")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 14
In silico Molecular Docking, DFT Analysis and ADMET Studies of Carbazole Alkaloid and Coumarins from Roots of Clausena anisata: A Potent Inhibitor for Quorum Sensing
Authors Eswaramoorthy R , Hailekiros H, Kedir F, Endale M
Received 18 November 2020
Accepted for publication 3 January 2021
Published 5 February 2021 Volume 2021:14 Pages 13—24
DOI https://doi.org/10.2147/AABC.S290912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Rajalakshmanan Eswaramoorthy, Hadgu Hailekiros, Fedlu Kedir, Milkyas Endale
Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, 1888, Ethiopia
Correspondence: Rajalakshmanan Eswaramoorthy
Associate Professor in Bio-Inorganic and Medicinal Chemistry, Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, 1888, Ethiopia
Email [email protected]
Milkyas Endale
Associate Professor in Organic Chemistry, Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, 1888, Ethiopia
Email [email protected]
Introduction: In modern drug design, in silico methods are largely used to understand drug-receptor interactions and quantum chemical properties. In the present study, a computational de novo design approach was used to confirm mode of binding for antibacterial activity, elucidating quantum chemical properties and ADMET-drug-likeness of carbazole alkaloid ( 1) and three coumarins ( 2– 4) isolated from roots of Clausena anisata.
Methods: Docking studies were performed with DNA-Gyrase (6F86) and LasR binding domain (2UV0) employing a flexible ligand docking approach using AutoDock Vina. SwissADME prediction and toxicological predictions were performed by ADMET. The optimized structures and molecular electrostatic potential surface of the isolated compounds were predicted by DFT analysis using B3LYP/6-31G basis levels.
Results and Discussion: The docking results revealed that compound 3 showed better docking scores against both DNA gyrase B and LasR binding domain compared with ciprofloxacin with potential as an inhibitor of bacterial DNA gyrase and quorum sensing LasR binding domain. The SwissADME prediction results showed that all the isolated compounds ( 1– 4) satisfy Lipinski’s rule of five with zero violations. Toxicological prediction results suggested that all compounds and ciprofloxacin are non-hepatotoxic, non-carcinogenic, non-irritant, immunogenic, and non-cytotoxic. The DFT analysis results revealed that compound 3 has large electronegativity (χeV), global softness (σ eV− 1), global electrophilicity (ωeV), and mutagenicity value closer to ciprofloxacin (with LD50 value of 480 mg/kg) suggesting better bioactivity and chemical reactivity with considerable intra-molecular charge transfer between electron-donor to electron-acceptor groups.
Conclusion: Overall, compound 3 may serve as a lead molecule that could be developed into a potent E. coli DNA gyrase B inhibitor and efficient inhibitor for quorum sensing auto-inducer LasR binding domain of Pseudomonas aeruginosa.
Keywords: alkaloids, coumarins, DNA gyrase, LasR binding domain, de novo DFT and docking studies
Introduction
Clausena anisata is one of the traditional medicinal plants that belong to the family of Rutaceae used for the treatment of bacterial and fungal infections.1–4 Recently, our group reported isolation and antibacterial activity of a carbazole alkaloid (1) and three coumarins (2–4) from the roots of Clausena anisata against strains of Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa and Bacillus subtilis.2 Additionally, the alkaloids and coumarins have been reported to be active against both Gram-positive and Gram-negative bacteria and fungi.2,3,5–13 However, the mechanism of action of these compounds has not been previously validated experimentally and little assessment was made on the potential “drug-likeness” of these compounds. In the discovery of new and complementary antibacterial agents, phytochemicals that show antibacterial activity can be examined for potential inhibition of bacterial target proteins such as peptide deformylase (PDF), topoisomerase IV (TopoIV), DNA gyrase B (GyrB), protein tyrosine phosphatase (Ptp), UDP-galactopyranosemutase (UGM), cytochrome P450 (CYP121), and NAD+-dependent DNA ligase, as well as phytochemical inhibitors of bacterial efflux pumps or quorum sensing proteins, or agents that enhance the immune system.
Pseudomonas aeruginosa is the most extensively studied strain due to the potentially fatal effects of its infections, that are responsible for many deaths each year worldwide. It is the most common Gram-negative bacterium found in hospital-acquired infections, being responsible for nosocomial pneumonia, urinary tract infections, and surgical wound or bloodstream infections. In general, many Gram-negative bacteria coordinate the activities of their populations through autoinducers.14 This is called quorum sensing and it can regulate pathogenic virulence factor production and antimicrobial resistance. Quorum sensing inhibitors can diminish the pathogenicity of P. aeruginosa.14–16 The emergence of LasR as a key regulator of P. aeruginosa pathogenesis has made its inhibition an interesting pharmaceutical goal. Additionally, DNA topoisomerases are crucial enzymes for maintaining the topological state of DNA.17–19 The ATP-dependent bacterial type II topoisomerases, DNA gyrase and DNA topoisomerase IV are responsible for the maintenance of the correct level of supercoiling in bacterial DNA. These enzymes have been well-validated as antibacterial targets as the fluoroquinolone antibiotics act at the DNA-cleavage site of these enzymes.20 The aminocoumarin antibiotics, including novobiocin, which was used clinically in the 1960s, target the ATP-binding site.21 Therefore, in the present study we examined the binding modes of reported inhibitors (coumarins and a carbazole alkaloid from the roots of Clausena anisata) within bacterial DNA-GyrB and LasR, considering their promising antibacterial activity along with DFT analysis for optimized structure, drug-likeness, pharmacokinetics, toxicity profile and compared with the clinically approved drug ciprofloxacin.
Materials and Methods
Preparation of Ligands
The 2D structures (.mol) of all compounds were drawn (Figure 1) and analyzed using ChemDraw 16.0. All the compounds (1–4) are converted to 3D structure (.pdb) using Chem3D 16.0. The 3D coordinates (.pdb) of each molecule were loaded on to Chem3D for energy minimization.
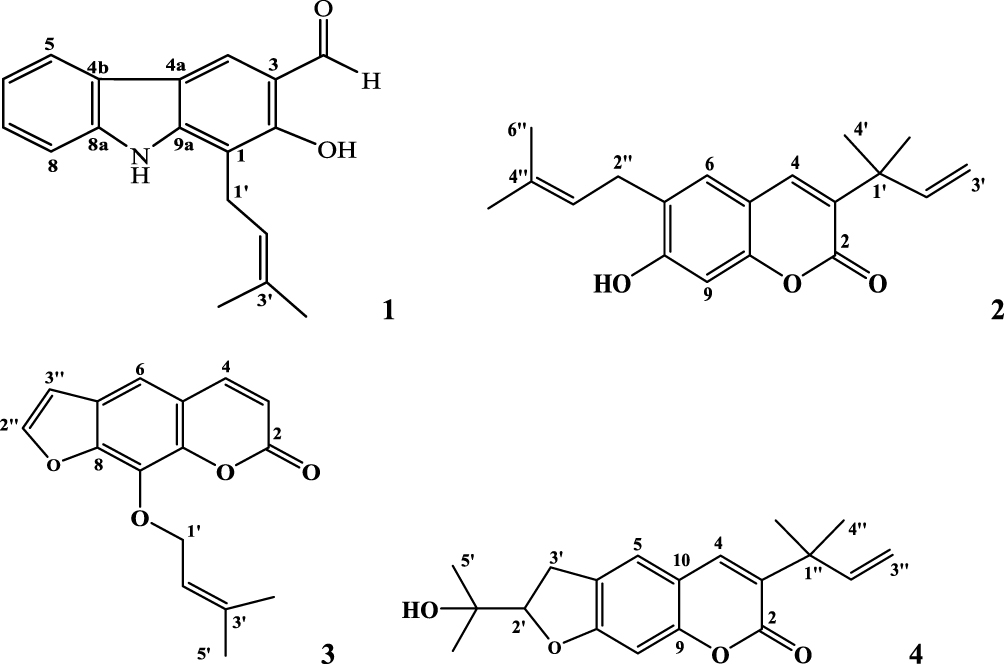 |
Figure 1 2D-ChemDraw structures of isolated compounds (1–4). |
Preparation of Macromolecule
The protein target, which is retrieved from the RCSB Protein Data Bank (PDB code 6F86 and 2UV0), serves as docking receptor.15,22 All the bound ligands and water molecules were removed from the active site of the receptor.
Molecular Docking Analysis (AutoDock Vina)
The molecular docking studies were carried out using AutoDockTools (ADT), which is a free graphic user interface (GUI) for the AutoDockVina program.23 AutoDockVina with standard protocol was used to dock the compounds (1–4) against the active site of protein (PDB ID: 6F86 and 2UV0).15,22,23 The grid box was constructed using 58, 58, and 40, pointing in x, y, and z directions, respectively, with a grid point spacing of 0.375 Å. The center grid box is of 14.527 Å, 56.689 Å and −5.122 Å. Nine different conformations were generated for each ligand scored using AutoDockVina scoring functions (Supplementary materials [Tables 1–8]) and ranked according to their binding energies (Supplementary materials). AutoDock Tools and PyMOL were used for the post-docking analyses. The conformations with the most favourable (least) free binding energy were selected for analysing the interactions between the target receptor and ligands by PyMOL.
In silico Drug-Likeness and Toxicity Predictions
Structures of compounds (1–4) were submitted to SwissADME tool and converted to their canonical simplified molecular input line entry system (SMILE), to estimate in silico pharmacokinetic parameters and other molecular properties based on the methodology reported by Amina et al., 2016.24 SwissADME predictor provided information on the numbers of hydrogen donors, hydrogen acceptors and rotatable bonds, total polar surface area and the synthetic accessibility of the compounds. The ligands were also subjected to Lipinski et al. screenings using SwissADME predictor. Drug-likeness is a prediction that determines whether a particular pharmacological agent has properties consistent with being an orally active drug. This prediction is based on an already established concept by Lipinski et al., called the Lipinski rule of five. The rule predicts that there is likely to be poor absorption or permeation when a compound possesses more than 5 H-bond donors, 10 H-bond acceptors, molecular weight greater than 500 and a calculated LogP (CLogP) greater than 5. The selection of compounds as drug candidates were determined by a parameter called drug score. The higher the drug score value, the higher the chance of the compound being considered as a drug candidate. The organ toxicities and toxicological endpoints of the ligands and their LD50 were predicted using Pro Tox II.25–27 The analyses of the compounds were compared with that of ciprofloxacin, and only compounds without violation of any of the screenings were used for the molecular docking analysis.
Quantum Computational Studies
The DFT (density functional theory) analysis of compounds (1–4) were performed using Gaussian 09 and visualized through Gauss view 5.0. The structural coordinates of the lead compounds were optimized using B3LYP/6-31 G (d,p) level basis set without any symmetrical constraints. The molecular electrostatic potential map and energies of the compounds were obtained from the optimized geometry. Koopman’s approximation was used to estimate the HOMO-LUMO energy gap and related reactive parameters (electronegativity, chemical potential, hardness, softness, electrophilicity).28
Results and Discussion
Molecular Docking Against DNA Gyrase B
Molecular docking analysis of isolated compounds showed better docking score within the active site of E. coli (6F86). Compounds 1–3 (−7.2, −7.7, and −7.6 kcal/mol, respectively) showed equal to better docking affinity than the preclinical drug ciprofloxacin (−7.2 kcal/mol) whereas compound 4 showed smaller docking affinity (−6.6 kcal/mol). All the molecules showed binding affinity within the binding sites of co-crystallized ligand (Figure 2). Compound 1 formed hydrogen bond interaction with Arg-76, Asn-46, Gly-77 and residual hydrophobic interaction with Ala-47, Glu-50, Ile-78, Val-167. Similarly, compound 2 showed one hydrogen bond (Asp-73) and residual hydrophobic interactions with amino acids Glu-50, Val-43, Val-47, Ile-78, Pro-79, Ile-94, Val-120, and Val-167. Compounds 3 and 4 both showed hydrogen bond interaction with Asn-46 whereas compound 4 showed an additional hydrogen bond with Arg-136 and hydrophobic residual interaction with Arg-76 and Pro-79. Compounds 1–3 demonstrated a similar binding affinity with residual amino acids compared with ciprofloxacin (hydrogen bond with residual amino acid Asp-73, Asn-46, Arg-76 and hydrophobic or pi-cation interaction with residual amino acids Glu-50, Ala-47, Gly-77, Ile-78, Pro-79, Ile-94, Ile-78). However, compounds 1 and 4 do not show the crucial interactions between the ligand and amino acid Asp-73. In silico docking results revealed that compounds 2 and 3 have least docked score (−7.7 and −7.6 kcal/mol, respectively) compared with other compounds and comparable residual interactions with that of ciprofloxacin within the binding pocket (Table 1), suggesting that these compounds are promising antibacterial agents against E. coli.6,8,29,30
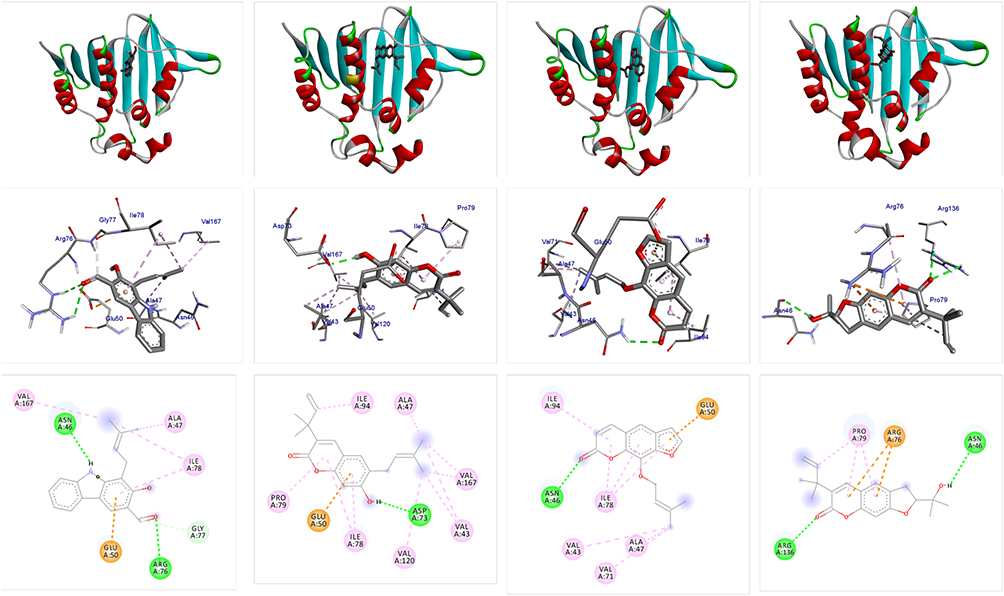 |
Figure 2 The 2D and 3D binding interactions of compounds (1–4) against DNA gyrase B (PDB ID: 6F86). Ribbon model shows the binding pocket structure of DNA gyrase B with compounds (1–4). Hydrogen bond between compounds and amino acids are shown as green dashed lines, hydrophobic interactions are shown as pink lines. |
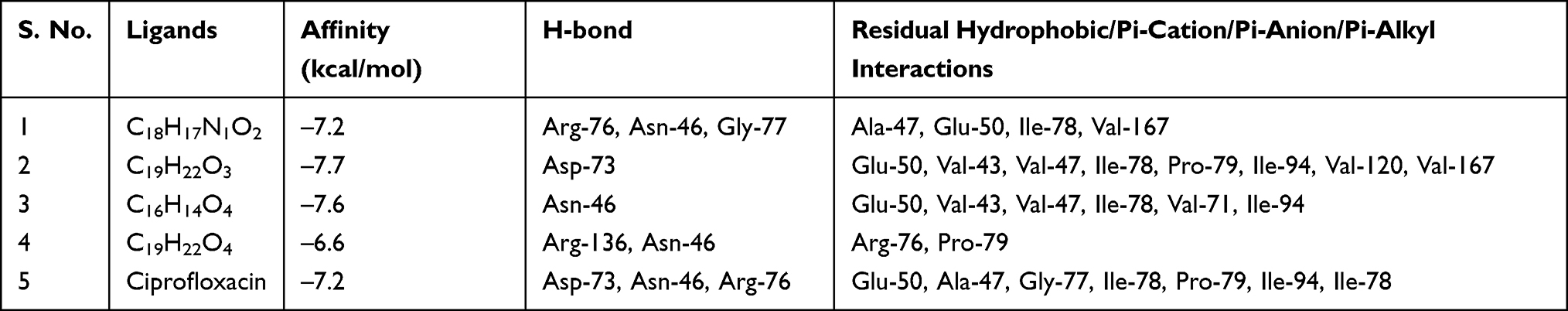 |
Table 1 Molecular Docking Scores and Residual Amino Acid Interactions of Compounds 1–4 Against E. coli DNA gyrase B (PDB ID 6F86) |
Molecular Docking Against Protein LasR Binding Domain
LasR is a key regulator of P. aeruginosa pathogenesis.31,32 Molecular docking of compounds against LasR binding domain displayed better docking scores toward P. aeruginosa (2UV0) compared with the DNA gyrase B docking results. Binding mode of active compounds (1–4) demonstrated docked scores of −8.9, −9.0, −9.3, and −9.2 kcal/mol, respectively. All isolated compounds showed docking interaction within the binding sites of co-crystallized quorum sensing auto-inducer molecules N-3-oxo-dodecanoyl-l-homoserine lactone (OHN) where hydrogen bonding interactions were observed with residual amino acids similar to OHN (Ser-129, Asp-73, Tyr-64, and Trp-60) and hydrophobic interactions with amino acid residues Leu-40, Tyr-47, Ala-50, Ala-70, Val-76, and Leu-125 (Figure 3). Compound 1 showed two hydrogen bonding interactions with amino acid residues Thr-75 and Thr-115 and residual hydrophobic interaction with amino acid residues Asp-73, Tyr-64, Val-76, Leu-36, Ile-52, Ala-50, Ala-70, Tyr-47, Cys-79, Leu-125, and Ala-127. Compounds 2 and 3 showed formation of hydrogen bonding with amino acids Ser-129, Thr-115 and hydrophobic interaction with amino acid residues Asp-73, Tyr-64, Leu-36, Ile-52, Ala-50, Tyr-47, and Ala-127 within the binding pocket (Table 2). In addition, compound 3 showed two more hydrogen bonding interactions with Tyr-47 and Thr-75. Compound 4 (−9.2 kcal/mol) showed one hydrogen bond interaction with amino acid Leu-110 and hydrophobic interaction with residual amino acids Asp-73, Tyr-64, Leu-36, Val-76, Ala-50, Ala-70, Tyr-47, and Ala-127. Molecular docking results showed that all the molecules have least docking score compared with co-crystallized ligand (OHN) and clinical drug (ciprofloxacin). The docking results are in good agreement with our previous in vitro antibacterial analysis where compounds 3 and 4 displayed better antibacterial activity against P. aeruginosa (14 ± 0.1 and 12 ± 0.2 mm zone of inhibition, respectively) compared with ciprofloxacin (15 ± 0.3 mm zone of inhibition)2,3,14,16,31 suggesting compounds 3 and 4 may be better antibacterial agents specifically through quorum sensing activity.
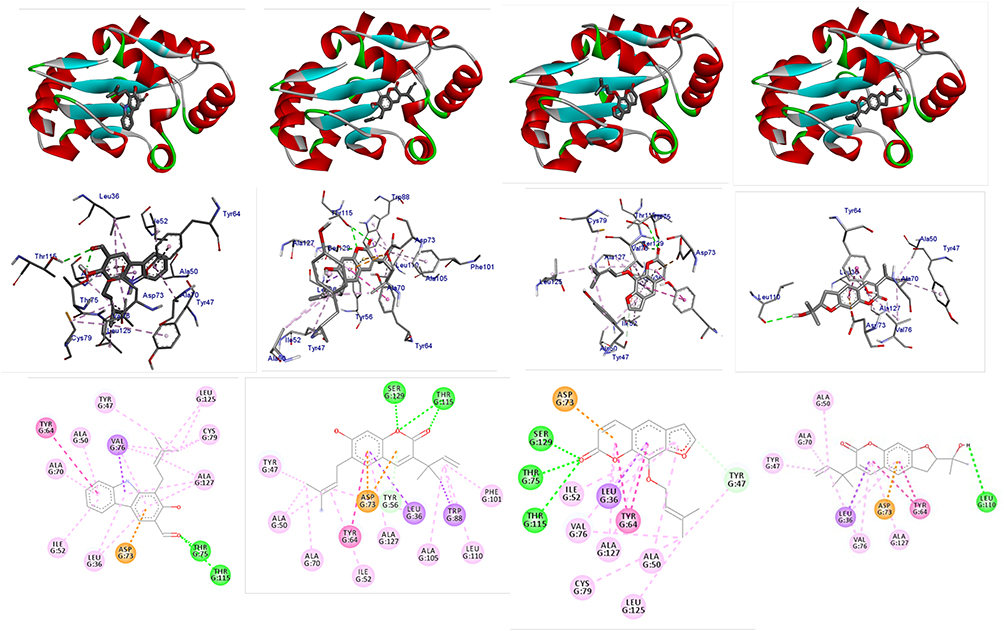 |
Figure 3 The 2D and 3D binding interactions of compounds (1–4) against LasR binding domain (PDB ID 2UV0). Ribbon model shows the binding pocket structure of LasR binding domain with compounds (1–4). Hydrogen bond between compounds and amino acids are shown as green dashed lines, hydrophobic interactions are shown as pink lines. |
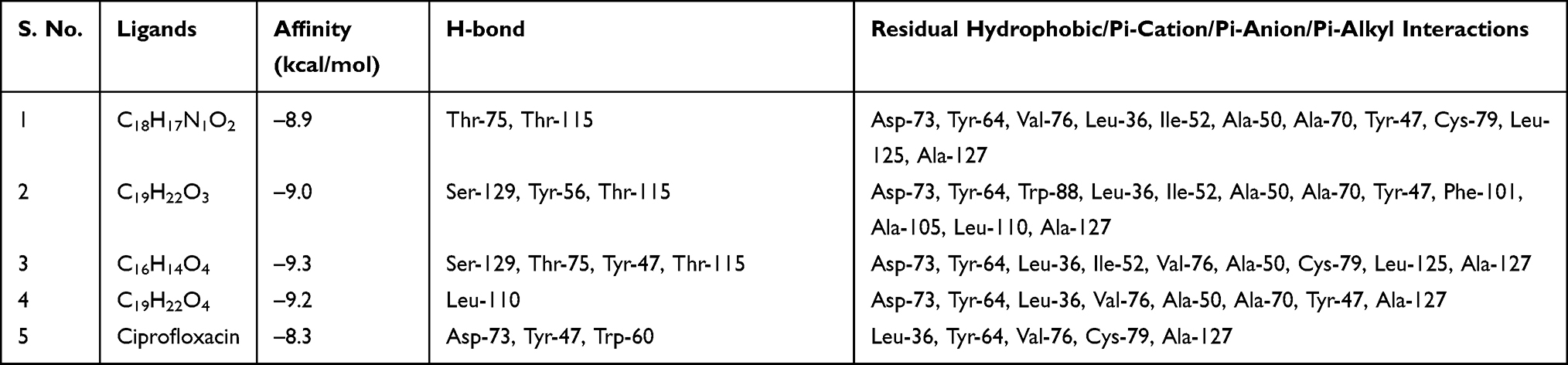 |
Table 2 Molecular Docking Scores and Residual Amino Acid Interactions of Compounds 1–4 Against LasR Binding Domain |
In silico Pharmacokinetics (Drug-Likeness) and Toxicity Analysis
The SwissADME computed results showed that all the isolated compounds (1–4) satisfy Lipinski’s rule of five with zero violations (Table 3). The Kp values of all molecules are within the range of –4.26 to –5.47 cm/s with ciprofloxacin (–8.98 cm/s) inferring low skin permeability. The predicted logP values revealed that they have optimal lipophilicity (ranging from 2.43 to 4.37). The SwissADME prediction parameters showed that all the compounds 1–4 have high gastrointestinal (GI) absorption, blood–brain barrier (BBB) permeation and no compounds are substrates of permeability glycoprotein (P-gp). The CYP’s interaction result showed that all the compounds are inhibitors for CYP1A2, CYP2C19, and CYP2C9. Compounds 1 and 4 were found to be potential inhibitors for CYP2D6 whereas compounds 2, 3, and ciprofloxacin were found to be non-inhibitors. For CYP3A4, compound 1 and ciprofloxacin were found to be non-inhibitors whereas compounds 2–4 were found to be potential inhibitors (Table 4).
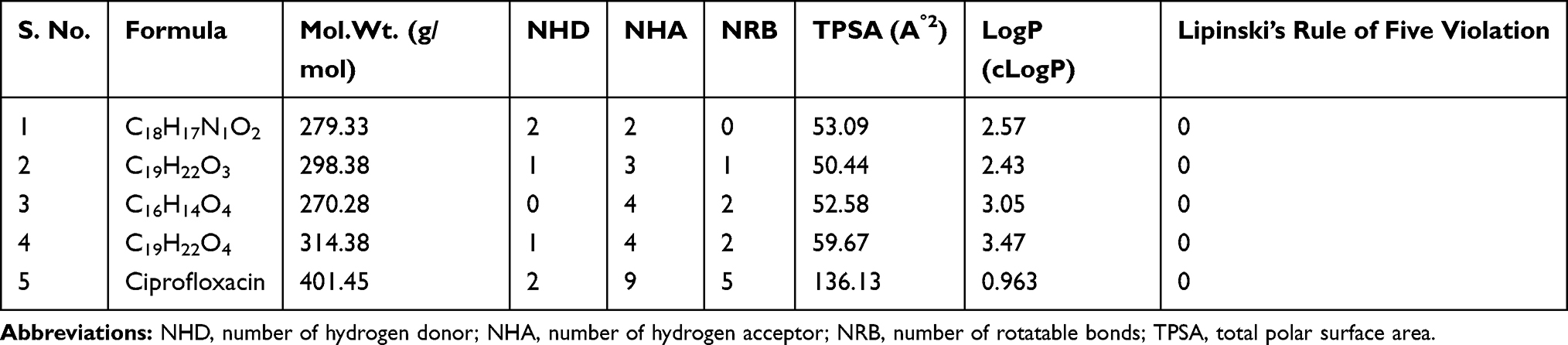 |
Table 3 Drug-Likeness Predictions of Compounds 1–4, Computed by SwissADME |
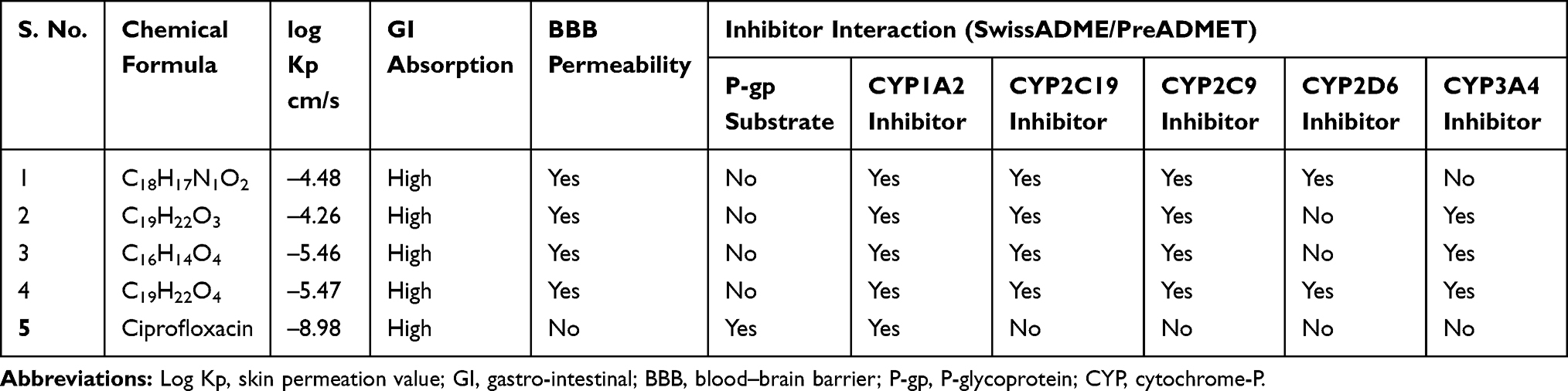 |
Table 4 ADME Predictions of Compounds 1–4, Computed by SwissADME and PreADMET |
Acute toxicity prediction results such as LD50 values and toxicity class classification [1 (toxic) to 6 (non-toxic)] revealed that none of the ligands demonstrated acute toxicity similar to ciprofloxacin with toxicity class classification 4 (harmful if swallowed) for compounds 3, 4 and ciprofloxacin. Toxicological prediction results suggested that all compounds and ciprofloxacin are non-hepatotoxic, non-carcinogenic, non-irritant, immunogenic and non-cytotoxic with the exception of compound 3 that displayed a mutagenicity value closer to ciprofloxacin (with LD50 value of 480 mg/kg) suggesting this compound can be the lead compound compared with other isolated compounds in the investigation (Table 5).
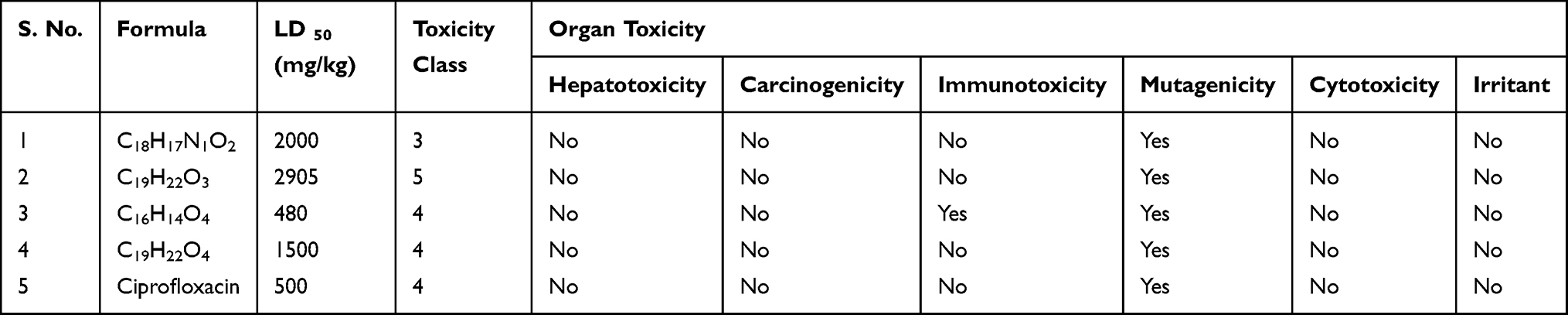 |
Table 5 Prediction of Toxicity of Compounds (1–4), Computed by Pro-Tox II and OSIRIS Property Explorer |
DFT Analysis for Optimized Structure and Quantum Chemical Parameters
Optimized geometries (Figure 4) and molecular electrostatic potential (MEP) of isolated compounds were computed using density functional theory (DFT) with the basis sets B3LYP/6-31 G (d,p). Bond lengths, bond angles, and dihedral angles were obtained from the optimized structure (Table 6). The determination of the MEP region is the best fit for identifying sites for intra- and inter-molecular interactions (Figure 7).33 Red/yellow regions indicate negative electrostatic potentials and the blue region shows positive, and green color designates neutral potential region.
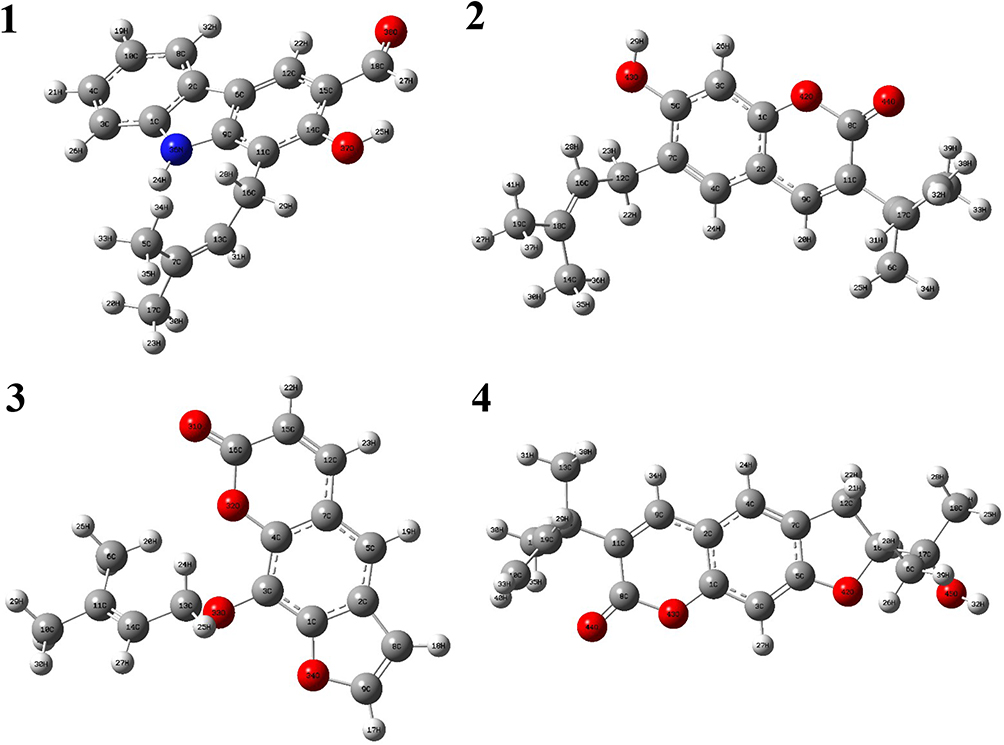 |
Figure 4 The optimized structures of compounds (1–4). |
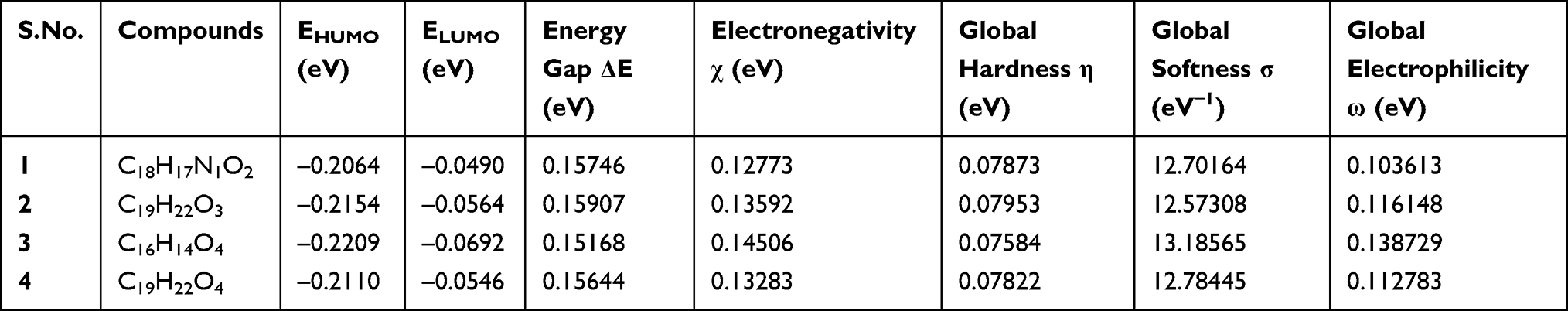 |
Table 6 The Various Quantum Chemical Parameters of Isolated Compounds (1–4) |
The DFT calculated Mulliken’s atomic charges (Figure 5) revealed charge distribution in individual atoms (Supplementary materials [Tables 9–16]). The HOMO, LUMO, and energy gap (ΔE) of the isolated compounds are calculated as –0.2064, –0.0490, and 0.15746 eV for 1, –0.2154, –0.0564 and 0.15907 eV for 2, –0.2209, –0.0692, and 0.15168 eV for 3, and –0.2110, –0.0546, and 0.15644 eV for 4 respectively (Figure 6). The results revealed that compound 3 showed the least energy gap (ΔE) compared with the other isolated compounds suggesting high chemical reactivity and considerable intramolecular charge transfer from electron donor (HOMO) to electron acceptor (LUMO) groups.34 Based on this results compound 3 has better bioactivity compared with other reported compounds herein. Additionally, compound 3 has large electronegativity (χeV), global softness (σ eV−1), and global electrophilicity (ωeV) compared with other compounds (Table 6).28
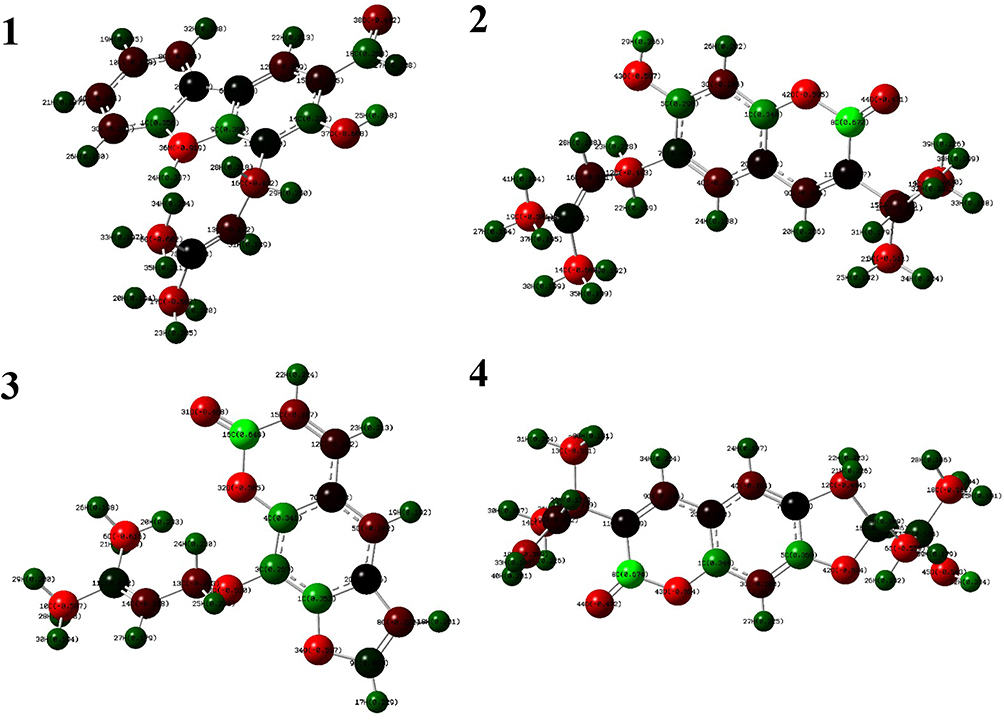 |
Figure 5 The DFT calculated Mulliken’s atomic charges of compounds (1–4). |
 |
Figure 6 Molecular orbitals and energies for the HOMO and LUMO of isolated compounds (1–4). |
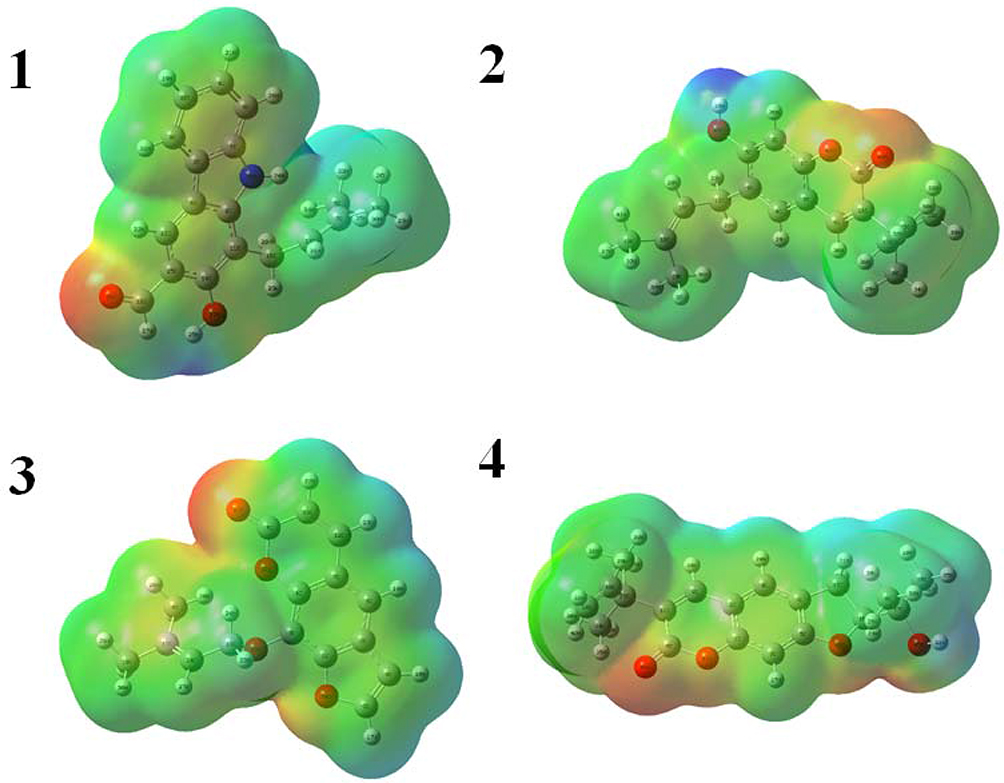 |
Figure 7 Molecular electrostatic potential surface of compounds (1–4). |
Conclusion
In the present work, a computational de novo approach was used to confirm mode of binding for antibacterial activity, elucidating quantum chemical properties and the ADMET-drug-likeness of a carbazole alkaloid (1) and three coumarins (2–4) isolated from roots of Clausena anisata. Compared with alkaloid (1), coumarins (2–4) displayed better docking score against both proteins and better docking scores than ciprofloxacin suggesting further work for functional group inclusion and modification to develop novel antibacterial agents with therapeutic activity. SwissADME results revealed that all the compounds are inhibitors for CYP1A2, CYP2C19, and CYP2C9. Compounds 1 and 4 were found to be potential inhibitors for CYP2D6 whereas compounds 2, 3, and ciprofloxacin were found to be non-inhibitors. For CYP3A4, compound 1 and ciprofloxacin were found to be non-inhibitors whereas compounds 2–4 were found to be potential inhibitors. Toxicological prediction results suggested that all compounds and ciprofloxacin are non-hepatotoxic, non-carcinogenic, non-irritant, immunogenic, and non-cytotoxic with the exception of compound 3 that displayed a mutagenicity value closer to ciprofloxacin (with LD50 value of 480 mg/kg). The DFT results revealed that compound 3 has better bioactivity and chemical reactivity with considerable intramolecular charge transfer between electron-donor and electron-acceptor groups. Based on the results of the present investigation, compound 3 may serve as a lead molecule and further work is recommended for functional group inclusion, modification, and SAR study to develop novel antibacterial agents with therapeutic activity against E. coli DNA gyrase B inhibitor and efficient inhibitors for quorum sensing auto-inducer LasR binding domain of Pseudomonas aeruginosa.
Supporting Information
Full data for Autodock Vina conformations of compounds 1-4 against LasR and DNA gyrase binding domain are included within supplementary materials.
Data Sharing Statement
AutoDock Vina conformations for compounds 1–4 against LasR and DNA gyrase B binding domain can be freely accessed from supplementary materials.
Acknowledgments
Authors would like to acknowledge Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, Ethiopia for funding support.
Disclosure
The authors declare no conflicts of interest.
References
1. Moses JP, Nattudurai G, Baskar K, Arokiyaraj S, Jayakumar M. Efficacy of essential oil from Clausena anisata and its impact on biochemical changes of Sitophilus oryzae. Environ Sci Pollut Res Int. 2020;27:23215–23221. doi:10.1007/s11356-020-08928-5
2. Tamene D, Endale M. Antibacterial activity of coumarins and carbazole alkaloid from roots of clausena anisata. Adv Pharmacol Sci. 2019;2019:5419854.
3. Agyepong N, Agyare C, Adarkwa-Yiadom M, Gbedema SY. Phytochemical investigation and anti-microbial activity of Clausena anisata (Willd)Hook. Afr J Tradit Complement Altern Med. 2014;11:200–209. doi:10.4314/ajtcam.v11i3.28
4. Mukandiwa L, Naidoo V, Katerere DR. The use of Clausena anisata in insect pest control in Africa: A review. J Ethnopharmacol. 2016;194:1103–1111. doi:10.1016/j.jep.2016.11.002
5. Pavela R, Maggi F, Lupidi G, et al. Clausena anisata and Dysphania ambrosioides essential oils: from ethno-medicine to modern uses as effective insecticides. Environ Sci Pollut Res Int. 2018;25:10493–10503. doi:10.1007/s11356-017-0267-9
6. Tankeo SB, Tane P, Kuete V. In vitro antibacterial and antibiotic-potentiation activities of the methanol extracts from Beilschmiedia acuta, Clausena anisata, Newbouldia laevis and Polyscias fulva against multidrug-resistant Gram-negative bacteria. BMC Complement Altern Med. 2015;15:412. doi:10.1186/s12906-015-0944-5
7. de Souza SM, Delle Monache F, Smania A
8. Yasunaka K, Abe F, Nagayama A, et al. Antibacterial activity of crude extracts from Mexican medicinal plants and purified coumarins and xanthones. J Ethnopharmacol. 2005;97:293–299. doi:10.1016/j.jep.2004.11.014
9. Boonsnongcheep P, Daodee S, Kitisripanya T, Putalun W. Increased carbazole alkaloid accumulation in clausena harmandiana callus culture by treatments of biocontrol agent, trichoderma harzianum and bacillus subtilis. Appl Biochem Biotechnol. 2019;189:871–883. doi:10.1007/s12010-019-03037-7
10. Manosalva L, Mutis A, Urzua A, Fajardo V, Quiroz A. Antibacterial activity of alkaloid fractions from berberis microphylla G. Forst and study of synergism with ampicillin and cephalothin. Molecules. 2016;21:76. doi:10.3390/molecules21010076
11. Solecka J, Rajnisz A, Laudy AE. A novel isoquinoline alkaloid, DD-carboxypeptidase inhibitor, with antibacterial activity isolated from Streptomyces sp. 8812. Part I: taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo). 2009;62:575–580. doi:10.1038/ja.2009.85
12. Torres YR, Berlinck RG, Nascimento GG, Fortier SC, Pessoa C, de Moraes MO. Antibacterial activity against resistant bacteria and cytotoxicity of four alkaloid toxins isolated from the marine sponge Arenosclera brasiliensis. Toxicon. 2002;40:885–891. doi:10.1016/S0041-0101(01)00286-0
13. Chakraborty A, Saha C, Podder G, Chowdhury BK, Bhattacharyya P. Carbazole alkaloid with antimicrobial activity from Clausena heptaphylla. Phytochemistry. 1995;38:787–789. doi:10.1016/0031-9422(94)00666-H
14. Weng L, Zhang Y, Yang Y, Wang L. Isolation of the autoinducer-quenching strain that inhibits LasR in Pseudomonas aeruginosa. Int J Mol Sci. 2014;15:6328–6342. doi:10.3390/ijms15046328
15. Bottomley MJ, Muraglia E, Bazzo R, Carfi A. Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J Biol Chem. 2007;282:13592–13600. doi:10.1074/jbc.M700556200
16. Chowdhury N, Bagchi A. Molecular insight into the activity of LasR protein from Pseudomonas aeruginosa in the regulation of virulence gene expression by this organism. Gene. 2016;580:80–87. doi:10.1016/j.gene.2015.12.067
17. Austin CA, Fisher LM. DNA topoisomerases: enzymes that change the shape of DNA. Sci Prog. 1990;74:147–161.
18. Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi:10.1146/annurev.biochem.70.1.369
19. Giaever GN, Snyder L, Wang JC. DNA supercoiling in vivo. Biophys Chem. 1988;29:7–15. doi:10.1016/0301-4622(88)87020-0
20. Hangas A, Aasumets K, Kekalainen NJ, et al. Ciprofloxacin impairs mitochondrial DNA replication initiation through inhibition of Topoisomerase 2. Nucleic Acids Res. 2018;46:9625–9636. doi:10.1093/nar/gky793
21. Jakopin Z, Ilas J, Barancokova M, et al. Discovery of substituted oxadiazoles as a novel scaffold for DNA gyrase inhibitors. Eur J Med Chem. 2017;130:171–184. doi:10.1016/j.ejmech.2017.02.046
22. Narramore S, Stevenson CEM, Maxwell A, Lawson DM, Fishwick CWG. New insights into the binding mode of pyridine-3-carboxamide inhibitors of E Coli DNA Gyrase. Bioorg Med Chem. 2019;27:3546–3550. doi:10.1016/j.bmc.2019.06.015
23. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi:10.1002/jcc.21334
24. Ahmed AH, Alkali YI. In silico pharmacokinetics and molecular docking studies of lead compounds derived from diospyros mespiliformis. PharmaTutor. 2019;7:31–37. doi:10.29161/PT.v7.i3.2019.31
25. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi:10.1093/nar/gky318
26. Banerjee P, Dehnbostel FO, Preissner R. Prediction Is a balancing act: importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front Chem. 2018;6:362. doi:10.3389/fchem.2018.00362
27. Drwal MN, Banerjee P, Dunkel M, Wettig MR, Preissner R. ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014;42:W53–W58. doi:10.1093/nar/gku401
28. Abu-Melha S. Design, Synthesis and DFT/DNP modeling study of new 2-amino-5-arylazothiazole derivatives as potential antibacterial agents. Molecules. 2018;23:434. doi:10.3390/molecules23020434
29. Gokgoz NB, Akbulut BS. Proteomics evidence for the activity of the putative antibacterial plant alkaloid (-)-roemerine: mainstreaming omics-guided drug discovery. OMICS. 2015;19:478–489. doi:10.1089/omi.2015.0056
30. Tada Y, Shikishima Y, Takaishi Y, et al. Coumarins and gamma-pyrone derivatives from Prangos pabularia: antibacterial activity and inhibition of cytokine release. Phytochemistry. 2002;59:649–654. doi:10.1016/S0031-9422(02)00023-7
31. Suneby EG, Herndon LR, Schneider TL. Pseudomonas aeruginosa LasR.DNA Binding Is Directly Inhibited by Quorum Sensing Antagonists. ACS Infectious Diseases. 2017;3:183–189. doi:10.1021/acsinfecdis.6b00163
32. Borlee BR, Geske GD, Blackwell HE, Handelsman J. Identification of synthetic inducers and inhibitors of the quorum-sensing regulator LasR in Pseudomonas aeruginosa by high-throughput screening. Appl Environ Microbiol. 2010;76:8255–8258. doi:10.1128/AEM.00499-10
33. Mumit MA, Pal TK, Alam MA, Islam MA, Paul S, Sheikh MC. DFT studies on vibrational and electronic spectra, HOMO-LUMO, MEP, HOMA, NBO and molecular docking analysis of benzyl-3-N-(2,4,5-trimethoxyphenylmethylene) hydrazinecarbodithioate. J Mol Struct. 2020;1220:128715. doi:10.1016/j.molstruc.2020.128715
34. Abu-Melha S. Pyridyl thiosemicarbazide: synthesis, crystal structure, DFT/B3LYP, molecular docking studies and its biological investigations. Chem Cent J. 2018;12:101. doi:10.1186/s13065-018-0469-3
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.