of liver cell necrosis was observed in the Cispt receiver group (Table 2, Figure 9). The severity of these lesions was significantly decreased in TPL-Cispt receiver rats compared to the PL-Cispt and Cispt groups.
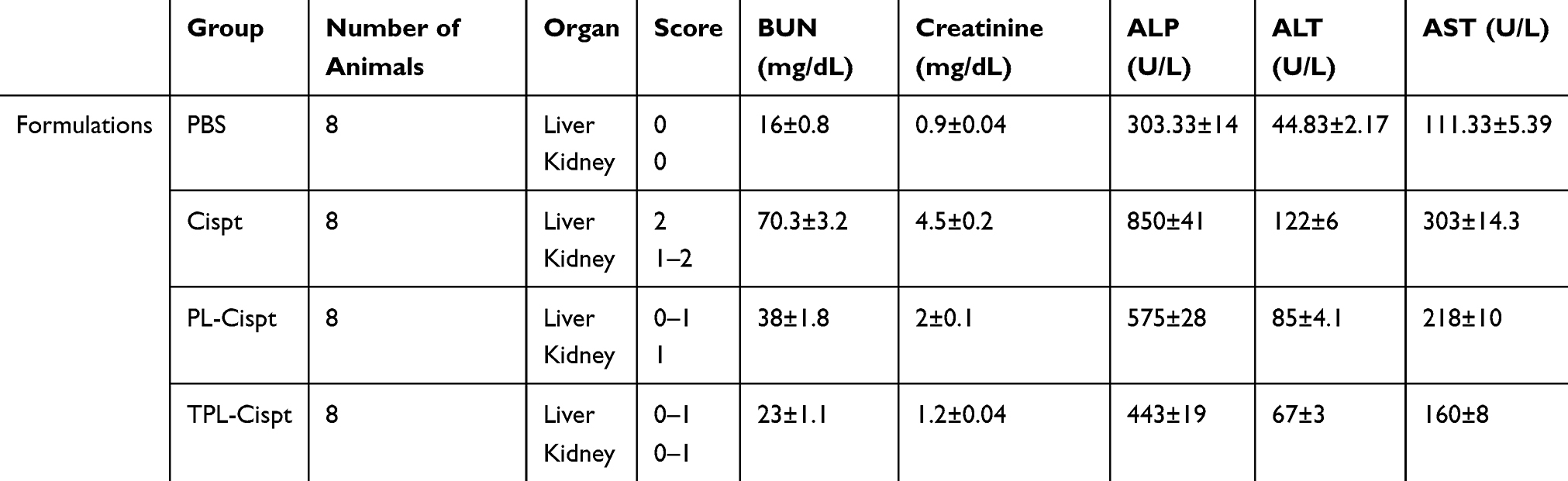 |
Table 2 Histopathological Assessment of Organ Toxicity and the Blood Concentrations of BUN and Creatinine (mg/dL) in Various Groups of Animals, Received Cispt, PL-Cispt and TPL-Cispt Compared to PBS Control Group |
 |
Figure 9 The results of toxicity evaluation of liver tissue using H&E staining in: (A) brain tumor-bearing rats, received Cispt and (B) healthy control group. The arrow shows cell necrosis due to the toxicity effects of Cispt (magnification size ×40, scale bar: 50 µm). |
Discussion
Brain drug delivery is a major challenge owing to the barriers generated by BBB. Despite the extensive researches during the past years, only a little progress has been achieved, especially in the field of nanomedicine.29 While many nanomedicines have shown promising therapeutic effects in preclinical studies, these results have failed to repeat in clinical trials. This may stem from several factors such as insufficient nanocarrier design, using preclinical disease models with low correlation with the human disease, and most importantly, selections and function of the targeted receptor to increase brain exposure.30 The TR has been widely investigated as a potential target for the brain drug delivery for more than 25 years. In this regard, William Pardridge was among the first researchers7,31–37 to show that brain delivery of various compounds is feasible using immunoliposome.
Liposome nanoparticles are used as drug carriers because of their easy synthesis, high biocompatibility, biodegradability, nonstimulation of the immune system and production on a large scale.38 This nanoparticles were able to increase the therapeutic effects of the encapsulated drug.21,39,40
In the current study, liposomes were synthesized using reverse phase evaporation method.17,21 In this method, liposomes with the high drug encapsulation efficiency are produced.41 For this purpose, cholesterol was used to increase the membrane packing and as a result decrease the membrane fluidity and permeability. Furthermore, DSPE-mPEG was used to increase the blood half-life and produce long-acting liposomes.42 Results of previous studies also demonstrated that PEGylation of liposomes caused an increase in the biological half-life of the encapsulated drug and as a result an increase in drug efficacy.43,44 In addition, DSPE-PEG-Mal linker was used to conjugate thiolated OX26 to the surface of liposomes as well. Maleimide (Mal) reacts rapidly and efficiently with the thiol group. This reaction is extensively used in bioconjugate chemistry.45,46 In the present study Cispt, as a powerful antitumor agent, was loaded into the liposomes. This drug is the first-line treatment for various types of malignancies such as ovarian and lung cancers. Cispt is also used as an adjuvant for the treatment of pediatric brain tumors.47 Herein, Cispt was encapsulated into PEGylated liposome nanoparticles and their therapeutic effects were evaluated on an orthotopic model of GBM, developed by C6 rat glioma cells.3
The formulations were characterized in an in vitro environment in terms of size, zeta potential, and drug loading efficiency. It was found that the size of PL-Cispt was larger than the size of blank liposome (142±8 vs 137±7 nm) indicating that Cispt was loaded into nanoparticles and the size was further increased in TLP-Cispt (157±8 vs 142±8 nm) confirming that OX26 antibody was conjugated into PL-Cispt nanoparticles. The zeta potential of nanoparticles was increased in PL-Cispt compared to that of blank liposomes (−4±0.2 vs −8±0.4 mV) due to the positive charge of Cispt.48 Nanoparticles with positive or negative charge in aqueous solutions with low ionic strength are colloidally stable.49 Furthermore, PL-Cispt and TPL-Cispt were morphologically evaluated using SEM and the results demonstrated that the formulations were formed as spherical nanoparticles with a smooth surface and without any obvious aggregation (Figure 1A and B). Moreover, the results of drug-encapsulation efficiency demonstrated that TPL-Cispt had approximately similar encapsulation efficiency to that of PL-Cispt (24±1.22 vs 25±1.23%), indicating that antibody conjugation had not adversely affected the drug-encapsulation efficiency.
In addition, the bioactivity of OX26-modified liposomal Cispt was evaluated by ELISA. The results demonstrated that the formulation maintained its bioactivity. Then the profile of drug release from PL-Cispt and TPL-Cispt was evaluated.
Drug release is an important factor to optimize the therapeutic effects of the loaded drug.1 Two modes of drug release have been reported including conventional and controlled releases. In conventional release systems such as tablets and capsules, pharmaceutical drugs are released very quickly once administered in the body. This results in in a sharp increase in blood concentration of the drug followed by a rapid decrease within a short period of time. Therefore, repeated dosing may be required to preserve the effective dose of drug.50 Such fluctuation in the plasma drug concentrations can cause toxic effects and/or insufficient drug efficacy. The repeated dosing may also result in patient inconvenience. Despite of conventional drug release, in controlled drug delivery systems, the kinetics of drug release and consequently drug therapeutic effects is increased. The blood–drug concentration is increased and then maintained in the effective range (ie, between minimum effective and maximum desired levels), in a sustained mode.50 Overall, controlled drug delivery systems compared to the conventional release systems provide various benefits such as maximal drug efficacy, minimal toxicity, lowered drug accumulation with chronic drug dosing, and minimal fluctuation in blood–drug concentration.50
In the present study, a burst release was observed in which 10% of the loaded drug was released in the first 30 min of the study (Figure 2). This could have resulted from the release of adsorbed drug onto the nanoparticles. Poy et al51 also reported a burst Cispt release from liposome, in which 15% of the loaded Cispt was released in the first hour of the study. There was no significant difference between the amount of drug release from PL-Cispt and TPL-Cispt (60 vs 64%), indicating that antibody conjugation into nanoparticles did not adversely affect the stability of liposomes, and as a result liposomes preserve a considerable amount of Cispt in the study. Moreover, incorporation of biodegradable materials such as PEG into nanoparticle construction, improves its aqueous solubility and assists sustained drug release at the target site for a few days or even weeks.15,52 Vázquez-Becerra et al,53 also reported that differences between the amount of drug release from PL-Cispt and TPL-Cispt (~5%) was slightly noticeable after 72 h. Overall, both formulations (PL-Cispt and TPL-Cispt) released Cispt in a slow-controlled manner, which can cause an increase in the therapeutic effects of the loaded drug.3,54
In designing drug delivery systems, inhibition of premature drug release is important to decrease drug toxicity. Nanoparticles as drug carriers have a crucial role in the production of formulations as they can enhance stability.49
In the present study, the results of stability evaluation demonstrated a decrease in size for both formulations (by 9%), resulting from the interaction of serum proteins with liposomes, condensing the liposomes and, therefore, decreasing their size (Figure 3A). Approximately an equal decrease in size was observed for both formulations, indicating that antibody conjugation into liposomes did not influence the liposomes stability. Both formulations demonstrated about a 17% decrease in size distribution, indicating that the interaction of liposomes with serum components caused a decrease in the liposomes homogeneity (Figure 3B). In addition, the results demonstrated that a decrease of approximately 26% occurred in the values of zeta potential for both nanoformulations (Figure 3C), indicating that these nanoformulations were stable in serum, although they had low amounts of zeta potentials (−4 and −6 mV). This might stem partially from the presence of PEG in the nanoparticle structure. PEG functions as a nanoparticle stabilizer.15 It reduces the adsorption of serum protein onto nanoparticles and decreases the capture rate of nanoparticles by the reticuloendothelial system.15 Overall, the stability results confirmed that both nanoformulations were stable and antibody conjugation into liposomes did not adversely influence the nanoparticles stability. Cispt is unstable in aqueous solution,55 therefore these formulations can be used to maintain the Cispt stability and as a result the drug therapeutic effects.
The cellular uptake of PL-Cispt and TPL-Cispt was calculated after two hours incubation using fluorescent dye coumarin-C6. The results demonstrated that TPL-Cispt compared to PL-Cispt had a significantly higher cellular uptake, in which the cellular uptake of TPL-Cispt was 1.43-fold higher than the PL-Cispt (Figure 4).
Biological membranes may function as a barrier to accumulation of therapeutic agents in the desired organelles in cells.56 As the endosome or lysosome degradative compartment may not be the ultimate therapeutic aim, thus development of new efficient strategies to deliver therapeutics across the biological membranes and thereafter protection from unfavorable hydrolytic condition of lysosome is mandatory.57 This goal can be attained by the use of new systems with high encapsulation efficiency and sustained drug release profile for targeted therapeutic delivery to specific cells or to specific intracellular components.58 In the present study, the results of cellular uptake indicated that modification of liposome with OX26 caused the cellular uptake of the particles to be increased by 1.43-fold compared to that of PL-Cispt (Figure 4). This increase in cellular uptake can result from the interaction of OX26 with TR, available on the plasma membrane of C6 cells.59
In addition, the BBB targeting effects of the formulations were evaluated and the results demonstrated that TPL-Cispt compared to PL-Cispt and the standard drug caused an increase in the cytotoxicity effects of Cispt (2.4- and 2.7-fold compared to PL-Cispt and Cispt, respectively) confirming that the potency of TPL-Cispt compared to PL-Cispt to cross the BBB is increased through transferrin-mediated transcytosis (Figure 5). Our results were supported with the study by Ulbrich et al,60 in which the transferrin-modified liposomes caused an increase in the loperamide delivery across the BBB through TR.60
Next, the efficacy of TPL-Cispt was evaluated in the brain tumor-bearing rats. The brain tumor was successfully developed (Figure 6). The results showed that the formulation caused a significant increase in the MST value compared to the standard Cispt receiver animals (25 vs 45 days, Figure 7). Cispt delivery to the brain tumor using polybutylcyanoacrylate (PBCA) nanoparticles has been also reported previously3 and the results of this study showed that the prepared PBCA nanoparticles were not efficient to transport Cispt across the BBB and its delivery to brain tumor, therefore the MST of the tumor-bearing animals did not increase compared to when Cispt was used.3 This shortcoming resulted from the large size of the PBCA nanoparticles (489 nm) which caused the particles' inability to cross the BBB.3 As a rule, the particle size below 100 nm is proper for crossing the BBB and brain penetration.61 However, there are various studies demonstrating that particles with a size bigger than 100 nm can cross the BBB and deliver their cargoes to the tumor.62–64 In the present study, TPL-Cispt was found to be potent to cross the BBB and deliver Cispt to the brain tumor due to its smaller size (157 nm vs 489 nm) and also targeting with OX26, resulting in improvement in the therapeutic effects compared to the Cispt-loaded PBCA nanoparticles. The TPL-Cispt compared to PL-Cispt also caused an increase in the MST values by 1.7-fold. This could result from the specific interaction of the OX26 antibody with TR and increase the brain penetration of the formulation via transcytosis.12,13 The measurement results of Cispt concentration in the brain tumor were confirmed by the potency of TLP-Cispt in brain–drug delivery as the Cispt concentration in brain tumor of rats received TPL-Cispt was 2.1 and 1.9-fold higher than that values in tumor-bearing rats, who received Cispt and PL-Cispt, respectively. Moreover, brain hemorrhage was observed in one tumor-bearing animal treated with Cispt, while this finding was not observed in animals treated with PL-Cispt or TPL-Cispt (Figure 8). This result might indicate that PL-Cispt and TPL-Cispt were more efficient compared to the standard Cispt in treatment of brain tumor and prevention of hemorrhage.
We also evaluated the toxicity effects of Cispt in standard and encapsulated forms. The results showed that Cispt encapsulation into liposomes caused a significant decrease in blood concentrations of ALT, AST, ALP, BUN and creatinine in the brain tumor-bearing rats who received PL-Cispt and TPL-Cispt receivers compared to Cispt receiver group. This confirms that Cispt encapsulation in liposomes is an efficient method to decrease the toxicity effects of the drug. The toxicity effects were also evaluated by histopathological studies. For this purpose, liver and kidney tissues were removed, processed, stained with H&E and evaluated with light microscopy. The results showed that Cispt encapsulation in liposomes caused a significant decrease in the histopathological effects (eg, liver cell necrosis) of Cispt (Figure 9, Table 2). These results were in agreement with the results of measurement of blood concentrations of ALT, AST, ALP, BUN and creatinine. One of the main features of nanoparticles is their potency to reduce the drug toxicity, which was confirmed in the present study. Researchers in various studies also showed that drug encapsulation into nanoparticles caused a reduction in drug toxicity effects.15,16
Conclusion
TPL-Cispt was successfully synthesized using OX26 monoclonal antibody, and its potency to increase the therapeutic effects of Cispt and decrease the toxicity effects of the drug, was evaluated in an in vivo environment. The in vitro characterization results showed that nanoparticles with a size of 157 nm and drug-loading efficiency of 24% were synthesized and that they were biologically active. The OX26 modification of the liposomes was found to be causing an increase in cellular uptake of the particles. The potency of the nanocarrier to increase the Cispt efficacy was increased when it was targeted with OX26 antibody (the MST value in TPL-Cispt group was increased by 1.7- and 1.8-fold, compared to the PL-Cispt and the standard drug receivers, respectively). This results from the fact that the brain penetration of the TPL-Cispt was increased compared to the other formulations due to interaction of OX26 antibody with TR and transcytosis of the formulation, resulting in the higher drug concentrations in brain tumor compared to that PL-Cispt (by 1.9-fold). Furthermore, TPL-Cispt caused a significant decrease in the toxicity effects of Cispt (P<0.001) in terms of measurement of blood concentrations of AST, ALT, ALP, BUN and creatinine. The results of toxicity effects were confirmed by histopathological studies. Overall, TPL-Cispt was found to be the best among all the formulations due to its higher potency to increase the therapeutic effects and decrease the toxicity effects of Cispt in brain tumor-bearing rats, suggesting that targeting PL-Cispt with OX26 monoclonal antibody is a promising approach to develop formulations with enhanced therapeutic efficacy and reduced toxicity for brain tumor therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lalatsa A, Leite DM, Figueiredo MF, O’Connor M. Nanotechnology in brain tumor targeting: efficacy and safety of nanoenabled carriers undergoing clinical testing. In: Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors. Elsevier; 2018:111–145.
2. ParvizHamidi M, Haddad G, Ostadrahimi S, et al. Circulating miR‐26a and miR‐21 as biomarkers for glioblastoma multiform. Biotechnol Appl Biochem. 2019;66(2):261–265. doi:10.1002/bab.1707
3. Ebrahimi Shahmabadi H, Movahedi F, Koohi Moftakhari Esfahani M, Koohi Moftakhari Esfahani M, et al. Efficacy of Cisplatin-loaded polybutyl cyanoacrylate nanoparticles on the glioblastoma. Tumor Biol. 2014;35(5):4799–4806. doi:10.1007/s13277-014-1630-9
4. Sezgin-bayindir Z, Ergin AD, Parmaksiz M, Elcin AE, Elcin YM, Yuksel N. Evaluation of various block copolymers for micelle formation and brain drug delivery: in vitro characterization and cellular uptake studies. J Drug Deliv Sci Technol. 2016;36:120–129. doi:10.1016/j.jddst.2016.10.003
5. Padhi S, Mazumder R, Development BS. Application of lipid nanotechnology on infectious diseases of CNS-current scenario. Ind J Pharm Educ Res. 2019;53(3):355–365. doi:10.5530/ijper.53.3.69
6. Paszko E, Senge M. Immunoliposomes. Curr Med Chem. 2012;19(31):5239–5277. doi:10.2174/092986712803833362
7. Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci. 1996;93(24):14164–14169. doi:10.1073/pnas.93.24.14164
8. Johnsen KB, Burkhart A, Melander F, et al. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci Rep. 2017;7(1):10396. doi:10.1038/s41598-017-11220-1
9. Malik IA, Naz N, Sheikh N, et al. Comparison of changes in gene expression of transferrin receptor-1 and other iron-regulatory proteins in rat liver and brain during acute-phase response. Cell Tissue Res. 2011;344(2):299. doi:10.1007/s00441-011-1152-3
10. Eavarone DA, Yu X, Bellamkonda RV. Targeted drug delivery to C6 glioma by transferrin‐coupled liposomes. J Biomed Mater Res. 2000;51(1):10–14. doi:10.1002/(SICI)1097-4636(200007)51:1<10::AID-JBM2>3.0.CO;2-R
11. Ji B, Maeda J, Higuchi M, et al. Pharmacokinetics and brain uptake of lactoferrin in rats. Life Sci. 2006;78(8):851–855. doi:10.1016/j.lfs.2005.05.085
12. Passeleu-le Bourdonnec C, Carrupt P-A, Scherrmann JM, Martel S. Methodologies to assess drug permeation through the blood–brain barrier for pharmaceutical research. Pharm Res. 2013;30(11):2729–2756.
13. Lalatsa A, Butt AM. Chapter 3 - physiology of the blood–brain barrier and mechanisms of transport across the BBB. In: Kesharwani P, Gupta U, editors. Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors. Academic Press; 2018:49–74.
14. Coluccia D, Figueiredo CA, Wu MY, et al. Enhancing glioblastoma treatment using cisplatin-gold-nanoparticle conjugates and targeted delivery with magnetic resonance-guided focused ultrasound. Nanomedicine. 2018;14(4):1137–1148. doi:10.1016/j.nano.2018.01.021
15. Alavi SE, Muflih Al Harthi S, Ebrahimi Shahmabadi H, Akbarzadeh A. Cisplatin-loaded polybutylcyanoacrylate nanoparticles with improved properties as an anticancer agent. Int J Mol Sci. 2019;20(7):1531. doi:10.3390/ijms20071531
16. Koohi Moftakhari Esfahani M, Alavi SE, Shahbazian S, Ebrahimi Shahmabadi H. Drug delivery of cisplatin to breast cancer by polybutylcyanoacrylate nanoparticles. Adv Polym Technol. 2018;37(3):674–678. doi:10.1002/adv.21709
17. Alavi SE, Koohi Moftakhari Esfahani M, Ghassemi S, Akbarzadeh A, Hassanshahi G. In vitro evaluation of the efficacy of liposomal and pegylated liposomal hydroxyurea. Ind J Clin Biochem. 2014;29(1):84–88. doi:10.1007/s12291-013-0315-2
18. Movahedi F, Ebrahimi Shahmabadi H, Alavi SE, Koohi Moftakhari Esfahani M. Release modeling and comparison of nanoarchaeosomal, nanoliposomal and pegylated nanoliposomal carriers for paclitaxel. Tumor Biol. 2014;35(9):8665–8672. doi:10.1007/s13277-014-2125-4
19. Najafzadeh H, Ghoreishi S, Mohammadian B, et al. Serum biochemical and histopathological changes in liver and kidney in lambs after zinc oxide nanoparticles administration. Veterinary World. 2013;6:8. doi:10.5455/vetworld.2013.534-537
20. Garcia-Garcia E, Andrieux K, Gil S, Couvreur P. Colloidal carriers and blood–brain barrier (BBB) translocation: a way to deliver drugs to the brain? Int J Pharm. 2005;298(2):274–292. doi:10.1016/j.ijpharm.2005.03.031
21. Koohi Moftakhari Esfahani M, Alavi SE, Movahedi F, Alavi F, Akbarzadeh A. Cytotoxicity of liposomal Paclitaxel in breast cancer cell line mcf-7. Ind J Clin Biochem. 2013;28(4):358–360. doi:10.1007/s12291-013-0296-1
22. Carland M, Tan KJ, White JM, et al. Syntheses, crystal structure and cytotoxicity of diamine platinum (II) complexes containing maltol. J Inorg Biochem. 2005;99(8):1738–1743. doi:10.1016/j.jinorgbio.2005.06.003
23. Ramalho MJ, Sevin E, Gosselet F, et al. Receptor-mediated PLGA nanoparticles for glioblastoma multiforme treatment. Int J Pharm. 2018;545(1):84–92. doi:10.1016/j.ijpharm.2018.04.062
24. Yue P-J, He L, Qiu S-W, et al. OX26/CTX-conjugated PEGylated liposome as a dual-targeting gene delivery system for brain glioma. Mol Cancer. 2014;13(1):191. doi:10.1186/1476-4598-13-191
25. Lu W, Tan Y-Z, Hu K-L, Jiang X-G. Cationic albumin conjugated pegylated nanoparticle with its transcytosis ability and little toxicity against blood–brain barrier. Int J Pharm. 2005;295(1–2):247–260. doi:10.1016/j.ijpharm.2005.01.043
26. Du J, Lu W-L, Ying X, et al. Dual-targeting topotecan liposomes modified with tamoxifen and wheat germ agglutinin significantly improve drug transport across the blood− brain barrier and survival of brain tumor-bearing animals. Mol Pharm. 2009;6(3):905–917. doi:10.1021/mp800218q
27. Miura FK, Alves MJF, Rocha MC, et al. Experimental model of C6 brain tumors in athymic rats. Arq Neuropsiquiatr. 2008;66(2A):238–241. doi:10.1590/S0004-282X2008000200019
28. Kakinuma K, Tanaka R, Takahashi H, Watanabe M, Nakagawa T, Kuroki M. Targeting chemotherapy for malignant brain tumor using thermosensitive liposome and localized hyperthermia. J Neurosurg. 1996;84(2):180–184. doi:10.3171/jns.1996.84.2.0180
29. Fullstone G, Nyberg S, Tian X, Battaglia G. From the blood to the central nervous system: a nanoparticle’s journey through the blood–brain barrier by transcytosis. Int Rev Neurobiol. 2016;130:41–72.
30. Johnsen K, Burkhart A, Andresen T, Moos T, Thomsen L. Nanomedicines for Brain Drug Delivery. Springer; 2017.
31. Shi N, Pardridge WM. Noninvasive gene targeting to the brain. Proc Natl Acad Sci. 2000;97(13):7567–7572. doi:10.1073/pnas.130187497
32. Shi N, Boado RJ, Pardridge WM. Receptor-mediated gene targeting to tissues in vivo following intravenous administration of pegylated immunoliposomes. Pharm Res. 2001;18(8):1091–1095. doi:10.1023/A:1010910523202
33. Shi N, Zhang Y, Zhu C, Boado RJ, Pardridge WM. Brain-specific expression of an exogenous gene after iv administration. Proc Natl Acad Sci. 2001;98(22):12754–12759. doi:10.1073/pnas.221450098
34. Huwyler J, Cerletti A, Fricker G, Eberle AN, Drewe J. By-passing of P-glycoprotein using immunoliposomes. J Drug Target. 2002;10(1):73–79. doi:10.1080/10611860290007559
35. Zhang Y-F, Boado RJ, Pardridge WM. Absence of toxicity of chronic weekly intravenous gene therapy with pegylated immunoliposomes. Pharm Res. 2003;20(11):1779–1785. doi:10.1023/B:PHAM.0000003375.13655.f9
36. Zhang Y, Schlachetzki F, Zhang Y-F, Boado RJ, Pardridge WM. Normalization of striatal tyrosine hydroxylase and reversal of motor impairment in experimental parkinsonism with intravenous nonviral gene therapy and a brain-specific promoter. Hum Gene Ther. 2004;15(4):339–350. doi:10.1089/104303404322959498
37. Zhang Y, Zhang Y-F, Bryant J, Charles A, Boado RJ, Pardridge WM. Intravenous RNA interference gene therapy targeting the human epidermal growth factor receptor prolongs survival in intracranial brain cancer. Clin Cancer Res. 2004;10(11):3667–3677. doi:10.1158/1078-0432.CCR-03-0740
38. Haghiralsadata F, Amoabedinyb G, Naderinezhadc S, Helderd MN, Kharanaghie EA, Zandieh-Doulabid B. Overview of preparation methods of polymeric and lipid-based (noisome, solid lipid, liposome) nanoparticles: a comprehensive review. Name Int J Polymeric Mater Polymeric Biomater. 2017.
39. Alavi SE, Koohi Moftakhari Esfahani M, Alavi F, Movahedi F, Akbarzadeh A. Drug delivery of hydroxyurea to breast cancer using liposomes. Ind J Clin Biochem. 2013;28(3):299–302. doi:10.1007/s12291-012-0291-y
40. Koohi Moftakhari Esfahani M, Alavi SE, Akbarzadeh A, et al. Pegylation of nanoliposomal paclitaxel enhances its efficacy in breast cancer. Trop J Pharm Res. 2014;13(8):1195–1198. doi:10.4314/tjpr.v13i8.1
41. Salimi A, Karami N, Moghimipour E. Liposomes as a novel drug delivery system: fundamental and pharmaceutical application. Asian J Pharm. 2018;12(01):S31–S42.
42. Song H, Su X, Yang K, et al. CD20 antibody-conjugated immunoliposomes for targeted chemotherapy of melanoma cancer initiating cells. J Biomed Nanotechnol. 2015;11(11):1927–1946. doi:10.1166/jbn.2015.2129
43. Alavi SE, Cabot PJ, Moyle PM. Glucagon-like Peptide-1 receptor agonists and strategies to improve their efficiency. Mol Pharm. 2019;16(6):2278–2295. doi:10.1021/acs.molpharmaceut.9b00308
44. Hatamihanza H, Alavi SE, Ebrahimi Shahmabadi H, Akbarzadeh A. Preparation, characterization and immunostimulatory effects of CRD2 and CRD3 from TNF Receptor-1 encapsulated into pegylated liposomal nanoparticles. Int J Pept Res Ther. 2019;1–9.
45. Billiet L, Gok O, Dove AP, Sanyal A, Nguyen L-T-T, Du Prez FE. Metal-free functionalization of linear polyurethanes by thiol-maleimide coupling reactions. Macromolecules. 2011;44(20):7874–7878. doi:10.1021/ma201323g
46. Biju V. Chemical modifications and bioconjugate reactions of nanomaterials for sensing, imaging, drug delivery and therapy. Chem Soc Rev. 2014;43(3):744–764. doi:10.1039/C3CS60273G
47. Timbie KF, Afzal U, Date A, et al. MR image-guided delivery of cisplatin-loaded brain-penetrating nanoparticles to invasive glioma with focused ultrasound. J Controlled Release. 2017;263:120–131. doi:10.1016/j.jconrel.2017.03.017
48. Ghaferi M, Amari S, Vivek Mohrir B, Raza A, Ebrahimi Shahmabadi H, Preparation AS. Characterization, and evaluation of cisplatin-loaded polybutylcyanoacrylate nanoparticles with improved in vitro and in vivo anticancer activities. Pharmaceuticals. 2020;13(44):1–16. doi:10.3390/ph13030044
49. Ghaferi M, Asadollahzadeh MJ, Akbarzadeh A, Ebrahimi Shahmabadi H, Alavi SE. Enhanced efficacy of pegylated liposomal cisplatin: in vitro and in vivo evaluation. Int J Mol Sci. 2020;21(2):559. doi:10.3390/ijms21020559
50. Kobayashi S, Müllen K. Encyclopedia of Polymeric Nanomaterials-With 2021 Figures and 146 Tables. Springer; 2015.
51. Poy D, Akbarzadeh A, Ebrahimi Shahmabadi H, et al. Preparation, characterization, and cytotoxic effects of liposomal nanoparticles containing cisplatin: an in vitro study. Chem Biol Drug Des. 2016;88(4):568–573. doi:10.1111/cbdd.12786
52. Alavi SE, Yap GY, Cabot PJ, Moyle PM. Optimised methods for the production and bioconjugation of site-specific, alkyne-modified glucagon-like peptide-1 (GLP-1) analogs to azide-modified delivery platforms using copper-catalysed alkyne-azide cycloaddition. Bioconjug Chem. 2020;31(7):1820–1834. doi:10.1021/acs.bioconjchem.0c00291
53. Vázquez-Becerra H, Pérez-Cárdenas E, Muñiz-Hernández S, Izquierdo-Sánchez V, Medina LA. Characterization and in vitro evaluation of nimotuzumab conjugated with cisplatin-loaded liposomes. J Liposome Res. 2017;27(4):274–282. doi:10.1080/08982104.2016.1207665
54. Alavi SE, Mansouri H, Koohi Moftakhari Esfahani M, Movahedi F, Akbarzadeh A, Chiani M. Archaeosome: as new drug carrier for delivery of paclitaxel to breast cancer. Ind J Clin Biochem. 2014;29(2):150–153. doi:10.1007/s12291-013-0305-4
55. Cubells MP, Aixela JP, Brumos VG, Pou SD, Flaque MV. Stability of cisplatin in sodium chloride 0.9% intravenous solution related to the container’s material. Pharm World Sci. 1993;15(1):34–36. doi:10.1007/BF02116167
56. Salatin S, Yari Khosroushahi A. Overviews on the cellular uptake mechanism of polysaccharide colloidal nanoparticles. J Cell Mol Med. 2017;21(9):1668–1686. doi:10.1111/jcmm.13110
57. Kong M, Park H, Cheng X, Chen X. Spatial–temporal event adaptive characteristics of nanocarrier drug delivery in cancer therapy. J Controlled Release. 2013;172(1):281–291. doi:10.1016/j.jconrel.2013.08.022
58. Zhang J, Chen XG, Peng WB, Liu CS. Uptake of oleoyl-chitosan nanoparticles by A549 cells. Nanomedicine. 2008;4(3):208–214. doi:10.1016/j.nano.2008.03.006
59. Shah N, Chaudhari K, Dantuluri P, Murthy R, Paclitaxel-loaded DS. PLGA nanoparticles surface modified with transferrin and Pluronic® P85, an in vitro cell line and in vivo biodistribution studies on rat model. J Drug Target. 2009;17(7):533–542. doi:10.1080/10611860903046628
60. Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin-and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood–brain barrier (BBB). Eur J Pharm Biopharm. 2009;71(2):251–256. doi:10.1016/j.ejpb.2008.08.021
61. Gao K, Jiang X. Influence of particle size on transport of methotrexate across blood brain barrier by polysorbate 80-coated polybutylcyanoacrylate nanoparticles. Int J Pharm. 2006;310(1–2):213–219. doi:10.1016/j.ijpharm.2005.11.040
62. Li H, Tong Y, Bai L, et al. Lactoferrin functionalized PEG-PLGA nanoparticles of shikonin for brain targeting therapy of glioma. Int J Biol Macromol. 2018;107:204–211. doi:10.1016/j.ijbiomac.2017.08.155
63. Petri B, Bootz A, Khalansky A, et al. Chemotherapy of brain tumour using doxorubicin bound to surfactant-coated poly (butyl cyanoacrylate) nanoparticles: revisiting the role of surfactants. J Controlled Release. 2007;117(1):51–58. doi:10.1016/j.jconrel.2006.10.015
64. Ambruosi A, Khalansky AS, Yamamoto H, Gelperina SE, Begley DJ, Kreuter J. Biodistribution of polysorbate 80-coated doxorubicin-loaded [14C]-poly(butyl cyanoacrylate) nanoparticles after intravenous administration to glioblastoma-bearing rats. J Drug Target. 2006;14(2):97–105. doi:10.1080/10611860600636135
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.