")
Back to Journals » International Journal of Nanomedicine » Volume 16
In vitro Study on Synergistic Interactions Between Free and Encapsulated Q-Griffithsin and Antiretrovirals Against HIV-1 Infection
Authors Minooei F, Fried JR, Fuqua JL, Palmer KE, Steinbach-Rankins JM
Received 22 October 2020
Accepted for publication 19 December 2020
Published 15 February 2021 Volume 2021:16 Pages 1189—1206
DOI https://doi.org/10.2147/IJN.S287310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Phong A Tran
Farnaz Minooei,1 Joel R Fried,1 Joshua L Fuqua,2– 4 Kenneth E Palmer,3– 5 Jill M Steinbach-Rankins2– 5
1Department of Chemical Engineering, University of Louisville Speed School of Engineering, Louisville, KY, USA; 2Department of Bioengineering, University of Louisville Speed School of Engineering, Louisville, KY, USA; 3Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY, USA; 4Center for Predictive Medicine, University of Louisville, Louisville, KY, USA; 5Department of Microbiology and Immunology, University of Louisville School of Medicine, Louisville, KY, USA
Correspondence: Jill M Steinbach-Rankins
Department of Bioengineering, University of Louisville Speed School of Engineering, 505 S. Hancock St., Room 623, Louisville, KY, 40202, USA
Tel +1 502-852-5486
Email [email protected]
Introduction: Human immunodeficiency virus (HIV) remains a persistent global challenge, impacting 38 million people worldwide. Antiretrovirals (ARVs) including tenofovir (TFV), raltegravir (RAL), and dapivirine (DAP) have been developed to prevent and treat HIV-1 via different mechanisms of action. In parallel, a promising biological candidate, griffithsin (GRFT), has demonstrated outstanding preclinical safety and potency against HIV-1. While ARV co-administration has been shown to enhance virus inhibition, synergistic interactions between ARVs and the oxidation-resistant variant of GRFT (Q-GRFT) have not yet been explored. Here, we co-administered Q-GRFT with TFV, RAL, and DAP, in free and encapsulated forms, to identify unique protein-drug synergies.
Methods: Nanoparticles (NPs) were synthesized using a single or double-emulsion technique and release from each formulation was assessed in simulated vaginal fluid. Next, each ARV, in free and encapsulated forms, was co-administered with Q-GRFT or Q-GRFT NPs to evaluate the impact of co-administration in HIV-1 pseudovirus assays, and the combination indices were calculated to identify synergistic interactions. Using the most synergistic formulations, we investigated the effect of agent incorporation in NP-fiber composites on release properties. Finally, NP safety was assessed in vitro using MTT assay.
Results: All active agents were encapsulated in NPs with desirable encapsulation efficiency (15– 100%), providing ∼ 20% release over 2 weeks. The co-administration of free Q-GRFT with each free ARV resulted in strong synergistic interactions, relative to each agent alone. Similarly, Q-GRFT NP and ARV NP co-administration resulted in synergy across all formulations, with the most potent interactions between encapsulated Q-GRFT and DAP. Furthermore, the incorporation of Q-GRFT and DAP in NP-fiber composites resulted in burst release of DAP and Q-GRFT with a second phase of Q-GRFT release. Finally, all NP formulations exhibited safety in vitro.
Conclusions: This work suggests that Q-GRFT and ARV co-administration in free or encapsulated forms may improve efficacy in achieving prophylaxis.
Keywords: griffithsin, nanoparticles, electrospun fibers, antiretrovirals, synergy, HIV-1 prevention, microbicide
Introduction
Human immunodeficiency virus (HIV) remains a major health challenge, impacting over 1.7 million people each year and ~38 million worldwide.1,2 Women are disproportionately affected by HIV infections, and more than half of the globally infected patients are women living in sub-Saharan Africa.3 In spite of eradication efforts, there is currently no cure or effective vaccine for HIV-1,1 hence a major emphasis has been placed on developing strategies to prevent and treat HIV-1 infection. Given the diversity and magnitude of these unmet reproductive health challenges, one focus has been to evaluate combinations of novel anti-HIV drugs delivered with multipurpose prevention technologies, to simultaneously target multiple sexually transmitted infections (STIs),4 such as herpes simplex virus 2 (HSV-2) and HIV-1.
One of the primary foci in preventing and treating HIV-1 infections has been the development of antiretroviral drugs (ARVs) that inhibit HIV at different stages of the viral infection cycle.5–9 Two ARVs currently used in clinical studies, tenofovir (TFV) and dapivirine (DAP), inhibit HIV-1 by blocking reverse transcriptase,7 while Raltegravir (RAL), another approved drug for HIV treatment, inhibits proviral DNA-strand transfer.10 Moreover, combinations of ARVs have demonstrated enhanced inhibition via different mechanisms of action.11 While ARVs are effective in preventing HIV-1 infection, they have been shown to elicit adverse effects including bone marrow suppression and hematologic effects,12 toxicity with long-term use, and an increased risk of antiviral resistance.13 These adverse effects are often further exacerbated by the poor oral bioavailability and frequent dosing required for many of these drugs, which have contributed to modest outcomes in clinical trials.14
To help overcome these challenges, new biological agents have been investigated to provide protection via different mechanisms of action.15 Antiviral lectins such as actinohivin, scytovirin, and microvirin have demonstrated efficacy against HIV-1 by binding to oligosaccharides linked to viral envelope glycoproteins; however, delivery of these lectins has been challenging due to low bioavailability and induction of an immune response.16 Furthermore, toxicities arising from acute immunological response, due to the administration of foreign proteins, are one of the most significant concerns in adopting protein-based active agents.17
In comparison to these biologics, an antiviral lectin, griffithsin, originally isolated from red alga Griffithsia sp.,18 has demonstrated potent anti-HIV activity in the picomolar range against both laboratory and primary HIV isolates, and is currently being evaluated in clinical trials. Griffithsin inhibits HIV-1 by binding to an envelope glycoprotein (gp120) and inactivating HIV-1 almost immediately upon contact. Studies have shown that GRFT is stable and maintains activity in buffered solutions (pH 4–8) and cervical vaginal lavage fluid at 25°C and 37°C for up to 1 week.19 Additionally, GRFT exhibits excellent safety profiles18,20–23 with negligible induction of pro-inflammatory cytokines23 and has demonstrated synergy with other antiretrovirals,14 suggesting the benefits of future co-administration strategies. Recently, an oxidation-resistant variant of GRFT, Q-GRFT, was shown to retain the biophysical, antiviral, and safety properties of wild-type GRFT, making it an even more suitable HIV microbicide candidate.24,25
Prophylactic and therapeutic strategies that use a combination of anti-HIV drugs to target infection have been shown to enhance the intracellular efficacy of each drug26,27 and decrease the transmission risk of antiviral-resistant viruses.14 One study demonstrated that a combination of multiple nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse-transcriptase inhibitors (NNRTIs), such as TFV and Efavirenz (EFV), provided synergistic inhibition of HIV-1, as well as an increase in intracellular drug efficacy.27 Additionally, GRFT has shown synergistic effect in combination with maraviroc, TFV, and EFV, requiring lower GRFT doses when used in combination. The strongest synergy was observed between GRFT and TFV (CI = 0.34) suggesting that a combination of GRFT and other active agents may provide a promising strategy to inhibit virus transmission and replication via different mechanisms of action.14
Regardless of activity, active agents require formulation into an acceptable dosage form that protects agent stability and potentially provides extended release. Current ARVs have been incorporated into short-acting or on-demand dosage forms such as tablets and gels, and long-acting sustained-release vehicles such as intravaginal rings (IVRs). However, oral ARVs incorporated in pre-exposure prophylaxis (PrEP) regimens have only been moderately successful in preventing, and to date do not cure HIV-1 infections. A variety of clinical studies28–33 have shown that the efficacy of oral PrEP ranges from 0% to 75%, depending in large part on user adherence.34
As an alternative to daily oral PrEP administration, topical intravaginal PrEP technologies (eg, gels, IVRs) have shown success in delivering active agents to the female reproductive tract (FRT). While clinical trials have demonstrated promising levels of protection for gels, efficacy has been shown to be significantly impacted or abbreviated by a lack of user adherence, due to the required frequent administration regimens and lack of retention attributed to product leakage.35 In comparison, IVRs can provide long-term protection against STIs and pregnancy (>1 month) by offering longer delivery durations and high durability in the FRT. However, clinical trials have reported some challenges regarding using IVRs such as vaginal discomfort, ease of product placement, low pliability, and hygiene issues,36 further emphasizing the need to design alternative delivery platforms with potentially increased convenience and ease of use.
Thus far, short-term safety studies with GRFT have demonstrated outstanding preclinical safety in vitro and in vivo23,37,38; however currently, there are no available delivery vehicles that prolong GRFT (or Q-GRFT) delivery to achieve efficacy beyond 72 hr post-administration. To begin to address these challenges, we and others have shown that polymeric nanoparticles (NPs) and electrospun fibers provide sustained-delivery, with the potential to safely incorporate and enhance the transport of biologics.39–61 Polymeric NPs have been investigated to encapsulate ARVs and biologics for HIV-1 treatment62–65 and have been shown to improve delivery to vaginal tissue by overcoming challenges such as agent instability; low cellular internalization and tissue distribution; and frequent dosing; while eliciting minimal immune response in the FRT.53,64–67 Polymeric fibers that encapsulate active agents have also been recently applied to intravaginal drug delivery, demonstrating sustained-delivery and the potential to safely incorporate biologics.47,68 Despite this, to date, there are no food and drug administration (FDA)–approved NP or fiber delivery platforms available to provide topical STI prevention.45,47,69 Moreover, a platform that incorporates the attributes of both platforms, such as NP-fibers, may improve delivery by providing a reservoir to retain NPs and decrease NP clearance during shedding.
Despite being well-established technologies used successfully as durable stents, scaffolds, and delivery reservoirs for long-term implantation and delivery in vivo,55,70,71 NP-fiber composites have only recently been explored for sustained-delivery to the FRT.69 Several groups have evaluated multilayered fibers for the delivery of active agents,72,73 and have shown that nano- (often hydrophilic) and micro-scale (often hydrophobic or higher MW polymers) can impart greater mechanical stability and flexibility compared to a single layer of fibers.74 Other work75–77 has investigated multilayered fibers and demonstrated time-regulated release of active agents. For intravaginal delivery specifically, an approach utilizing stacked fibers was evaluated for short-term (60 hr) intravaginal delivery, in part to demonstrate how individual layers pressed together post-spinning may modulate release.59 Recently, we developed a multilayered NP-fiber composite, that incorporates poly(lactic-co-glycolic) acid (PLGA) NPs, that sustains the delivery of GRFT in vitro, relative to sustained-delivery from NPs over ~7 days. In addition to the potential for sustained-delivery in the order of months, this NP-fiber architecture demonstrated efficacy against HIV-1 infections in vitro and protection against a lethal dose of HSV-2 in a murine model. Based on these studies, NP-fiber composites can be used to tailor the release of active agents and provide long-term efficacy.
In this work, we sought to examine the delivery of Q-GRFT and three different ARVs (eg, TFV, RAL, and DAP) to identify unique protein-drug synergies between Q-GRFT and these active agents delivered in free or encapsulated forms. We selected these ARVs due to their different solubilities and mechanisms of action against HIV-1. TFV is a hydrophilic drug commonly used in vaginal gels; DAP is a hydrophobic agent that is widely used in IVRs against both HIV-1 and HSV-2 infections, and RAL is a small hydrophilic molecule that is used late in pregnancy to reduce the risk of transferring HIV from mother to child.78 We demonstrated that the co-administration of free or encapsulated active agents with different mechanisms of action may increase the prophylactic effect and decrease the dose needed to attain efficacy against HIV-1 infection. Moreover, given the potential of NP-based platforms to modulate active agent release, we sought to preliminarily assess the release of different active agent agents from NPs and selected the most synergistic NP formulations for incorporation in multilayered NP-fiber composites.
Materials and Methods
Materials
Poly(lactic-co-glycolic) acid used for synthesizing NPs (50:50, 0.55–0.75 dL/g, 31–57k MW) was purchased from Lactel Absorbable Polymers (Cupertino, CA). Polycaprolactone (PCL, MW=80,000 Da), polyethylene oxide (PEO, MW=600,000 Da), TFV and DAP were purchased from Sigma-Aldrich (St Louis, MO), while RAL was provided from Selleckchem (Houston, TX). Q-GRFT was kindly provided by Dr. Fuqua (University of Louisville, KY). Dichloromethane (DCM) and keratinocyte serum-free medium (KSFM) were purchased from Thermo Fisher (Waltham, MA). Other chemicals, including dimethyl sulfoxide (DMSO), acetonitrile HPLC grade (ACN), trifluoroacetic acid (TFA), and thiazolyl blue tetrazolium bromide (MTT), were obtained from Sigma Aldrich.
Nanoparticle Fabrication
Blank NPs and NPs loaded with Q-GRFT, TFV, RAL or DAP were fabricated using either single- or double-emulsion technique. For each formulation, 100 mg PLGA was dissolved in 2 mL of DCM and incubated overnight. For Q-GRFT, TFV, and RAL NPs, 10 mg of the active agent was dissolved in 200 μL Tris-EDTA (TE) buffer. For DAP NPs, 10 mg of DAP was dissolved in 200 μL DCM. The active agent solution was added dropwise to the polymer solution while the solution was vortexed. Then, the active agent-polymer solution was sonicated and added to 5% PVA in Milli-Q water (2 mL), vortexed and sonicated again. The emulsified solution was then added to a larger volume of aqueous solution (50 mL of 0.3% PVA) and incubated for 3 hr while stirring to evaporate DCM. Then, NPs were centrifuged (25,364 ×g for 10 min at 4°C), washed twice using 30 mL Milli-Q water, suspended in 5 mL Milli-Q water and stored at −80°C for 2 hr. Finally, NPs were lyophilized to obtain solid particles and were subsequently stored at −20°C.
NP-Fiber Composite Fabrication
To prepare the electrospinning solution, PEO and PCL were dissolved in DI water (5% w/w) and TFE (11% w/w), respectively, and incubated at 37°C overnight. Prior to electrospinning, 10% w/w of PLGA NPs were added to the PEO solution and the solution was electrospun with a flow rate of 0.3 mL/hr and voltage of 25 kV. Pre-cut NP-PEO fibers (3–5 mg) were placed on the freshly electrospun PCL layer followed by electrospinning another layer of PCL fiber on top to make a sandwich-shaped structure. The electrospinning conditions for PCL layers were set to 15 cm needle to collector distance, flow rate of 2.2 mL/hr, and voltage of 20 kV. The NP-fiber composite was desiccated overnight and stored at 4°C.
NP Characterization: Size, Loading, and Release Size
The size and morphology of unhydrated NPs were determined using scanning electron microscopy (SEM). First, samples were sputter-coated with a palladium/gold alloy layer with a thickness of 15 to 18 nm, and imaged using a Supra 35 SEM (Zeiss, Oberkochen, Germany). The average size of unhydrated NPs was evaluated with ImageJ software (NIH, Bethesda, Maryland) by measuring the diameter of 50 NPs per image and 3 images for each formulation.
Loading
To assess the loading of NPs, 1–2 mg of NPs were dissolved in 100 μL DMSO and diluted with TE buffer to the final volume of 1 mL. Serial 1:2 sample dilutions were then made in TE buffer. To quantify the amount of TFV, RAL, and DAP, spectrophotometric absorbance was measured at 260, 300 and 290 nm, respectively, on a Synergy HT reader (BioTek, Winooski, VT, USA). To quantify the amount of Q-GRFT, an enzyme-linked immunosorbent assay (ELISA) was used. Briefly, 96-well Nunc Maxisorp plates were incubated with 100 µL of 250 ng/mL gp120 in 1x phosphate-buffered saline (PBS) as a coating buffer and stored at 4°C overnight. Then, plates were blocked with 300 μL of 3% w/v bovine serum albumin (BSA) in PBS containing 0.05% Tween-20 (1x PBST) and incubated at room temperature for 2 hr. Plates were then washed using a Gardner Denver Multiwash III plate washer (Milwaukee, WI) followed by adding 100 μL of the samples. Afterwards, 100 μL of rabbit anti-GRFT (1:10,000) and goat anti-rabbit IgG-HRP (1:20,000) were subsequently added after 1 hr incubation at 37°C to detect gp120-bound Q-GRFT. Finally, 100 µL of SureBlue TMB microwell peroxidase substrate (Sera Care, Milford, MA, USA) was added to each well and the reaction was quenched by adding 100 µL of 1 N H2SO4 (Thermo Fisher) followed by reading the absorbance at 450 nm.
In vitro Release
The release of Q-GRFT, TFV, RAL, and DAP loaded PLGA NPs was determined over 30 days in simulated vaginal fluid (SVF, pH 4.5). Three to five milligrams of NPs were dispersed in 1 mL of SVF in 1.5 mL centrifuge tubes and incubated at 37°C with continuous shaking (150 rpm). At each time point, samples were centrifuged at 18,500 ×g at 4°C for 10 min, the eluates were collected, and NPs were resuspended in 1mL of fresh SVF. Similarly, the release of the active agent from NP-fiber constructs was evaluated by placing a 5 mg fiber composite piece in a 5-mL microcentrifuge tube filled with 1 mL SVF. The samples were incubated on a shaker at 150 rpm at 37°C, under sink conditions. At each time point, the solution was replaced with fresh SVF and release from NP-fiber composites was assessed for up to 90 days. The release of Q-GRFT, from NPs and NP-fiber composites, was quantified via ELISA as mentioned before. For quantifying DAP released from NP-fiber composites, reverse phase high-performance liquid chromatography with UV detection (HPLC-UV) was used (Agilent, CA, USA). First, the composite eluates in SVF were diluted with ACN to obtain a 1:1 SVF:ACN v:v ratio. Then, 50 µL of sample was injected into a Jupiter C18 300 A-C18 column (250 ×4.6 mm; Agilent Technologies, CA, USA). The mixture of 65% of 0.01 v/v% TFA and 35% ACN was used as mobile phase with the flow rate of 1 mL/min and the running time for DAP was 22 min. Finally, DAP was detected via absorbance at 290 nm and the concentration of DAP in unknown samples was determined by plotting the area under the curve versus concentration. The standard curve over the range of 0.08 to 10 µg/mL was determined to be linear.
Cell Lines, Virus and Cell Culture
TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program (ARRRP) and were used for HIV infection. These cells are engineered HeLa cells that express CD4, CCR5, and CXCR4 receptors and have Tat-responsive reporter genes for firefly luciferase (Luc) that allows accurate measuring of HIV-1 infection.26 TZM-bl cells were maintained in Dulbecco’s Modified Eagle medium (DMEM, from VWR) supplemented with 10% fetal bovine serum (FBS), 25 mM HEPES buffer, and 50 μg/mL gentamicin (Thermo Fisher). Vaginal epithelial (VK2/E6E7), End1/E6E7 (End1), and ectocervical, Ect1/E6E7 (Ect1) cell lines were provided courtesy of Dr. Kenneth Palmer from the University of Louisville, and were originally from ATCC (Rockville, MD). VK2, End1, and Ect1 cells were cultured in Keratinocyte Serum-Free (KSFM) media containing recombinant human epidermal growth factor (0.1 ng/mL), bovine pituitary extract (50 μg/mL), calcium chloride (0.4 mM) (Thermo Fisher), and 1% penicillin and streptomycin. HEK-293T/17 cells (ATCC, Rockville, MD) were used for HIV production and were cultured in minimum essential medium (MEM) supplemented with 10% FBS, and 1% penicillin (100 µg/mL) and streptomycin (100 µg/mL) (VWR Radnor, PA). The HIV-1 pseudovirus was made in house by transfecting HEK293T/17 cells with envelope (env)-expressing plasmid (CCR5-tropic clade A strain, Q769.h5), env-deficient HIV-1 backbone vector (HIV-1pNL4-3.Luc.R-E-, NIH AIDS Reagent Program, 11884 and 3418) and Mirus reagent (Mirus, Madison, WI). After incubating cells with transfection complex for 72 hr at 37°C, the cells were centrifuged (200 ×g, 5 min) and the supernatant containing viral particles was collected, filtered using a 0.45-μm membrane filter (VWR), and stored at −80°C.
In vitro HIV Inhibition
To evaluate the anti-HIV-1 activity of anti(retro)viral NPs, an in vitro HIV inhibition assay was conducted by infecting TZM-bl cells with HIV-1 pseudovirus (CCR5-using clade A strain Q769.h5) and quantifying the luciferase activity of cell lysates. Briefly, the desired amount of free or encapsulated active agent was dissolved in 1 mL of DMEM, followed by 1:2 serial dilutions to a final volume of 50 μL. Then, 100 μL of cell solution (106 cells per plate) was added to each well. After incubating the cells and treatment for 1 hr at 37°C, 50 μL of virus solution was added to each well. Untreated cells only and virus-infected cells were used as negative and positive controls of infection, respectively. After 48 hr incubation at 37°C, 100 µL culture medium was replaced with 100 µL Bright-Glo reagent solution (Promega Corporation, Madison, WI) and luminescence was measured after 3 min. HIV-1 infection was determined based on the luminescence deviations from the virus-infected cell control.
In vitro Cytotoxicity
An MTT assay was used to determine the cytotoxicity of blank and anti(retro)viral NPs (0.1 and 1 mg/mL) administered to VK2, End1, and Ect1/E6E7 cell lines. First, cells were plated at a density of 150,000 cells/well in 24-well plates and incubated for 24 hr at 37°C, followed by adding treatments in triplicate. Media only (no treatment) and 10% DMSO were used as viable and non-viable cell controls. After 24, 48, and 72 hr incubation at 37°C, 55 µL of MTT reagent was added to each well and incubated at 37°C for 4 hr, followed by adding 100 µL of lysis buffer containing 10% sodium dodecyl sulfate (Fisher Scientific) and 0.01 M hydrochloric acid. After overnight incubation, the absorbance was read at 570 nm and normalized to cell-only absorbance to attain the relative percent of cell viability.
Combination Effect
The activity of the active agents in combination was determined using the combination index (CI). First, the half-maximal inhibitory concentration (IC50) value of each drug in free and encapsulated form was measured with the HIV inhibition assay. Then, each drug was mixed with either Q-GRFT or Q-GRFT NPs in an equipotent IC50 ratio (1:1 ratio of IC50). For NPs, the equipotent IC50 value was determined based on NP loading. After measuring the IC50 values of each active agent in combination, the Chou–Talalay method was used to measure CI.79–81
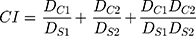
When Q-GRFT and the drug are in free form, Ds1 and Ds2 represent the IC50 of free Q-GRFT and the free drug applied separately, while DC1 and DC2 are the IC50 values of free Q-GRFT and free drug applied in combination, respectively. When Q-GRFT NPs are co-administered with free drug, Ds1 and Ds2 represent the IC50 of Q-GRFT NPs and the free drug when applied separately, while DC1 and DC2 are the IC50 values of Q-GRFT NPs and free drug when they are co-administered. Finally, in the case of Q-GRFT NPs and encapsulated drug co-administration, Ds1 and Ds2 represent the IC50 of Q-GRFT NPs and drug NPs, while DC1 and DC2 are the IC50 values of Q-GRFT NPs and drug NPs in combination, respectively. Values of CI > 1, CI = 1, and CI < 1 determine additive, antagonistic, and synergistic interactions, respectively.
Statistical Analysis
All experiments were done in triplicate and GraphPad Prism (GraphPad Software, La Jolla, CA) was used for data analysis. Unless otherwise noted, three independent replicates were assessed for each sample and statistical significance was evaluated using a one-way ANOVA test (Bonferroni), and p-values ≤0.05 were considered statistically significant.
Results
NP Characterization
PLGA NPs formulated with 100 µg of ARVs (TFV, RAL, or DAP) or Q-GRFT per mg of NP were successfully synthesized with high yield (>70%) and spherical morphology. SEM images of ARV and Q-GRFT NPs are shown in Figure 1, demonstrating NP sizes ranging from 98.9 to 107.2 nm. Furthermore, nanoparticle loading efficiency varied with the active agent (Table 1); Q-GRFT, TFV, RAL, and DAP NPs were loaded with 48.9 ± 12.7, 15.4 ± 3.3, 103.5 ± 11.9, and 60.2 ± 2.7 µg active agent/mg NP, respectively, directly corresponding to the encapsulation efficiency.
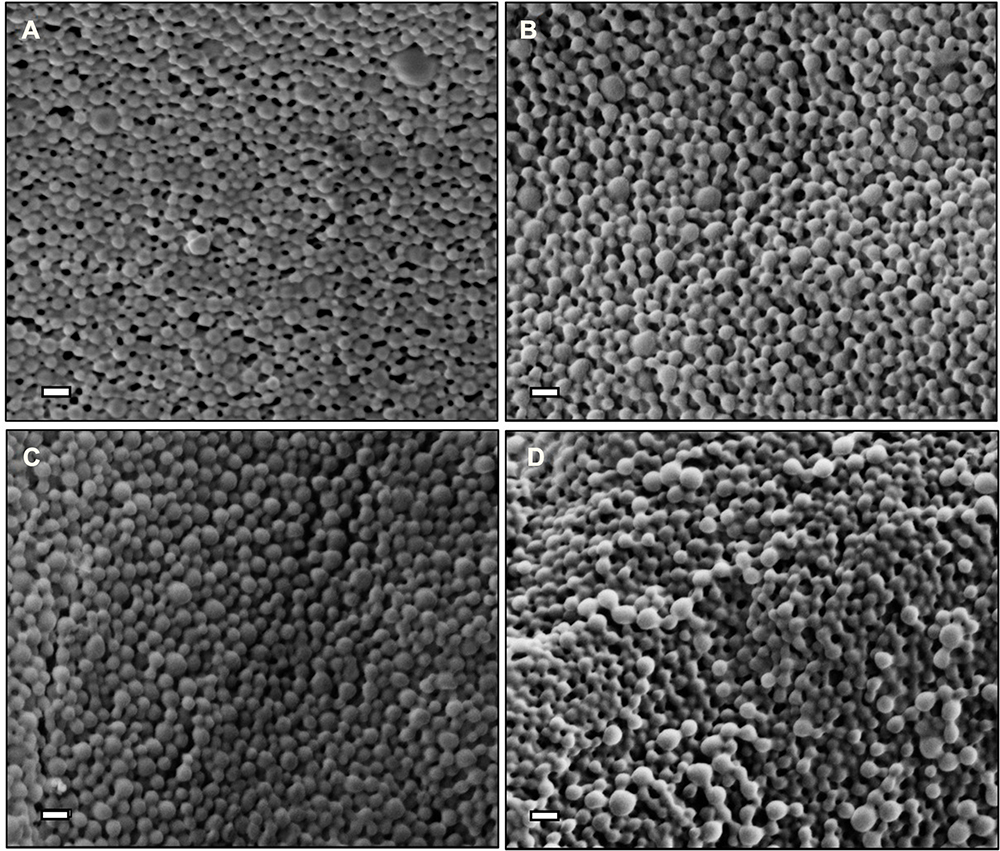 |
Figure 1 Scanning electron microscopy images of PLGA nanoparticles loaded with 10% w/w (A) Q-GRFT, (B) TFV, (C) RAL, and (D) DAP. Scale bars represent 200 nm. |
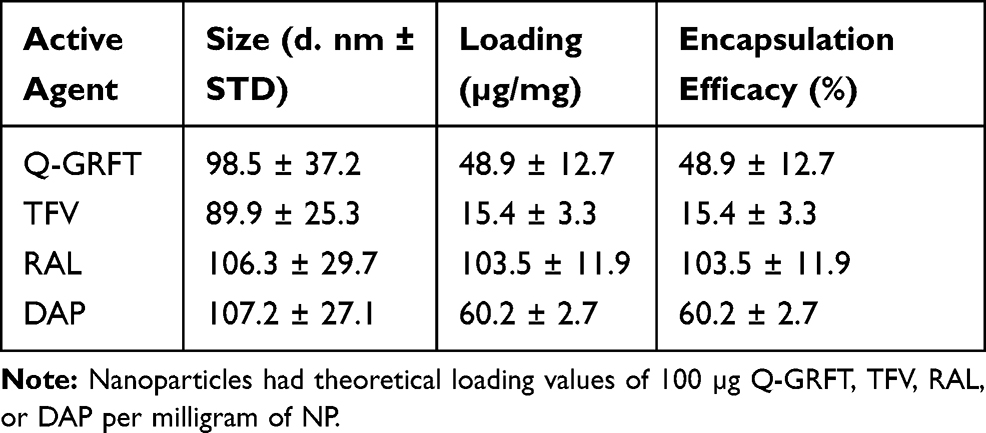 |
Table 1 Size, Loading, and Encapsulation Efficacy of PLGA Nanoparticles |
In vitro Release from NPs
The cumulative release of each active agent as a function of mass and percent total loading, from PLGA NPs over 4 weeks in SVF is shown in Figure 2. For all formulations, a burst release was observed within the first 8 hr, followed by a more gradual release over 2 weeks. Specifically, 2.51, 1.72, 8.96, and 8.99 µg Q-GRFT, TFV, RAL, and DAP per mg of NPs (representing 5%, 11%, 9%, and 15% of actual loading) were released after 8 hr. Over 2 weeks, the total cumulative release of Q-GRFT, TFV, RAL, and DAP from their respective NP formulations was 4.64, 2.64, 11.84, and 13.41 µg/mg, resulting in less than 20% release of each active agent.
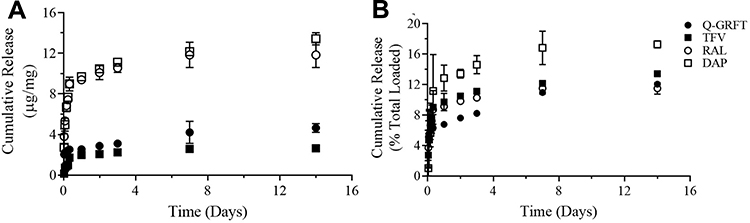 |
Figure 2 The cumulative release of Q-GRFT, TFV, RAL, and DAP from PLGA nanoparticles as a function of (A) total active agent release or (B) the percent of total loading, after exposure to SVF for up to 14 d. Release values are shown as the mean ± standard deviation of three independent NP batches. Please note panel B y-axis is scaled to 20% to more easily visualize differences in release. |
In vitro HIV-1 Inhibition Studies
Individual Administration of Q-GRFT or ARVs in Free or Encapsulated Form
The ability of individual active agents to protect against infection in free and encapsulated forms was evaluated by the administration of free active agent to TZM-bl cells for 1 hr, followed by virus administration. The IC50 values of free Q-GRFT, TFV, RAL, and DAP were 57.2 ± 13.8, 382.3 ± 39.6, 3.3 ± 0.2, and 0.034 ± 0.008 ng/mL, respectively, while the encapsulation of individual in agents in NPs generally decreased the IC50 values to 14.0 ± 4.1, 273.7 ± 78.8, 3.0 ± 0.5, and 0.043 ± 0.0007 ng/mL (Figure 3). For all ARVs, the IC50 values of free and encapsulated agents (the latter, based on release after 48 hr in congruence with administration duration in the pseudovirus assay) were similar (p > 0.05), while the IC50 values of Q-GRFT NPs were ~4-fold lower than observed for free Q-GRFT (p ≤ 0.05) (Table 2).
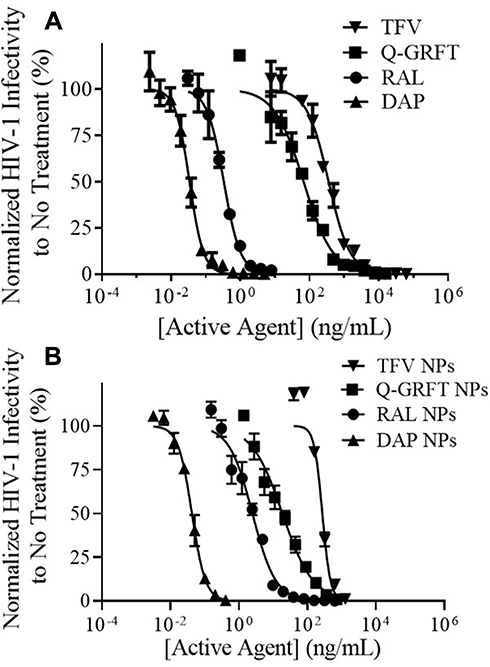 |
Figure 3 The IC50 curves of (A) free and (B) NP-encapsulated active agents after administration to TZM-bl cells 1 hr prior to HIV-1 pseudovirus infection. Infectivity values are normalized to uninfected cells and are shown as the mean ± standard deviation of three NP batches. |
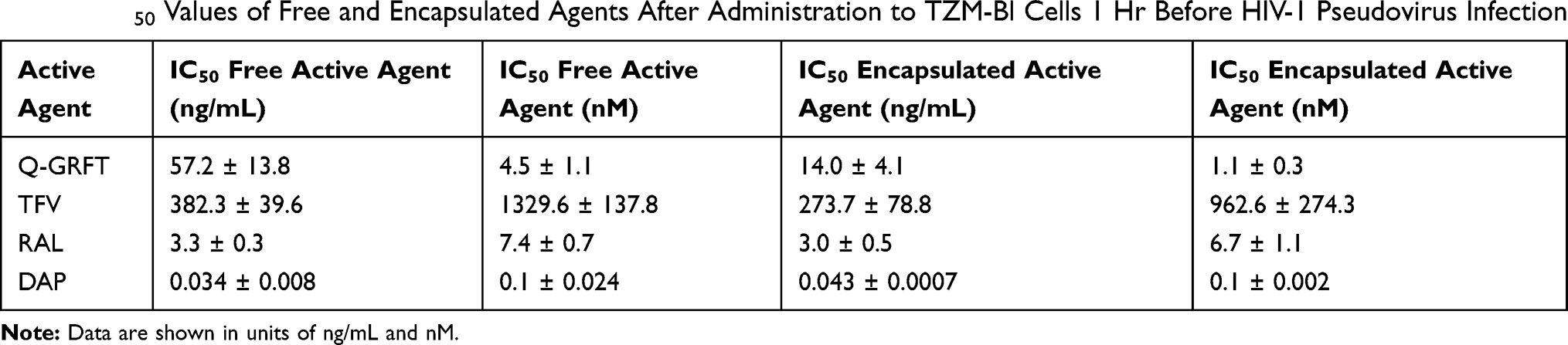 |
Table 2 IC50 Values of Free and Encapsulated Agents After Administration to TZM-Bl Cells 1 Hr Before HIV-1 Pseudovirus Infection |
Combinations of Q-GRFT with Each ARV
Q-GRFT was subsequently co-administered with each ARV at an equipotent IC50 value ratio to compare the combined efficacy with each active agent. As shown in Figure 4, when free Q-GRFT was co-administered with free ARVs, all IC50 curves were shifted to the left, demonstrating that a lower dose of each agent is able to achieve similar efficacy. When free Q-GRFT was co-administered with free TFV, RAL, or DAP, the IC50 of Q-GRFT decreased by 6.3-, 4.4-, and 3.7-fold, respectively, relative to the administration of Q-GRFT alone (p ≤ 0.05). Similarly, the IC50 values of free TFV, RAL, or DAP co-administered with free Q-GRFT decreased by 2.8-, 5.6-, and 4.6-fold, respectively, relative to the administration of free drug alone (p ≤ 0.05).
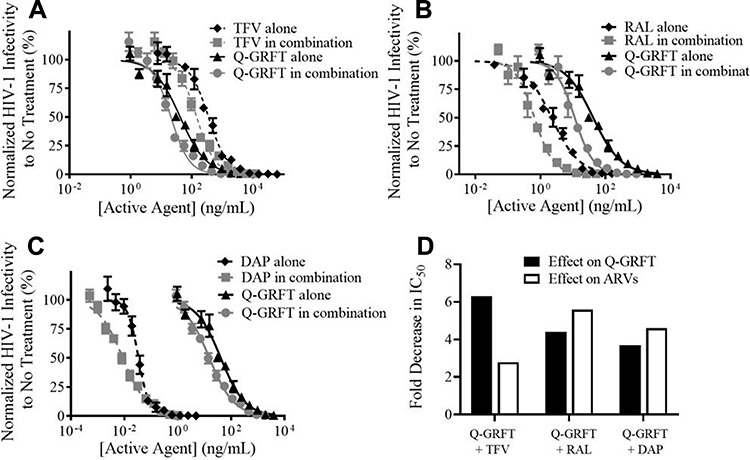 |
Figure 4 The IC50 curves for free ARVs and free Q-GRFT co-administration demonstrate synergistic interactions. (A) Free TFV + free Q-GRFT, (B) free RAL + free Q-GRFT and (C) free DAP + free Q-GRFT. (D) Fold decrease in the IC50 values of Q-GRFT and ARVs after co-administration to TZM-bl cells 1 hr prior to HIV-1 pseudovirus infection. The normalized infectivity values are shown as the mean ± standard deviation of three independent samples. Please note differences in log scale on the x-axis. |
Combinations of Q-GRFT NPs with Each ARV
The activity Q-GRFT NPs with each free ARV was assessed using the equipotency ratio of Q-GRFT NPs to each ARV, with the IC50 of Q-GRFT NPs here expressed based on the 48 hr release value. As shown in Figure 5, the IC50 values of Q-GRFT NPs decreased by 6.6, 4.3, and 2.2-fold when co-administered with free TFV, RAL, or DAP, respectively, relative to the administration of Q-GRFT NPs alone. The IC50 values of free TFV and RAL co-administered with free Q-GRFT NPs decreased by 1.6 and 1.4-fold, relative to free TFV and RAL; however, minimal reduction (~0.5-fold) was observed in the IC50 value of DAP.
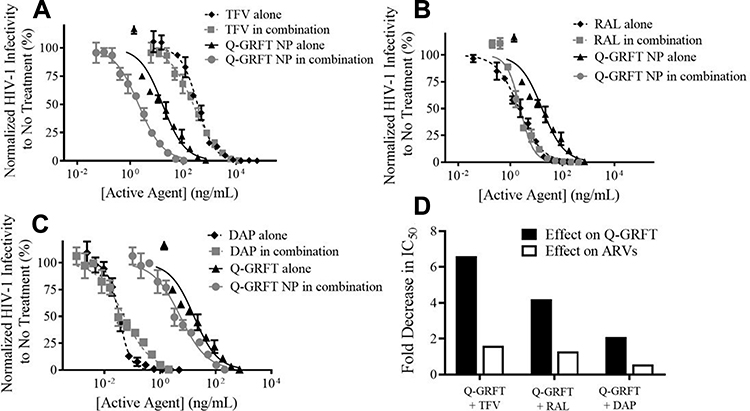 |
Figure 5 The IC50 curves for free drugs and Q-GRFT NP co-administration demonstrate synergistic interactions. (A) Free TFV + Q-GRFT NPs, (B) free RAL + Q-GRFT NPs and (C) free DAP + Q-GRFT NPs. (D) Fold decrease in the IC50 values of Q-GRFT NPs and ARVs after co-administration to TZM-bl cells 1 hr prior to HIV-1 pseudovirus infection. The normalized infectivity values are shown as the mean ± standard deviation of three independent samples. Please note differences in log scale on the x-axis. |
Combinations of Q-GRFT NPs with ARV NPs
Lastly, the efficacy of Q-GRFT NPs and individual ARV NP groups was determined using the equipotency ratio of Q-GRFT NPs to each ARV NP, with the IC50s of Q-GRFT NPs and ARV NPs expressed based on respective 48 hr release values. A significant shift was observed in the IC50 curves after Q-GRFT NP and ARV NP co-administration (Figure 6). Specifically, a 2.5, 2.7, and 6.0-fold reduction in Q-GRFT NP IC50 values was observed when they were co-administered with TFV NPs, RAL NPs, or DAP NPs. In addition, co-administration of TFV NPs, RAL NPs, or DAP NPs with Q-GRFT NPs resulted in 3.3, 2.3, and 8.6-fold reduction in the IC50 of each encapsulated drug in combination, relative to each encapsulated drug alone. In general, although all NPs showed synergistic interactions when administered at an equipotency ratio, the most synergistic interactions were observed for DAP NPs and Q-GRFT NPs, resulting in 8.6 and 6.1-fold decreases in IC50 respectively, relative to individual DAP NPs and Q-GRFT NPs. The summary of IC50 values of different active agents administered individually, relative to the IC50 values after co-administration in free and encapsulated forms, is shown in Table 3.
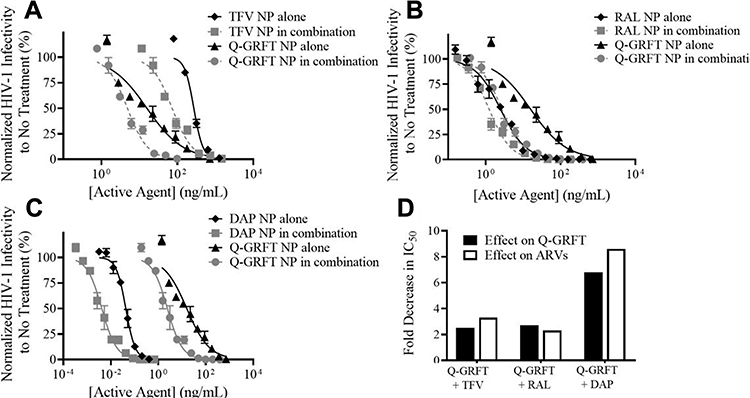 |
Figure 6 The IC50 curves for ARV NP and Q-GRFT NP co-administration. (A) TFV NPs + Q-GRFT NPs, (B) RAL NPs + Q-GRFT NPs and (C) DAP NPs + Q-GRFT NPs. (D) Fold decrease in the IC50 values of Q-GRFT NPs and ARV NPs after co-administration to TZM-bl cells 1 hr prior to HIV-1 pseudovirus infection. The normalized infectivity values are shown as the mean ± standard deviation of three independent samples. Please note differences in log scale on the x-axis. |
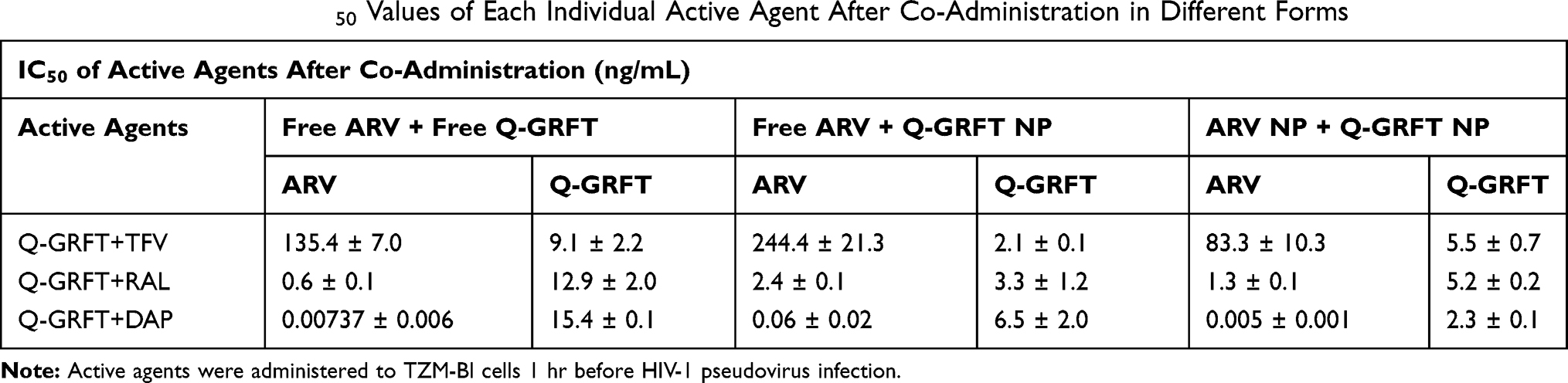 |
Table 3 Summary of the IC50 Values of Each Individual Active Agent After Co-Administration in Different Forms |
Evaluation of Synergistic Interactions Based on CI Calculations
The CI value was calculated using the Chou–Talalay method based on the ratio of IC50 values for each active agent when administered separately and in combination (Figure 7).
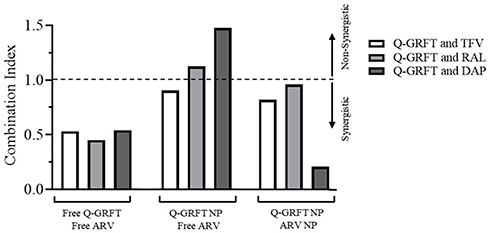 |
Figure 7 Combination indices of free Q-GRFT and free ARVs, Q-GRFT NPs and free ARVs, and Q-GRFT NPs and ARV NPs co-administered to TZM-bl cells. |
Combinations of Q-GRFT with Each ARV
When Q-GRFT was co-administered with free TFV, RAL, or DAP, the corresponding CI values were 0.53, 0.45, and 0.54, indicating similarly strong synergistic interactions between all protein-drug combinations.
Combinations of Q-GRFT NPs with Each ARV
When Q-GRFT NPs were administered in combination with free TFV, RAL, or DAP, the calculated CI values were 0.9, 1.12, and 1.47 showing synergistic interactions between Q-GRFT NPs and TFV.
Combinations of Q-GRFT NPs with ARV NPs
Co-administration of Q-GRFT NPs with ARV NPs demonstrated strong synergistic interactions with DAP NPs (CI = 0.21) and more modest synergistic interactions with TFV NPs and RAL NPs (CI = 0.82 and 0.96, respectively). Among the different combined treatment groups, the strongest synergistic interaction was observed for Q-GRFT NPs when administered with DAP NPs (CI = 0.21).
Assessment of NP-Fiber Composites
Based on the most synergistic formulation evaluated above, nanoparticles containing 10% of either Q-GRFT or DAP were successfully incorporated into the PEO portion of multilayered fibers to demonstrate the release of active agents over a longer duration. Q-GRFT and DAP were added in a ratio of 1:10 w:w NP:PEO fiber (1 mg NP:10 mg fiber) to obtain a final concentration of 10 µg active agent/mg NP-fiber composite (based on 100 µg active agent/mg NP and 100 µg NP/mg PEO fiber loading). The actual loading of Q-GRFT and DAP in the NP-fiber composite was 6.9 ± 1.9 and 7.1 ± 0.46 µg/mg, respectively. As shown in Figure 8, DAP was fully released within the first few days, with negligible release over 3 months. For Q-GRFT NP-fiber composites, a burst release of 0.68 µg Q-GRFT/mg PEO was observed in the first day, followed by minimal release over 42 days. A second phase of release was observed between days 42 to 63, followed by a gradual release for up to 90 days. The release values from most of the time points, for a 5 mg fiber-composite piece was within the in vitro IC50 range of free Q-GRFT and DAP (57 and 0.03 ng/mL, in TZM-bl cells), with the exception of the 8 and 72 hr time points for Q-GRFT (30 and 16 ng/mL) and 14 d, 63 to 84 d, in which the release was below the detection limit (78 ng/mL).
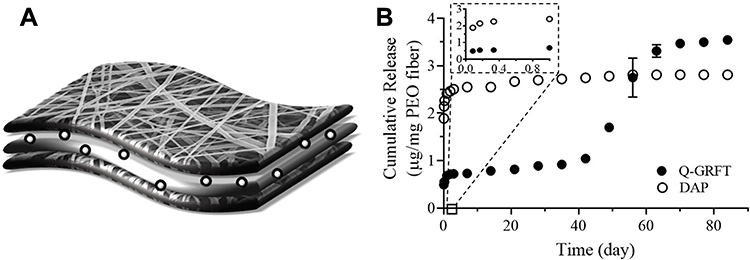 |
Figure 8 (A) Schematic of NP-fiber composites in which spheres may depict Q-GRFT NPs or DAP NPs. (B) The cumulative release of Q-GRFT and DAP from NP-fiber composites with an initial theoretical loading of 10 µg active agent per mg PEO fiber. Release values are shown as the mean ± standard deviation of three independent NP-fiber batches. |
In vitro Cytotoxicity Studies
Blank NPs, as well as Q-GRFT and ARV NPs were evaluated for cytotoxicity at the highest concentrations used for cell inhibition assays (1 mg/mL). No significant decreases in viability were observed in the viability of VK2, Ect1, and End1/E6E7 cell lines, after treatment with Q-GRFT, TFV, or RAL NPs, relative to untreated cells. These groups showed greater than 92% viability after 24, 48, and 72 hr NP administration; however, a significant decrease in cell viability was observed in VK2 cells treated with 1 mg/mL DAP NPs after 48 and 72 hr (p ≤ 0.01). In contrast, the negative control for viability (10% DMSO) resulted in dramatically decreased cell viability spanning 7% to 18% across different cell lines (Figure 9, p ≤ 0.05).
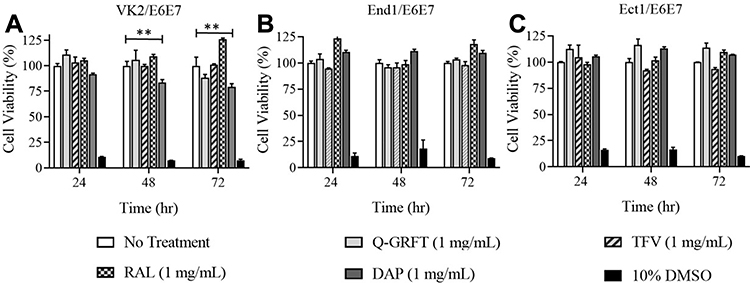 |
Figure 9 In vitro cytotoxicity of NPs encapsulating different agents administered to (A) VK2/E6E7 (B) End1/E6E7, and (C) Ect1/E6E7 cell lines using MTT assay. Viabilities are shown as the mean ± standard deviation from administration of three independent samples. Statistical significance between experimental groups, as calculated by one-way ANOVA, is represented by **p ≤ 0.01. |
Discussion
The co-administration of multiple active agents offers the potential to address some of the challenges surrounding PrEP, including low user adherence, high toxicity, and modest efficaciousness, by enabling virus targeting via different mechanisms of action and a corresponding decrease in the required dose and adaptation to drug resistance. Due to these benefits, synergistic interactions between a variety of ARVs have been studied to evaluate the effectiveness of a multi-agent approach to treat STIs such as HIV-1.14,57,82–84 While the efficacy of TFV, RAL, or DAP in combination with other active agents including lamivudine, emtricitabine, and maraviroc has been investigated in clinical trials,85–87 the combination of these ARVs with biologics, such as Q-GRFT, has been lesser explored.83 To date, there are only two studies that have evaluated the potential synergy between GRFT, a potent antiviral biologic, and ARVs for HIV-1 prevention.14,83 Furthermore, a new oxidation-resistant variant of GRFT (Q-GRFT) was developed which has not been investigated in combination with other active agents. Lastly, only one study has evaluated the impact of wild-type GRFT (relative to Q-GRFT) and ARV NP co-administration. Therefore, the goal of this study was to evaluate the synergy between the free and encapsulated forms of Q-GRFT and a few key “model” ARVs, which are used in oral PrEP and span different hydrophilicities and mechanisms of action.
While most ARVs inhibit virus post-cell entry, Q-GRFT is a biologic entry inhibitor with the potential to inhibit virus at the source of infection, prior to mucus penetration and cell entry. Due to the different mechanisms of action and the potential for early intervention with Q-GRFT, co-administration of Q-GRFT with established ARVs may enhance prophylactic and therapeutic outcomes.
Yet despite the promise of Q-GRFT and other antiviral agents, challenges such as the lack of consistent user adherence – often requiring frequent administrations and high dosing – and inadequate transport to target tissue, limit the utility of these agents in free form in clinical translation. To address these challenges, different dosage forms have been used to provide combinational delivery of these agents for HIV treatment. As two examples, polymeric films and IVRs have been used to deliver combinations of different active agents including TFV, DAP, and maraviroc85–88 for rapid versus sustained-delivery applications. For applications requiring delivery to physiologically complex environments, such as the FRT, polymeric NPs have been shown to dramatically enhance the transport of biologics and other active agents, and to enhance agent instability and cellular internalization.64,66,89 A recent study in our lab showed that PLGA NPs can encapsulate GRFT with high loading and showed promising efficacy against HIV-1 in vitro and HSV-2 in vivo.69 A recent study external to our group83 evaluated the synergy between GRFT and DAP in free, encapsulated, and co-encapsulated forms, demonstrating strong synergy between GRFT and DAP co-encapsulated in PLGA NPs at an equipotent IC50 value ratio, against HIV-1.83 To expand our knowledge to a variety of ARVs and to obtain a better understanding of synergistic interactions between active agent in free and encapsulated forms, we sought to evaluate the synergistic interactions between an oxidation-resistant variant of GRFT (Q-GRFT) and three different ARVs co-administered in free form, free and NP form, or NP form.
In this study, individual active agent encapsulation efficacy in NPs varied between 15% and 100%, based on the properties of the encapsulated agent. Overall, NPs that encapsulated Q-GRFT, RAL, or DAP obtained encapsulation efficiencies higher than 49%, while NPs provided lower loading (15%) of the hydrophilic ARV, TFV (Table 1). Similar observations have been reported in previous studies, demonstrating only 16% TFV loading in PLGA NPs.62 In contrast with TFV, Q-GRFT NPs had relatively high loading (49%), which was similar to other studies that reported 41% and 42% loading of wild-type GRFT in PLGA NPs,69,83 indicating similar loading potential for GRFT and Q-GRFT. This high level of loading may be attributed to the hydrophilicity of Q-GRFT and potential interactions of the mannose-binding sites which may interact via polar, electrostatic, or hydrogen bonding with hydrophobic residues of the PLGA matrix. In addition, protein surface absorption on polymeric NPs may also contribute to the higher loading observed with Q-GRFT, which may be further enhanced in future work by optimizing the concentration of protein and the use of stabilizers.90 Interestingly, and despite the hydrophilicity and smaller size of RAL (eg, relative to Q-GRFT), in our formulations, RAL obtained the highest loading, which was significantly higher than previously reported (55%).84 Similarly, hydrophobic DAP achieved high NP encapsulation, which was in agreement with other studies that reported 70% loading of DAP in PLGA NPs. The high loading levels observed with DAP may be attributed to the efficiency with which the double-emulsion technique encapsulates hydrophobic molecules.83
In addition to evaluating active agent loading, the release of each active agent from PLGA NPs was evaluated over 2 weeks. Release results showed that all NP formulations provided modest and similar trends in sustained-release over the course of 2 weeks in SVF (<20%, Figure 2). For Q-GRFT specifically, 11% of the total loading was released after 7 d, which is similar to that reported for similarly loaded wild-type GRFT from PLGA NPs (11%83 and 20%69). For TFV NPs, ~17% release was achieved over 14 d, similar in amount released, to that observed under acidic pH conditions in another study.62 Due to the relatively low loading and similarly low release of TFV observed from blended PLGA NPs in other work, formulations have moved to incorporate a more hydrophobic prodrug of TFV, tenofovir disoproxil fumarate, to obtain more desirable loading (~57%), and increased release (~80% release after 48 hr)91. For RAL NPs, similar release (~11%) to Q-GRFT was achieved, based on total loading; however, the release was lower than a previous report which showed ~60% release after 7 d from PLGA NPs (in PBS) with similar 100 µg/mg theoretical loading.26 Similarly, in our study, DAP release (16%) was lower than previously reported results (69% and 65%);83,92 however, the theoretical loading was not reported, which is known to have a significant impact on release. Overall, similar ranges of release, as a function of overall loading, were observed for the active agents, which may be attributed to active agent entrapment in the NP, due to non-covalent binding between the agent and polymer. However, RAL and DAP NPs surprisingly exhibited higher levels of release as a function of mass active agent, which was unexpected due to their hydrophobicity. Previous studies have illustrated that encapsulant–polymer interactions and compatibilities have a significant impact on the particle structure, loading, and release characteristics.93 In future work, these interactions may be modulated by integrating different materials and processing parameters such as polymer and drug concentration, polymer molecular weight, aqueous and organic phase volume, and drug content.94 In addition, assessing the release of agents from NPs under more basic pH conditions is also recommended due to the increase in vaginal pH in the presence of semen. A previous study showed that GRFT release from PLGA NPs increased in PBS (pH= 7.4), relative to SVF (pH=4.5), while less DAP released from PLGA NPs in more basic pH conditions.83
Prior to conducting synergy studies, the IC50 of each individual free agent against HIV-1 in vitro was determined relative to each corresponding NP-encapsulated agent (Figure 3). The IC50 of free Q-GRFT (57.2 ng/mL, 4.5 nM) was slightly higher than the values for GRFT reported in previous studies (24 and 0.51 ng/mL), when virus was added either 1 hr post-incubation51 or simultaneous to treatment of TZM-bl cells.83 In comparison, the IC50s of free TFV and RAL in this study were1.3 µM (382 ng/mL) and 7.42 nM (3.3 ng/mL) which were similar to previous studies (517 nM57 and 1.5 nM26 for TFV and RAL, respectively) using the same cell line and administration time. For DAP, an IC50 of 0.1 nM (0.034 ng/mL) was observed here, which was lower than previous reports of 4.7 nM. However, the higher IC50 value may be attributed to differences in treatment regimen. In our study, cells were infected with the virus 1 hr post-administration, while in a previous work, TZM-bl cells were infected with the virus immediately after drug administration. While TZM-bl cells have been extensively used to evaluate the efficacy of active agents against HIV-1, primary human macrophages and T cells may be considered in future work to further assess the efficacy of our delivery platforms in different target cells; however, similar efficacy results would be expected. In addition, it has been shown that the virus subtype impacts the IC50 of DAP (and other active agents), and based on this, these values can vary between 0.8 and 3.5 nM.95 When these active agents were encapsulated in NPs, similar IC50 values to those with free agent administration were observed, with the exception of Q-GRFT NPs in which the IC50 was 4.1-fold lower than observed with free Q-GRFT. For Q-GRFT NPs, the lower IC50 values are in agreement with another study that showed the IC50 of wild-type GRFT in PLGA NPs decreases, relative to free GRFT,69 possibly by enhancing protein stability and activity.
To evaluate the potential of Q-GRFT and ARV combinations to synergistically prevent HIV-1 infection, free Q-GRFT was co-administered with free ARVs, Q-GRFT NPs with free ARVs, and Q-GRFT NPs with ARV NPs, and the IC50 values of the individual agents were compared to their combinations in each form (Table 3). When free Q-GRFT was co-administered with each free ARV, synergistic interactions were observed for all co-administration groups with a 3.7 to 6.3-fold decrease in the IC50 of free Q-GRFT and a 2.8- to 5.6-fold reduction in the IC50 values of free ARVs (Figure 4). These results are in agreement with previous studies that assessed the impact of co-administering equipotent ratios (1:1 IC50 ratios) of free GRFT and TFV (CI = 0.6) in CD4+ MT-4 cells.14 Another study demonstrated very strong synergy (CI < 0.1) between free GRFT and free DAP co-administered to TZM-bl cells in equipotent ratios.83
More interestingly, when Q-GRFT and ARVs were each encapsulated in NPs, the synergistic interactions observed between Q-GRFT and DAP incorporated in NPs significantly improved relative to free Q-GRFT and free DAP co-administration (Figure 6). In fact, among all of the co-administration groups, the most synergistic interaction was observed between Q-GRFT NPs and DAP NPs with 6.0 and 8.6-fold improvements in activity for Q-GRFT and DAP, respectively. This observation is in agreement with a previous study that showed strong synergy between GRFT NPs and DAP NPs (CI = 0.08) using TZM-bl cells and equipotent active agent ratios.83 The lower synergy observed in our study, between Q-GRFT NPs and TFV NPs or Q-GRFT NPs and RAL NPs may be due to the higher IC50 value for each ARV, which requires higher drug concentrations to be released from NPs to exert efficacy. Co-administration of active agents with different mechanisms of action in NPs may increase prophylactic effect, decrease the dose needed to attain efficacy against HIV-1 infection, and reduce the chance of developing drug resistance. Furthermore, previous work has shown that encapsulating active agents in NPs may improve synergistic interactions by enhancing drug uptake and cell internalization.57 Moreover, NPs may overcome chemical incompatibilities between co-administered agents.57 While synergy was observed in all NP formulations, in future studies, different molar ratios of Q-GRFT NPs:ARV NPs can be explored to seek the most synergistic ratios. Moreover, the administered ratios may be adjusted based on the toxicity of each agent to balance toxicity with efficacy. In addition, the time frame of agent administration prior to virus exposure may be of interest to investigate. Since each active agent has a different mechanism of action and inhibits the virus at different stages of infection, administration time can play a role in obtaining efficacy and synergistic interactions between agents.
In addition to free Q-GRFT/free ARV and Q-GRFT NP/ARV NP co-administration, the efficacy of co-administration of Q-GRFT NPs with free ARVs was evaluated. Overall, no reduction in the IC50 values of free DAP was observed; however, there was a modest decrease of 1.6- and 1.4-fold in the IC50 values of free TFV and free RAL (Figure 5). Less synergistic interactions between free ARVs and encapsulated Q-GRFT may be attributed to the lower release of Q-GRFT, relative to available free TFV, RAL, and DAP, which may not provide the most synergistic ratio.
In addition to efficacy, the safety of NPs encapsulating each of the active agents was evaluated in vitro, and showed that encapsulated Q-GRFT, TFV, RAL, and DAP at the maximum dose used in efficacy studies (1 mg/mL), are non-toxic to vaginal cells. Previous studies have also demonstrated the safety of PLGA NPs loaded with GRFT,69 TFV,63 RAL,84 and DAP83,92 in vitro and in vivo69,83 further validating that co-administration can enhance the efficacy of these active agents while maintaining their safety. It should be mentioned that the relatively higher toxicity of DAP NPs in VK2/E6E7 cells may be mitigated with the administration of lower doses via synergistic and targeted NP delivery. Moreover, due to the high synergy between Q-GRFT NPs and DAP NPs, a lower dose of active agent may be considered to obtain strong efficacy against HIV-1. Additionally, the co-administration of two or three drugs with Q-GRFT would be expected to result in lower IC50 values and higher efficacy, relative to two-agent co-administration. In future work, these more complex dosing regimens may be explored.
In line with promising efficacy and safety, another important criterion is to provide a practical long-term prevention strategy in an effective delivery platform. The NP co-administration strategy evaluated here inhibited HIV-1 infection in vitro, and with one exception (ie, TFV), released efficacious concentrations within the IC50 range of each active agent in TZM-bl cells, for up to 7 days. However, this release may be improved, by either improving the NP formulation, or creating a hybrid delivery platform similar to previous work.69 In this study, we preliminarily evaluated NP-fiber composites to prolong the release of our most synergistic NP formulations, by incorporating Q-GRFT NPs and DAP NPs. In our NP-fiber composite platform, DAP, a small hydrophobic molecule, showed a burst release followed by a minimal release that was still in the IC50 range of DAP observed in TZM-bl cells. On the other hand, the Q-GRFT release curve followed an “S” shape, indicating two distinct release phases on day 1 and days 42 through 60, each of which was followed by more gradual release. We attribute DAP release within the first few hours to the small size of DAP, while Q-GRFT, due to its larger size, may have been entrapped within fiber layers, providing slightly more gradual release through day 42. The second phase of release is attributed to NP-fiber polymer degradation, resulting in another burst release. These release profiles are in agreement with our previous work with that incorporated wild-type GRFT in an NP-fiber composite.69 Furthermore, the release profile may be optimized by changing the NP content and altering the outer layer thickness.69
In addition to modulating the release, NP-fiber composites like NPs alone may also enable virus inhibition at different stages by providing a burst release (here of DAP and NNRTI), which can provide an on-demand rapid protection against HIV-1, followed by more prolonged release of Q-GRFT, an entry inhibitor, for long-term prevention and treatment. In addition to virus inhibition via the use of active agents, previous studies in our group have shown that the electrospun fibers can also physically trap the virus, which may lend increased protection through physical interactions.47 Moreover, lower doses of active agents are released from these platforms which may result in less toxicity. In the future, NP-fiber composites may serve as a multipurpose delivery platform to target multiple infection types by encapsulating different agents and may enhance NP retention relative to free NP administration, to promote user adherence if release can be tuned for specific agents. Furthermore, in future work, the distribution of these active agent combinations in more complex ex vivo and in vivo models may be assessed to provide a more therapeutically relevant information regarding release profiles.
Conclusion
In this work, NPs encapsulating Q-GRFT, TFV, RAL, and DAP were successfully developed with high loading and efficacy against HIV-1 pseudovirus infection in vitro. In addition, the synergistic interactions between free Q-GRFT and free ARVs, Q-GRFT NPs and free ARVs, and Q-GRFT NPs and ARV NPs were assessed. Significant enhancements in efficacy were observed when free Q-GRFT and free ARVs were co-administered, relative to the administration of individual active agents. Moreover, synergy was observed between free Q-GRFT and all ARVs when they were individually encapsulated in PLGA NPs. For Q-GRFT and DAP in particular, synergistic interactions improved upon co-administration of Q-GRFT NPs with DAP NPs. Overall, this work shows that the potent HIV-1 inhibitor, Q-GRFT, can be co-administered with different ARVs, including NRTIs and integrase inhibitors, to improve antiviral efficacy against HIV-1, and that modest, therapeutically relevant sustained-release can be achieved over 14 days. Furthermore, the co-administration of Q-GRFT with ARVs may be further considered in future studies to impart long-term protection by modulating multi-agent release in hybrid platforms such as NP-fiber composites.
Abbreviations
ARV, antiretroviral; DAP, dapivirine; Ect1, ectocervix, epithelial HPV-16 E6/E7 transformed; End1, endocervix, epithelial HPV-16 E6/E7 transformed; FRT, female reproductive tract; GRFT, griffithsin; HIV-1, human immunodeficiency virus type 1; HSV, herpes simplex virus; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium; NP, nanoparticle; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, non-nucleoside reverse transcriptase inhibitor; PCL, polycaprolactone; PEO, poly(ethylene oxide); PLGA, poly(lactic-co-glycolic) acid; RAL, raltegravir; TFV, tenofovir; TZM-bl, CXCR4-positive HeLa cell clone, engineered to express CD4 and CCR5; VK2, vaginal keratinocyte2 cell line.
Data Sharing Statement
Underlying data will be available from the corresponding author upon reasonable request.
Ethics Approval and Informed Consent
Not applicable.
Consent for Publications
Not applicable.
Acknowledgments
We thank Ian Santisteban for quantifying the release of active agents via HPLC. We also thank Dr. Stuart Williams at the University of Louisville for the use of his electrospinning instrument.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the NIH NIGMS P20GM125504, Jewish Heritage Fund for Excellence (G2803) and NIH NIAID R01AI139671 grants.
Disclosure
The authors declare they have no competing interests.
References
1. Garcia-Vidal E, Badia R, Pujantell M, et al. Dual effect of the broad spectrum kinase inhibitor midostaurin in acute and latent HIV-1 infection. Antiviral Res. 2019;168:18–27. doi:10.1016/j.antiviral.2019.05.003
2. Gama L, Koup RA. New-generation high-potency and designer antibodies: role in HIV-1 treatment. Annu Rev Med. 2018;69:409–419.
3. Baeten JM, Palanee-Phillips T, Brown ER, et al. Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. N Engl J Med. 2016;375:2121–2132.
4. Boonstra H, Barot S, Lusti-Narasimhan M. Making the case for multipurpose prevention technologies: the socio-epidemiological rationale. BJOG. 2014;121 Suppl 5:23–26. doi:10.1111/1471-0528.12851
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830–839.
6. Rupp R, Rosenthal SL, Stanberry LR. VivaGel(™) (SPL7013 Gel): A candidate dendrimer – microbicide for the prevention of HIV and HSV infection. Int J Nanomedicine. 2007;2:561–566.
7. Prokofjeva MM, Kochetkov SN, Prassolov VS. Therapy of HIV infection: current approaches and prospects. Acta Naturae. 2016;8:23–32.
8. Broder S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res. 2010;85:1–18.
9. Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. 2012;2:a007161.
10. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48(7):931–939. doi:10.1086/597290
11. Mamo T, Moseman EA, Kolishetti N, et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine. 2010;5(2):269–285. doi:10.2217/nnm.10.1
12. Richman DD, Fischl MA, Grieco MH, et al. The toxicity of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. N Engl J Med. 1987;317(4):192–197. doi:10.1056/NEJM198707233170402
13. Sheldon J, Camino N, Rodés B, et al. Selection of hepatitis B virus polymerase mutations in HIV-coinfected patients treated with tenofovir.. Antivir Ther. 2005;10(6):727.
14. Ferir G, Palmer KE, Schols D. Synergistic activity profile of griffithsin in combination with tenofovir, maraviroc and enfuvirtide against HIV-1 clade C. Virology. 2011;417(2):253–258. doi:10.1016/j.virol.2011.07.004
15. Chupradit K, Moonmuang S, Nangola S, et al. Current peptide and protein candidates challenging HIV therapy beyond the vaccine era. Viruses. 2017;9(10):281. doi:10.3390/v9100281
16. Mitchell CA, Ramessar K, O’Keefe BR. Antiviral lectins: selective inhibitors of viral entry. Antiviral Res. 2017;142:37–54.
17. Baker MP, Carr FJ. Pre-clinical considerations in the assessment of immunogenicity for protein therapeutics. Curr Drug Saf. 2010;5:308–313.
18. Mori T, O’Keefe BR, Sowder RC
19. Emau P, Tian B, O’keefe B, et al. Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti‐HIV microbicide. J Med Primatol. 2007;36:244–253.
20. O’Keefe BR, Giomarelli B, Barnard DL, et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J Virol. 2010;84:2511–2521. doi:10.1128/JVI.02322-09
21. O’Keefe BR, Vojdani F, Buffa V, et al. Scaleable manufacture of HIV-1 entry inhibitor griffithsin and validation of its safety and efficacy as a topical microbicide component. Proc Natl Acad Sci U S A. 2009;106:6099–6104. doi:10.1073/pnas.0901506106
22. Barton C, Kouokam JC, Lasnik AB, et al. Activity of and effect of subcutaneous treatment with the broad-spectrum antiviral lectin griffithsin in two laboratory rodent models. Antimicrob Agents Chemother. 2014;58:120–127. doi:10.1128/AAC.01407-13
23. Kouokam JC, Huskens D, Schols D, et al. Investigation of griffithsin’s interactions with human cells confirms its outstanding safety and efficacy profile as a microbicide candidate. PLoS One. 2011;6(e22635). doi:10.1371/journal.pone.0022635
24. Günaydın G, Edfeldt G, Garber DA, et al. Impact of Q-Griffithsin anti-HIV microbicide gel in non-human primates: in situ analyses of epithelial and immune cell markers in rectal mucosa. Sci Rep. 2019;9:1–12.
25. Corman JM, Hamorsky KT, Shepherd JW, Hiatt E, Fuqua JL, Palmer KE. Stability of plasmid and viral banks supporting the cGMP manufacture of Q-Griffithsin from a TMV-based viral vector. J Biotechnol. 2020;320:74–76.
26. Jiang Y, Cao S, Bright DK, et al. Nanoparticle-based ARV drug combinations for synergistic inhibition of cell-free and cell–cell HIV transmission. Mol Pharm. 2015;12:4363–4374.
27. Feng JY, Ly JK, Myrick F, et al. The triple combination of tenofovir, emtricitabine and efavirenz shows synergistic anti-HIV-1 activity in vitro: a mechanism of action study. Retrovirology. 2009;6:44.
28. Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367:399–410.
29. Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, double-blind, placebo-controlled Phase 3 trial. The Lancet. 2013;381:2083–2090.
30. Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587–2599.
31. Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372:509–518.
32. Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367:423–434.
33. Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367:411–422.
34. Haberer JE. Current Concepts for PrEP Adherence: in The PrEP revolution; from clinical trials to routine practice. Curr Opin HIV AIDS. 2016;11:10.
35. Rohan LC, Sassi AB. Vaginal drug delivery systems for HIV prevention. AAPS J. 2009;11:78. doi:10.1208/s12248-009-9082-7
36. Guthrie KM, Vargas S, Shaw JG, et al. The promise of intravaginal rings for prevention: user perceptions of biomechanical properties and implications for prevention product development. PLoS One. 2015;10:e0145642.
37. Emau P, Tian B, O’Keefe B, et al. Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti-HIV microbicide. J Med Primatol. 2007;36:244–253. doi:10.1111/j.1600-0684.2007.00242.x
38. Nixon B, Stefanidou M, Mesquita PM, et al. Griffithsin protects mice from genital herpes by preventing cell-to-cell spread. J Virol. 2013;87:6257–6269. doi:10.1128/JVI.00012-13
39. Steinbach JM. Protein and oligonucleotide delivery systems for vaginal microbicides against viral STIs. Cell Mol Life Sci. 2015;72:469–503. doi:10.1007/s00018-014-1756-3
40. Steinbach JM, Seo YE, Saltzman WM. Cell penetrating peptide-modified poly(lactic-co-glycolic acid) nanoparticles with enhanced cell internalization. Acta Biomater. 2015. doi:10.1016/j.actbio.2015.11.029
41. Steinbach JM, Weller CE, Booth CJ, Saltzman WM. Polymer nanoparticles encapsulating siRNA for treatment of HSV-2 genital infection. J Control Release. 2012;162:102–110. doi:10.1016/j.jconrel.2012.06.008
42. Aniagyei SE, Sims LB, Malik DA, et al. Evaluation of poly(lactic-co-glycolic acid) and poly(dl-lactide-co-epsilon-caprolactone) electrospun fibers for the treatment of HSV-2 infection. Mater Sci Eng C Mater Biol Appl. 2017;72:238–251. doi:10.1016/j.msec.2016.11.029
43. Ball C, Krogstad E, Chaowanachan T, Woodrow KA. Drug-eluting fibers for HIV-1 inhibition and contraception. PLoS One. 2012;7:e49792. doi:10.1371/journal.pone.0049792
44. Blakney AK, Ball C, Krogstad EA, Woodrow KA. Electrospun fibers for vaginal anti-HIV drug delivery. Antiviral Res. 2013;100 Suppl:S9–16. doi:10.1016/j.antiviral.2013.09.022
45. Duan JH, Steinbach-Rankins JM. Adaptable Griffithsin delivery from polymer blend nanoparticles and electrospun fibers. Aids Res Hum Retrov. 2016;32:218.
46. Ensign LM, Tang BC, Wang YY, et al. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci Transl Med. 2012;4:138ra179. doi:10.1126/scitranslmed.3003453
47. Grooms TN, Vuong HR, Tyo KM, et al. Griffithsin-modified electrospun fibers as a delivery scaffold to prevent HIV infection. Antimicrob Agents Ch. 2016;60:6518–6531. doi:10.1128/Aac.00956-16
48. Huang C, Soenen SJ, Rejman J, et al. Stimuli-responsive electrospun fibers and their applications. Chem Soc Rev. 2011;40:2417–2434. doi:10.1039/c0cs00181c
49. Huang C, Soenen SJ, van Gulck E, et al. Electrospun cellulose acetate phthalate fibers for semen induced anti-HIV vaginal drug delivery. Biomaterials. 2012;33:962–969. doi:10.1016/j.biomaterials.2011.10.004
50. Mallipeddi R, Rohan LC. Nanoparticle-based vaginal drug delivery systems for HIV prevention. Expert Opin Drug Deliv. 2010;7:37–48. doi:10.1517/17425240903338055
51. Tyo KM, Steinbach-Rankins JM. Electrospun polymer nanofibers for long-term protection against HIV and HSV-2. Aids Res Hum Retrov. 2016;32:223.
52. Wheeler LA, Trifonova R, Vrbanac V, et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J Clin Invest. 2011;121:2401–2412. doi:10.1172/JCI45876
53. Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8:526–533. doi:10.1038/nmat2444
54. Wu Y, Navarro F, Lal A, et al. Durable protection from Herpes Simplex Virus-2 transmission following intravaginal application of siRNAs targeting both a viral and host gene. Cell Host Microbe. 2009;5:84–94.
55. Zamani M, Prabhakaran MP, Ramakrishna S. Advances in drug delivery via electrospun and electrosprayed nanomaterials. Int J Nanomedicine. 2013;8:2997–3017. doi:10.2147/IJN.S43575
56. Krogstad EA, Woodrow KA. Manufacturing scale-up of electrospun poly(vinyl alcohol) fibers containing tenofovir for vaginal drug delivery. Int J Pharm. 2014;475:282–291. doi:10.1016/j.ijpharm.2014.08.039
57. Chaowanachan T, Krogstad E, Ball C, Woodrow KA. Drug synergy of tenofovir and nanoparticle-based antiretrovirals for HIV prophylaxis. PLoS One. 2013;8:e61416. doi:10.1371/journal.pone.0061416
58. Carson D, Jiang Y, Woodrow KA. Tunable release of multiclass anti-HIV drugs that are water-soluble and loaded at high drug content in polyester blended electrospun fibers. Pharm Res. 2016;33:125–136. doi:10.1007/s11095-015-1769-0
59. Blakney AK, Krogstad EA, Jiang YH, Woodrow KA. Delivery of multipurpose prevention drug combinations from electrospun nanofibers using composite microarchitectures. Int J Nanomedicine. 2014;9:2967–2978. doi:10.2147/IJN.S61664
60. Ball C, Woodrow KA. Electrospun solid dispersions of Maraviroc for rapid intravaginal preexposure prophylaxis of HIV. Antimicrob Agents Chemother. 2014;58:4855–4865. doi:10.1128/AAC.02564-14
61. Ball C, Chou SF, Jiang Y, Woodrow KA. Coaxially electrospun fiber-based microbicides facilitate broadly tunable release of maraviroc. Mater Sci Eng C Mater Biol Appl. 2016;63:117–124. doi:10.1016/j.msec.2016.02.018
62. Zhang T, Sturgis TF, Youan -B-B. Ph-Responsive Nanoparticles Releasing Tenofovir Intended for the Prevention of HIV Transmission Eur J Pharm Biopharm. 2011;79:526–536.
63. Machado A, Cunha-Reis C, Araújo F, et al. Development and in vivo safety assessment of tenofovir-loaded nanoparticles-in-film as a novel vaginal microbicide delivery System Acta Biomaterialia. 2016;44:332–340.
64. Ensign LM, Cone R, Hanes J. Nanoparticle-based drug delivery to the vagina: a review. J Controlled Release. 2014;190:500–514. doi:10.1016/j.jconrel.2014.04.033
65. Ensign LM, Tang BC, Wang -Y-Y, et al. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci Transl Med. 2012;4:138ra179–138ra179.
66. Ensign LM, Tang BC, Wang -Y-Y, et al. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci Transl Med. 2012;4. doi:10.1126/scitranslmed.3003453.
67. Das Neves J, Araújo F, Andrade F, Amiji M, Bahia MF, Sarmento B. Biodistribution and pharmacokinetics of dapivirine-loaded nanoparticles after vaginal delivery in mice. Pharm Res. 2014;31:1834–1845.
68. Hua D, Liu Z, Wang F, et al. pH responsive polyurethane (core) and cellulose acetate phthalate (shell) electrospun fibers for intravaginal drug delivery. Carbohydr Polym. 2016;151:1240–1244. doi:10.1016/j.carbpol.2016.06.066
69. Tyo KM, Lasnik AB, Zhang L, et al. Sustained-release Griffithsin nanoparticle-fiber composites against HIV-1 and HSV-2 infections. J Controlled Release. 2020;321:84–99.
70. Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 2012;41:2545–2561. doi:10.1039/c2cs15327k
71. Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–347.
72. GeunHyung K, Hyeon Y, YunKyung P. Drug release from various thicknesses of layered mats consisting of electrospun polycaprolactone and polyethylene oxide micro/nanofibers. Appl Phys a Mater Sci Process. 2010;100:1197–1204. doi:10.1007/s00339-010-5785-y
73. Wang Y-F, Guo H-F, Ying D-J. Multilayer scaffold of electrospun PLA-PCL-collagen nanofibers as a dural substitute. J Biomed Mater Res B Appl Biomater. 2013;101:1359–1366. doi:10.1002/jbm.b.32953
74. Liu L, Kamei K, Yoshioka M, et al. Nano-on-micro fibrous extracellular matrices for scalable expansion of human ES/iPS cells. Biomaterials. 2017;124:47–54.
75. Okuda T, Tominaga K, Kidoaki S. Time-programmed dual release formulation by multilayered drug-loaded nanofiber meshes. J Controlled Release. 2010;143:258–264.
76. Vakilian S, Mashayekhan S, Shabani I, Khorashadizadeh M, Fallah A, Soleimani M. Structural stability and sustained release of protein from a multilayer nanofiber/nanoparticle composite. Int J Biol Macromol. 2015;75:248–257.
77. Kidoaki S, Kwon IK, Matsuda T. Mesoscopic spatial designs of nano- and microfiber meshes for tissue-engineering matrix and scaffold based on newly devised multilayering and mixing electrospinning techniques. Biomaterials. 2005;26:37–46.
78. Westling K, Pettersson K, Kaldma A, Navér L. Rapid decline in HIV viral load when introducing raltegravir-containing antiretroviral treatment late in pregnancy. AIDS Patient Care STDS. 2012;26:714–717.
79. Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446.
80. Houdaihed L, Evans JC, Allen C. Codelivery of paclitaxel and everolimus at the optimal synergistic ratio: a promising solution for the treatment of breast cancer. Mol Pharm. 2018;15:3672–3681.
81. Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55.
82. Schader SM, Colby-Germinario SP, Schachter JR, Xu H, Wainberg MA. Synergy against drug-resistant HIV-1 with the microbicide antiretrovirals, dapivirine and tenofovir, in combination. AIDS. 2011;25:1585–1594.
83. Yang H, Li J, Patel SK, Palmer KE, Devlin B, Rohan LC. Design of poly (lactic-co-glycolic acid)(PLGA) nanoparticles for vaginal co-delivery of griffithsin and dapivirine and their synergistic effect for HIV prophylaxis. Pharmaceutics. 2019;11:184.
84. Date AA, Shibata A, Goede M, et al. Development and evaluation of a thermosensitive vaginal gel containing raltegravir+ efavirenz loaded nanoparticles for HIV prophylaxis. Antiviral Res. 2012;96:430–436.
85. Chen BA, Panther L, Marzinke MA, et al. Phase 1 safety, pharmacokinetics, and pharmacodynamics of dapivirine and maraviroc vaginal rings: a double-blind randomized trial. J Acquir Immune Defic Syndr. 2015;70:242.
86. Bani-Sadr F, Palmer P, Scieux C, Molina J. Ninety-six—week efficacy of combination therapy with lamivudine and tenofovir in patients coinfected with HIV-1 and Wild-Type Hepatitis B Virus. Clin Infect Dis. 2004;39:1062–1064.
87. Lennox JL, DeJesus E, Lazzarin A, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. The Lancet. 2009;374:796–806.
88. Dezzutti CS, Shetler C, Mahalingam A, et al. Safety and efficacy of tenofovir/IQP-0528 combination gels–a dual compartment microbicide for HIV-1 prevention. Antiviral Res. 2012;96:221–225.
89. Steinbach JM, Weller CE, Booth CJ, Saltzman WM. Polymer nanoparticles encapsulating siRNA for treatment of HSV-2 genital infection. J Controlled Release. 2012;162:102–110. doi:10.1016/j.jconrel.2012.06.008
90. Pagels RF, Prud’Homme RK. Polymeric nanoparticles and microparticles for the delivery of peptides, biologics, and soluble therapeutics. J Controlled Release. 2015;219:519–535.
91. Shailender J, Ravi PR, Saha P, Dalvi A, Myneni S. Tenofovir disoproxil fumarate loaded PLGA nanoparticles for enhanced oral absorption: effect of experimental variables and in vitro, ex vivo and in vivo evaluation. Colloids Surf B Biointerfaces. 2017;158:610–619.
92. Das Neves J, Sarmento B. Precise engineering of dapivirine-loaded nanoparticles for the development of anti-HIV vaginal microbicides. Acta Biomater. 2015;18:77–87.
93. Jäger A, Jäger E, Giacomelli FC, et al. Structural changes on polymeric nanoparticles induced by hydrophobic drug entrapment. Colloids Surf a Physicochem Eng Asp. 2018;538:238–249.
94. Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336:367–375.
95. Fletcher P, Harman S, Azijn H, et al. Inhibition of HIV-1 infection by the candidate microbicide, dapivirine, a non-nucleoside reverse transcriptase inhibitor. Antimicrob Agents Ch. 2008.
96. Tomoda K, Terashima H, Suzuki K, Inagi T, Terada H, Makino K. Enhanced transdermal delivery of indomethacin-loaded PLGA nanoparticles by iontophoresis. Colloids Surf B Biointerfaces. 2011;88:706–710.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.