")
Back to Journals » Drug Design, Development and Therapy » Volume 11
In silico discovery and in vitro activity of inhibitors against Mycobacterium tuberculosis 7,8-diaminopelargonic acid synthase (Mtb BioA)
Authors Billones JB , Carrillo MCO, Organo VG, Sy JBA, Clavio NAB, Macalino SJY, Emnacen IA, Lee AP, Ko PKL, Concepcion GP
Received 16 August 2016
Accepted for publication 6 December 2016
Published 2 March 2017 Volume 2017:11 Pages 563—574
DOI https://doi.org/10.2147/DDDT.S119930
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Georgios Panos
Junie B Billones,1,2 Maria Constancia O Carrillo,1 Voltaire G Organo,1 Jamie Bernadette A Sy,1 Nina Abigail B Clavio,1 Stephani Joy Y Macalino,1 Inno A Emnacen,1 Alexandra P Lee,1 Paul Kenny L Ko,1 Gisela P Concepcion3
1OVPAA-EIDR Program, “Computer-Aided Discovery of Compounds for the Treatment of Tuberculosis in the Philippines”, Department of Physical Sciences and Mathematics, College of Arts and Sciences, University of the Philippines Manila, Manila, Philippines; 2Institute of Pharmaceutical Sciences, National Institutes of Health, University of the Philippines Manila, Manila, Philippines; 3Marine Science Institute, College of Science, University of the Philippines Diliman, Quezon City, Philippines
Abstract: Computer-aided drug discovery and development approaches such as virtual screening, molecular docking, and in silico drug property calculations have been utilized in this effort to discover new lead compounds against tuberculosis. The enzyme 7,8-diaminopelargonic acid aminotransferase (BioA) in Mycobacterium tuberculosis (Mtb), primarily involved in the lipid biosynthesis pathway, was chosen as the drug target due to the fact that humans are not capable of synthesizing biotin endogenously. The computational screening of 4.5 million compounds from the Enamine REAL database has ultimately yielded 45 high-scoring, high-affinity compounds with desirable in silico absorption, distribution, metabolism, excretion, and toxicity properties. Seventeen of the 45 compounds were subjected to bioactivity validation using the resazurin microtiter assay. Among the 4 actives, compound 7 ((Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid) displayed inhibitory activity up to 83% at 10 µg/mL concentration against the growth of the Mtb H37Ra strain.
Keywords: CADDD, ADMET, TOPKAT, BioA inhibitor, structure-based pharmacophore, pharmacophore, molecular docking, resazurin microtiter assay
Introduction
The traditional process of drug discovery and development is generally viewed as extremely time-consuming and labor-intensive. Typically, it takes about 10–15 years,1 and about US $800 million is required to complete the entire drug discovery and development timeline.2 This traditional process generally involves the synthesis of huge compound libraries and high-throughput screenings to determine potential compounds with bioactivities.3 However, this process has been proven to be quite unsuccessful, as hit rates were low4 and approximately 50% of the late-stage failures are attributed to unsatisfactory absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties.5 Computer-aided drug discovery and development (CADDD) helps in reducing the time and cost of drug discovery by applying computational power that can consequently streamline the whole process.6
CADDD can be applied to a variety of diseases, especially to those that cause major public health concerns and require immediate development of effective and safe treatments. For example, one serious malady that is afflicting one-third of the world population is tuberculosis or TB,7 a respiratory disease caused by the bacterium Mycobacterium tuberculosis (Mtb). Hitherto, there is no concrete cure for this disease due to the continuing resurgence of drug-resistant strains, which calls for newer and more effective antitubercular drugs.
We have applied CADDD approaches in discovering possible leads against druggable targets in Mtb.8–17 An interesting drug target in Mtb is the enzyme 7,8-diaminopelargonic acid (DAPA) aminotransferase, more commonly known as Mtb BioA. BioA is involved in biotin biosynthesis pathway, one of the most important survival pathways of Mtb.18 More specifically, BioA is the second enzyme in the pathway which catalyzes the conversion of 7-keto-aminopelargonic acid (KAPA) to DAPA.19 Mtb is dependent on biotin synthesis for survival during latency. In the absence of biotin, disruption of the biotin synthesis pathway results in cell death rather than growth arrest.20,21 BioA has been implicated in long-term survival of mycobacteria,22 more specifically for optimal stationary-phase growth in rich, biotin-replete medium.23 Additionally, biotin synthesis is unique to plants and microorganisms.24 The fact that humans lack this enzyme, as they are not capable of synthesizing biotin endogenously, makes it even more an ideal drug target for antitubercular drug discovery.19
A known potent inhibitor of BioA is amiclenomycin (ACM),25 specifically its cis isomer.26 It is a mechanism-based inhibitor which works through covalent modification of the pyridoxal phosphate (PLP) cofactor of BioA via aromatization.26,27 Its simplified amino-alcohol analog, ACM-OH, is known to have better whole-cell activity against Mtb. Although ACM is an excellent inhibitor, it has poor chemical stability due to spontaneous aromatization,26 which makes it a weak potential drug candidate. Thus, it is highly instructive to further search for compounds against BioA which may pave the way for the development of a new class of antituberculosis drugs.
In this study, we employed CADDD approaches in an effort to discover new leads against TB. Accordingly, Enamine REAL database was screened based on a pharmacophore generated from the active site of BioA. The high-scoring compounds were then subsequently docked into BioA’s active site, and the high-affinity hits were also subjected to in silico ADMET evaluation. The inhibitory activity of the compounds that passed the ADMET filter was then tested experimentally against Mtb H37Ra using the resazurin microtiter assay (REMA).
Materials and methods
Computational screening
All computational screening methods were performed using the Discovery Studio 4.0 (DS4.0; BIOVIA-Dassault Systèmes, formerly Accelrys) running on Windows 7 operating system in a machine with an Intel® Core™ i7-3770 3.40GHz quad core processor.
Generation of structure-based pharmacophore model
A three-dimensional (3D) crystal structure of DAPA synthase (BioA: Rv1568) (PDB ID: 3TFU)27 was retrieved from the Protein Data Bank (www.rcsb.org).28 The structure was then prepared and optimized using the Prepare Protein and Minimization protocols of DS4.0. Structure preparation such as insertion of missing atoms, optimization of side chain conformation, removal of alternate conformations, protonation of titratable residues, as well as modeling of missing loop regions was done in order to prime the enzyme target for further computational screening. After optimization, a binding-site sphere was subsequently defined on the enzyme around the location of the invariant residue, Lys283, which was covalently linked to the PLP cofactor.20 Based on the chemical features (hydrophobic, H-donor, H-acceptor) of the generated binding-site sphere, a pharmacophore model was then generated by employing the Interaction Generation protocol of DS4.0.
Virtual screening of compound libraries
A total of 4.5 million compounds from the Enamine REAL database (parts 1–9) (http://www.enamine.net) were screened. By using the Prepare Ligands protocol, several conformations of the compounds were generated and were subsequently compiled into a single library using the Build 3D Database protocol of DS4.0. The ligand library was then screened against the pharmacophore model by rigid and flexible fitting methods run in succession. The high-scoring compounds (fit value scores >3.0) were subsequently subjected to molecular docking studies.
Molecular docking
Prior to docking the high-scoring compounds from the pharmacophore-based screening, validation of the CDOCKER docking protocol was performed by docking KAPA and ACM, known substrate and inhibitor of BioA, respectively. After confirming interaction profile reproducibility, the virtual screening hits were docked into the BioA active site. The binding affinity calculation was done using the Calculate Binding Energies protocol of DS4.0. The compounds with better binding energy values than ACM were subjected to another round of elimination using ADMET filters.
In silico ADMET prediction
The compounds selected for further screening were subjected to ADMET calculations. Parameters such as aqueous solubility, absorption, plasma protein binding, cytochrome P450 2D6 inhibition, and hepatotoxicity were all determined using the ADMET protocol in DS4.0 (Table 1). Moreover, the toxicity potential (ie, carcinogenicity and mutagenicity) of the compounds was also predicted using the TOPKAT (TOxicity Prediction by Komputer Assisted Technology) protocol in DS4.0 (Table 2).
 | Table 1 ADMET descriptor values in DS4.0 and their corresponding interpretations |
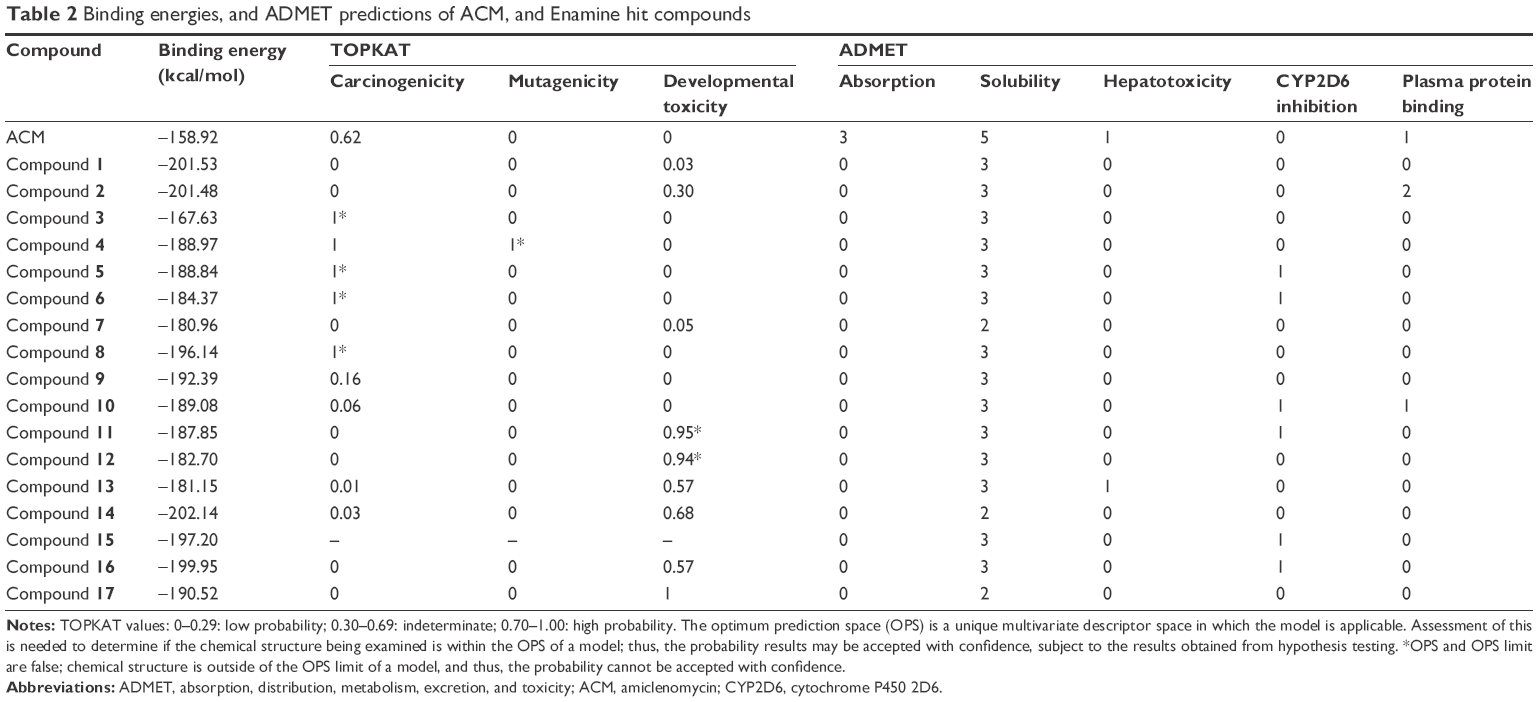 | Table 2 Binding energies, and ADMET predictions of ACM, and Enamine hit compounds |
Protein ligand interaction diagram
Two-dimensional and 3D ligand interaction diagrams for the compounds that showed H37Ra growth-inhibitory activities, as well as that of KAPA, ACM, and ACM-OH, were generated in order to examine the pertinent amino acid interactions of these compounds with the protein active site.
Resazurin-based microtiter plate assay
Bacterial strains and culture conditions
Mtb H37Ra (ATCC 25177) bacterial stocks were provided by the Marine Natural Products Laboratory, Marine Science Institute, University of the Philippines. Bacterial stocks were thawed and plated on Middlebrook 7H11 agar supplemented with 10% oleic acid–albumin–dextrose–catalase (Titan Media, Delhi, India). Plates were then incubated at 37°C for 3–4 weeks, and then subsequently inoculated with Middlebrook 7H9 broth supplemented with 10% albumin–dextrose–catalase (Titan Media). Broth tubes were incubated in a shaking incubator at 37°C and 150 rpm for an additional 3–4 weeks.
Compound preparation
Hit compounds from the Enamine database were all procured from Enamine Ltd, Kiev, Ukraine. All compounds were solubilized in dimethyl sulfoxide (DMSO) at a stock concentration of 2 mg/mL, aliquoted, and stored at −20°C. Rifampicin (Sigma-Aldrich, St Louis, MO, USA) was used as the assay positive control.
REMA
A McFarland No 1 (A625 nm ≈0.25) culture of H37Ra was first prepared as the assay inoculum. The adjusted culture was then diluted further to a 1:49 mixture of culture:M7H9 broth. Test solutions of Enamine compounds and rifampicin were prepared in DMSO at final concentrations of 1 mg/mL (high) and 0.01 μg/mL (low). Since each test well had a final volume of 200 μL, and 2 μL of the compound was added per test well, the final well concentrations of the drug and rifampicin were 10 μg/mL and 0.1 μg/mL. Each sample was tested in quadruplicates.
The assay proper was performed in 96-well, flat-bottomed microtiter plates. The well plates were then incubated at 37°C and 150 rpm for 7 days in a shaking incubator. At day 7, 20 μL of 0.02% resazurin (Sigma-Aldrich) was added to all the wells and then incubated for an additional 24 hours. The fluorescence readings of all the wells were then read at an excitation filter of 530 nm and an emission filter of 590 nm.
Results and discussion
The crystal structure of BioA (PDB ID: 3TFU)27 (Figure 1) was prepared and optimized prior to virtual screening experiment.18 The optimization step, however, may impose changes in the conformation of the protein that might result in large deviations that can adversely affect the computational results. Delightfully, the root-mean-square deviation value of 0.70 Å, calculated via superimposition of the C-alpha pairs of the raw and the minimized protein structures, was highly satisfactory (Figure 1). The binding-site sphere (x, y, z: 41.37, 6.1, −22.3; r=10 Å) was defined around the location of residue Lys283, which was covalently linked to the cofactor, PLP.20 The pharmacophore model with 25 features (9 hydrophobic, 9 donors, and 7 acceptors; Figure 2) was the basis in the virtual screening of database ligands.
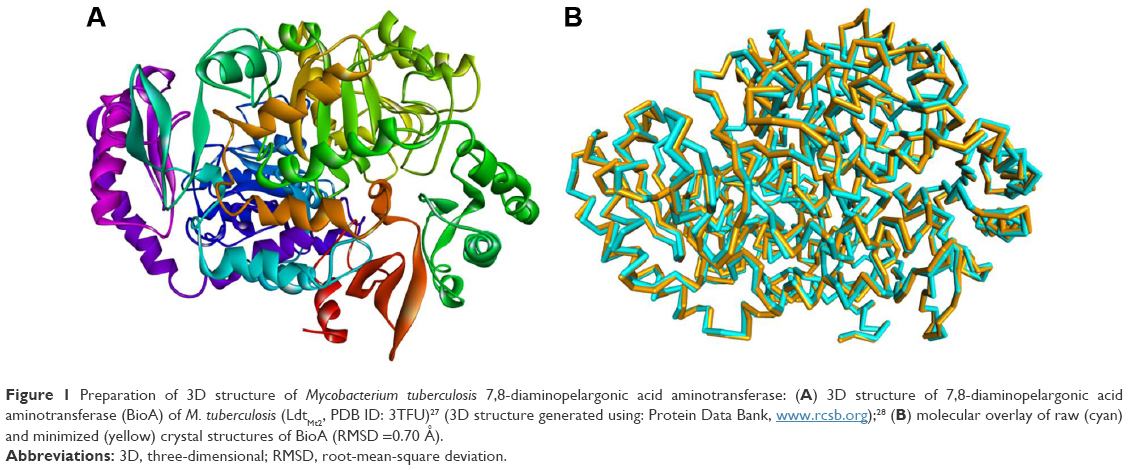 | Figure 1 Preparation of 3D structure of Mycobacterium tuberculosis 7,8-diaminopelargonic acid aminotransferase: (A) 3D structure of 7,8-diaminopelargonic acid aminotransferase (BioA) of M. tuberculosis (LdtMt2, PDB ID: 3TFU)27 (3D structure generated using: Protein Data Bank, www.rcsb.org);28 (B) molecular overlay of raw (cyan) and minimized (yellow) crystal structures of BioA (RMSD =0.70 Å). |
 | Figure 2 Structure-based pharmacophore model of BioA. This pharmacophore was modeled based on BioA’s active site, showing 9 hydrophobic (cyan), 9 donor (magenta), and 7 acceptor (green) features. |
In this study, a total of 4.5 million compounds from the Enamine REAL database were prepared to remove duplicate structures, enumerate isomers and tautomers, and generate 3D conformations.29 The Lipinski filter was turned off since a number of accounts showed that some compounds, including many natural products and natural product-like molecules, have become successful candidate drugs, despite violating the Lipinski’s Rule of Five.30,31 Virtual screening was done by rigid and flexible fitting methods with reference to a pharmacophore model generated based on the structure of the target. This step efficiently reduces the number of compounds that was included in the subsequent molecular docking studies.
To validate the applicability of the docking protocol, the reference compounds, KAPA and ACM, were docked in silico to BioA. As desired, the docked BioA–KAPA and BioA–ACM complexes involved interactions with similar residues as reported in previous studies.20,26 The van der Waals (vdW) interactions of KAPA with Tyr25, Trp64, and Phe402 were consistent with observations from the study of Dey et al.20 In comparison to a more recently released crystal structure of BioA complexed with KAPA (PDB ID: 4CXQ),32 the docked complex displayed similar hydrogen bonding between KAPA and Tyr157, and hydrophobic interaction with Trp64. The docked complex also predicted additional interactions that are not found in other studies, such as the charge–pi interaction with Trp64 and Phe402 of BioA, which are consistent with the experimental results observed by Dey et al.20 However, the covalent bond formation between ACM and PLP has been represented as polar interaction in the docking results. The results also showed vdW forces with Tyr25, Tyr157, and Lys283, a conserved residue in BioA.22
The docking experiment with ACM returned a binding energy of −158.81 kcal/mol, which was set as the threshold value in our screening. Out of over 4 million compounds, 45 turned out to have better binding energies than ACM, with acceptable predicted toxicity properties and favorable pharmacokinetic profiles. The calculated properties of 17 accessible compounds are detailed in Table 2. The mycobacterial activity of these hit compounds was subsequently tested in vitro using the H37Ra strain of Mtb.
In this study, the REMA was employed in whole-cell bioactivity measurements. Although target-based screenings usually employ inhibition assays against the target, whole-cell assay was utilized here because some compounds that have been active in target-based screens were often found to be inactive on the whole bacterium.33,34 REMA is considered as one of the most rapid, simple, yet inexpensive methods for drug susceptibility testing.35 Moreover, since resazurin is a relatively inexpensive dye, the REMA can be feasibly applied in low-resource settings.36 Out of 45 high-scoring, high-binding hits, only 17 (Table S1) were available for acquisition and subjected to in vitro assay against the Mtb H37Ra strain. Each compound was tested at a high-dose concentration (10 μg/mL) and a low-dose concentration (0.1 μg/mL). The percentages of inhibition for each compound are displayed in Figure 3.
 | Figure 3 Comparison of the percentages of inhibition of H37Ra growth for 4 Enamine test compounds and rifampicin (positive control) at 10 μg/mL and 0.1 μg/mL. Results are shown as averages of ±SD of 4 independent experiments. |
Figure 3 shows the percentages of H37Ra growth inhibition for 4 (Figure 4) of the 17 compounds tested. Although predicted to bind favorably with BioA, the remaining 13 compounds (not shown) demonstrated minuscule activities. These results demonstrate the utility of whole-cell assay in confirming the bioactivity of hit compounds obtained from structure-based methods. Possibly, the high-affinity hits were not able to penetrate the characteristic thick, waxy lipid layer of the mycobacterium. Compound 7 ((Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid) was shown to be the most bioactive among all the compounds tested. Although it apparently lost its bioactivity at 0.1 μg/mL or at 0.27 μM, it exhibited the highest percentage of H37Ra growth inhibition (82.55%) at 10 μg/mL or at 26.85 μM. In subsequent experiments involving 6 fivefold dilutions starting from 125 μM to 0.04 μM, the minimum inhibitory concentration of 7 was found to be ~25 μM. The other active hits include compounds 9 (N1-(2,3-dimethylcyclohexyl)-N2-(2-methyl-5-(methylcarbamoyl)phenyl)oxalamide), 11 (isopropyl 3-(2-(cyclopentanecarboxamido)propanamido)-3-(p-tolyl)propanoate), and 5 (N-(1-isobutyl-1H-pyrazol-5-yl)-5-((2-methoxyphenoxy)methyl)furan-2-carboxamide), in order of decreasing activity.
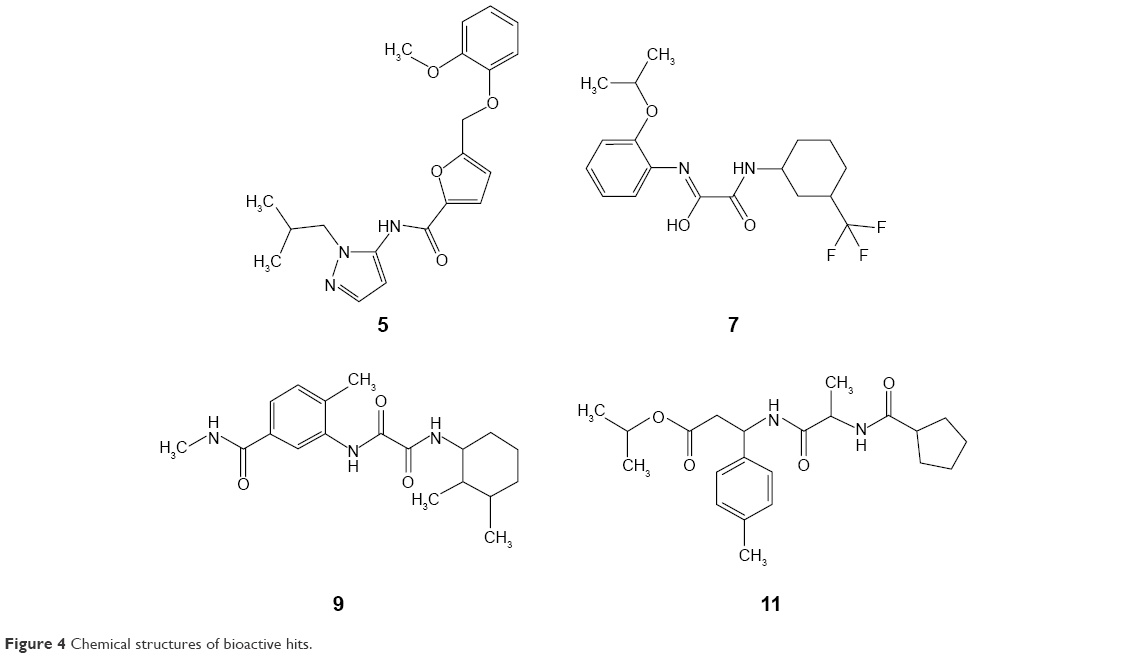 | Figure 4 Chemical structures of bioactive hits. |
The most active hit, compound 7 (Figures 5 and 6), notably exhibited interaction with PLP cofactor and with prominent residues at the active site, namely Trp64, Trp65, Tyr157, Lys283, Arg400, and Phe402. Additionally, vdW interactions were observed between 7 and Trp65, Lys283, Tyr157, Phe402, and PLP cofactor. Other interactions that largely contributed to its binding affinity were the hydrogen bonding with the side chain of Tyr407 and the pi–pi bonding with Trp64. Furthermore, the polar interaction shown between compound 7 and Arg400 may represent the salt bridge that forms between ACM and Arg400.20
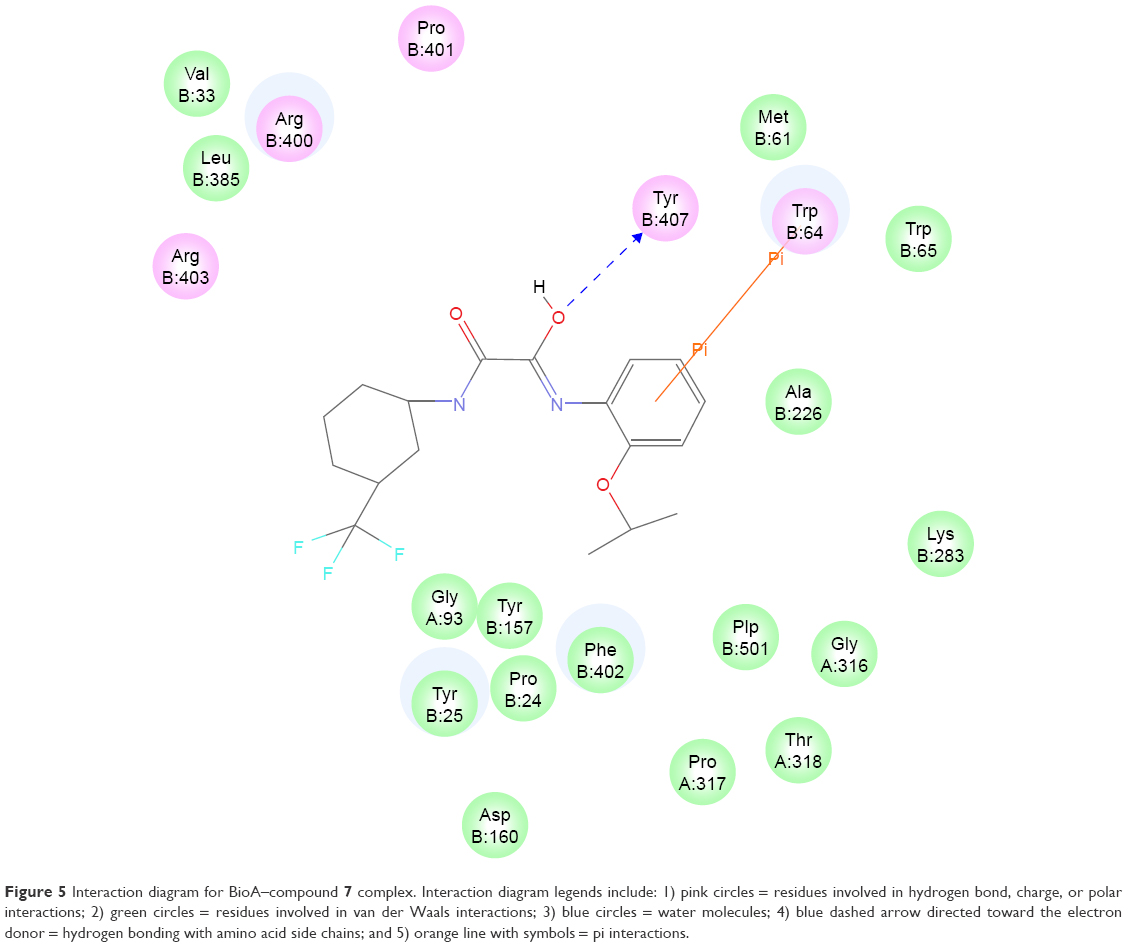 | Figure 5 Interaction diagram for BioA–compound 7 complex. Interaction diagram legends include: 1) pink circles = residues involved in hydrogen bond, charge, or polar interactions; 2) green circles = residues involved in van der Waals interactions; 3) blue circles = water molecules; 4) blue dashed arrow directed toward the electron donor = hydrogen bonding with amino acid side chains; and 5) orange line with symbols = pi interactions. |
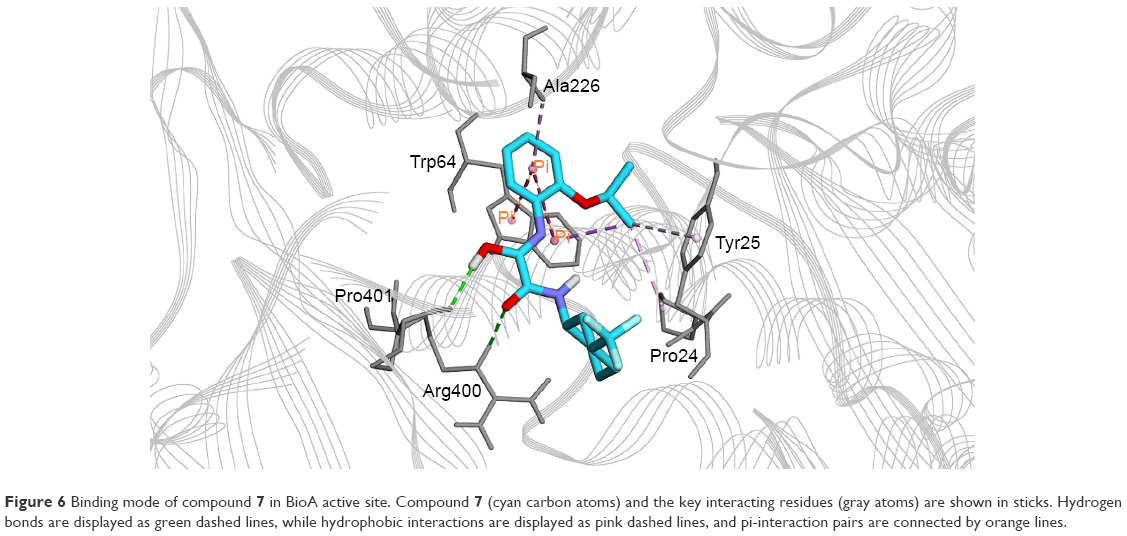 | Figure 6 Binding mode of compound 7 in BioA active site. Compound 7 (cyan carbon atoms) and the key interacting residues (gray atoms) are shown in sticks. Hydrogen bonds are displayed as green dashed lines, while hydrophobic interactions are displayed as pink dashed lines, and pi-interaction pairs are connected by orange lines. |
Conclusion
Computer-aided approaches to drug discovery were utilized in this study in order to discover potential novel compounds against TB. Specifically, virtual screening, molecular docking, and in silico ADMET calculations were performed on Enamine REAL database containing 4.5 million compounds. Out of 45 high-scoring, high-binding compounds with favorable in silico drug-like properties, 17 compounds were tested for their in vitro bioactivities against Mtb H37Ra using REMA. Four compounds displayed encouraging bioactivities, with compound 7 ((Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid) being the most active. The docking studies showed that 7 interacts with several pertinent residues in the active site of BioA, particularly forms hydrogen bonding with the side chain of Tyr407 and pi–pi bonding with Trp64. These encouraging results should prompt follow-up lead optimization studies to further improve the potency of the identified hits.
Acknowledgment
This study was supported by the Office of the Vice President for Academic Affairs (OVPAA), University of the Philippines System under the Emerging Inter-Disciplinary Research (EIDR) program (OVPAA-EIDR 12-001-121102).
Disclosure
The authors report no conflicts of interest in this work.
References
Workman P. How much gets there and what does it do?: the need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Curr Pharm Des. 2003;9(11):891–902. | ||
DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22(2):151–185. | ||
Song CM, Lim SJ, Tong JC. Recent advances in computer-aided drug design. Brief Bioinform. 2009;10(5):579–591. | ||
Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today. 2006;11(13–14):580–594. | ||
Hou T, Xu X. Recent development and application of virtual screening in drug discovery: an overview. Curr Pharm Des. 2004;10(9):1011–1033. | ||
Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem Biol Interact. 2008;171(2):165–176. | ||
WHO [webpage on the Internet]. Global tuberculosis report 2015. Geneva: WHO; 2015. Available from: http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf?ua=1. Accessed November 6, 2015. | ||
Billones JB, Carrillo MCO, Organo VG, et al. Toward antituberculosis drugs: in silico screening of synthetic compounds against Mycobacterium tuberculosis l,d-transpeptidase 2. Drug Des Dev Ther. 2016;10:1147–1157. | ||
Billones JB, Carrillo MC, Organo V, Macalino SJY, Emnacen I, Sy JB. Virtual screening against M. tuberculosis 7,8-diaminopelargonic acid synthase (MtbBioA) and in silico toxicity evaluation of top hits. Curr Enzym Inhib. 2014;10(2):105–112. | ||
Billones JB, Carrillo MCO, Organo VG, Macalino SJY, Emnacen IA, Sy JBA. Virtual screening against Mycobacterium tuberculosis lipoate protein ligase B (MtbLipB) and in silico ADMET evaluation of top hits. Orient J Chem. 2013;29(4):1457–1468. | ||
Uy VCC, Billones JB. Towards antituberculosis drugs: virtual screening for potential inhibitors of pantothenate synthetase of Mycobacterium tuberculosis. Phil Sci Lett. 2012;5(2):122–130. | ||
Yang CTM, Billones JB. Towards antituberculosis drugs: molecular docking of curcumin and its analogues to pantothenate synthetase. Phil J Sci. 2012;141(2):187–196. | ||
Billones JB, Valle AMF. Structure-based design of inhibitors against maltosyltransferase GlgE. Orient J Chem. 2014;30(3):1137–1145. | ||
Clavio NAB, Billones JB. Virtual screening against Mycobacterium tuberculosis isocitrate lyase and in silico ADME-Tox evaluation of top hits. J Chem Pharm Res. 2014;6(10):727–738. | ||
Cordova DLM, Abuel RJD, Galingana MO, Villanueva LA, Billones JB. Piggyback drug development: molecular docking of Entacapone analogues as direct M. tuberculosis InhA inhibitors. J Chem Pharm Res. 2015;7(5):636–642. | ||
Sampaco III AB, Billones JB. Virtual screening of natural products, molecular docking and dynamics simulations on M. tuberculosis S-adenosyl-L-homocysteine hydrolase. Orient J Chem. 2015;31(4):1859–1865. | ||
Billones JB. Reverse docking study unravels the potential Mycobacterium tuberculosis enzyme targets of Agelasine F. Orient J Chem. 2016;32(2):851–858. | ||
Bhor VM, Dev S, Vasanthakumar GR, Surolia A. Spectral and kinetic characterization of 7,8-diaminopelargonic acid synthase from Mycobacterium tuberculosis. IUBMB Life. 2006;58(4):225–233. | ||
Käck H, Sandmark J, Gibson K, Schneider G, Lindqvist Y. Crystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymes. J Mol Biol. 1999;291(4):857–876. | ||
Dey S, Lane JM, Lee RE, Rubin EJ, Sacchettini JC. Structural characterization of the Mycobacterium tuberculosis biotin biosynthesis enzymes 7,8-diaminopelargonic acid synthase and dethiobiotin synthetase. Biochemistry. 2010;49(31):6746–6760. | ||
Woong Park S, Klotzsche M, Wilson DJ, et al. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 2011;7(9):e1002264. | ||
Mann S, Ploux O. 7,8-Diaminoperlargonic acid aminotransferase from Mycobacterium tuberculosis, a potential therapeutic target: characterization and inhibition studies. FEBS J. 2006;273(20):4778–4789. | ||
Keer J, Smeulders MJ, Gray KM, Williams HD. Mutants of Mycobacterium smegmatis impaired in stationary-phase survival. Microbiology. 2000;146(Pt 9):2209–2217. | ||
Rendina AR, Taylor WS, Gibson K, et al. The design and synthesis of inhibitors of dethiobiotin synthetase as potential herbicides. Pestic Sci. 1999;55(3):236–247. | ||
Okami Y, Kitahara T, Hamada M, Naganawa H, Kondo S. Studies on a new amino acid antibiotic, amiclenomycin. J Antibiot (Tokyo). 1974;27(9):656–664. | ||
Sandmark J, Mann S, Marquet A, Schneider G. Structural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycin. J Biol Chem. 2002;277(45):43352–43358. | ||
Shi C, Geders TW, Park SW, et al. Mechanism-based inactivation by aromatization of the transaminase BioA involved in biotin biosynthesis in Mycobacterium tuberculosis. J Am Chem Soc. 2011;133(45):18194–18201. | ||
Berman HM. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. | ||
Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment. San Diego: Dassault Systèmes; 2015. | ||
Murray CW, Rees DC. The rise of fragment-based drug discovery. Nat Chem. 2009;1(3):187–192. | ||
Petit J, Meurice N, Kaiser C, Maggiora G. Softening the Rule of Five – where to draw the line? Bioorg Med Chem. 2012;20(18):5343–5351. | ||
Dai R, Wilson DJ, Geders TW, Aldrich CC, Finzel BC. Inhibition of Mycobacterium tuberculosis transaminase BioA by aryl hydrazines and hydrazides. Chembiochem. 2014;15(4):575–586. | ||
Pethe K, Sequeira PC, Agarwalla S, et al. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010;1:57. | ||
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130(5):797–810. | ||
Palomino JC, Martin A, Camacho M, Guerra H, Swings J, Portaels F. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46(8):2720–2722. | ||
Palomino JC, Martin A, Portaels F. Rapid drug resistance detection in Mycobacterium tuberculosis: a review of colourimetric methods. Clin Microbiol Infect. 2007;13(8):754–762. |
Supplementary material
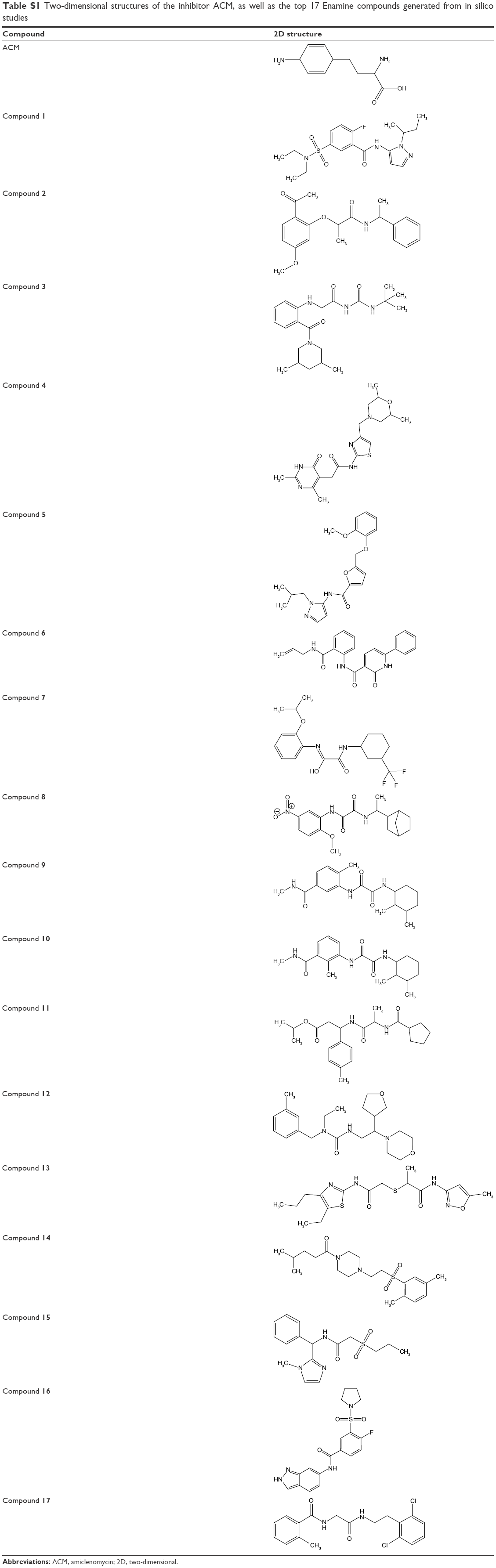 | Table S1 Two-dimensional structures of the inhibitor ACM, as well as the top 17 Enamine compounds generated from in silico studies |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.