Back to Journals » OncoTargets and Therapy » Volume 15
In vitro Characterization of Enhanced Human Immune Responses by GM-CSF Encoding HSV-1-Induced Melanoma Cells
Authors Delic M ![]() , Boeswald V, Goepfert K, Pabst P, Moehler M
, Boeswald V, Goepfert K, Pabst P, Moehler M ![]()
Received 19 November 2021
Accepted for publication 23 September 2022
Published 27 October 2022 Volume 2022:15 Pages 1291—1307
DOI https://doi.org/10.2147/OTT.S350136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Maike Delic, Veronika Boeswald, Katrin Goepfert, Petra Pabst, Markus Moehler
University Medical Center of the Johannes Gutenberg University Mainz, 1st Department of Internal Medicine, Mainz, Germany
Correspondence: Maike Delic, University Medical Center of the Johannes Gutenberg University Mainz, 1st Department of Internal Medicine, Langenbeckstrasse 1, Mainz, 55131, Germany, Tel +49 6131 179803, Fax +49 6131 179657, Email [email protected]
Purpose: We studied the innate and adaptive immune response against melanoma cells after JS-1 (wild-type herpes simplex virus 1, wt HSV-1) or Talimogene laherparepvec (T-VEC) infection and evaluated the antitumoral efficacy in human melanoma cells. We analyzed the putative synergistic biological and immunological effects of JS-1 or T-VEC combined with cytostatic drugs in human tumor and immune cells. T-VEC is a genetically modified strain of HSV-1. Genetic modifications (insertion of the granulocyte-macrophage colony-stimulating factor (GM-CSF) gene) were made to attenuate the virus and increase selectivity for cancer cells. In addition to the direct oncolytic effect, we investigated the immune stimulatory effects of T-VEC by comparing it with JS-1. JS-1 is identical T-VEC except for the inserted GM-CSF gene.
Materials and Methods: We analyzed the effects of T-VEC and JS-1 with cytostatic drugs in human tumor-immune cell coculture experiments. After coculture, the surface markers CD80, CD83 and CD86 were measured by fluorescence-activated cell sorting and the cytokines, interleukin (IL)-2, IL-6, tumor necrosis factor (TNF)-α and GM-CSF, by enzyme-linked immunosorbent assays. Furthermore, we analyzed the potential of the viruses to induce T cell activation, measured on the basis of CD4, CD8 and CD69. Analysis of these markers and cytokines allows for conclusions to be drawn concerning the maturation of dendritic cells (DCs) and the immunostimulatory effects of the treatment.
Results: We documented increased activation of human cytotoxic T lymphocytes after infection by both HSV-1 strains and treatment with cytostatic drugs without significant differences between T-VEC and JS-1.
Conclusion: We demonstrated an immune response as a result of infection with both viruses, but T-VEC was in vitro not stronger than JS-1. The immunostimulatory effects of the viruses could be partially increased by chemotherapy, providing a rationale for future preclinical studies designed to explore T-VEC in combined regimens.
Keywords: oncolytic virotherapy, immunotherapy, dendritic cells, T-VEC, immunostimulatory effect
Introduction
Since oncolytic viruses are a relatively new therapeutic method and only two viruses have been approved worldwide so far, their use is mainly limited to clinical studies.1,2 Compared with conventional tumor therapies such as radiation therapy and poly-chemotherapy, oncolytic viruses are generally better tolerated and safer due to increased tumor specificity.3 In contrast to targeted therapies, they are not dependent on specific tumor-expressed receptors or antigens, which mean that tumor resistance can be reduced.4
Current studies follow the principle of combination therapy each having different approaches. The effectiveness in combination with standard therapy should always be checked for new treatment options. An increased response has already been shown for oncolytic viruses when combined with radiation in vitro5 and valproic acid in vivo and in vitro.6 Chemotherapeutic agents such as doxorubicin7 and oxaliplatin8 showed an induction of immunogenic cell death, which is also induced by oncolytic viruses. A synergistic combination could enable a dose reduction of the cytotoxic agents with consequently fewer side effects. Such synergism has already been proven for the combination of gemcitabine and H-1PV,9 cisplatin and vaccinia virus GLV-1h6810 as well as doxorubicin with adenovirus SG511-BECN.11 The combination treatment of melanoma cells with cisplatin or vincristine and H-1PV also showed an increased maturation of dendritic cells (DCs) with increased cytokine release compared to cytostatic therapy alone.12
Despite the approved promising immunotherapeutic substances, patients with advanced melanoma are often progressive in course of the disease and can no longer be treated with standard therapies.13 New, potent combination treatment to reduce the dose without compromising the success of the therapy are needed. A synergistic or at least additive combination treatment with other active substances would be desirable. For example, a spontaneously mutated strain of HSV-1, HF10, showed cytolytic effects after infection in an in vitro study that revealed murine and human melanoma tumor cells.14 In addition, Rigvir, an unmodified picornavirus (melanoma-adapted ECHO-7 virus) acts as an oncotropic and oncolytic virus for treatment of melanoma.15
The advantage of oncolytic viruses such as T-VEC is the release of new antigens that can re-enhance the immune system.12,16,17
Talimogene laherparepvec (T-VEC) is a genetically modified strain of herpes simplex virus 1 (HSV-1). HSV belongs to Herpesviridae family.18 Genetic modifications were made to the virus in order to attenuate it and increase selectivity for cancer cells. The gene encoding the neurovirulence factor ICP34.5 was deleted in the JS-1 strain and the virus exhibits tumor-selective replication and gene expression.19 The HSV-1 isolate JS-1 served as the basic structure for the development of T-VEC, which per se already has an increased oncolytic activity compared with clinical HSV-1 isolates. Furthermore, the deletion of the ICP47 gene blocks antigen processing and enhances immune stimulating properties of the virus.20 In addition, to create T-VEC, the granulocyte-macrophage colony-stimulating factor (GM-CSF) gene was inserted into the JS-1/34.5-/47-vector backbone. This causes differentiation of progenitor cells into DCs and is a potent immune stimulator. Thus, the insertion of the human GM-CSF gene into the viral genome enhances the antitumor response both locally and at sites distant to where the virus is injected. Expression of GM-CSF in the local tumor environment serves to achieve several biologic goals, the induction of local inflammation, enhancement of DC activity, and an increase in HLA class I expression.21
Due to the genetic modifications, T-VEC is able to efficiently infect highly proliferative, malignant cells without attacking healthy cells, to generate an increased cytopathic effect and to intensify a tumor-directed immune response. T-VEC is therefore designed to selectively target and destroy cancer cells and may thus also activate DC maturation and cytotoxic T lymphocytes (CTLs) by T-VEC induced tumor cell lysates. New combinations with other immunostimulatory agents may further overcome tumor mediated immune silencing mechanisms and may further improve the therapeutic outcome of the oncolytic T-VEC. T-VEC was tested in a Phase I/II study in combination with cisplatin-based chemoradiotherapy for squamous cell carcinomas of the head and neck.22 In addition to good tolerability, locoregional tumor control was achieved in all treated patients, which showed that a combination of oncolytic viruses and chemotherapy has no clinical disadvantages.
Therefore, the oncolytic potential and immune stimulating properties of T-VEC in human melanoma cells make it an attractive candidate for the use in cancer therapy and an extension of the immunotherapy armamentarium.
Materials and Methods
Human Melanoma Cell Lines
Human melanoma cell line SK29MEL HLA-A2-positive and its HLA-A2-negative clone SK29MEL1.22 (both cell lines were gifts of T. Woelfel’s group, University Medical Center Mainz) were used. The Ethics Committee of the Rhineland-Palatinate Medical Association approved the use of the described cell lines. The melanoma line was established at the MSKCC in the 1970s from the patient SK29. Woelfel et al cloned the cells and derived the (HLA-A2.1 loss) variant −1.22 from the clone SK-MEL-29.1 via immune selection. They made fingerprint analysis with bacteriophage M13 to ensure that the cells came from the patient SK29.23,24 Cells were cultivated as previously described.25
Viral Infection
0.1*106 melanoma cells per well were cultivated for one day before virus infection. For infection with JS-1 (wt HSV-1) or JS-1 hGM-CSF (T-VEC) cell medium was removed and an infection was performed with different multiplicities of infection (MOIs) from 0 to 10 plaque-forming units (PFU)/cell for both viruses, for different incubation times (24 hours (h) to 48 h). The initial incubation period was 1 h and 2.5 mL of fresh medium was supplemented to the cells of 6-well plates. Where indicated, cytostatic drugs were added after 1 h of infection and a supplementary incubation period of 1 or 2 days with the combined treatment was performed.
Cytostatic Drugs
One hour after virus infection, respective concentrations of cytostatic drugs diluted in medium were added (Pharmacy of the University Medical Center Mainz, Mainz, Germany). Concentrations of cytostatic drugs used were: SK29MEL: cisplatin, inhibitory concentration causing 20% growth inhibition (IC20) 0.07 µg/mL, vincristine IC20 17.5 µg/mL, doxorubicin IC20 0.13 µg/mL; SK29MEL1.22: cisplatin IC20 2.5 µg/mL, vincristine IC20 22.7 µg/mL, doxorubicin IC20 0.13 µg/mL. Inhibitory concentration values (IC20) of cytostatic drugs were calculated using the CalcuSyn application (Biosoft, Cambridge, UK) from the dose response curves.
DC Differentiation and Maturation
Monocytes were isolated by adherence to HLA-A2-positive human buffy coats from healthy blood donors obtained through the Department of Transfusion Medicine, University Medical Center Mainz (Mainz, Germany) as previously described.26,27 Monocytes were treated with 500 U interleukin (IL)-4 (ImmunoTools, Friesoythe, Germany) and 500 U GM-CSF (Berlex; Bayer Healthcare Pharmaceuticals, Leverkusen, Germany) for 6 days to obtain immature DCs (iDCs).16 A cytokine cocktail containing tumor necrosis factor (TNF)-α, IL-6, IL-1-β (all Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and PGE2 (Sigma-Aldrich Chemie GmbH, Munich, Germany) led to maturation of DCs (mature DCs [mDCs]).
Coculture Model
For coculture experiments, melanoma cells (SK29MEL; SK29MEL1.22) were seeded in six-well plates and treated with viruses or cytostatic drugs as already described. DCs were isolated as described and seeded in six-well plates in a 5:1 ratio (melanoma cells: DCs). After 3 days, the maturation status was determined by fluorescence-activated cell sorting (FACS) and enzyme-linked immunosorbent assays (ELISAs). In a first approach (DC coculture), melanoma cells were seeded and infected with JS-1 or T-VEC after 24 h. After 1 h of incubation, the tumor cells were treated with the cytostatic drugs doxorubicin, cisplatin or vincristine. As a positive control, iDCs were matured by adding a cytokine cocktail as previously described.28 The cells were cocultured for 48 h before staining for FACS analysis.
T Cells
The Pan T cell isolation kit human from Miltenyi was used to separate T cells in a magnetic-activated cell sorting (MACS) system according to the manufacturer’s instructions. For use in coculture, monocytes and T cells from the same donor were used. The collected fraction contained the unlabeled CD4+ and CD8+ T cells. These T cells were used for coculture. For CTL experiments, melanoma cells were seeded, infected after 24 h and after 24 h, iDCs were added at a ratio of 1:5 and cocultivated, as described, further. T cells were added 24 h later in a ratio of 1:1 to the cocultivated melanoma cell lysates and stimulated with 100 IU/mL of Proleukin 2. T cells, stimulated with 1 μL/mL CD3 and CD28, served as positive controls. After 48 h, the cells were stained and the activation was analyzed by FACS.
Luminescence Assays
The cytotoxic effect of JS-1 and T-VEC and/or cytostatic drug-treated melanoma cells was determined by time-dependent measurement of cell growth. For MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) viability assays, cells were seeded in 96-well plates (Sigma, München, Germany) and assays performed as previously described.17 For single treatments in MTT assays, serial dilutions of cytostatic drugs or a dilution of MOIs were generated and added to the different cell lines. Percentage viability was defined as relative absorbance of treated vs untreated control cells.
Enzyme-Linked Immunosorbent Assay (ELISA)
The supernatant of cocultivated cells was collected before harvesting and storage at −80°C. Cytokine analyses of IL-2, IL-6, TNF-α and GM-CSF were performed as per the protocols of ELISA kits (Invitrogen, Karlsruhe). Plates were read in an ELISA reader (ELISA Reader, Bio-Tek Instruments, Bad Friedrichshall, Germany) at 450 nm, and values at 570 nm were subtracted. ELISA reader creates calibration curves with standards purchased at Invitrogen, Karlsruhe. Results were expressed as means of duplicate wells.
Flow Cytometry
In DC coculture experiments with tumor lysates, cells were harvested and stained with anti-CD80 APC, -CD83 PE and -CD86 PE antibodies (BD Biosciences Pharmingen, Heidelberg, Germany). Fluorescence was measured with a minimum of 15,000 events per sample and was analyzed on a FACSCalibur flow cytometry system (BD Biosciences, Heidelberg, Germany) according to the manufacturer’s instructions. FACS staining was performed as recommended by the manufacturer’s data sheets. In DC-CTL coculture experiments, cells were harvested and stained with anti-CD4 PE and -CD8 FITC (Invitrogen, Karlsruhe) and anti-CD69 APC antibodies (eBioscience, San Diego, USA). Data analysis was performed using Cell FlowJo (BD Biosciences, Heidelberg, Germany).
Statistics
All data shown are given as mean ± standard deviation. Differences between the groups were calculated with Student’s t-test. A distinction is made according to *P ≤ 0.05; **P ≤ 0.01, and ***P ≤ 0.001. The P-values of ≤ was considered to be significant.
Results
Cell Viability After Infection with JS-1 or T-VEC and Treatment with Cytostatic Drugs
The effect of cytostatic drugs was compared to that of combination with virus infection. T-VEC and JS-1 reduced SK29MEL viability in a MOI-dependent manner (data not shown). For infection with T-VEC a MOI of 0.1 and for JS-1 a MOI of 0.01 with 2 days of incubation were chosen for further experiments.
After infection for 24 h or 48 h, T-VEC (JS-1 hGM-CSF) and JS-1 reduced cell viability in a MOI-dependent manner. Both viruses induced a reduction of viability, in a time- and concentration-dependent manner. Combinations of both viruses with cytotoxic agents caused a stronger reduction of cell viability than the virus alone. A longer incubation period resulted in a stronger reduction of viable cells (shown exemplarily for SK29MEL in combination with doxorubicin and viruses after 24 h; Figure 1A and Figure 1). The respective conditions with the strongest effect were chosen for further experiments.
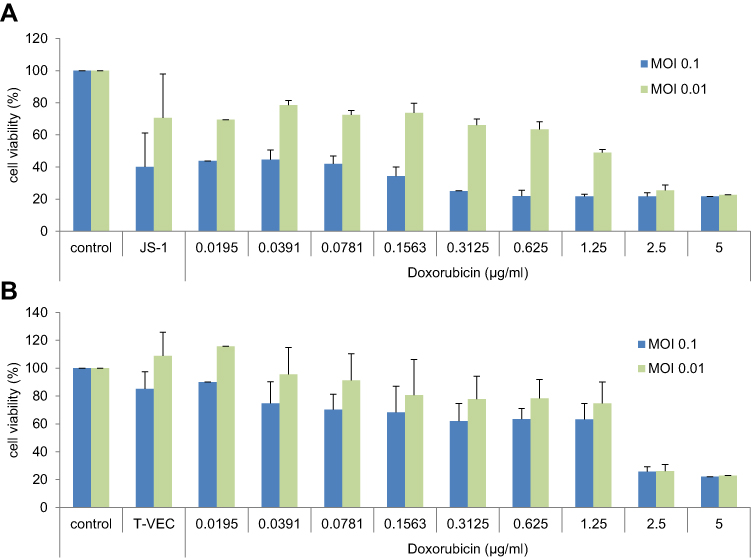 |
Figure 1 (A and B) Influence of cytostatic drugs and virus infection on cell viability. Abbreviation: MOI, multiplicity of infection. Notes: Cell viability of SK29MEL cells after infection with (A) JS-1 or (B) T-VEC (MOI 0.1 and 0.01) and treatment with doxorubicin (μg/mL), incubation for 24 h. The effects of JS-1 and T-VEC on the viability were measured by MTT assay. Graphs show the percentage of living cells after virus infection vs untreated cell control (=100% viability). Data are shown as average values from three independent experiments. |
Coculture Experiments
In order to illustrate the interaction of differently generated tumor cell lysates with immune cells in vitro, cocultures were formed from tumor cell lysates and HLA-corresponding immune cells. To analyze the immunomodulatory potential of the viruses, the maturation of the DCs was examined by flow cytometry using the markers CD80, CD83 and CD86. After maturation, these proteins are increasingly expressed on the surface of the DCs and thus allow conclusions to be drawn about their degree of maturation.28 CD80 and CD86 serve as coligands for the interaction with T lymphocytes, CD83 is a direct marker of DC maturation and is hardly present on iDCs.
Expression of the Surface Markers CD80, CD83 and CD86
Figure 2A shows the proportion of CD80/CD86 and CD86/83 positive cells in the coculture. This proportion corresponds to the number of mature DCs that express these co-ligands to an increased extent. The maturation by means of a cytokine cocktail led to an expression of CD80+/CD86+ of 43.0% (blue bar). The iDCs cultivated without a maturation stimulus shows only 6.7% CD80+/CD86+, which represents a lack of maturation.
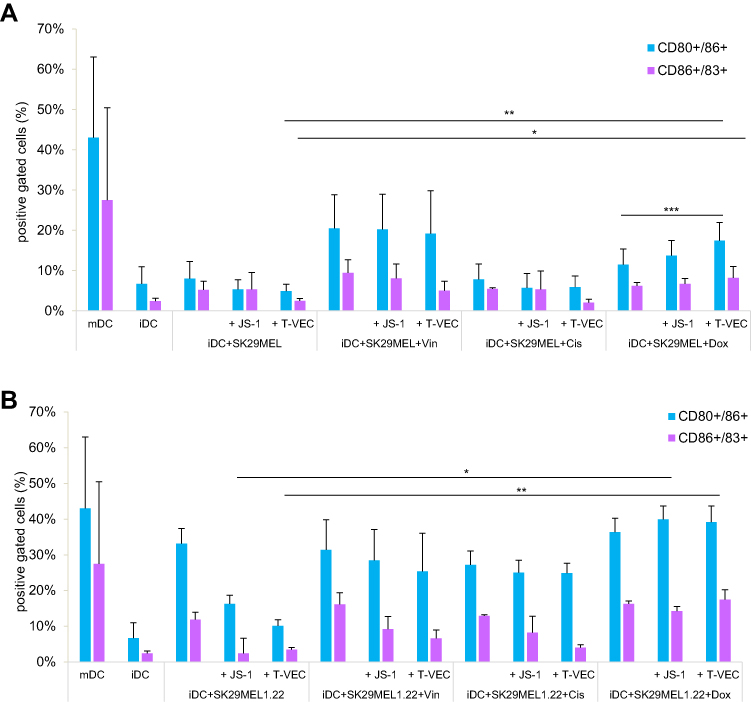 |
Figure 2 (A and B) Coculture of virus-infected and/or chemotherapy-induced TCLs with iDCs. Abbreviations: DCs, dendritic cells; FACS, fluorescence-activated cell sorting; TCL, tumor cell lysate; mDC, mature dendritic cells; iDCs, immature dendritic cells. Notes: FACS analysis of the expression of CD80 and CD86 on DCs, mDCs matured by cytokine cocktail and on iDCs after coculture with melanoma cell lysates of lines SK29MEL (A) and SK29MEL1.22 (B) generated by various treatments. The infection with JS-1 or T-VEC and the treatment with vincristine (Vin), cisplatin (Cis) and doxorubicin (Dox) were examined alone and in combination with the oncolytic viruses. The mean values and standard deviations of three measurements carried were shown. DCs matured by adding a cytokine cocktail served as a positive control. *P ≤ 0.05; **P ≤ 0.01, and ***P ≤ 0.001. |
For the SK29MEL cells, there was no increased DC maturation due to infection with JS-1 or T-VEC. The treatment of the melanoma cells with vincristine led to an increased expression of both coligands, but the value of 20.5% after monotherapy could not be exceeded by the additional infection with JS-1 or T-VEC. In monotherapy and combination therapy, cisplatin did not show any effect on maturation different from that of untreated cells. Doxorubicin alone was able to slightly increase the expression of the maturity markers, which could be further improved by virus infection. After doxorubicin monotherapy, 11.5% CD80+/CD86+ cells were detected, which was increased to 13.7% by JS-1 and to 17.5% by T-VEC.
The purple bars show the coexpression of the markers CD83 and CD86. The proportion of mature, thus CD83+ cells among all CD86+ cells are shown. Among the DCs induced to mature by a cytokine cocktail, an average of 27.5% CD83+/CD86+ was represented. Unstimulated, non-cocultivated iDCs did not mature as expected and reached an expression density of both markers of 2.4%. After cocultivation of iDCs and untreated SK29MEL, a CD83+/86+ proportion of 5.2% was found, which was not increased by infection with JS-1 or T-VEC. Vincristine alone was able to increase maturation up to 9.4%, but the combination with oncolytic viruses reduced it. For example, iDCs, which were cocultivated with SK29MEL lysates generated by vincristine and T-VEC achieved a maturation of 5.0%. The combination of cisplatin and T-VEC reduced the expression of CD83 and CD86 even to 2.1%, which is in the range of unstimulated iDCs. The doxorubicin treatment was also unable to induce maturation more strongly than untreated melanoma cells, but the combination with T-VEC showed a minimal effect, 8.2% of the cells expressed the two examined markers.
Figure 2B shows the reaction of the DCs cocultivated with SK29MEL1.22 lysates. Infection with oncolytic viruses alone reduced expression of CD80+/CD86+ (blue bars) to 10.1% by T-VEC. Vincristine and cisplatin showed no increased induction of DC maturation in comparison to untreated melanoma cells and their combination with JS-1 or T-VEC also had no positive influence. The doxorubicin monotherapy led to a CD80+/CD86+ expression of 36.3%, which was increased to about 39% by additional virus infection. A relevant difference between the two virus strains could not be identified.
Untreated SK29MEL1.22 cells induced the CD83+/CD86+ expression (purple bars) of the DCs by 11.8%. The infection with JS-1 or T-VEC reduced the expression rate in this cell series to a greater extent, so that after T-VEC infection only 3.4% CD83+/CD86+ were present. Vincristine and cisplatin showed effects that were comparable to one another: the monotherapy had a slight positive influence on maturation (16.1% and 13.0%, respectively), but the combination with JS-1 reduced this to lower values than for untreated SK29MEL1.22 (9.2% and 8.2%) and in combination with T-VEC showed even lower maturation rates (6.6% and 4.1%). Doxorubicin induced maturation to a slightly greater extent in monotherapy and combination therapy and was the only cytostatic agent that showed a tendency to improve maturation to 17.4% when combined with T-VEC.
Cytokine Secretion by DC
In addition to examining the surface markers, the release of immune-stimulating cytokines, which are increasingly produced by mDCs, was examined. Signs of DC maturation include increased amounts of IL-6.28
Figure 3 shows the measured IL-6 concentration in pg/mL in the supernatants of the coculture from melanoma cell lysates and iDCs. The DCs (mDCs) stimulated by the cytokine cocktail showed an increased IL-6 production to 731 pg/mL and thus maturation could be assumed. By contrast, unstimulated and uncultivated iDCs and untreated melanoma cells produced almost no IL-6, as expected. For SK29MEL (red square), no increased IL-6 production was shown by virus treatment. On the contrary, IL-6 levels for JS-1 (38.5 pg/mL) and T-VEC (22.2 pg/mL) infected cells tended to be lower than that of the iDCs cocultivated with untreated SK29MEL (56.7 pg/mL). Treatment with cisplatin alone or in combination with JS-1 or T-VEC, as in the FACS, did not show any effects beyond that of untreated melanoma cells. Treatment with vincristine increased the IL-6 production of the DCs to 94.6 pg/mL, with no additional effect being observed due to the combination with oncolytic viruses. Doxorubicin alone led to a twice as high IL-6 production compared with untreated melanoma cell lysates (115 pg/mL vs 56 pg/mL) and IL-6 level could be enhanced due to the infection with JS-1 (166.9 pg/mL) and T-VEC (170.0 pg/mL). There was no relevant difference between the two viruses. The SK29MEL lysates generated by the combination of doxorubicin and T-VEC were the most potent stimulators of IL-6 production.
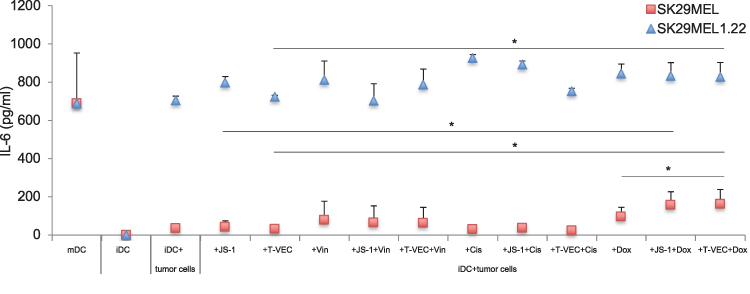 |
Figure 3 IL-6 production by DCs after coculture with different treated SK29MEL and SK29MEL1.22. Abbreviations: DCs, dendritic cells; ELISA, enzyme-linked immunosorbent assay; iDCs, immature dendritic cells. Notes: Analysis of IL-6 production by ELISA by iDCs, mDCs matured by cytokine cocktail and iDCs after coculture with SK29MEL (red square) or SK29MEL1.22 (blue triangle) differently treated. The infection with JS-1 or T-VEC and the treatment with vincristine, cisplatin and doxorubicin were examined individually and in combination with the oncolytic viruses. The mean values and standard deviations of three measurements are shown. *P ≤ 0.05. |
In the coculture of SK29MEL1.22 (blue triangle) lysates and iDCs, a constantly increased IL-6 concentration was found compared with SK29MEL. The iDCs cocultured with untreated SK29MEL1.22 secreted 646.9 pg/mL, which was increased by JS-1 (828.8 pg/mL) and not relevantly influenced by T-VEC (664.7 pg/mL).
Figure 4 shows the concentration of GM-CSF in the supernatant of the cocultures from melanoma cell lysates of both cell lines (SK29MEL, orange bars and SK29MEL1.22, yellow bars) and iDCs after treatment with JS-1 or T-VEC and the three cytotoxic agents. Melanoma cells alone (SK29MEL and SK29MEL1.22) did not show an increased GM-CSF level (92 pg/mL and 46pg/mL) (data not shown). There is an increased production by mDCs (9645 pg/mL) matured with the cytokine cocktail. Non-cocultivated iDCs produced only very small amounts of GM-CSF (55 pg/mL). iDCs cocultured with untreated SK29MEL produced a small amount of GM-CSF at 359 pg/mL. The infection with JS-1 did not lead to an increase. Melanoma cell lysates generated by T-VEC infection induced the maturation of the DCs, which can be seen from the increase in the GM-CSF concentration to 9962 pg/mL. Treatment with cytostatic drugs alone or in combination with JS-1 did not increase GM-CSF production. T-VEC in combination with cisplatin reached an amount of 10,136 pg/mL, comparable with the pure T-VEC treatment. Combining T-VEC with vincristine (4191 pg/mL) and especially with doxorubicin (519 pg/mL) reduced the secretion of GM-CSF compared with monotherapy with T-VEC. A similar picture emerged in the coculture of HL-A2-negative SK29MEL1.22 with iDCs. The greatest increase in GM-CSF production was induced to a comparable extent by the pure T-VEC treatment (9583 pg/mL) and the combination of T-VEC with cisplatin (9569 pg/mL). The combination of all three cytotoxic agents with T-VEC showed increased concentrations of GM-CSF compared with cytotoxic agents alone. The combination of T-VEC and doxorubicin, however, showed a significantly lower GM-CSF production than the individual treatment with T-VEC (2519 pg/mL vs 9583 pg/mL).
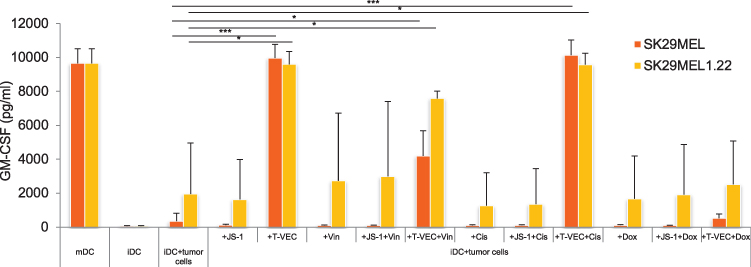 |
Figure 4 GM-CSF production by DCs after coculture with differently treated SK29MEL and SK29MEL1.22 using ELISA. Abbreviations: ELISA, enzyme-linked immunosorbent assay; GM-CSF, granulocyte-macrophage colony-stimulating factor; iDCs, immature dendritic cells. Notes: Analysis of GM-CSF production by iDCs, mDCs matured by cytokine cocktail and iDCs after coculture with differently treated SK29MEL (orange bars) and SK29MEL1.22 (yellow bars). The infection with JS-1 or T-VEC and the treatment with vincristine, cisplatin and doxorubicin were examined individually and in combination with the oncolytic viruses. The mean values and standard deviations of three measurements carried out independently of one another are shown. *P ≤ 0.05; ***P ≤ 0.001. |
Expression of the Surface Markers CD4, CD8 and CD69
To investigate the T-cell reaction, the melanoma cells SK29MEL and SK29MEL1.22 were infected with JS-1 or T-VEC and additionally treated with the three cytotoxic agents. The degree of activation of the T lymphocytes was examined in the FACS using the surface markers CD4, CD8 and CD69. CD4 and CD8 characterize the subtype of T lymphocytes (CD4+ Th or CD8+ CTL) and CD69 is considered to be an early T-cell activation marker. When activated, it is upregulated within 30 minutes and remains measurable for up to 72h.29
Figure 5A shows the activation of the helper T (Th) cells, which is characterized by the simultaneous expression of CD4 and CD69 on their surface. The figure shows the percentage of cells in the coculture that express both surface molecules. T cells stimulated by adding CD3 and CD28 showed a Th cell activation of 53% (CD4+CD69+cells) and 39% (CD8+CD69+cells). In contrast, non-stimulated and non-cocultivated T cells expressed both markers in only 4% and 5% of cells.
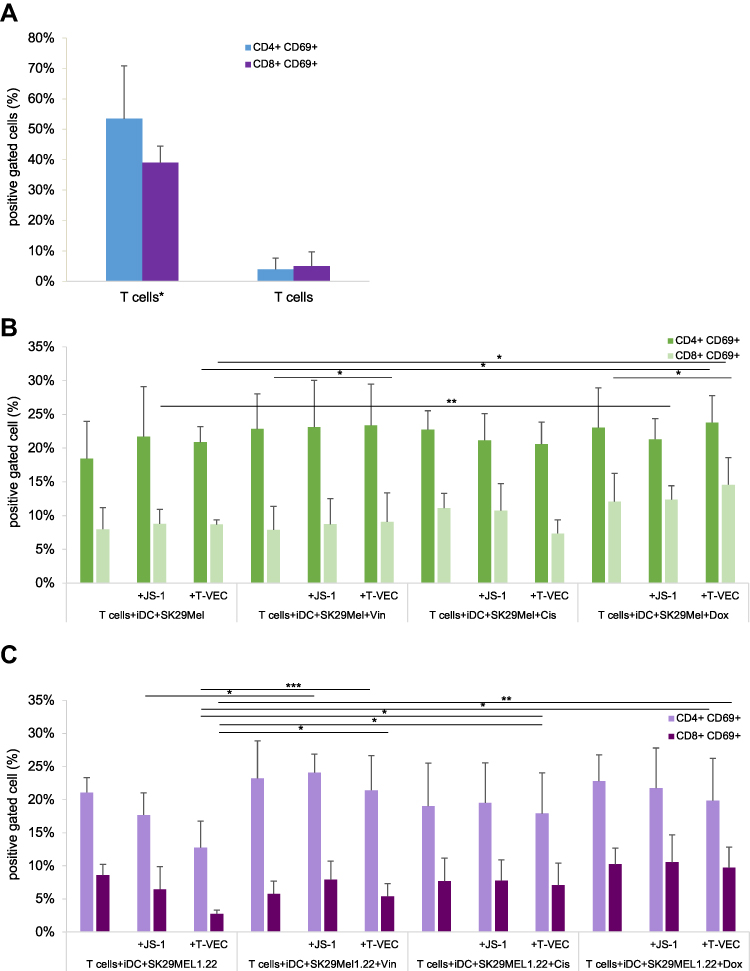 |
Figure 5 (A–C) Expression of activation markers on T lymphocytes. Abbreviations: *T cells stimulated with CD3 and CD28; CTL, cytotoxic T lymphocytes; iDCs, immature dendritic cells; TCL, tumor cell lysates. Notes: Cocultivation of virally or drug-induced TCLs with iDCs and CTLs. Activation markers CD69 and also CD4 and CD8 were stained and FACS analysis was performed. For the CTL experiment melanoma cells were seeded, infected after 24 h and after 24 h, iDCs were added at a ratio of 1:5 and cocultivated as described further. T cells were added 24 hours later in a ratio of 1:1 to the cocultivated melanoma cell lysates. (A) Expression of activation markers on stimulated *And unstimulated T cells. (B) CTL experiment with SK29MEL and with SK29MEL1.22 (C). Experiments were performed independently three times. *P ≤ 0.05; **P≤ 0.01, and ***P ≤ 0.001. |
Figure 5B shows that there was a pronounced increase in Th activation to 18% through coculture with iDCs and untreated melanoma cells of the SK29MEL line. Infection of the melanoma cells with JS-1 or T-VEC induced an increase in CD4+CD69+ expression to 22% and 21%, respectively. Treatment with vincristine alone (23%) and in combination with JS-1 or T-VEC (23%) did not show any relevant effects beyond the untreated SK29MEL. The same could be shown for the treatment with cisplatin and doxorubicin. The combination of cisplatin and viruses did not cause increased Th activation. Melanoma cell lysates generated by cisplatin alone achieved an increased expression of CD4 and CD69 on the T cells of 23%. Treatment of SK29MEL with doxorubicin in combination with viruses showed a similar tendency (green bars).
The light green bars show the proportion of cells in the coculture, which expressed CD8 and CD69 on their surface. The coculture of T cells with iDCs and untreated SK29MEL cells induced 8% CTL activation, which was not increased by JS-1 and T-VEC. Vincristine treatment alone and in combination with oncolytic viruses was unable to induce activation more strongly than untreated SK29MEL. The tumor cell lysates generated by cisplatin and doxorubicin led to an increased CTL activation of 11% and 12%. Through the combination of T-VEC and doxorubicin, 15% of the cells were CD8+/CD69+. In combination with cisplatin and T-VEC, the activation rate (7%) was reduced.
Figure 5C shows the CD4+CD69+ expression after coculture with lysates of the SK29MEL1.22 cell line (violet bars). The untreated melanoma cells induced the activation of Th cells by 21%. Treatment of the melanoma cells with vincristine alone (23%) and in combination with JS-1 (24%) led to a slight increased expression of CD4+ and CD69+ compared with untreated tumor cells. The combination with cisplatin showed no increased expression in each case. The melanoma cell lysates generated by doxorubicin induced 23% activation of Th cells, which was not increased by additional infection with JS-1 (22%) or T-VEC (20%).
T cells which were cocultivated with untreated SK29MEL1.22 and iDCs expressed the two measured markers CD8+ and CD69+ to 9%. Combination with vincristine alone achieved 6%. Additional infection with JS-1 reached 8% and in combination with T-VEC 5%. Treatment with cisplatin alone or in combination with viruses did not increase CD8+CD69+ expression. Doxorubicin treatment alone or in combination with JS-1 and T-VEC increase CD8+CD69+ expression slightly up to 10% and 11%.
Evaluation of CTL Activation by the Release of Proinflammatory Cytokines
Activated CTLs increasingly secrete immunostimulatory cytokines and thus induce their own proliferation and differentiation.30 Their concentration should therefore be examined as an additional sign of CTL activation. We analyzed the IL-2 concentration in the supernatant after coculture of melanoma cell lysates, iDCs and T lymphocytes (data not shown). The coculture of T cells and iDCs with melanoma cells resulted in an increase in IL-2 production in both cell lines compared with non-cocultivated T cells. A difference in IL-2 production between untreated and infected melanoma cells or those treated with cytotoxic agents could not be determined in either cell line. This value was not exceeded by any of the treatments tested. No effect of the virus and cytotoxic treatment on IL-2 production and thus CTL activation could be demonstrated. Furthermore, the expected increase in TNF-α secretion after virus infection alone did not occur in this study (data not shown).
Discussion
After promising results in pre-clinical studies, specific oncolytic viruses are currently investigated in clinical studies.31–34 The T-VEC virus used in our study was able to enhance durable response rate of melanoma patients with an increased response of the local and metastatic lesions.35,36 It is a promising therapeutic agent because of the good clinical effect while having a low level of undesirable side effects.37
A key mechanism of action of cytostatic agents is the induction of programmed cell death (apoptosis). However, permanent cures for tumors are prevented by tumoral resistance mechanisms to cytostatic agents and the immune system. The systemic chemotherapy of these substances could be enhanced efficiently if synergistically combined with locally acting, cell-death-inducing viruses, which simultaneously stimulate the immune system. The central aim of this combination was to overcome the apoptotic resistance of the tumors. Therefore, we investigated the increased apoptosis in tumors with suitable combinations and, on the other hand, the better activated tumor-directed immune defense. For both viruses, a reduction of viability, in a time-and concentration-dependent manner could be shown. The combination of JS-1 and T-VEC with cytostatic drugs caused a stronger reduction of cell viability than the viruses alone (Figure 1A and Figure 1).
For SK29MEL1.22 melanoma cells similar results were observed (data not shown). Further treatment schedules of 24 h or 48 h of viral infection and treatment with cytostatic drugs were tested and showed similar results (data not shown). For combined treatment with H-1PV and cytostatic drugs similar results could be shown.9,12,16,38 Doxorubicin can enhance the antitumor effect of an oncolytic adenovirus (CEA‐regulated CD55‐TMn) by promoting cell apoptosis in live cancer cells and a mouse model.39 Furthermore, combination therapy with doxorubicin and oncolytic adenovirus synergistically enhances cytotoxicity in human chronic myeloid leukemia cells in vitro.11 Kellish et al observed increased small cell lung cancer survival following intrapulmonary administration of myxoma virus (MYXV) that was enhanced by combined treatment with cisplatin.40
Oncolytic viruses have a multimodal mechanism of action that consists of direct oncolysis, antivascular properties and the generation of a tumor-directed immune response.41 For various oncolytic viruses, increased maturation of DCs after infection with increased secretion of proinflammatory cytokines has already been demonstrated.17,42–45 The effect of an infection with unchanged viruses on DCs shows a dependency on the virus group: reoviruses stimulate DCs directly,44 while measles viruses inhibit DC function and trigger cell death46 and adenoviruses are neutral towards DCs.47 The type of tumor cell and its immunogenicity also influence the consequences of the infection.48
In this work, we have shown that untreated melanoma cell lysates can stimulate the maturation of DCs and especially the subsequent activation of the CTLs. This corresponds to the results of Remmel et al, that various melanoma cells induce the phenotypic maturation of DCs in vitro.49 Infection of the melanoma cells with the HSV-1 derivatives JS-1 and T-VEC should further intensify this maturation.
For parvovirus H-1, we have shown in a melanoma model that the infection of cells increased the expression of CD80, CD83 and CD86 as well as the secretion of TNF-α and IL-6.12,50 The same could also be observed in colon cancer cells.16 In the melanoma model, T-VEC induces immunogenic cell death with time-dependent release of damage-associated molecular patterns (DAMPs) and cytokine production,51 which can serve as a maturation stimulus for DCs. With regard to CD83/86 expression, the tumor cell lysates were able to induce DC maturation to varying degrees. Observed for CD80/86, the combination of doxorubicin and T-VEC and for SK29MEL additionally vincristine was shown to be the most potent stimuli in this study. Cisplatin did not induce increased maturation and its immunomodulatory effects tended to be inhibited when combined with oncolytic viruses. The tumor cell lysates generated by cytostatic treatment and virus infection were thus able to induce DC maturation to varying degrees. A clear immunostimulatory effect of the examined viruses alone was not shown. Something similar has already been observed for adenovirus-infected melanoma cells,52 and a vaccinia virus did not induce any increased cytokine secretion in vitro despite the DAMP release being detected.53
Schierer et al described that human iDCs phagocytosed adenovirus-generated melanoma cell lysates in vitro, but subsequent maturation did not take place.52 An additional stimulus was required, delivered in the form of a cytokine mix or CD40L coding in the virus genome. It could also be observed that the infected melanoma cell lysates alone are not a sufficient stimulus for DC maturation. It is possible that the GM-CSF expression was not high enough and that consequently the DCs were not given sufficient stimulus in vitro by both virus strains, or that an unknown activating component was missing in the model.
TNF-α was examined in this work and increased secretion was expected after coculture of DCs with melanoma cell lysates generated by oncolytic virus infection, as previously observed for a Newcastle disease virus (NDV).42 TNF-α has antivascular properties, which, together with the direct antivascular capacity of HSV-1,54 could develop a synergistic pronecrotic effect on tumor cells. It is also known that TNF-α stimulates the maturation of DCs.55 The expected increase in TNF-α secretion after virus infection alone did not occur in this study (data not shown).
In addition to antitumor and DC-stimulating properties,28 IL-6 also has the ability to inhibit Treg, which suppresses the tumor immune defense.56 It promotes the survival of naïve T cells57 and induces a CD8 response.58 In the present study, a slight increase in IL-6 secretion could be observed through the untreated SK29MEL lysates, but not additionally through virus infection of the cell line. The fact that the infection of tumor cells with oncolytic viruses in DC coculture does not increase these two cytokines has already been reported for a vaccinia virus.53 In addition, IL-6 could suppress DC activation and maturation by activation of the STAT3 signaling pathway.59 Such proteins have not been described for HSV-1; it is therefore possible that the lack of increased expression in our study is most likely due to the general lack of maturation of the DCs after virus infection. This is consistent with the unobserved clear increase in expression of the CD molecules.
Mature DCs activate T cells, which are essential for effective tumor defense. By coculture of isolated T lymphocytes with iDCs and the infected melanoma cell lysates of both lines, a possible T-cell activating effect of the HSV-1 derivatives was investigated in this work. The most potent stimuli for SK29MEL were found to be the combination treatment of doxorubicin and T-VEC and for SK29MEL1.22 doxorubicin with JS-1. These results are consistent with those of CTL activation. The activation of CTLs (CD8/CD69) can thus be induced by melanoma cell lysates. For SK29MEL, doxorubicin with T-VEC and for SK29MEL1.22, the untreated melanoma cells showed the strongest CTL stimulation and the combination of doxorubicin and JS-1. Overall, a stronger CD4 than CD8 response was shown in all approaches due to the coculture of melanoma cell lysates, iDCs and T lymphocytes.
Since DC maturation and cross-presentation are of fundamental importance for the activation of the T-cell response,60 no strong induction could be expected with regard to CTL activation as a result of the low DC maturation rate observed after virus infection. The strength of the T-cell response depends on the amount of antigen presented, the duration of cell contact, and the DC maturity stimulus.61 However, we showed an increased activation of the T lymphocytes by both HSV-1 strains in comparison to unstimulated T cells, which has already been reported for H-1PV.50
Hirooka et al observed an increased infiltration of the tumor area with CD8+ and CD4+ lymphocytes in patients after treatment with the herpes virus HF10.62 Miller et al found the contribution of the immune response to the therapeutic effect of HSV-1 in a study on immunodeficient mice with intracranial melanoma.63 In contrast to immunocompetent conspecifics, their survival could not be significantly prolonged by the virus, which was attributed to the lack of immune reaction. Moesta et al reported something similar when, due to depletion of CD8+ lymphocytes in mice, the local and systemic effect of T-VEC did not occur and survival was not prolonged either.64 A positive effect on the immune response through infection of the melanoma cells with HSV-1 derivatives could therefore be relevant for the clinical success of T-VEC.
As an immunostimulatory cytokine, GM-CSF attracts immune effector cells into the tumor microenvironment (TME), induces the maturation of DCs, improves the antigen presentation and promotes the development of a specific immune response.65 It is therefore relevant for generating a humoral and cellular antitumor response. Nevertheless, the combination of GM-CSF with immunotherapeutic agents, cytostatic agents and as an adjuvant after surgery for melanoma was able to extend survival in several studies.66,67 The new aspect of combining oncolysis and immunotherapy has good prospects to further enhance cancer therapy.68
Therefore, the use of GM-CSF in oncolytic viruses to strengthen their immune induction is justified. The GM-CSF expression in T-VEC supports DC maturation and thus the T cell response, but this effect has not yet been proven preclinically.
In this work, we could show by ELISA that increased GM-CSF production takes place through T-VEC infection of melanoma cells. JS-1 showed no relevant effect on the GM-CSF concentration in comparison with iDCs cocultivated with untreated cells, neither in monotherapy nor in combination with cytotoxic agents. The increased GM-CSF production in all cocultures containing T-VEC-induced lysates can be explained by the genetic modifications of T-VEC. The concentration of the cytokine was 86-fold higher after coculture of the T-VEC-induced SK29MEL lysates with iDCs than after JS-1 infection. This agrees with the results of Liu et al, who first described the virus.20 Kemp et al examined a GM-CSF-coding reovirus in comparison with an unchanged reovirus, which also shows an increased production of the cytokine.69
Burke et al found no clear advantage of the cytokine gene on the immune cell activation of PBMCs for a GM-CSF-coding NDV.42 The influence of GM-CSF only became apparent through the selection of CD14+ monocytes. Kim et al also observed that the insertion of GM-CSF into the genome of the vaccinia virus JX594 had no effect on tumor cell lysis.70 In this work, too, DC maturation, as assessed using the surface markers by FACS and IL-6 by ELISA, even tended to be lower after T-VEC infection than after JS-1 infection, despite the strong GM-CSF secretion. Thus, an advantage of GM-CSF gene insertion could not be clearly demonstrated. Liu et al were unable to show any improvement in local tumor regression in a mouse model due to the GM-CSF gene in the T-VEC strain.20 However, an increased decline in non-injected distant metastases was observed, which was attributed to the systemic tumor immune defense.
The lymphocyte activation marker CD69 was upregulated in CD4+ and CD8+ lymphocytes after T-VEC infection, ie, the T cells were activated, but there was no increased expression of it compared with the JS-1 infection. Kemp et al reported that the insertion of GM-CSF into the virus genome had no influence on CD4+ and CD8+ in the local tumor environment and attributed it to the low immunogenicity of the tumor.69 In vitro, Cerullo et al observed that an adenovirus supplemented with GM-CSF did not affect the oncolytic capacity, but the formation of a tumor-specific memory with immunity to new tumors of the same type was reinforced by the additional GM-CSF.71
So far, the cytotoxic effect of the combination of oncolytic viruses and chemotherapy has mainly been considered, with less attention given to the immunomodulatory effect. In this work, the effect of T-VEC and JS-1 in combination with doxorubicin, cisplatin and vincristine in terms of immunomodulation was investigated. The cytostatic drugs alone already partially represented a maturation stimulus for the immune cells. A synergism between the examined herpes viruses and the cytostatic drugs could not be explicitly proven either. However, there were tendencies according to which specific combinations could potentiate immune cell activation in the melanoma model, on which further studies should be based. The combination of JS-1 and T-VEC with cytotoxic agents showed predominantly stronger immunomodulatory effects than the pure virus infection, the combination with doxorubicin in particular showing a positive effect. Since cytotoxic agents attack the integrity of the host cells, it is questionable whether a stronger immune effect could have been seen through combination with oncolytic viruses at a lower dose.
Treatment of DCs with vincristine alone in vitro induced a 150% increase in IL-2 production, which indicates T-cell activation.72 In contrast, Kaneno et al reported that although vincristine phenotypically induced the maturation of the DCs, this did not improve the antigen presentation.73 This T-cell activating effect of vincristine could not be confirmed in this work by increased expression of CD69 on CD4+ and CD8+ T cells. Although the DCs showed maturation, as in Kaneno et al, there was no enhanced T-cell response. The combination of vincristine and oncolytic viruses has not been studied frequently. Since vincristine inhibits microtubule polymerization, which HSV-1 depends on for its transport into the cell nucleus, a possible negative interaction due to reduced viral replication has already been suspected.74 Cinatl et al, however, demonstrated an increased cytotoxicity of vincristine with the oncolytic virus G207 and showed that the combination in vivo can lead to a complete remission in embryonic rhabdomyosarcoma.74 Moehler et al were able to observe an additive increase in toxicity due to the vincristine combination in comparison to monotherapy in melanoma cells of the SK29MEL line infected with H-1PV.12 After combination treatment, the melanoma cell lysates led to increased immune activation in the form of DC maturation. Like vinblastine, which is a similar compound, vincristine shows antiangiogenic properties in tumors.75
Modulatory effects of cisplatin on the TME and the immune system have been described many times.7 Buttiglieri et al investigated the effect of tumor cell lysates generated by cisplatin on DC function and maturation and could not demonstrate any stimulatory influence.76 The DCs only took up small amounts of tumor cell lysates and did not secrete increased amounts of IL-12, so that there was no relevant cross-presentation and T-cell activation. This was shown by the lack of an increase in the concentration of IFN-γ in an ELISA. Also, cisplatin was not able to induce a type I IFN response in vitro, which would indicate an immunogenic mode of action of the cytostatic agent.77 After cisplatin therapy, there was also no increased secretion of IL-6 and TNF-α.78
This lack of immune activation by cisplatin can be confirmed in our work. As monotherapy, it did not induce any maturation of DCs in cell culture beyond the untreated melanoma cells, neither with regard to CD80/83/86 expression nor to cytokine secretion. However, there was an increased infiltration of tumors with CD4+ and CD8+ lymphocytes after cisplatin treatment.79 Treatment with cisplatin and paclitaxel synergistically induced the increased secretion of IL-2 and IFN-γ, the main cytokines of CTLs. In addition to stimulating a T-cell response, cisplatin also sensitizes the tumor to CTLs.80 An increase in CD4+ and CD8+ lymphocyte activation could also be shown in our work for cisplatin-generated lysates of the SK29MEL line, which slightly increase CD69 expression. The combination of cisplatin with various oncolytic viruses has so far been examined mainly with regard to cytotoxicity. Preclinically and clinically, depending on the virus and tumor origin, there were sometimes synergistic or at least additive effects10,12,81,82 and sometimes no additional cytotoxic effects.83 Herpes viruses, as they were also used for this work, showed in several studies an oncolytic effect that could be potentiated by cisplatin. The combination of differently modified herpes viruses and cisplatin achieved in models of non-small cell lung cancer84 and pancreatic carcinoma85 increased therapeutic efficacies compared with the respective monotherapy. The ICP34.5-deleted HSV-1 strain NV1066 showed synergistic or additive cytotoxic effects such as increased viral replication in vitro in combination with cisplatin in various tumor cells.86 Moehler et al were able to show an intensification of DC maturation by combination treatment compared with cytostatic monotherapy.12 Pandha et al, by contrast, in addition to increased cytotoxicity, described an inhibitory effect on cytokine secretion after treatment of melanoma cells with cisplatin and a reovirus in vivo and in vitro.87 Here, the expression of CD80, CD83 and CD86 was not higher than in iDCs cocultured with SK29MEL and SK29MEL1.22 lysates after cisplatin treatment either alone or in combination with the viruses in untreated melanoma cells. On the contrary, the maturation was even lower after combination therapy than with pure cisplatin treatment, which is consistent with the observations of Pandha et al.87
Doxorubicin has multiple proven immunostimulatory effects7 and induces the release of HSP70 in vitro, a protein functioning as a danger signal, which leads to the maturation of DCs and thus to the induction of a T-cell response.76,77 Shurin et al showed the increased DC antigen presentation induced by low-dose doxorubicin with upregulation of CD80, CD86, CD40, MHC II and increased IL-12 secretion.72 Hu et al also reported an increased expression of CD80 after doxorubicin treatment.78 An increased expression of the coligands CD80 and CD86 after treatment with doxorubicin alone could be confirmed in this work. An increased CTL activation by doxorubicin-generated tumor cell lysates could be confirmed in this work, too.
A combination of doxorubicin and various oncolytic viruses has shown additive and sometimes synergistic cytotoxic effects in several studies.76,83 Bolyard et al demonstrated a synergism of doxorubicin and an ICP34.5-deleted herpes virus with increased tumor apoptosis and longer survival compared with the respective monotherapy for ovarian cancer in a mouse model.88 A clinical phase I/II study on patients with breast cancer showed that T-VEC enhanced response to neoadjuvant chemotherapy with a response rate of 55% and it was feasible at the approved dose, with manageable toxicity.89 An immune effect of the combination, as it was investigated in this work, could not yet be shown.90 In this context, we must consult the different conditions in the used tumor model and in patients, where additional factors play a role in the immune response concerning to the tumor microenvironment. Overall, a positive effect of the combination on the immune response could be observed here. DC maturation was enhanced by infection with HSV-1 derivatives in addition to doxorubicin treatment. Compared with doxorubicin monotherapy, there was an increased expression of the surface markers; an increased secretion of IL-6 and also, T-cell activation could be partially increased.
Conclusions
In addition to the already proven direct oncolytic effect,20 the immune effects of modified HSV-1 viruses were examined in vitro in this work. Our thesis that the GM-CSF-encoding T-VEC strain improves the immune effects in the human melanoma model could not be confirmed in this study. There was indeed an immune response as a result of the infection with both tested viruses, but T-VEC did not induce a stronger response in vitro than JS-1. The combination of oncolytic viruses with chemotherapy has also been clinically tested and has shown positive results.91,92 Studies with T-VEC previously suggested a combination therapy impact with, for example, immune checkpoint inhibitors93 on the cytotoxic and immunomodulatory effect of treatment. Malignant human melanoma is a highly immunogenic tumor that can be therapeutically injected easily in most cases and therefore meets the basic requirements for successful oncolytic virus therapy.94 Oncolytic viruses in the form of T-VEC are already a promising therapeutic option, especially for patients with stage IIIB to IVM1a tumors,37 which can be optimized as far as possible through combinations.35 Combinatorial studies of oncolytic viruses and immunotherapy will be significant for the future.95 By further investigation virus entry, host-virus interactions after systemic virus application and virus bio-distribution, the application modalities of OVs could be improved. In addition, by increased immunotherapy in clinical oncology, there is a gain in importance of induction of anti-tumor immunity by viral oncolysis.96
In order to be able to use T-VEC optimally in the future, preclinical studies as well as proof of concept (POC) studies in mouse models should be carried out. With a better understanding of the exact mechanisms of action, patient- and tumor-related influencing factors could be defined which modulate the therapeutic response. The indication could possibly be extended to diagnose other types of tumors, and thus more patients could benefit from this therapeutic agent T-VEC.
In our study, we are able to show that the immunostimulatory effects of the tested viruses could be partially increased by chemotherapy. We recommended that this approach is pursued in preclinical studies, exploring T-VEC mainly in combination. The combination of doxorubicin and T-VEC has given the clearest positive results, and this approach can be recommended for further investigations.
Acknowledgments
Aspects of this article are part of the doctoral thesis of V Boeswald. Our thanks go to Amgen for supporting material and further conceptual discussion. The project was partially supported by an educational grant of Amgen.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Garber K. China approves World’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. doi:10.1093/jnci/djj111
2. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5(1):e1115641. doi:10.1080/2162402x.2015.1115641
3. Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. 2014;4:74. doi:10.3389/fonc.2014.00074
4. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. 2018;6(1):140. doi:10.1186/s40425-018-0458-z
5. Geletneky K, Hartkopf AD, Krempien R, Rommelaere J, Schlehofer JR. Improved killing of human high-grade glioma cells by combining ionizing radiation with oncolytic parvovirus H-1 infection. J Biomed Biotechnol. 2010;2010:350748. doi:10.1155/2010/350748
6. Li J, Bonifati S, Hristov G, et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol Med. 2013;5(10):1537–1555. doi:10.1002/emmm.201302796
7. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21(1):15–25. doi:10.1038/cdd.2013.67
8. Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482–491. doi:10.1038/onc.2009.356
9. Angelova AL, Grekova SP, Heller A, et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J Virol. 2014;88(10):5263–5276. doi:10.1128/jvi.03688-13
10. Yu YA, Galanis C, Woo Y, et al. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1h68. Mol Cancer Ther. 2009;8(1):141–151. doi:10.1158/1535-7163.Mct-08-0533
11. Li L, You LS, Mao LP, Jin SH, Chen XH, Qian WB. Combing oncolytic adenovirus expressing Beclin-1 with chemotherapy agent doxorubicin synergistically enhances cytotoxicity in human CML cells in vitro. Acta Pharmacol Sin. 2018;39(2):251–260. doi:10.1038/aps.2017.100
12. Moehler M, Sieben M, Roth S, et al. Activation of the human immune system by chemotherapeutic or targeted agents combined with the oncolytic parvovirus H-1. BMC Cancer. 2011;11:464. doi:10.1186/1471-2407-11-464
13. Michielin O, van Akkooi ACJ, Ascierto PA, Dummer R, Keilholz U; [email protected] EGCEa. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up dagger. Ann Oncol. 2019;30(12):1884–1901. doi:10.1093/annonc/mdz411
14. Watanabe D, Goshima F, Mori I, Tamada Y, Matsumoto Y, Nishiyama Y. Oncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10. J Dermatol Sci. 2008;50(3):185–196. doi:10.1016/j.jdermsci.2007.12.001
15. Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: the Rigvir(R) story. Eur J Pharmacol. 2018;837:117–126. doi:10.1016/j.ejphar.2018.08.042
16. Heinrich B, Goepfert K, Delic M, Galle PR, Moehler M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer cells. Onco Targets Ther. 2013;6:1119–1127. doi:10.2147/ott.S49371
17. Heinrich B, Klein J, Delic M, et al. Immunogenicity of oncolytic vaccinia viruses JX-GFP and TG6002 in a human melanoma in vitro model: studying immunogenic cell death, dendritic cell maturation and interaction with cytotoxic T lymphocytes. Onco Targets Ther. 2017;10:2389–2401. doi:10.2147/ott.S126320
18. Smiley JR. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol. 2004;78(3):1063–1068. doi:10.1128/jvi.78.3.1063-1068.2004
19. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. 2017;18(1):1–15. doi:10.1007/s40257-016-0238-9
20. Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. doi:10.1038/sj.gt.3301885
21. Wong HH, Lemoine NR, Wang Y. Oncolytic viruses for cancer therapy: overcoming the obstacles. Viruses. 2010;2(1):78–106. doi:10.3390/v2010078
22. Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16(15):4005–4015. doi:10.1158/1078-0432.Ccr-10-0196
23. Livingston PO, Shiku H, Bean MA, Pinsky CM, Oettgen HF, Old LJ. Cell-mediated cytotoxicity for cultured autologous melanoma cells. Int J Cancer. 1979;24(1):34–44. doi:10.1002/ijc.2910240107
24. Wölfel T, Klehmann E, Müller C, Schütt KH, Meyer Zum Büschenfelde KH, Knuth A. Lysis of human melanoma cells by autologous cytolytic T cell clones. Identification of human histocompatibility leukocyte antigen A2 as a restriction element for three different antigens. J Exp Med. 1989;170(3):797–810. doi:10.1084/jem.170.3.797
25. Goepfert K, Dinsart C, Rommelaere J, Foerster F, Moehler M. Rational combination of parvovirus H1 with CTLA-4 and PD-1 checkpoint inhibitors dampens the tumor induced immune silencing. Front Oncol. 2019;9:425. doi:10.3389/fonc.2019.00425
26. Jonuleit H, Kuhn U, Muller G, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27(12):3135–3142. doi:10.1002/eji.1830271209
27. Jonuleit H, Giesecke A, Kandemir A, Paragnik L, Knop J, Enk AH. Induction of tumor peptide-specific cytotoxic T cells under serum-free conditions by mature human dendritic cells. Arch Dermatol Res. 2000;292(7):325–332. doi:10.1007/s004030000144
28. Adams S, O’Neill DW, Bhardwaj N. Recent advances in dendritic cell biology. J Clin Immunol. 2005;25(3):177–188. doi:10.1007/s10875-005-4086-2
29. Marzio R, Mauël J, Betz-Corradin S. CD69 and regulation of the immune function. Immunopharmacol Immunotoxicol. 1999;21(3):565–582. doi:10.3109/08923979909007126
30. Borish LC, Steinke JW. 2. Cytokines and chemokines. J Allergy Clin Immunol. 2003;111(2Suppl):S460–S475. doi:10.1067/mai.2003.108
31. Breitbach CJ, Arulanandam R, De Silva N, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013;73(4):1265–1275. doi:10.1158/0008-5472.Can-12-2687
32. Geletneky K, Hajda J, Angelova AL, et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first Phase I/Iia glioblastoma trial. Mol Ther. 2017;25(12):2620–2634. doi:10.1016/j.ymthe.2017.08.016
33. Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. doi:10.1038/nm.3089
34. Kiyohara E, Tanemura A, Nishioka M, et al. Intratumoral injection of hemagglutinating virus of Japan-envelope vector yielded an antitumor effect for advanced melanoma: a phase I/IIa clinical study. Cancer Immunol Immunother. 2020;69(6):1131–1140. doi:10.1007/s00262-020-02509-8
35. Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. doi:10.1200/jco.2014.58.3377
36. Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–730. doi:10.1245/s10434-009-0809-6
37. Andtbacka RHI, Collichio F, Harrington KJ, et al. Final analyses of OPTiM: a randomized Phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J Immunother Cancer. 2019;7(1):145. doi:10.1186/s40425-019-0623-z
38. Sieben M, Herzer K, Zeidler M, et al. Killing of p53-deficient hepatoma cells by parvovirus H-1 and chemotherapeutics requires promyelocytic leukemia protein. World J Gastroenterol. 2008;14(24):3819–3828. doi:10.3748/wjg.14.3819
39. Xiao B, Ying C, Chen Y, et al. Doxorubicin hydrochloride enhanced antitumour effect of CEA-regulated oncolytic virotherapy in live cancer cells and a mouse model. J Cell Mol Med. 2020;24(22):13431–13439. doi:10.1111/jcmm.15966
40. Kellish P, Shabashvili D, Rahman MM, et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J Clin Invest. 2019;129(6):2279–2292. doi:10.1172/JCI121323
41. Marchini A, Scott EM, Rommelaere J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses. 2016;8(1). doi:10.3390/v8010009
42. Burke S, Shergold A, Elder MJ, et al. Oncolytic Newcastle disease virus activation of the innate immune response and priming of antitumor adaptive responses in vitro. Cancer Immunol Immunother. 2020;69(6):1015–1027. doi:10.1007/s00262-020-02495-x
43. Enderlin M, Kleinmann EV, Struyf S, et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009;16(2):149–160. doi:10.1038/cgt.2008.62
44. Errington F, Steele L, Prestwich R, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180(9):6018–6026. doi:10.4049/jimmunol.180.9.6018
45. Guillerme JB, Boisgerault N, Roulois D, et al. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res. 2013;19(5):1147–1158. doi:10.1158/1078-0432.CCR-12-2733
46. Grosjean I, Caux C, Bella C, et al. Measles virus infects human dendritic cells and blocks their allostimulatory properties for CD4+ T cells. J Exp Med. 1997;186(6):801–812. doi:10.1084/jem.186.6.801
47. Schierer S, Hesse A, Müller I, et al. Modulation of viability and maturation of human monocyte-derived dendritic cells by oncolytic adenoviruses. Int J Cancer. 2008;122(1):219–229. doi:10.1002/ijc.23074
48. Liu T, Zhang Y, Cao Y, et al. Optimization of oncolytic effect of Newcastle disease virus Clone30 by selecting sensitive tumor host and constructing more oncolytic viruses. Gene Ther. 2020. doi:10.1038/s41434-020-0145-9
49. Remmel E, Terracciano L, Noppen C, et al. Modulation of dendritic cell phenotype and mobility by tumor cells in vitro. Hum Immunol. 2001;62(1):39–49. doi:10.1016/s0198-8859(00)00221-4
50. Moehler MH, Zeidler M, Wilsberg V, et al. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum Gene Ther. 2005;16(8):996–1005. doi:10.1089/hum.2005.16.996
51. Bommareddy PK, Zloza A, Rabkin SD, Kaufman HL. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology. 2019;8(7):1591875. doi:10.1080/2162402X.2019.1591875
52. Schierer S, Hesse A, Knippertz I, et al. Human dendritic cells efficiently phagocytose adenoviral oncolysate but require additional stimulation to mature. Int J Cancer. 2012;130(7):1682–1694. doi:10.1002/ijc.26176
53. Ma J, Ramachandran M, Jin C, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020;11(1):48. doi:10.1038/s41419-020-2236-3
54. Benencia F, Courreges MC, Conejo-García JR, et al. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum Gene Ther. 2005;16(6):765–778. doi:10.1089/hum.2005.16.765
55. Brunner C, Seiderer J, Schlamp A, et al. Enhanced dendritic cell maturation by TNF-alpha or cytidine-phosphate-guanosine DNA drives T cell activation in vitro and therapeutic anti-tumor immune responses in vivo. J Immunol. 2000;165(11):6278–6286. doi:10.4049/jimmunol.165.11.6278
56. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299(5609):1033–1036. doi:10.1126/science.1078231
57. Teague TK, Schaefer BC, Hildeman D, et al. Activation-induced inhibition of interleukin 6-mediated T cell survival and signal transducer and activator of transcription 1 signaling. J Exp Med. 2000;191(6):915–926. doi:10.1084/jem.191.6.915
58. Vanden Bush TJ, Buchta CM, Claudio J, Bishop GA. Cutting Edge: importance of IL-6 and cooperation between innate and adaptive immune receptors in cellular vaccination with B lymphocytes. J Immunol. 2009;183(8):4833–4837. doi:10.4049/jimmunol.0900968
59. Park SJ, Nakagawa T, Kitamura H, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173(6):3844–3854. doi:10.4049/jimmunol.173.6.3844
60. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193(2):233–238. doi:10.1084/jem.193.2.233
61. Adams S, O’Neill DW, Bhardwaj N. Recent advances in dendritic cell biology. J Clin Immunol. 2005;25(2):87–98. doi:10.1007/s10875-005-2814-2
62. Hirooka Y, Kasuya H, Ishikawa T, et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer. 2018;18(1):596. doi:10.1186/s12885-018-4453-z
63. Miller CG, Fraser NW. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol Ther. 2003;7(6):741–747. doi:10.1016/s1525-0016(03)00120-5
64. Moesta AK, Cooke K, Piasecki J, et al. Local delivery of OncoVEX(mGM-CSF) generates systemic antitumor immune responses enhanced by cytotoxic T-lymphocyte-associated protein blockade. Clin Cancer Res. 2017;23(20):6190–6202. doi:10.1158/1078-0432.CCR-17-0681
65. Shi Y, Liu CH, Roberts AI, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16(2):126–133. doi:10.1038/sj.cr.7310017
66. Eroglu Z, Kong KM, Jakowatz JG, Samlowski W, Fruehauf JP. Phase II clinical trial evaluating docetaxel, vinorelbine and GM-CSF in stage IV melanoma. Cancer Chemother Pharmacol. 2011;68(4):1081–1087. doi:10.1007/s00280-011-1703-z
67. Hodi FS, Lee S, McDermott DF, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744–1753. doi:10.1001/jama.2014.13943
68. Deng L, Yang X, Fan J, et al. An oncolytic vaccinia virus armed with GM-CSF and IL-24 double genes for cancer targeted therapy. Onco Targets Ther. 2020;13:3535–3544. doi:10.2147/OTT.S249816
69. Kemp V, van den Wollenberg DJM, Camps MGM, et al. Arming oncolytic reovirus with GM-CSF gene to enhance immunity. Cancer Gene Ther. 2019;26(9–10):268–281. doi:10.1038/s41417-018-0063-9
70. Kim M, Nitschké M, Sennino B, et al. Amplification of oncolytic vaccinia virus widespread tumor cell killing by sunitinib through multiple mechanisms. Cancer Res. 2018;78(4):922–937. doi:10.1158/0008-5472.Can-15-3308
71. Cerullo V, Pesonen S, Diaconu I, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010;70(11):4297–4309. doi:10.1158/0008-5472.Can-09-3567
72. Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. 2009;183(1):137–144. doi:10.4049/jimmunol.0900734
73. Kaneno R, Shurin GV, Tourkova IL, Shurin MR. Chemomodulation of human dendritic cell function by antineoplastic agents in low noncytotoxic concentrations. J Transl Med. 2009;7:58. doi:10.1186/1479-5876-7-58
74. Cinatl J, Cinatl J, Michaelis M, et al. Potent oncolytic activity of multimutated herpes simplex virus G207 in combination with vincristine against human rhabdomyosarcoma. Cancer Res. 2003;63(7):1508–1514.
75. Park KJ, Yu MO, Park DH, Park JY, Chung YG, Kang SH. Role of vincristine in the inhibition of angiogenesis in glioblastoma. Neurol Res. 2016;38(10):871–879. doi:10.1080/01616412.2016.1211231
76. Buttiglieri S, Galetto A, Forno S, De Andrea M, Matera L. Influence of drug-induced apoptotic death on processing and presentation of tumor antigens by dendritic cells. Int J Cancer. 2003;106(4):516–520. doi:10.1002/ijc.11243
77. Sistigu A, Yamazaki T, Vacchelli E, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. doi:10.1038/nm.3708
78. Hu J, Kinn J, Zirakzadeh AA, et al. The effects of chemotherapeutic drugs on human monocyte-derived dendritic cell differentiation and antigen presentation. Clin Exp Immunol. 2013;172(3):490–499. doi:10.1111/cei.12060
79. Tsuchikawa T, Miyamoto M, Yamamura Y, Shichinohe T, Hirano S, Kondo S. The immunological impact of neoadjuvant chemotherapy on the tumor microenvironment of esophageal squamous cell carcinoma. Ann Surg Oncol. 2012;19(5):1713–1719. doi:10.1245/s10434-011-1906-x
80. Bergmann-Leitner ES, Abrams SI. Differential role of Fas/Fas ligand interactions in cytolysis of primary and metastatic colon carcinoma cell lines by human antigen-specific CD8+ CTL. J Immunol. 2000;164(9):4941–4954. doi:10.4049/jimmunol.164.9.4941
81. Wennier ST, Liu J, McFadden G. Bugs and drugs: oncolytic virotherapy in combination with chemotherapy. Curr Pharm Biotechnol. 2012;13(9):1817–1833. doi:10.2174/138920112800958850
82. Sei S, Mussio JK, Yang QE, et al. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol Cancer. 2009;8:47. doi:10.1186/1476-4598-8-47
83. Passer BJ, Castelo-Branco P, Buhrman JS, Varghese S, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus vectors and taxanes synergize to promote killing of prostate cancer cells. Cancer Gene Ther. 2009;16(7):551–560. doi:10.1038/cgt.2009.10
84. Toyoizumi T, Mick R, Abbas AE, Kang EH, Kaiser LR, Molnar-Kimber KL. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancer. Hum Gene Ther. 1999;10(18):3013–3029. doi:10.1089/10430349950016410
85. Kasuya H, Nishiyama Y, Nomoto S, et al. Suitability of a US3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther. 2007;14(6):533–542. doi:10.1038/sj.cgt.7701049
86. Adusumilli PS, Chan MK, Chun YS, et al. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol Ther. 2006;5(1):48–53. doi:10.4161/cbt.5.1.2237
87. Pandha HS, Heinemann L, Simpson GR, et al. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res. 2009;15(19):6158–6166. doi:10.1158/1078-0432.CCR-09-0796
88. Bolyard C, Yoo JY, Wang PY, et al. Doxorubicin synergizes with 34.5ENVE to enhance antitumor efficacy against metastatic ovarian cancer. Clin Cancer Res. 2014;20(24):6479–6494. doi:10.1158/1078-0432.Ccr-14-0463
89. Soliman H, Hogue D, Han H, et al. A Phase I trial of talimogene laherparepvec in combination with neoadjuvant chemotherapy for the treatment of nonmetastatic triple-negative breast cancer. Clin Cancer Res. 2021;27(4):1012–1018. doi:10.1158/1078-0432.CCR-20-3105
90. Simpson GR, Relph K, Harrington K, Melcher A, Pandha H. Cancer immunotherapy via combining oncolytic virotherapy with chemotherapy: recent advances. Oncolytic Virother. 2016;5:1–13. doi:10.2147/ov.S66083
91. Opyrchal M, Aderca I, Galanis E. Phase I clinical trial of locoregional administration of the oncolytic adenovirus ONYX-015 in combination with mitomycin-C, doxorubicin, and cisplatin chemotherapy in patients with advanced sarcomas. Methods Mol Biol. 2009;542:705–717. doi:10.1007/978-1-59745-561-9_35
92. Karapanagiotou EM, Roulstone V, Twigger K, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18(7):2080–2089. doi:10.1158/1078-0432.Ccr-11-2181
93. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36(17):1658–1667. doi:10.1200/JCO.2017.73.7379
94. Dharmadhikari N, Mehnert JM, Kaufman HL. Oncolytic virus immunotherapy for melanoma. Curr Treat Options Oncol. 2015;16(3):326. doi:10.1007/s11864-014-0326-0
95. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer. 2020;8(2). doi:10.1136/jitc-2020-001486
96. Nettelbeck DM, Leber MF, Altomonte J, et al. Virotherapy in Germany—recent activities in virus engineering, preclinical development, and clinical studies. Viruses. 2021;13(8):1420.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.