")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 15
In silico Drug Repurposing of Anticancer Drug 5-FU and Analogues Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics Simulation, Pharmacokinetics and Chemical Reactivity Studies
Authors Matondo A , Dendera W , Isamura BK, Ngbolua KTN, Mambo HVS, Muzomwe M, Mudogo V
Received 12 March 2022
Accepted for publication 5 August 2022
Published 15 August 2022 Volume 2022:15 Pages 59—77
DOI https://doi.org/10.2147/AABC.S366111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Alex MacKerell
Aristote Matondo,1 Washington Dendera,2 Bienfait Kabuyaya Isamura,1– 3 Koto-te-Nyiwa Ngbolua,4 Hilaire VS Mambo,1 Mayaliwa Muzomwe,1 Virima Mudogo1
1Department of Chemistry, University of Kinshasa, Kinshasa, Democratic Republic of the Congo; 2Department of Chemistry, Rhodes University, Makhanda, Eastern Cape, South Africa; 3Research Center for Theoretical Chemistry and Physics in Central Africa, Department of Chemistry, University of Kinshasa, Kinshasa, Democratic Republic of the Congo; 4Department of Biology, University of Kinshasa, Kinshasa, Democratic Republic of the Congo
Correspondence: Aristote Matondo, Email [email protected]
Background: Since the last COVID-19 outbreak, several approaches have been given a try to quickly tackle this global calamity. One of the well-established strategies is the drug repurposing, which consists in finding new therapeutic uses for approved drugs. Following the same paradigm, we report in the present study, an investigation of the potential inhibitory activity of 5-FU and nineteen of its analogues against the SARS-CoV-2 main protease (3CLpro).
Material and Methods: Molecular docking calculations were performed to investigate the binding affinity of the ligands within the active site of 3CLpro. The best binding candidates were further considered for molecular dynamics simulations for 100 ns to gain a time-resolved understanding of the behavior of the guest-host complexes. Furthermore, the profile of druggability of the best binding ligands was assessed based on ADMET predictions. Finally, their chemical reactivity was elucidated using different reactivity descriptors, namely the molecular electrostatic potential (MEP), Fukui functions and frontier molecular orbitals.
Results and Discussion: From the calculations performed, four candidates (compounds 14, 15, 16 and 18) show promising results with respect to the binding affinity to the target protease, 3CLpro, the therapeutic profile of druggability and safety. These compounds are maintained inside the active site of 3CLpro thanks to a variety of noncovalent interactions, especially hydrogen bonds, involving important amino acids such as GLU166, HIS163, GLY143, ASN142, HIS172, CYS145. Molecular dynamics simulations suggest that the four ligands are well trapped within the active site of the protein over a time gap of 100 ns, ligand 18 being the most retained.
Conclusion: In line with the findings reported herein, we recommend that further in-vitro and in-vivo investigations are carried out to shed light on the possible mechanism of pharmacological action of the proposed ligands.
Keywords: drug repurposing, 5-fluorouracil, SARS-CoV-2, molecular docking, molecular dynamics simulation, ADMET
Introduction
Over the last two decades, two major outbreaks have emerged in the world; Severe Acute Respiratory Syndrome Coronavirus-1 (SARS-CoV-1) which was first reported in November 2002 in Guangdong, China, and Middle Eastern Respiratory Syndrome (MERS-CoV) which appeared in Saudi Arabia in 2012.1 The current outbreak of the novel coronavirus disease (COVID-19) started in December 2019 in Wuhan, China, and is caused by a novel Severe Acute Respiratory Syndrome, named SARS-CoV-2.2 On the 22nd of June 2022, Worldometers reported that 6,343,924 people had died from the disease.3 To date, some drugs have been approved for the COVID-19 but the disease remains a global pandemic with few low-cost treatment options. Despite the emergence of the first generation of COVID-19 vaccines and their approval by several countries, some reported problems for a special type of vaccines such as thrombosis in vaccinated individuals4 have increased the population reluctance against the anti-COVID-19 vaccines, already observed in several parts of the world, especially in Africa.5 Such reluctance against anti-COVID-19 vaccines reminds us of the urgent need to develop anti-COVID-19 agents, especially at low cost to counteract the pandemic and stop viral infections.
In order to tackle the virus, scientists are coming up with different strategies. One of the legendary approaches relies on assessing the efficiency of plant secondary metabolites and other nutraceuticals against SARS-CoV-2 proteins.6,7 So, many natural products from plant biodiversity have been identified as promising SARS-CoV-2 Mpro inhibitors.8 Since SARS-CoV-2 encodes several proteins, previous reported computational studies were applied to identify potent compounds using other targets of SARS-CoV-2, including RNA-dependent RNA polymerase (RdRp),9 papain-like proteases (PLpro),10 spike glycoprotein,11 angiotensin converting enzyme 2 (ACE2)12 and the receptor-binding domain (RDB).13
Another sound and widely employed strategy is the so-called drug repurposing, which consists in ascribing new therapeutic uses for approved/available drugs.14,15 Drug repurposing is an acknowledged protocol in the scientific community, drawing its uniqueness from the ability to reduce the duration of a typical drug discovery pipeline, ie from a lead compound to a marketable drug, while also cutting down on the overall cost associated with the multiple maneuvers giving rise to an effective drug as compared to de novo drug discovery.16 It is worth noting that the drug repurposing technique follows two paradigms: on-target and off-target. In on-target concept, a single drug molecule is applied to a new therapeutic indication with the same biological target. The off-target flavor means that a single drug molecule or a drug candidate interacts with one or multiple new targets, out of the original scope, for new therapeutic indications. In recent years, the drug repurposing strategy has allowed many pharmaceutical companies to develop new drugs based on the discovery of novel biological targets.
Since the COVID-19 outbreak, the drug repurposing approach is increasingly being used in an attempt to find out an effective treatment against the disease. For instance, several drugs with antimalarial17 or antiviral18 properties have been found to have an anti-COVID effect. Recently, various drug repurposing studies that looked at anticancer drugs and their possible effect against COVID-19 revealed that many of them could be used in the fight against COVID-19.19–21 Furthermore, according to a hypothesis formulated by Ahmad, the combination of the anticancer drug 5-fluorouracil (5-FU) with deoxyribonucleosides and deoxyribose could represent therapeutic options for the coronavirus disease.22 Acting as anticancer drug, 5-FU is converted to 5-fluoro-2’-deoxyuridine-5’-monophosphate (FdUMP) which then forms a complex with the target enzyme thymidylate synthase (TS).23 With this in mind, we have turned our interest towards the anticancer drug 5-fluorouracil (5-FU) and nineteen of its analogues previously reported.24 For the sake of clarity, these compounds have been labelled from 1 to 20 as shown in Figure 1.
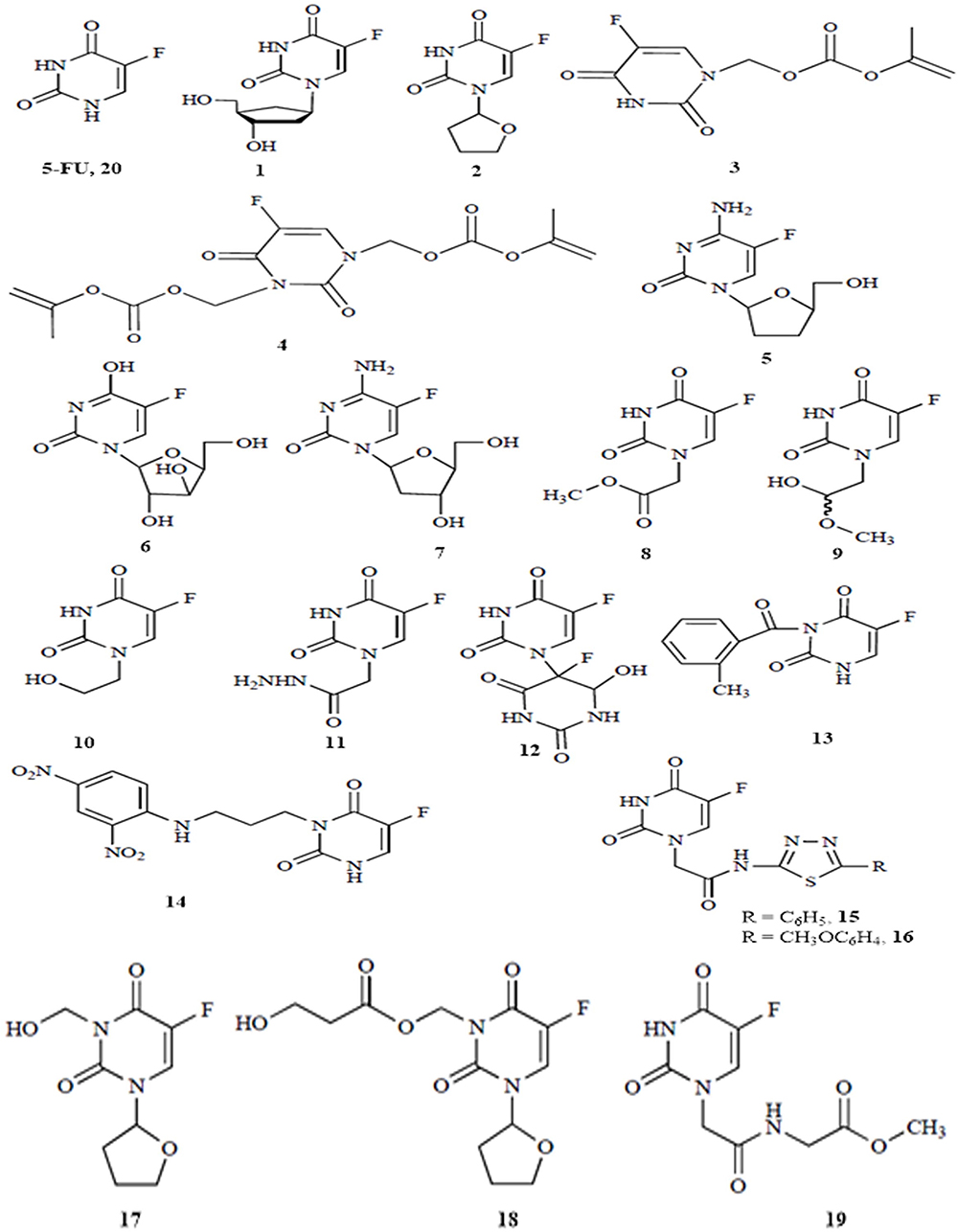 |
Figure 1 2D structures of 5-FU and its selected analogues. |
With regard to the scope of this work, it would have been unpractical to cover the vast and bewildering literature on compounds 1–20, though, we have managed to provide the reader with the big lines. First, when compared to the parent 5-FU molecule, compounds 1 and 2 exhibit enhanced pharmacological and pharmacokinetic properties. In the same rationale, supramolecular assemblies incorporating compound 3 and compound 4 into the main chain of certain polymers were found to increase their anticancer activity.25 Several nucleoside derivatives of 5-FU have also shown interesting results against cancer, but also against human immunodeficiency virus (HIV) and hepatitis B virus (HBV). This is the case of compounds 5, 6 and 7.26 Even though it is true that fluorine is quite rich in the earth crust, halogenated organic compounds are rare in nature. However, to the best of our knowledge, only thirteen fluorine-containing secondary metabolites have been isolated from plants and microbes. In 2003, Xu et al discovered and extracted five natural fluorine-containing compounds in the study of bioactive substances in the tissues of the sea toad,27 including compounds 8, 9, 10 and 11. Compound 12 is effective against cancer cells under radiation, and compound 13 is a benzoyl derivative of 5-FU that is more stable and effective than 5-FU.24 Compound 14 was actually not found cytotoxic since alkylation of 5-FU with non-labile groups resulted in the formation of the inactive compound. Be reminded that 1,3,4-thiadiazole derivatives are well-known agents with broad antitumor activity. In this vein, the combination of their core fragment with 5-FU has led to several 5-FU derivatives, such as compounds 15 and 16.28 Compounds 17 and 18 are substitutional derivatives of tegafur (compound 2),29 while compound 19 is a peptide analog of 5-FU with anticancer activity.30
In the present study, we report the interaction of anticancer drug 5-FU and nineteen of its analogues with the COVID-19 main protease (Mpro) or the 3-Chymotrypsin-Like protease (3CLpro) using molecular docking and molecular dynamics simulations. In silico predictions of pharmacokinetics properties31 of the best candidates based on docking scores are then performed to establish their ADMET (absorption, distribution, metabolism, excretion and toxicity) profile. Our investigation goes on to elucidate the chemical reactivity of the best candidates as one way to pave the road for further studies on these molecules.
Materials and Methods
Preparation of Ligands
The 2D structures of 5-FU and its analogues as shown in Figure 1 were drawn using ChemDraw Pro 16.0 software, then converted into 3D structures and saved as.sdf format. Further, the 3D structures of selected compounds were subjected to full geometry optimization by semi empirical method PM6 (Parametrization Method 6).32 It is worth noting that energy minimization process of ligands at this step is essential as it contributes to the removal of unfavorable contacts, which may affect the binding ability of the ligand. All the optimized structures were visualized using Chemcraft software33 and are shown in Figure 2.
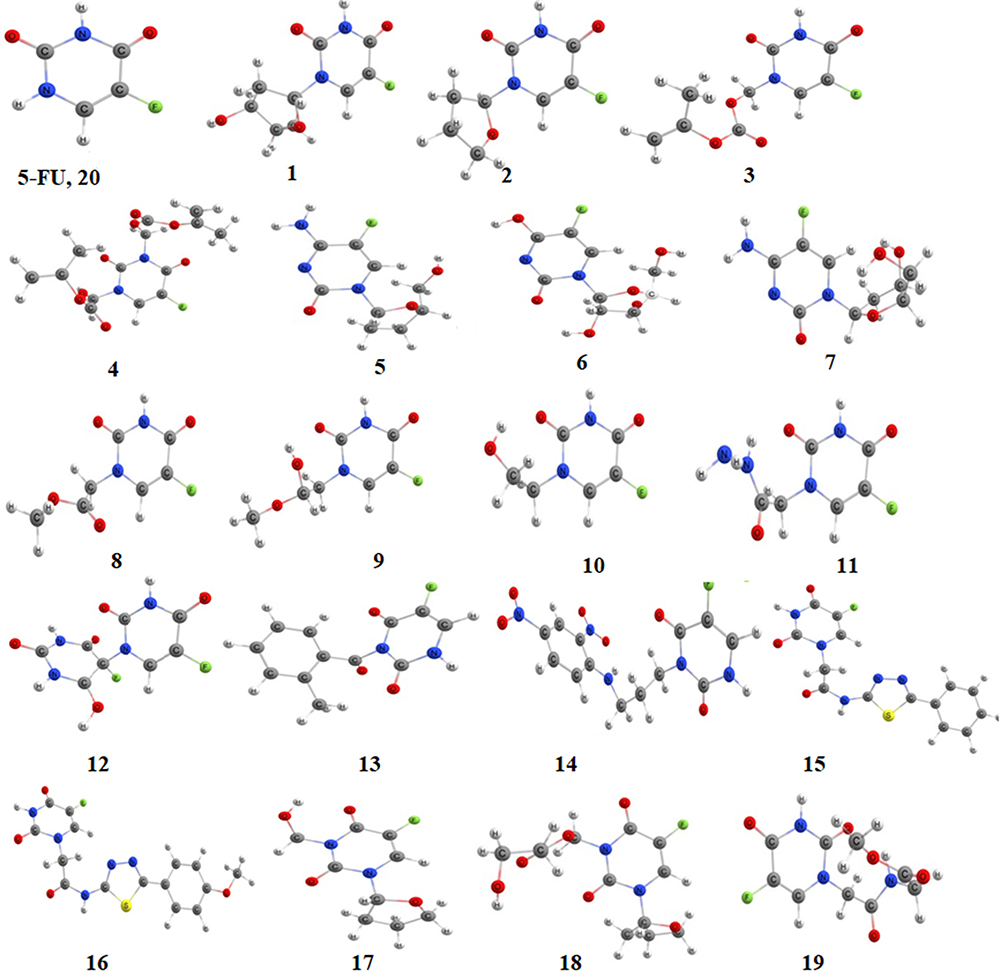 |
Figure 2 3D structures of 5-FU and its selected analogues. |
Preparation of the Target Protein, 3CLpro
The coronavirus-2 main protease (SARS-CoV-2 Main protease, Mpro) or 3-chymotrypsin like-protease (3CLpro) is one of the most vital enzymes of the novel coronavirus (COVID-19) and a crucial target for drug discovery. The coronavirus-2 or SARS-CoV-2 genome translates four structural proteins, namely small envelope protein (E), spike glycoprotein (S), matrix glycoprotein (M) and nucleocapsid protein (N).34 Next to the four structural genes, there are two proteases, namely the papain-like protease (PLpro) and the 3-chymotrypsin-like protease (3CLpro). The latter is required for coronavirus maturation, which plays a prime role for the viral life cycle and makes it an interesting target for anti-COVID drug development.35 3CLpro is a 33.8 kDa cysteine protease responsible for proteolytic cleavage of viral replicase polyproteins at 11 interdomain sites to produce several non-structural proteins essential for viral activity.36 It is a homodimer with 306 amino acids (AA) in each protomer and consists of three functional domains, namely Domain I, II and III; and a catalytic dyad of HIS41 and CYS145.
In the present study, the three-dimensional crystal structure of the target protein 3CLpro complexed with the Michael acceptor inhibitor N337 was extracted from the Protein Data Bank (PDB Id: 6LU7)38 and imported into Chimera to visualize the binding domain of the complex and identify the amino acids in the binding pocket. Discovery Studio39 features were used to clean and prepare the protein. This step involved removing the N3 inhibitor, minimizing the energy of the protein, and removing solvation water molecules. The optimized protein was saved in.pdbqt format and imported into PyRx Autodock Vina40 for molecular docking. The schematic structure of SARS-CoV-2 Mpro showing its three different domains I, II and III is presented in Figure 3A. The active site is situated between the cleft of domains I and II. The structural features are shown by color, where domain-I is in green, domain-II in light pink, domain-III in purple blue and an extended loop in magenta. The complex formed between the SARS-CoV-2 Mpro and the N3 inhibitor is shown in figure 3B.
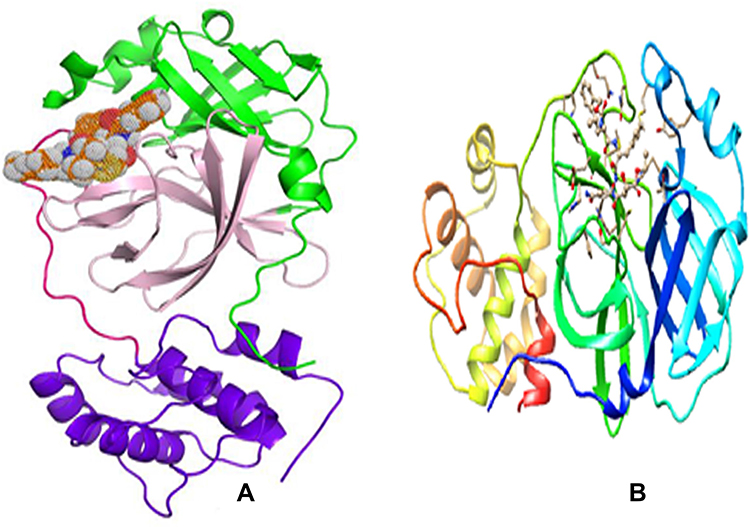 |
Figure 3 The 3-D crystal structure of the SARS-CoV-2 Mpro (PDB ID: 6LU7); (A) Domain-I: green, Domain-II: light pink, Domain-III: purple blue and an extended loop in magenta. A catalytic dyad of the conserved residues HIS41 and CYS145 is in blue inside the substrate-binding site that is shaped by a cleft amongst domains I and II; (B) The complex formed between the SARS-CoV-2 Mpro and the N3 inhibitor. |
Molecular Docking and MD Simulations
All the prepared ligands were docked to the target protein 3CLpro using Autodock Vina. This program uses grids to pre-calculate the binding interactions at different positions within the binding site where values are stored in look-up tables and accessed automatically. This docking program executes rigid-body docking by considering both the protein and ligand onto grids.40 The binding pocket of the target protein was decided using a grid size of 50 x 50×50 Å3 with the help of an auto grid centered at x = 43.730, y = 12.702 and z = 59.371.
The docking was run five times and average binding energies for each complex were retained. The docking results were visualized using Discovery Studio Visualizer 4.0.
For each of the best-docked ligands, molecular dynamics simulations of the associated complex were performed for a duration of 100 ns at 300 K in the framework of the NTP ensemble using the Schrodinger suite of code. Prior to any simulation, the system was adequately prepared to include water molecules (10399) and counter ions (32 Na+ and 29 Cl−). The equilibration of the protein and the trapped ligand were monitored in terms of fluctuations of their respective RMSDs over the simulation time. To measure the ‘extendedness’ of a given ligand throughout the simulation, its radius of gyration (rGyr) was computed at every step of the simulation. Note that rGyr is equivalent to its principal moment of inertia.
Reactivity Study
Understanding the chemical reactivity of drug molecules is of paramount importance. Indeed, a drug can lose its effectiveness over time, notably due to resistance problems and/or side effects. To remedy this, one of the avenues is to proceed by structural modifications or to combine the molecule with another to act in synergy. These techniques require an understanding of the chemical reactivity of the drug molecules41–43 among many others the molecular electrostatic potential for electrostatic interactions modelling,44 Fukui functions f+ and f– for predicting preferential active sites for nucleophilic and electrophilic attacks, respectively;45,46 and frontier molecular orbitals (FMOs), ie the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) to understand the kinetic stability of compounds.47 More importantly, HOMO and LUMO orbitals are mainly responsible for biological interactions between ligands and proteins in a complex.45,48–50 Indeed, before the work of Pang et al,51 there was no clear molecular mechanism to explain the protein–ligand interaction based on the electronic structure of a protein. However, by combining the calculation of the complete electronic structure of a protein with its hydrophobic pocket and perturbation theory, Pang et al discovered two rules about protein–ligand interactions. The first rule is that the interaction occurs only between the lowest unoccupied molecular orbitals (LUMO) of a protein and the highest occupied molecular orbital (HOMO) of its ligand, not the opposite. Accordingly, on the one hand, knowing the pattern of interaction between a ligand and a protein, and on the other hand, by plotting the HOMO of a ligand, one can locate the LUMO of the protein. This means that only residues or atoms located in both the LUMOs of a protein and a surface pocket of a protein are active residues or active atoms of the protein and the corresponding pocket is the ligand-binding site, and that is the second rule of Pang. These two rules are derived from the energy-level structure of a protein and could be one of the important criteria for drug design.
Gaussian 0952 software was used to refine the PM6 equilibrium geometries of the best binding candidates at the B3LYP/6-311++G(d,p) level of theory. This level of theory has met required accuracy in numerous previous studies.53,54 The predicted stationary points were confirmed by vibrational frequency calculations to be real minima on their potential energy surface. The MultiWFN code55 was used to generate FMOs, which were then visualized with Gaussview.56
Results and Discussion
Energetics and Geometries
The binding energy (BEs) of complexes formed between 5-FU and nineteen of its analogues with the SARS-CoV-2’ protease 3CLpro are shown in Figure 4. The latter also depicts the 3D representation of interactions of the most binding complexes. The binding energies range from −4.8 to −7.9 kcal/mol. These negative values suggest an overall favorable and spontaneous packing of the ligands in the inhibitory site. 5-FU, taken as a prototype ligand, is the compound that forms the least stable complex with the enzyme 3CLpro. Another ligand that is automatically a bad candidate for the inhibition of 3CLpro is ligand 12, which occupies, together with 5-FU the last position with binding affinity equal to −4.8 kcal/mol.
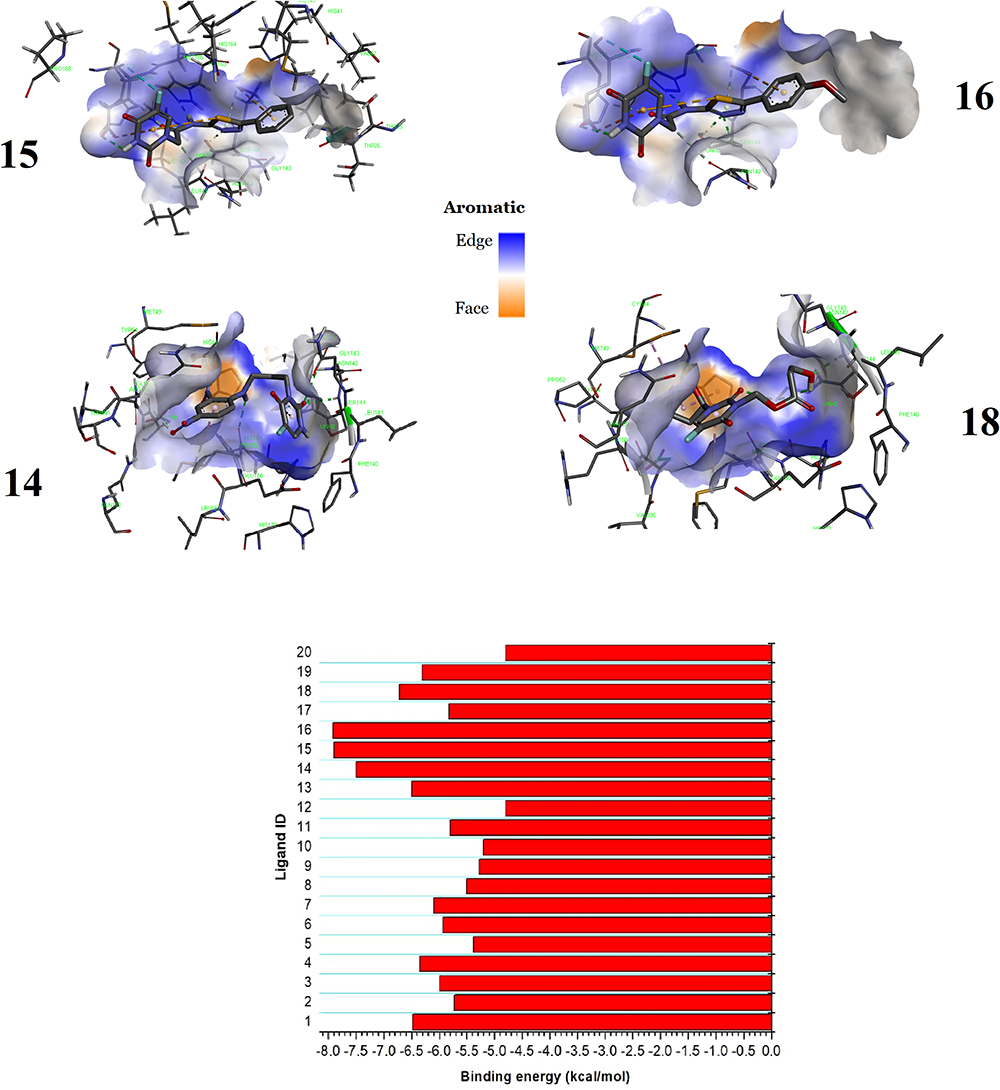 |
Figure 4 Binding energy of all ligands with the 3CLpro and the 3D representation of interactions of the most binding complexes. |
Indeed, it has been demonstrated that potential inhibitors of the main protease of the SARS-CoV-2 virus should have binding energies greater than −6.5 kcal/mol.57,58 Taking this value as a reference, only 4 ligands, namely compounds 15, 16, 14 and 18, can be considered at this stage as potential inhibitors of the 3-chymotrypsin like protease. Figure 4 shows that ligands 15 and 16 each forms a complex with the 3CLpro target with a binding energy of −7.9 kcal/mol; followed by ligand 14 which comes in second position with a binding energy of −7.5 kcal/mol. Ligand 18 comes last with a binding energy of −6.7 kcal/mol. Nevertheless, a molecular docking study using Glide Schrodinger Guide tool suggested that, despite the small binding affinity of −4.4 kcal/mol, withanone (PubChem Id.: 21679027) should be regarded as a potential inhibitor of the protease 3CLpro.59 This insight was revealed by post-docking studies including, on the one hand, calculations of ligand-receptor affinities by the MM/GBSA method which gave a binding energy of −34.51 ± 9.63 kcal/mol, and on the other hand, molecular dynamics simulations which deciphered several stabilizing interactions between the protein-ligand complex.59 This illustration evidences that post-docking results are often of paramount importance. Noteworthy is that withanone was engaged into a hydrogen bond with the catalytic CYS145 residue and established hydrophobic π-π interactions with key residues of the protease.
In the present study, MD simulations were performed for ligands 15, 16, 14 and 18, which exhibit the most favorable binding affinities. However, ligands 1 and 13 (−6.5 kcal/mol), 4 and 19 (−6.3 kcal/mol), 7 (−6.1 kcal/mol) and 3 (−6.0 kcal/mol) are also fairly good candidates since their binding energies are not only close to that of the N3 ligand which is −7.4 kcal/mol,6 but also slightly lower than the reference value of −6.5 kcal/mol.
Interaction Analyses
The activity of enzymes is linked to the presence in their structure of a particular site called the active site, which has the shape of a cavity or groove. Molecules or ligands bind to the active site of the target enzyme by forming interactions with the surface of the active site cavity, or better with some of the amino acid residues that form the active site cavity. In the particular case of the crystal structure of 3CLpro extracted from PDB, the binding cavity is defined by a series of important residues, namely THR24, THR25, PHE140, ASN142, GLY143, CYS145, HIS41, HIS163, HIS164, GLU166 and HIS172.6,8,60 Ligands interacting with this protease are expected to develop a variety of interaction with some of these amino acids.
Close inspection of the binding mode of the four top-ranked ligands (Figure 5) with the main protease 3CLpro, conventional hydrogen bonding, unconventional hydrogen bonding with carbon and π-electrons as fragment A (A–H⋯B), van der Waals (vdW) interactions, and hydrophobic interactions are the main driving forces for the stability of the complexes. The two most stable complexes show remarkable similarity in binding energy and binding mode. This is not surprising because molecules 15 and 16 are differentiated only by the R group (Figure 1). However, from their structures, one would expect ligand 16 to form a slightly more stable complex with 3CLpro than ligand 15, due to the presence of an extra oxygen atom in ligand 16 whose unpaired electrons insinuates a nucleophilic character and subsequently the ability to form stabilizing contacts with electron-deficient sites in the protease’s cavity. However, this atom was not involved in any H-bonded interaction.
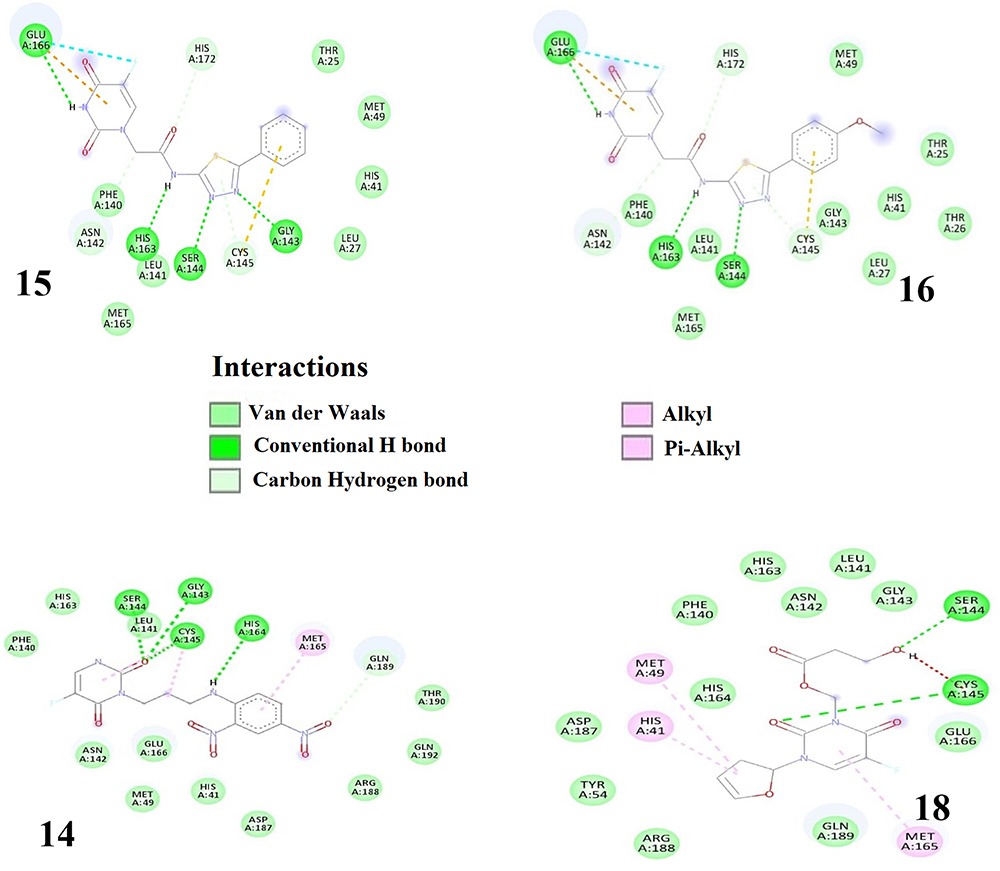 |
Figure 5 2D representation of the interaction of top 4 ranked compounds with 3CLpro. |
Ligands 15 and 16 form eight hydrogen bonds (4 conventional for ligand 15 and 3 for ligand 16) with the amino acid (AA) residues GLU166, HIS163, SER144, GLY143, ASN142, HIS172, CYS145 et LEU141. The stability of these two complexes is also enhanced by the presence of a halogen bond with the interacting residue GLU166, the presence of non-conventional H-bonds with π-electrons as the donor moiety, and hydrophobic interactions. It can be seen that six (in bold) of the eight AA residues involved in the hydrogen bonding interactions in ligands 15 and 16 are part of the AA residues of the active site of the 3CLpro protease. Note also that the active site residues PHE140 and THR25 are involved in the vdW interactions in the complexes of ligands 15 and 16. Also interacting with these ligands are residues MET49, HIS41, LEU141, alongside LEU27 and THR26 for ligand 16.
As far as ligands 14 and 18 are concerned, docking calculations suggest that the former is maintained by four strong hydrogen bonds with interacting AA residues SER144, GLY143, CYS145, and HIS164, as well as weak H-bonds (GLN189, LEU141, etc.) and hydrophobic interactions (MET165 and CYS145). On the other hand, compound 18 forms only two strong hydrogen bonds with residues SER144 and CYS145, several vdW interactions but no weak H-bonds.
To sum up, there is no doubt hydrogen bonding interactions contribute substantially to the total stability of the complexes between 3CLpro and ligands 14–16, 18 since an increased number of hydrogen bonds leads to higher stability of the complexes. Another interaction that enhances ligand-receptor binding affinity stems from van der Waals interactions. Hydrophobic interactions, although present, contribute only slightly to the overall stabilization of these protein-ligand complexes.
Physicochemical Properties and ADME-T Profile
In drug design, the main challenge is to come up with one or more compounds with the desired drug properties. In addition to high affinity for the target protein, the compound must have an appropriate selectivity profile, be able to reach the site of action and have acceptable drug-like properties. That’s one of the major objectives of the clinical phase in drug discovery, aiming to guarantee that the compound under trial meets the required criteria and a good therapeutic profile of druggability. The pharmacokinetic profile of a compound defines its properties of absorption, distribution, metabolism and excretion (ADME). In addition to the ADME, a good candidate must be not carcinogenic and hepatotoxic, that is, non-toxic. In the present investigation, the pharmacokinetic parameters and the toxicity assessment of the four best docked ligands were estimated using online SwissADME61 and the pkCSM server,62 respectively. Canonical SMILES of the top four candidates were used to calculate their physicochemical properties and predict their ADME-T profile. The results obtained are collected in Table 1.
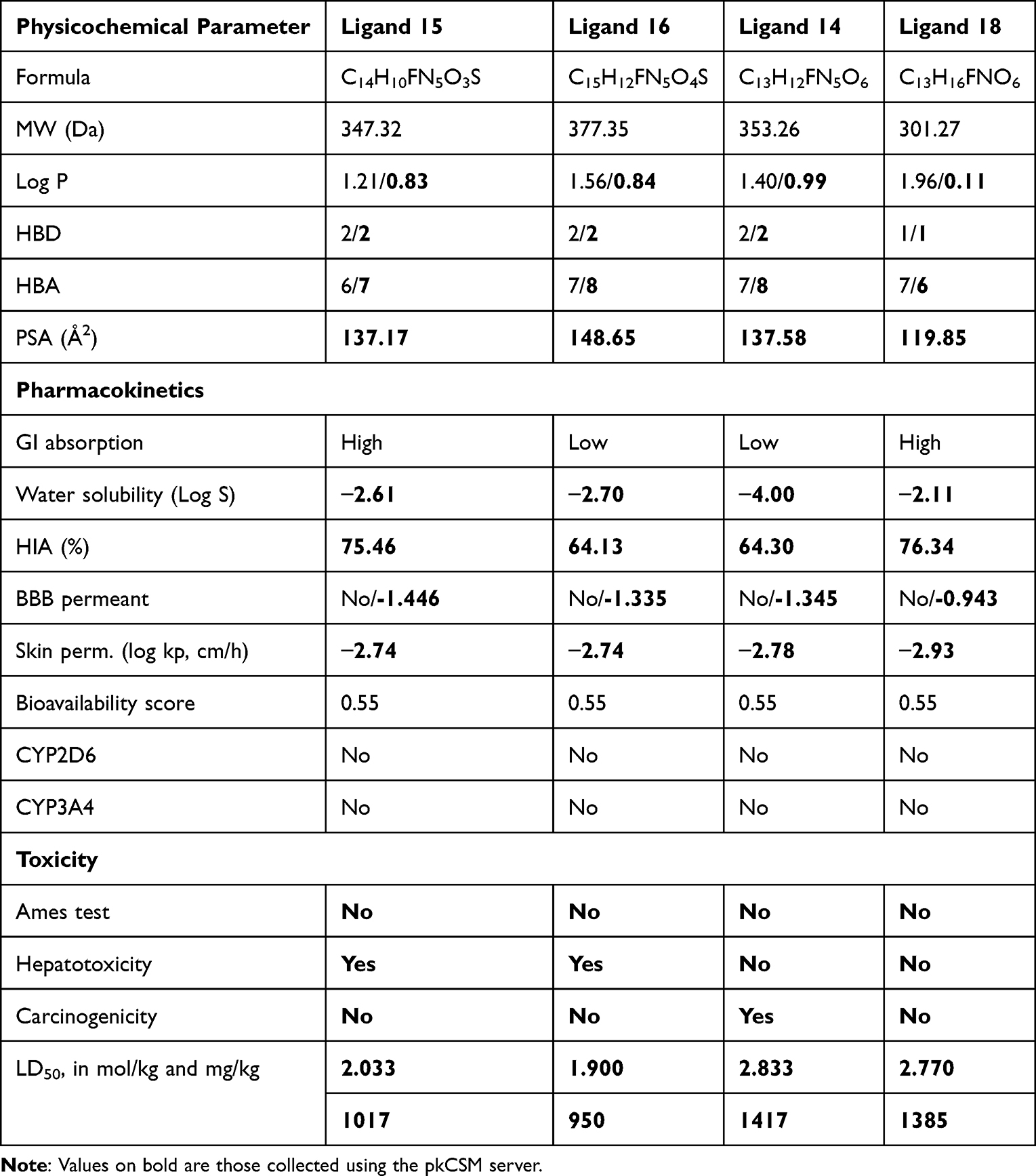 |
Table 1 Physicochemical Parameter and ADME-T Profiles of the Four Ranked Ligands |
Based on physicochemical parameters, druglikeness properties based on oral administration were evaluated according to Lipinski’s rule-of-five: molecular weight < 500 Daltons (Da), calculated lipophilicity (Log P) < 5, number of hydrogen-bond acceptors (HBA) < 10 and number of hydrogen bond donors (HBD) < 5.63 The results in Table 1 reveal that the four high dock-scoring ligands exhibit good bioavailability and appropriate druglikeness properties as no violation to the Lipinski’s rule-of-five was observed. This is reflected by their molecular weight, which range between 301 to 307 g/mol, the number of H-bond donors (HBD between 1 and 2), that of H-bond acceptors (HBA between 6 and 8), and the octanol-water partition coefficient (log P) between 1 and 2.
The polar surface area (PSA) is an extremely instructive parameter for optimal drug absorption. Predicted values of PSA remain lower than the threshold value of 140 Å2, except for ligand 16 (148.65 Å2). It is well known that physicochemical descriptors of drug molecules are often inadequate for predicting their oral bioavailability.64 Although parameters such as drug solubility can affect oral bioavailability, in most cases the main determining factors are likely to be metabolism and absorption at the intestinal level. Metabolism may occur prior to absorption, as in the case of peptides, or during absorption, particularly due to the activity of the intracellular enzyme CYP3A4.
Gastrointestinal (GI) absorption of a drug is another relevant factor especially when the drug is intended for oral administration. The data in Table 2 reveal that only two of the best-docked 5-FU analogues are expected to be rapidly absorbed from the GI tract (ligands 15 and 18). However, based on the water solubility data of the ligands obtained from the pkCSM server, only ligand 14 is poorly soluble, with a log S value lower than −4. Similarly, the human intestinal absorption (HIA) values reveal that compounds 15 and 18 have a higher probability of being absorbed by the human intestine (76%) as compared to compounds 16 and 14 (64%).
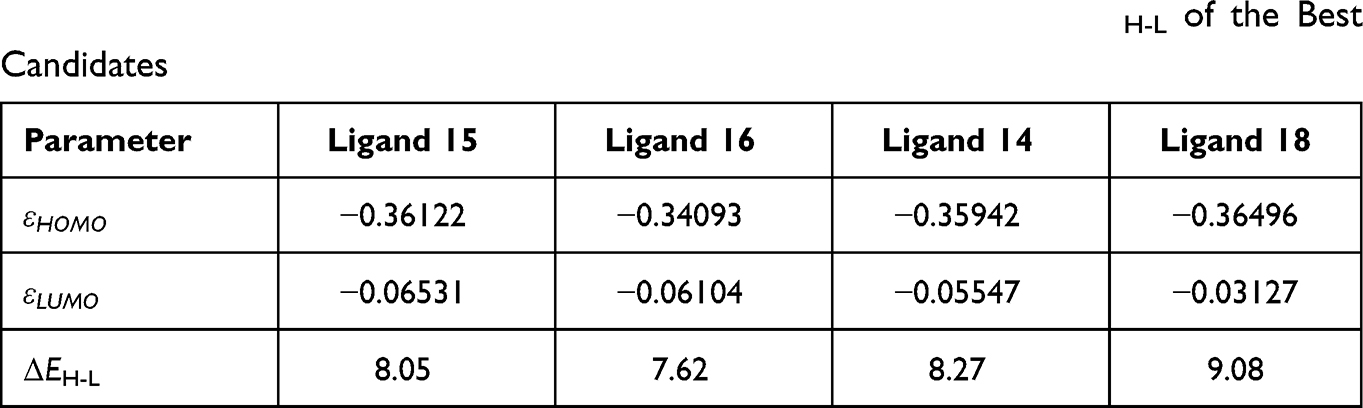 |
Table 2 The HOMO and LUMO Energies (in a.u.) and Their Gap ΔEH-L of the Best Candidates |
Moreover, blood-brain barrier permeability (BBB permeability) determines whether a molecule will act beneficially or adversely on the brain. The passage of conventional anti-cancer molecules across the BBB can cause significant destruction of brain neurons, leading to severe neurological consequences.65 The BBB permeability values for the four selected molecules are all negative. This suggests that these compounds should not produce neurotoxicity in the brain, since they will not cross the BBB. The bioavailability score evaluated at 0.55 confirms that the four ligands have good absorption and distribution in tissues since all potential candidates can have more than 10% bioavailability in rats.66
Turning next to metabolism, drugs can be metabolized by oxidation, reduction, hydrolysis, hydration, conjugation, condensation, or isomerization; whatever the process, the goal is to make the drug easier to excrete. Enzymes involved in metabolism are present in many tissues, but are generally most concentrated in the liver. It has been reported that the cytochrome enzymes CYP2D6 and CYP3A4 are primarily responsible for drug metabolism. Interestingly, the top four candidates are not inhibitors of the cytochrome enzymes CYP2D6 and CYP3A4. As such, these ligands may be metabolized before attaining optimal plasma concentrations.67 In other words, metabolism may occur before a drug reaches the systemic circulation, leading to low bioavailability, often encountered with oral forms of poorly water-soluble and slowly absorbed drugs.
Toxicity results are quite encouraging as none of the four compounds is likely to induce mutagenicity, and all are non-carcinogenic with the exception of ligand 14. With respect to hepatotoxicity, the two best docked ligands are predicted to be capable of conferring hepatotoxicity. However, this hepatotoxicity is often not due to the compound itself, but rather to the metabolic process. Indeed, the rate of drug metabolism varies among patients. Some patients metabolize a drug so rapidly that therapeutically effective blood and tissue levels are not achieved; whereas in others, metabolism may be so slow that toxic effects occur with usual doses.67 Therefore, a drug may be predicted to be non-hepatoxic, yet shows liver toxicity in some patients, and vice-versa. In either case, theoretical results must be validated by experiments. Furthermore, skin toxicity is also an issue in drug development. The recommended value for skin permeability of a drug molecule is set at to −2.5 cm/h or higher.62 From Table 2, it can be seen that the four compounds will produce little or no skin toxicity since they do not penetrate the skin layer very easily.
Finally, estimated values for the oral rat acute toxicity expressed in terms of 50% of lethal dose (LD50) are all classified in class 4, which means that after administration, these compounds might be only slightly toxic (Globally Harmonized System: 300 < Category 4 ≤ 2000) and can thus be considered as safe.
It should be noted that the therapeutic profile discussed here is for oral administration. Since the absorption of a drug is determined not only by its physicochemical properties, but its galenic formulation and route of administration as well, the druggability of compounds can be increase by monitoring different galenic formulations (eg, tablets, capsules, solutions) and other routes of administration (intravenous route, parenteral, inhalation, etc).
Chemical Reactivity of Ligands
Molecular Electrostatic Potentials (ESP or MEP)
Among reactivity descriptors, electrostatic potential is widely used in site prediction and relative reactivity for electrophilic attacks, in studies of biological recognition and hydrogen bonding interactions. The negative electrostatic potential (in red) represents the attraction of the proton by the region of high electron concentration in the molecule, while the positive electrostatic potential (in blue) corresponds to the repulsion of the proton by the atomic nuclei. Figure 6 shows the ESP maps for ligands 15, 16, 14, and 18.
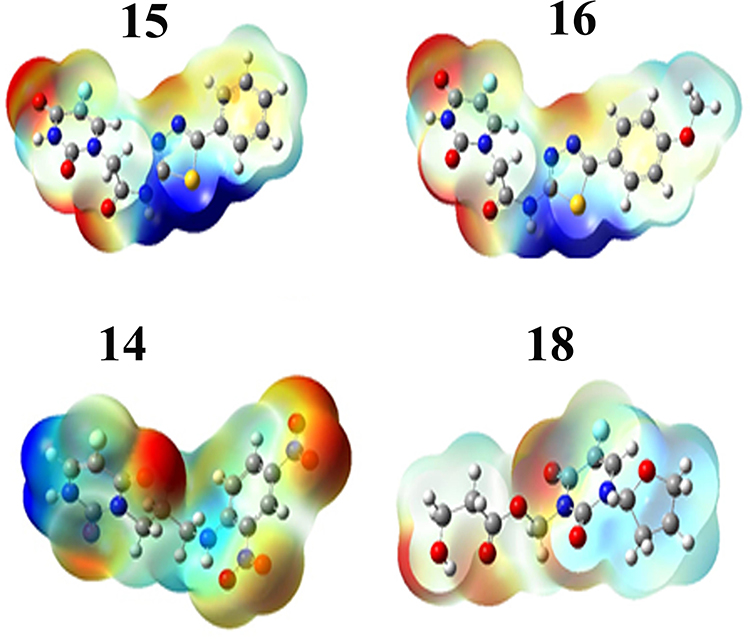 |
Figure 6 Molecular electrostatic potentials of the 4 best docked ligands. |
The MEP maps are consistent with the hydrogen-bonding interactions observed between the amino acid residues of the protease and the four ligands. For instance, they clear indicate why the oxygen atom of the methoxy group in ligand 16 is unlikely to engage into H-bonding interactions as mentioned above. The ESP also shows non-polar regions (in cyan), which are very present in ligand 18, concentrated on the imidazole ring. Similar regions are also predicted in ligand 14, but with high concentration from the middle to the right of the molecular surface. These non-polar regions are in perfect agreement with the hydrophobic interactions observed in ligands 14 and 18.
Fukui Functions f – and f +
It is well-known that Fukui functions are essential indicators in the study of the regioselectivity of a molecule whose predictive may outperform that of ESPs in some situations. Fukui functions are defined in the context of the conceptual density functional theory.68 These functions quantify the response of a molecular system to the change of its electron density distribution with respect to the total number of electrons. Their original and widely admitted definition assumes that the external potential stemming from the nuclei is constant. This condition implies that anionic and cationic states resulting from the addition or removal of one electron from a neutral system should have the same geometry. To comply with this, anionic and cationic states of ligands 14, 15, 16, and 18 were generated by single point calculations from the optimized geometries of the neutral molecules. The Fukui function exists in two main flavors: f – is associated with electrophilic attacks, while f + indicates good candidate sites for nucleophilic attacks. These two functions for the 4 ligands are shown in Figure 7.
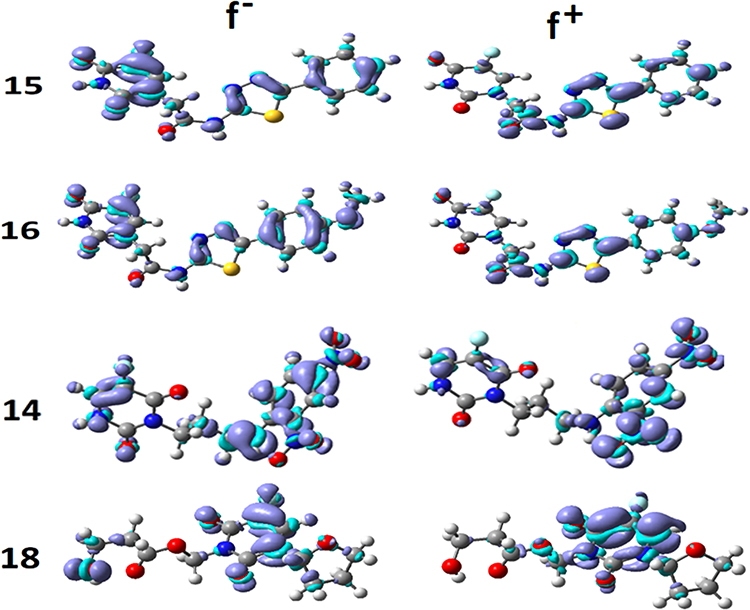 |
Figure 7 Fukui functions f – and f + of the 4 best docked ligands. |
The most active sites, susceptible to electrophilic attack, in agreement with the ESPs, are located throughout the molecule, but with a high concentration in the left side for ligands 15 and 16, and in the right side for ligands 14 and 18. The absence of a negative Fukui function on the sulfur atom in compounds 15 and 16, is consistent with its non-reactivity (Figure 5). Notice however that this atom is predicted to be capable of nucleophilic attack by the positive Fukui function.
It is important to point out some inconsistencies between the ESPs and the negative Fukui functions. It is surprising to see on the one hand the absence of the negative Fukui function on the carbonyl group of ligand 14, which disagrees with its ability to form stable H-bonds as suggested by docking results (Figure 5). On the other hand, the MEP map of ligand 18 suggests a slightly negative region on the carbonyl group outside the aromatic ring, while the negative Fukui function does not show any density concentration on this site. Be reminded that no electrostatic interaction was predicted between this site and the protein, thus supporting the Fukui function.
Frontier Molecular Orbitals
The HOMO (top of the figure) and LUMO (bottom of the figure) orbitals of the 4 best-ranked ligands are shown in Figure 8. Table 2 lists the energies of the HOMO and LUMO orbitals, and their gaps.
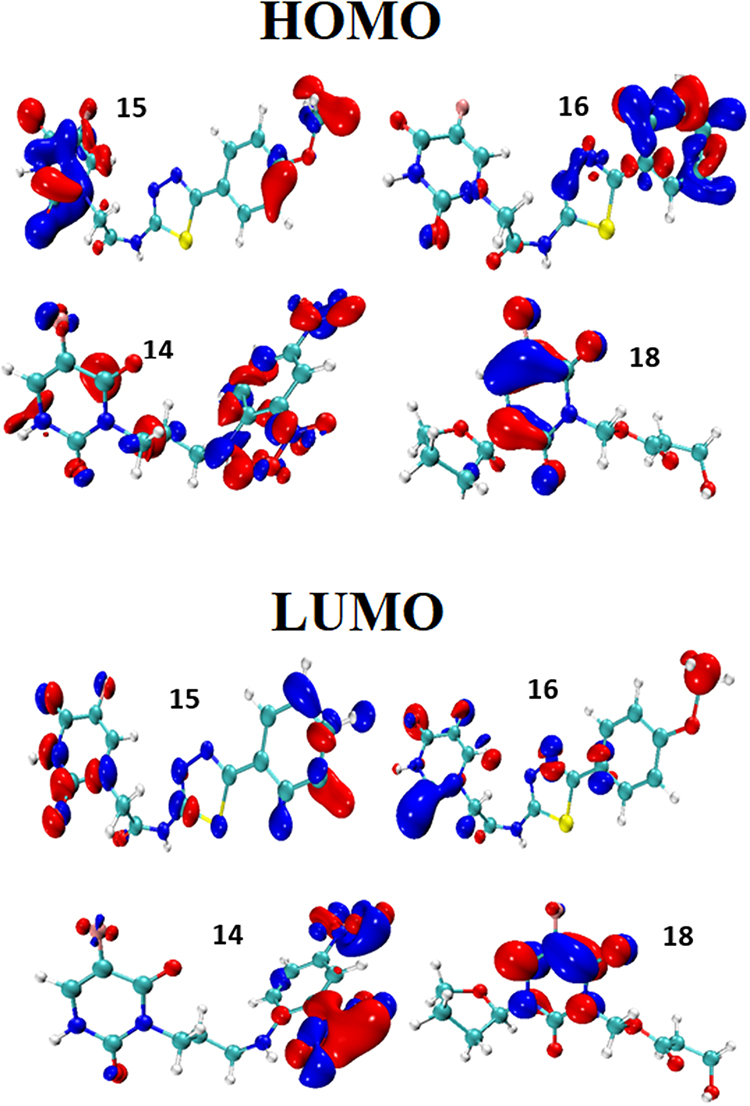 |
Figure 8 Frontier molecular orbitals HOMO (top) and LUMO (bottom). |
The HOMO orbitals identify the active sites of the ligands selected as best candidates, the same as those identified by the negative Fukui function. This finding supports the idea that these sites are actually reacting with the amino acid residues of the 3CLpro protein, in support of Figure 5. Although we did not compute the electronic structure of 3CLpro, one should expect based on the previous finding combined with Pang’s rules, that the electron density of the LUMO must be concentrated on the residues that we identified to be involved in the interaction with the best binding ligands.
The energy gap between the HOMO and LUMO is often related to chemical reactivity or kinetic stability. A rule of thumb is that a higher HOMO-LUMO barrier is indicative of lower reactivity or better kinetic stability. Therefore, the following kinetic stability order can be inferred from Table 2: ligand 16 > ligand 15 > ligand 14 > ligand 18. This order corroborates with the computed binding affinities. With the largest ΔEH-L gap (9.08 eV), ligand 18 is confirmed to be the least reactive compound among the four, followed by compound 14 (8.27 eV) and the two most binding ligands respectively.
Molecular Dynamics Simulation
Figure 9 displays the superimposed RMSDs for the MD simulation of the complexes formed between 3CLpro and each of the four best binding ligands, namely ligand 14 (Figure 9A), ligand 15 (Figure 9B), ligand 16 (Figure 9C) and ligand 18 (Figure 9D). The ligands radius of gyration over the simulation time are shown in Figure 10.
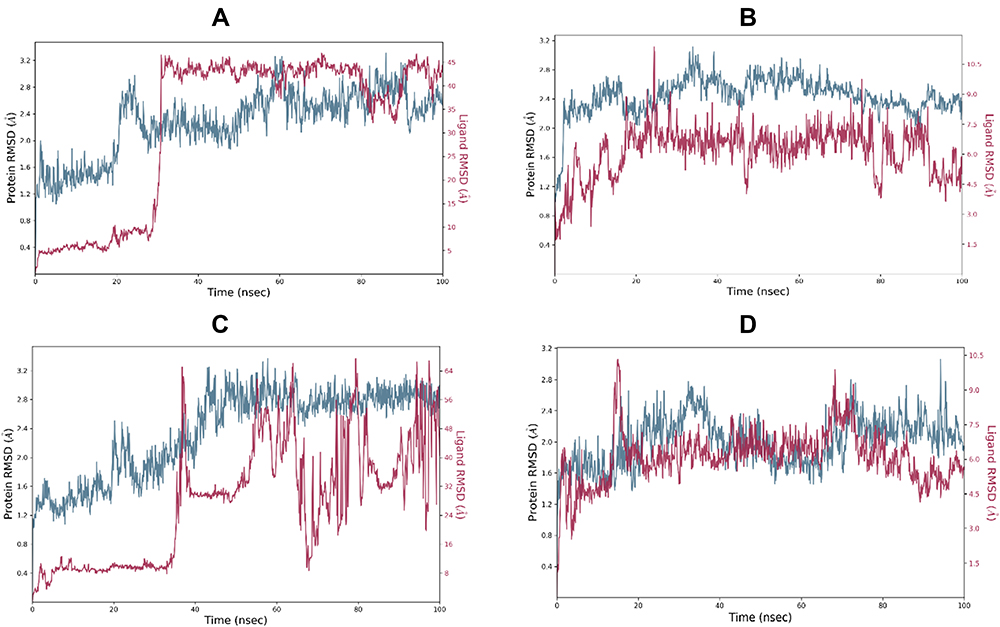 |
Figure 9 RMSD of four best docked ligands and the 3CLpro protein over a simulation time of 100 ns. Subplots A, B, C and D correspond to four protein-ligand complexes involving ligands 14, 15, 16 and 18 respectively. |
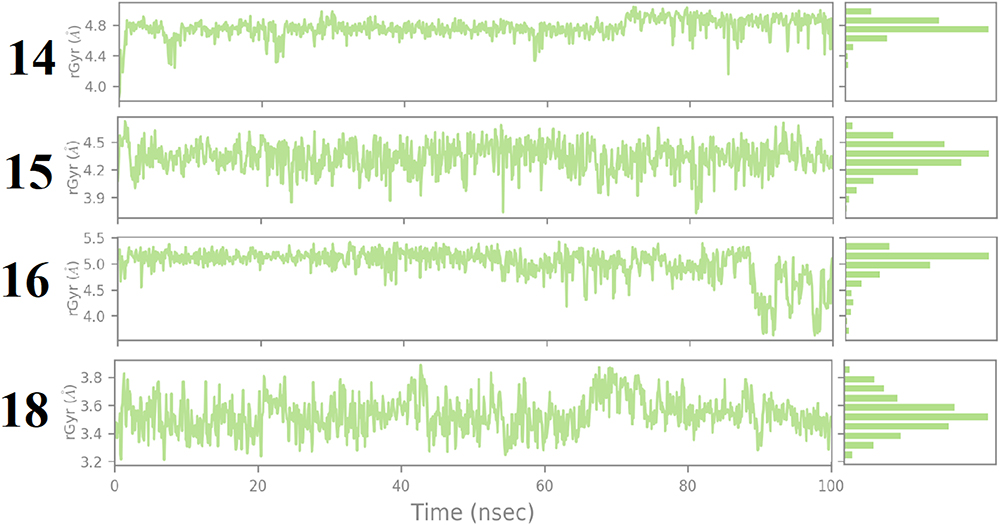 |
Figure 10 Radius of gyration of the ligands over the simulation time. |
From Figure 9A, it is clear that the receptor assumes a partial stability at a range of 1.2–2.0Å for the first 20 ns this fluctuates to stabilize the protein at a range of 1.8–3.2 Å for the remaining part of the simulation. The ligand on the other hand makes a jump from 1.0 Å to temporarily stabilize at a range of 0.3–0.5 Å following which a jump happens to allow the ligand to stabilize 10ns after the protein stabilizes. This jump can be traced to the shift in the interactions from range 4–12 to 4 to 10. This ripple effect can be witnessed in the sharp fluctuation in the radius of gyration, which remains stable for the first 60ns except for the fluctuations in the 10th and 20th ns period. After the fluctuation in the 60th ns period, the compactness is then maintained in the range of 4.6 to 4.8 Å until the simulation lapses. Our docking results capture the best pose in which SER144, GLY143, CYS145, HIS164 and MET 165 in which the first four and the last residues interact with the ligand via H-bonds and π-Alkyl bonds respectively. In the Md simulation, our trajectory begins with interactions between the receptor via THR26, HIS41, CYS 44, ASN142, HIS164, GLU166 with interaction totals ranging from 4–12. This declines to range 4–10 after 20 ns then from 30ns to 60 ns the interactions are maintained between 2 and 7. In the time between 70 and 80 ns, they vary from 0–4 until the simulation ends with 2.
Figure 9B shows that the protein begins to stabilize after 2 ns, after this point the protein fluctuates in a range of 2.0 to 2.8 Å. Eighteen nanoseconds after the protein stabilizes, the ligand stabilizes in the 1.2 to 2.2 Å range. Instances of RMDS spikes around the 40th, 50th, 70th, 80th and the 90th ns mark could be traced to interactions ≥ 4. After 90ns the peaks for the ligand drop slightly in their range to 1.0 to 1.7 Å. At this point, it can be resolved that the ligand maintains its complex with the host throughout the simulation, keeping the total number of interactions in the active site in the range of 1–9. Out of these interactions, LYS70 and ALA70 manage to keep their interactions H-bonds, Water bridges, hydrophobic, for more than 50% of the simulation time.
As far as the 3CLpro … ligand 16 complex is concerned, Figure 9C reveals that 3CLpro begins to stabilize after 50 ns. Around the same period the ligand, partially stabilizes, only to fluctuate through the remaining parts of the simulation until 90 ns when there is an indication of stability. A longer simulation would have allowed for a more conclusive result on the stability status of the ligand. Nevertheless, the distance maintained by the ligand despite its fluctuation show that the ligand does not leave the active site. The contact of residues essential to maintaining the stability of the complex changes through the course of the simulation. In the docking results, H-bonds are observed on GLU166, HIS163 and SER144. This transitioning of H-bonds from residue to residue could suggest why the ligand takes long to stabilize. Notably after 2 ns the ligand maintains contact with Glu166 and GLN189 and spatially with ARG188 for the next 30 ns. At distances between 0.4 and 2.0 Å the ligand keeps constant. Meanwhile the protein gradually approaches stability. Although GLU166, GLN189, ARG188 and MET235, (which show the highest number of interactions per residue) contribute to the larger percentage of H-bond and water bridges contact time, they all happen in the first half of the simulation time. The final interactions consist of GLU 270 and ASN 274. In summary, the total number of interactions throughout the timeline maintains a steady range of between 2 to 6 in the first 30ns then picks up after 40ns only to decline at 60 ns where range shifts to between 0 and 5, after this the interactions will change to a 0–6 range between the 80th and 90th ns. When the simulation ends, there is no interaction owing to a need for a longer simulation time. This constant drop of interactions after 60 ns could explain the reason why the RMSD values keeps fluctuating with taller peaks. The radius of gyration keeps constant through the simulation, but fluctuates after 70 ns. At the point where the ligand begins to show signs of stability in the active site, a non-consistent increase in the number of interactions in the active site is observed. These quick alternating residue interactions might have effect on the radius of gyration documented. However, the interactions are short lived as the simulation terminates with no interaction.
Finally, monitoring the RMSD of ligand 18 and the protein over 100 ns of simulation indicates that the protein quickly stabilizes after 2 ns followed by the ligand 5ns later, however this is short-lived as a sharp spike in the 18th ns shows the ligand partially shifting then regaining its stability around the 20th ns. This sharp shift can be traced to the ligand losing most of its interactions. Throughout the simulation, the ligand maintains its distance in a range of 1.2 to 2.0Å showing that it does not leave the active site. This can be accredited to the GLU47, GLU166, GLN189, H-bonds, water bridges and polar interactions (GLU166) which are maintained for more than 20% of the simulation time. These interactions vary in total from 1–8 through the 100ns period with more fluctuations around the 3rd, 18th, 54–55th, 79th and 99th. ns. Relative to ligands 14, 15 and 16, ligand 18 manages to preserve its position within the active site at closer distances to the protein 1.0 Å or less.
Conclusion
This study was carried out to investigate the interaction of the FDA approved anticancer drug 5-fluorouracil and nineteen of its analogues with the first resolved COVID-19 crystal structure, 3CLpro or SARS-CoV-2 main protease, using molecular docking and dynamics simulations, with the hope that they can act as anticancer compounds by inhibiting the COVID-19 protease. The result of this drug repurposing study showed that 5-FU and most of its analogues cannot act as antiviral drug to tackle the COVID-19. Our study revealed that from a set of twenty compounds, only four compounds, namely ligands 15, 16, 14 and 18, are found to be potential candidates since each formed a considerably stable complex with the target protease, 3CLpro. Molecular dynamics simulations results suggest that the four ligands are maintained within the active site of the protein during the simulation, ligand 18 being the most retained. More importantly, the pharmacokinetic behavior of the four best docked ligands revealed that they generally have good therapeutic profile of druggability and are safe. Chemical reactivity mainly molecular electrostatic potential, Fukui functions and frontier molecular orbitals were considered to discuss the global and local reactivity of the four best candidates. Further in-vitro and in-vivo investigations are needed to shed light on the possible mechanism of pharmacological action of the proposed ligands.
Acknowledgment
The authors are grateful to the Department of Chemistry at Rhodes University, South Africa, and the Center for High Performance Computing for having provided some of the computing resources used to carry out this study.
Disclosure
The authors declare no conflict of interest.
References
1. Hui DS, Azhar EI, Kim YJ, et al. Middle East respiratory syndrome coronavirus: risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect Dis. 2018;18:e217– 27. doi:10.1016/S1473-3099(18)30127-0
2. Di W, Tiantian W, Qun L, Zhicong Y. The SARS-CoV-2 outbreak: what we know. Int J Infect Dis. 2020;94:44–48. doi:10.1016/j.ijid.2020.03.004
3. Woldometer. Report coronavirus cases: 6,343,924 deaths from COVID-19 virus pandemic-Woldometer; 2022 [Updated June 22,2022]. Available from: www.woldometers.info/coronavirus.
4. Abdulla ZA, Al-Bashir SM, Al-Salih NS, et al. A Summary of the SARS-CoV-2 vaccines and technologies available or under development. Pathogens. 2021;10:788–809. doi:10.3390/pathogens10070788
5. Ketterer F, Trefois P, Miermans M-C, Vanmeerbeek M, Giet D. Reluctance to vaccinate: an approach to the phenomenon through data from literature. Rev Med Liège. 2013;68:74–78.
6. Mpiana PT, Ngbolua KN, Tshibangu DST, et al. Identification of potential inhibitors of SARS-CoV-2 main protease from Aloe vera compounds: a molecular docking study. Chem Phys Lett. 2020;754:137751. doi:10.1016/j.cplett.2018.09.074
7. Bhuiyan FR, Howlader S, Raihan T, Hasan M. Plants metabolites: possibility of natural therapeutics against the COVID-19 pandemic. Front Med. 2020;7:444–469. doi:10.3389/fmed.2020.00444
8. Lokhande KB, Nawani N, Swamy KV, Pawar S. Biflavonoids from Rhus succedanea as probable natural inhibitors against SARS-CoV-2: a molecular docking and molecular dynamics approach. J Biomol Struct Dyn. 2020. doi:10.1080/07391102.2020.1858165
9. Pandey K, Lokhande KB, Swamy KV, Nagar S, Dake M. In silico exploration of phytoconstituents from Phyllanthus emblica and Aegle marmelos as potential therapeutics against SARS-CoV-2 RdRp. Bioinf Biol Insights. 2021;15:1–13. doi:10.1177/11779322211027403
10. Swargiary A, Mahmud S, Saleh MA. Screening of phytochemicals as potent inhibitor of 3-chymotrypsin and papain-like proteases of SARS-CoV2: an in silico approach to combat COVID-19. J Biomol Struct Dyn. 2020;1–15. doi:10.1080/07391102.2020.1835729
11. Lokhande KB, Apte GR, Shrivastava A, et al. Sensing the interactions between carbohydrate binding agents and N-linked glycans of SARS-CoV-2 spike glycoprotein using molecular docking and simulation studies. J Biomol Struct Dyn. 2020. doi:10.1080/07391102.2020.1805019
12. Abdelli I, Hassani F, Bekkel Brikci S, Ghalem S. In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. J Biomol Struct Dyn. 2020;38:1–17. doi:10.1080/07391102.2020.1763199
13. Lokhande KB, Banerjee T, Swamy KV, et al. An in silico scientific basis for LL-37 as a therapeutic for Covid-19.. Proteins. 2021;90:1029–1043. doi:10.1002/prot.26198
14. Pulakuntla S, Lokhande KB, Padmavathi P, et al. Mutational analysis in international isolates and drug repurposing against SARS-CoV-2 spike protein: molecular docking and simulation approach. Virus Dis. 2021;32:690–702. doi:10.1007/s13337-021-00720-4
15. Singh TU, Parida S, Lingaraju MC, et al. Drug repurposing approach to fight COVID‑19. Pharmacol Rep. 2020;72:1479–1508. doi:10.1007/s43440-020-00155-6
16. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nature reviews. Drug Discov. 2014;4:673–683.
17. Schlagenhauf P, Grobusch MP, Maier JD, Gautret P. Repurposing antimalarials and other drugs for COVID-19. Trav Med Infect Dis. 2020;34:101658. doi:10.1016/j.tmaid.2020.101873
18. Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob Agents Chemother. 2020;64:e00819–e00820. doi:10.1128/AAC.00819-20
19. El Bairi K, Trapani D, Petrillo A, et al. Repurposing anticancer drugs for the management of COVID-19. Eur J Cancer. 2020;141:40–61. doi:10.1016/j.ejca.2020.09.014
20. Ciliberto G, Mancini R, Paggi MG. Drug repurposing against COVID-19: focus on anticancer agents. J Exp Clin Cancer Res. 2020;39:86–94. doi:10.1186/s13046-020-01590-2
21. Saini KS, Lanza C, Romano M, et al. Repurposing anticancer drugs for COVID-19 induced inflammation, immune dysfunction, and coagulopathy. Br J Cancer. 2020;1–4. doi:10.1038/s41416-020-0948-x
22. Ahmad SI. 5-Fluorouracil in combination with deoxyribonucleosides and deoxyribose as possible therapeutic options for the Coronavirus, COVID-19 infection. Med Hypotheses. 2020;142:142109754. doi:10.1016/j.mehy.2020.109754
23. Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16(4):215–237. doi:10.2165/00003088-198916040-00002
24. Pan X, Wang C, Wang F, et al. Development of 5-fluorouracil derivatives as anticancer agents. Curr Med Chem. 2011;18:4538–4556. doi:10.2174/092986711797287584
25. Liu Z, Rimmer S. Synthesis and release of 5-fluorouracil from poly (Nvinylpyrrolidinone) bearing 5-fluorouracil derivatives. J Control Release. 2002;81:91–99. doi:10.1016/S0168-3659(02)00048-2
26. Tzioumaki N, Manta S, Tsoukala E, et al. Synthesis and biological evaluation of unsaturated keto and exomethylene D-arabinopyranonucleoside analogs: novel 5-fluorouracil analogs that target thymidylate synthase. Eur J Med Chem. 2011;46:993–1005. doi:10.1016/j.ejmech.2011.01.005
27. Xu XH, Yao GM, Li YM, et al. 5-fluorouracil derivatives from the Sponge Phakellia fusca. J Nat Prod. 2003;66:285–288. doi:10.1021/np020034f
28. Zheng KB, He J, Zhang J. Synthesis and antitumor activity of N1-acetylamino-(5-alkyl/aryl-1,3,4-thiadiazole-2-yl)-5-fluorouracil derivatives. Chin Chem Lett. 2008;19:1281–1284. doi:10.1016/j.cclet.2008.09.021
29. Engel D, Nudelman A, Tarasenko N, et al. Novel prodrugs of tegafur that display improved anticancer activity and antiangiogenic properties. J Med Chem. 2008;51:314–323. doi:10.1021/jm7009827
30. Yin P, Hu ML, Hu LC. Synthesis, structural characterization and anticarcinogenic activity of a new Gly–Gly dipeptide derivative: methyl 2-(2-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)acetate. J Mol Struct. 2008;882:75–79. doi:10.1016/j.molstruc.2007.09.017
31. Ngbolua KTN, Kilembe JT, Matondo A, et al. In silico studies on the interaction of four cytotoxic compounds with angiogenesis target protein HIF‑1α and human androgen receptor and their ADMET properties. Bull Nat Res Centre. 2022;46:101–112. doi:10.1186/s42269-022-00793-1
32. Rezac J, Fanfrlık J, Salahub D, Hobza P. Semiempirical quantum chemical PM6 method augmented by dispersion and H-bonding correction terms reliably describes various types of noncovalent complexes. J Chem Theory Comput. 2009;5:1749–1760. doi:10.1021/ct9000922
33. Zhurko GA, Zhurko DA. Chemcraft program, academic version 1.8; 2014. Available from: http://www.Chemcraftprogram.com.
34. Reddy AD, Suh SB, Ghaffari R, et al. Bioinformatics analysis of SARS proteins and molecular dynamics simulated structure of an alpha-helix motif. Bull-Korean Chem Soc. 2003;24:899–900.
35. Xia B, Kang X. Activation and maturation of SARS-CoV main protease. Protein Cell. 2011;2:282–290. doi:10.1007/s13238-011-1034-1
36. Fan K, Wei P, Feng Q, et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J Biol Chem. 2004;279:1637–1642. doi:10.1074/jbc.M310875200
37. Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi:10.1371/journal.pbio.0030324
38. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARSCoV- 2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi:10.1038/s41586-020-2223-y
39. Biovia DS. Discovery Studio Visualizer. San Diego, CA, USA: Dassault Systèmes; 2017.
40. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi:10.1002/jcc.21334
41. Thomas R, Mary YS, Resmi KS, et al. Synthesis and spectroscopic study of two new pyrazole derivatives with detailed computational evaluation of their reactivity and pharmaceutical potential. J Mol Struct. 2018;1156:210–215. doi:10.1016/j.molstruc.2019.01.014
42. Mary YS, Miniyar PB, Mary YS, et al. Synthesis and spectroscopic study of three new oxadiazole derivatives with detailed computational evaluation of their reactivity and pharmaceutical potential. J Mol Struct. 2018;1173:469–480. doi:10.1016/j.molstruc.2018.07.026
43. Costa RA, Junior ESA, Lopes GBP, et al. Structural, vibrational, UV–vis, quantum-chemical properties, molecular docking and anti-cancer activity study of annomontine and N-hydroxyannomontine β-carboline alkaloids: a combined experimental and DFT approach. J Mol Struct. 2018;1171:682–695. doi:10.1016/j.molstruc.2018.06.054
44. Kasende OE, Matondo A, Muya JT, Scheiner S. Interactions between temozolomide and guanine and its S and Se-substituted analogues. Int J Quantum Chem. 2017;117:157–169. doi:10.1002/qua.25294
45. Muya JT, Mwanangombo DT, Hoeil C, et al. Conceptual DFT study of the chemical reactivity of four natural products with anti‑sickling activity. SN Appl Sci. 2019;1:1457–1474. doi:10.1007/s4245
46. Matondo A, Thomas R, Tsalu PV, et al. α-methylation and α-fluorination electronic effects on the regioselectivity of carbonyl groups of uracil by H and triel bonds in the interaction of U, T and 5FU with HCl and TrH3 (Tr = B, Al). J Mol Graphics Model. 2019;88:237–246. doi:10.1016/j.jmgm.2019.02.006
47. Mary YS, Mary YS, Resmi KS, et al. Modeling the structural and reactivity properties of hydrazono methyl-4H-chromen-4-one derivatives-wavefunction-dependent properties, molecular docking, and dynamics simulation studies. J Mol Model. 2021;27:186–198. doi:10.1007/s00894-021-04800-6
48. Honorio KM, Da Silva ABF. An AM1 study on the electron-donating and electron-accepting character of biomolecules. Int Quantum Chem. 2003;95:126–132. doi:10.1002/qua.10661
49. Da Silva RR, Ramalho TC, Santos JM, et al. On the limits of highest occupied molecular orbital driven reactions: the frontier effective for reaction molecular orbital concept. J Phys Chem. 2006;110:1031–1040. doi:10.1021/jp054434y
50. Maltarollo VG, Silva DC, Honório KM. Advanced QSAR studies on PPARδ ligands related to metabolic diseases. J Braz Chem Soc. 2012;23:85–95. doi:10.1590/S0103-50532012000100013
51. Pang X, Zhou L, Zhang M, et al. Two rules on the protein-ligand interaction. Open Conf Proc J. 2012;3:70–80. doi:10.2174/2210289201203010070
52. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09 (C.01). Wallingford CT: Gaussian, Inc.; 2009.
53. Isamura BK, Lobb KA, Muya JT. Regioselectivity, chemical bonding and physical nature of the interaction between imidazole and XAHs (X=H, F, Cl, Br, CH3, and A=S, Se, Te). Mol Phys. 2022;120. doi:10.1080/00268976.2022.2026511
54. Matondo A, Mukeba CT, Muzomwe M, et al. Unravelling syn- and anti- orientation in the regioselectivity of carbonyl groups of 5-fluorouracil an anticancer drug toward proton donors. Chem Phys Lett. 2018;712:196–207. doi:10.1016/j.cplett.2018.09.074
55. Lu T, Chen F. Multiwfn: a multifunctional wave function analyzer. J Comput Chem. 2012;33:580–592. doi:10.1002/jcc.22885
56. Dennington R, Keith T, Millan J. GaussView 5.0.8. Shawnee Mission, KS: Semichem. Inc.; 2009.
57. Mengist HM, Dilnessa T, Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front Chem. 2021;9:622898. doi:10.3389/fchem.2021.622898
58. Cherrak SA, Merzouk H, Mokhtari-Soulimane N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PLoS One. 2020;15:e0240653. doi:10.1371/journal.pone.0240653
59. Kumar V, Dhanjal JK, Kaul SC, Wadhwa R, Sundar D. Withanone and caffeic acid phenethyl ester are predicted to interact with main protease M(pro) of SARS-CoV-2 and inhibit its activity. J Biomol Struct Dyn. 2020;75:1–13.
60. Kandeel M, Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. doi:10.1016/j.lfs.2020.117627
61. Daina A, Olivier M, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi:10.1038/srep42717
62. Pires DE, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using Graph-Based Signatures. J Med Chem. 2015;58:4066–4072. doi:10.1021/acs.jmedchem.5b00104
63. Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Method. 2000;44:235–249. doi:10.1016/S1056-8719(00)00107-6
64. Barthea L, Woodley J, Houina G. Gastrointestinal absorption of drugs: methods and studies. Fundam Clin Pharmacol. 1999;13:154–168. doi:10.1111/j.1472-8206.1999.tb00334.x
65. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi:10.1602/neurorx.2.1.3
66. Martin YC. A bioavailability score. J Med Chem. 2015;48:3164–3170. doi:10.1021/jm0492002
67. MSD. Drug metabolism-clinical pharmacology; 2022 [Updated February 11, 2022]. Available from: https://www.msdmanuals.com/fr/professional/.
68. Geerlings P, De Proft F, Conceptual DFT. the chemical relevance of higher response functions. Phys Chem Chem Phys. 2008;10:3028–3042. doi:10.1039/b717671f
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.