")
Back to Journals » International Journal of Nanomedicine » Volume 15
Improvements in the Oral Absorption and Anticancer Efficacy of an Oxaliplatin-Loaded Solid Formulation: Pharmacokinetic Properties in Rats and Nonhuman Primates and the Effects of Oral Metronomic Dosing on Colorectal Cancer
Authors Pangeni R , Subedi L, Jha SK, Kweon S, Kang SH, Chang KY, Choi JU , Byun Y, Park JW
Received 12 June 2020
Accepted for publication 8 September 2020
Published 9 October 2020 Volume 2020:15 Pages 7719—7743
DOI https://doi.org/10.2147/IJN.S267424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Rudra Pangeni,1,* Laxman Subedi,2,* Saurav Kumar Jha,2 Seho Kweon,3 Seo-Hee Kang,4 Kwan-Young Chang,4 Jeong Uk Choi,5 Youngro Byun,3 Jin Woo Park1,2
1College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Muan-gun, Jeonnam, 58554, Republic of Korea; 2Department of Biomedicine, Health & Life Convergence Sciences, BK21 Four, Mokpo National University, Muan-gun, Jeonnam, 58554, Republic of Korea; 3Department of Molecular Medicine and Biopharmaceutical Science, Graduate School of Convergence Science and Technology, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; 4Global R&D Center, IcureBNP, Seoul 08511, Republic of Korea; 5College of Pharmacy, Chonnam National University, Gwangju 61186, Republic of Korea
*These authors contributed equally to this work
Correspondence: Youngro Byun
College of Pharmacy, Seoul National University, 1 Gwanak-Ro, Seoul 08826, Republic of Korea
Tel +82 2 880 7866
Fax +82 2 872 7864
Email [email protected]
Jin Woo Park
College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, 1666 Youngsan-Ro, Muan-gun, Jeonnam 58554, Republic of Korea
Tel +82 61 450 2704
Fax +82 61 450 2689
Email [email protected]
Objective: The anticancer efficacy of orally administered chemotherapeutics is often constrained by low intestinal membrane permeability and oral bioavailability. In this context, we designed a solid oral formulation of oxaliplatin (OP), a third-generation cisplatin analog, to improve oral bioavailability and investigate its application in metronomic chemotherapy.
Methods: An ion-pairing complex of OP with a permeation enhancer, Nα-deoxycholyl-l-lysyl-methylester (DLM), was successfully prepared and then mixed with dispersing agents (including poloxamer 188 and Labrasol) to form the solid, amorphous oral formulation OP/DLM (OP/DLM-SF; hereafter, ODSF).
Results: The optimized powder formulation was sized in the nanoscale range (133± 1.47 nm). The effective permeability of OP increased by 12.4-fold after ionic complex formation with DLM and was further increased by 24.0-fold after incorporation into ODSF. ODSF exhibited respective increases of 128% and 1010% in apparent permeability across a Caco-2 monolayer, compared to OP/DLM and OP. Furthermore, inhibition of bile acid transporters by actinomycin D and caveola-mediated uptake by brefeldin in Caco-2 cell monolayers reduced the apparent permeability values of ODSF by 58.4% and 51.1%, respectively, suggesting predominant roles for bile acid transporters and caveola-mediated transport in intestinal absorption of ODSF. In addition, macropinocytosis and paracellular and transcellular passive transport significantly influenced the intestinal permeation of ODSF. The oral bioavailabilities of ODSF in rats and monkeys were 68.2% and 277% higher, respectively, than the oral bioavailability of free OP. In vivo analyses of anticancer efficacy in CT26 and HCT116 cell-bearing mice treated with ODSF demonstrated significant suppression of tumor growth, with respective maximal tumor volume reductions of 7.77-fold and 4.07-fold, compared to controls.
Conclusion: ODSF exhibits therapeutic potential, constituting an effective delivery system that increases oral bioavailability, with applications to metronomic chemotherapy.
Keywords: oxaliplatin, bile acid transporter-mediated permeation, oral bioavailability, metronomic dosing, colorectal cancer
Introduction
Conventional chemotherapeutics have been widely used in clinical practice, with the goal of achieving complete eradication of tumors. However, these agents have several limitations that affect the efficacy of anticancer therapy, including high toxicity, limited aqueous solubility, lack of selectivity, and induction of multiple drug resistance.1 In addition, most conventional chemotherapeutic agents are administered by means of intravenous (IV) injection at the maximum tolerated dose, which requires extended drug-free periods to allow the recovery of normal host cells. This use of drug-free periods may lead to the regrowth of tumor cells through the mobilization of circulating endothelial progenitor cells, thereby resulting in tumor neovascularization.2,3 To improve treatment efficacy and prolong the survival of cancer patients, there is a need for intensive research regarding new targets and treatment strategies to fight cancer. In particular, low-dose chronic administration of conventional chemotherapeutic drugs without drug-free intervals (ie, metronomic chemotherapy) shows strong anti-angiogenic activity, along with reduction of toxic effects and inhibition of vascular rebound.1,3 In addition, metronomic chemotherapy has the potential to promote the initiation or persistence of a tumor-targeting immune response by activation of natural killer cells and preferential targeting of both myeloid-derived suppressor cells and T-regulatory cells.4 However, successful implementation of metronomic administration is facilitated by oral administration alone, not by daily IV injections. Administration of chemotherapeutic agents via the oral route has several strengths: it minimizes the limitations of conventional chemotherapy, preserves quality of life, and modulates drug release from the dosage form, thereby yielding favorable pharmacokinetics during prolonged treatment.5,6 However, oral administration of chemotherapeutic agents presents multiple challenges due to the physicochemical properties of the drugs and biological barriers to drug delivery, including low aqueous solubility, poor intestinal membrane permeability, high P-glycoprotein (P-gp) efflux, low or variable bioavailability, gastrointestinal side effects, limited dose options, novel toxicity profiles, and potential difficulty with treatment adherence.5,7,8 Notably, several novel strategies are currently under investigation to overcome these challenges and improve oral bioavailability.9–13
Platinum-based chemotherapeutics are the cornerstones of cancer treatment and have been widely used in clinical practice. The third-generation platinum analogue, oxaliplatin (cis-[(1R,2R)-1,2-cyclohexanediamine-N,N’] [oxalato(2)-O,O´] platinum; OP) is used for the treatment of advanced colorectal cancer. Structurally, OP consists of a central platinum atom surrounded by an oxalate ion and 1,2-diaminocyclohexane in the trans position; this drug exhibits its cytotoxic activity by forming intra- and inter-strand crosslinks between two adjacent guanine residues or a guanine or an adenine, thereby interfering with DNA replication and transcription machinery through nuclear DNA adduct formation.14,15 Although OP has a better safety profile than other platinum-containing compounds, such as cisplatin and carboplatin, its therapeutic use remains limited because of its toxicity profile and unfavorable pharmacokinetics.16,17 The US Food and Drug Administration approved OP as first-line chemotherapy, in combination with 5-fluorouracil, for the treatment of advanced colorectal cancer.18 OP has also been identified as a promising anticancer drug for patients unresponsive to other platinum compounds and 5-fluorouracil; importantly, it has fewer side effects than the other platinum compounds.19,20 In addition, treatment of colorectal cancer in mice with OP evokes the presentation of damage-associated molecular patterns within cancer cells and induces immunogenic cell death; thus, OP exhibits immunostimulatory potential.21,22 Moreover, the immunostimulatory efficacy of OP in mice depends on gram-positive bacteria in the intestinal microbiota, as evidenced by the suppression of proinflammatory genes in the absence of microbiota.23,24 However, given its low aqueous solubility and poor intestinal membrane permeability, the oral bioavailability of OP is low.25 To address these issues, several strategies have been studied, including ion-pairing complex formation with bile acid derivatives, coadministration with biodegradable polymers or solubility and permeation enhancers, and formulation of thermosensitive liposomes and nanoparticles.19,20,26,27
Among the abovementioned strategies, Urbanska et al developed an oral formulation of OP for the treatment of colorectal cancer, whereby OP was first loaded into nanoparticles consisting of polymeric molecules and further encapsulated in microsized alginate-based particles; this resulted in pH-sensitive, mucoadhesive, chitosan-coated, alginate microspheres. The study demonstrated significant suppression of tumor growth progression and improvement of morbidity, compared to the control group, in an orthotopic mouse model of colorectal cancer.28
Bile salts and their derivatives have been studied as potential absorption enhancers due to their atypical physicochemical properties and biocompatibility.29,30 In addition, bile acid acts as a solubilizer for drug molecules, helps to overcome barriers of the gastrointestinal route, and assists in the transport of complexed molecules (physical or chemical) across biological membranes.29,30 Several studies have suggested that the conjugation of bile acids with amino acids may reduce the precipitation of bile acids and increase the hydrophilicity of bile acid derivatives; it may also reduce the cytotoxic and membranolytic properties of bile acids.31,32 Previously, we reported that a positively charged bile acid derivative, Nα-deoxycholyl-
Here, we focus on the design of a stable oral powder formulation of OP by incorporating the ion-pairing complex of OP and DLM. To achieve this goal, OP was ionically complexed with DLM in combination with Labrasol and poloxamer 188 (P188) as dispersants, thus forming ODSF. This powder formulation was expected to increase the oral bioavailability of OP due to the enhanced lipophilic properties of OP, formation of stable self-assembling micelles in aqueous solution, dispersant activity of P188, opening of intestinal membrane tight junctions (TJs), and change in the membrane fluidity by Labrasol; these effects were presumed to enhance oral absorption of the drug. Artificial membrane and Caco-2 cell monolayer permeability of ODSF were evaluated to confirm the elevated oral absorption of OP. In addition, the intestinal transport pathways followed by ODSF to improve oral absorption were studied in Caco-2 cell monolayers, using specific pharmacological inhibitors. Finally, the oral bioavailability of ODSF in rats and monkeys, and tumor growth inhibition during oral metronomic chemotherapy of mice bearing mouse and human colon cancer cells were evaluated.
Materials and Methods
Materials
Oxaliplatin (OP) was purchased from Jiangsu Hengrui Medicine Co., Ltd. (Jiangsu Province, China). Caprylocaproyl macrogol-8-glycerides (Labrasol) were obtained from Gattefossé (Saint Priest, France). Polyoxyethylene (160) polyoxypropylene (30) glycol (poloxamer 188, P188) was provided by BASF (Ludwigshafen, Germany). DC, ethyl chloroformate, N-methylmorpholine, Nε-Boc-
Animals
Sprague–Dawley rats (males, 6–7 weeks old, 200–250 g) and BALB/c mice (females, 6–7 weeks old, 20–25 g) were purchased from Orientbio (Gwangju, Republic of Korea). The animals were housed under standard conditions in terms of temperature (23±2°C), relative humidity (RH; 55±10%), and light (12/12 h light/dark cycle). The animals had ad libitum access to a standard laboratory diet (Nestlé Purina PetCare Research, St. Louis, MO, USA) and ion-sterilized tap water. Ethical approval for this study was obtained from the Institutional Animal Care and Use Committee of Mokpo National University (Jeonnam, Republic of Korea; approval no. MNU-IACUC-2019–014).
The pharmacokinetic study on cynomolgus monkeys (Macaca fascicularis; nonnaïve males, 41–54 months old, 3.3–3.7 kg; Khan 7 Makara, Phnom Penh, Cambodia) was performed by Genia (a subsidiary non-clinical testing contract research organization [CRO] of Orientbio; Gyeonggi-do, Republic of Korea), which was approved by the Institutional Animal Care and Use Committee of Orientbio (Gyeonggi-do, Republic of Korea; approval no. ORIENT-IACUC-19,154). All animal experiments were performed in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and the guidelines of our Institutional Animal Care and Use Committee.
Preparation of ODSF
In the present study, DLM was used as an oral absorption enhancer and was synthesized by the conjugation of positively charged
To further enhance the intestinal membrane permeability of OP, an oral powder formulation of OP/DLM was prepared using the solid dispersion technique. Briefly, 12.5 mg of OP was dissolved in 12.5 mL of P188 solution (125 mg of P188 in 10 mL of deionized water) containing 93.75 µL of Labrasol. Next, 36 mg of DLM was dissolved in 10 mL of deionized water and added dropwise to the OP solution at a molar ratio of 1:2, with continuous stirring. The mixture was then freeze-dried at –70°C to completely remove the water and obtain an OP/DLM powder formulation.
Separately, to determine the role of DLM in enhancing the intestinal membrane permeability of OP, an oral powder formulation of OP without DLM was prepared, using Labrasol and P188 as dispersants. Briefly, 12.5 mg of OP was dissolved in 12.5 mL of P188 solution (10 mg/mL in deionized water) containing 93.75 µL of Labrasol. The mixture was vortexed for 5 min and freeze-dried at –70°C to completely remove water and obtain OP-SF in powder form.
Physical State and Physicochemical Characterization of ODSF
To confirm the formation of an OP–DLM ionic complex, the crystalline properties of OP; DLM; P188; OP/DLM; ODSF; and a physical mixture of OP, DLM, and P188 were compared using powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC). PXRD peaks were measured using a diffractometer (D8 Advance; Bruker AXS GmbH, Karlsruhe, Germany) operated at 40 mA and 40 kV; the Cu-Kα1 line at λ=1.5418 Å served as the radiation source. Powder samples were packed and prepared in an aluminum specimen holder. Each sample was scanned via step-scan mode in a 2-theta (2θ) range of 3°≤2θ≤50° at a scan rate of 0.02°/s, using an automatic divergence slit assembly with a proportional detector. Sample scanning was performed at 25°C. Thermal analyses were performed using a DSC instrument (Q1000 V9.9 Build 303; TA Instruments Inc., New Castle, DE, USA). Approximately 2 mg of each powder was transferred to an aluminum crucible and non-hermetically sealed. The DSC scans ran from 25°C to 350°C at 5°C/min in a nitrogen atmosphere.
Next, the average particle sizes and polydispersity indices (PDIs) of the optimum oral powder formulation of OP and individual excipients (eg, DLM, P188, and Labrasol) were measured based on the dynamic light scattering technique; the zeta potentials of OP/DLM and ODSF were measured using an analyzer (Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK) at 25°C. The ODSF was dispersed in deionized water (1:20, w/v), then sonicated for 1 min to minimize multiple scattering effects. In addition, the shape, surface morphology, and particle size of ODSF were determined by high-resolution transmission electron microscopy. The optimum formulation was diluted with deionized water (1:100, v/v), dropped onto a copper grid, and then negatively stained with 2% (w/v) phosphotungstic acid solution. Excess solution was removed with filter paper and the grid was observed using a transmission electron microscope (JEM-200; JEOL, Tokyo, Japan).
In vitro Artificial Intestinal Membrane Permeability of ODSF
Permeability studies of free OP, OP/DLM, OP-SF, and ODSF through an artificial membrane were performed using a parallel artificial membrane permeability assay (PAMPA; BD Biosciences, San Jose, CA, USA) in the same manner as described previously.38 Briefly, 300 µL phosphate-buffered saline (pH 6.8) was added to the acceptor compartment, and 200 µL free OP, OP/DLM, OP-SF, or ODSF diluted to a concentration of 200 µg/mL in phosphate-buffered saline (pH 6.8) was added to each donor compartment. The acceptor and donor plates were sandwiched, while ensuring that the underside of the membrane was in contact with the acceptor solution. Then, the entire system was incubated for 5 h at 25°C. Following incubation, the PAMPA plate sandwich was separated and samples were withdrawn from both the donor and acceptor plates. The concentrations of OP or OP/DLM permeated through the artificial membrane were measured via HPLC using a C18 column (4.6×250 mm, 5 µm, 100 Å) at 40°C. The mobile phase consisted of water (pH 3.0, adjusted with phosphoric acid)–acetonitrile (99:1, v/v) and ran at a flow rate of 1.2 mL/min. The injection volume was 20 µL; measurement was performed using a UV detector at 210 nm. The concentration data were used to calculate the effective permeability (Pe) of each drug as follows: Pe (×10−6 cm/s)=–ln[1–CA(t)/Ceq]/[A×(1/VD+1/VA)×t]×10−6, where Pe is the effective permeability (cm/s), A is the filter insert area (0.228 cm2), VD is the volume of the donor well (0.2 mL), VA is the volume of the receptor well (0.3 mL), t is the total incubation time in seconds, CA(t) denotes the concentration of drug in the receptor well at time t, and Ceq was calculated using the following equation: [CD(t)×VD+CA(t)×VA]/(VD+VA), where CD(t) denotes the concentration of drug in the donor well at time t.
In vitro Permeability Across a Caco-2 Monolayer ODSF
The in vitro human intestinal Caco-2 cell monolayer permeability assay was performed as described previously.26 Briefly, aliquots of approximately 1×105 cells/well (Caco-2 cells; ATCC® HTB-37™) were seeded onto each 24-well Transwell® filter insert (pore size 0.4 μm, surface area 0.33 cm2; Corning Incorporated, Corning, NY, USA) in Dulbecco’s modified Eagle’s medium (Lonza, Basel, Switzerland) containing 10% (v/v) fetal bovine serum (Gibco/Thermo Fisher Scientific) and 1% penicillin/streptomycin (Gibco/Thermo Fisher Scientific). Media in both the apical and basolateral chambers were changed on alternate days up to day 8, then daily from day 9 onwards. Cells were allowed to grow and differentiate; at day 16, the integrity of the Caco-2 cell monolayer was evaluated by measurement of transepithelial electrical resistance (TEER) using an Epithelial Volt-Ohm Meter (Millicell® ERS-2; Merck, Darmstadt, Germany). Caco-2 cell monolayers with TEER values >350 Ω·cm2 were used for experiments to measure the permeabilities of free OP, OP/DLM, OP-SF, and ODSF. Prior to the assay, media on both apical and basolateral compartments were removed and replaced with 0.1 mL and 0.6 mL prewarmed (37°C) Hanks’ balanced salt solution (HBSS), respectively, and equilibrated for 20 min in an incubator at 37°C. Next, HBSS in the basolateral compartment was removed and replaced with 0.6 mL fresh HBSS; the medium in the apical compartment was replaced with 0.1 mL free OP, OP/DLM, OP-SF, or ODSF diluted in HBSS (equivalent to 100 µg/mL OP). Then, the plates were incubated at 37°C and samples (100 µL) were taken from the basolateral compartment after 0.5, 1, 2, 3, 4, and 5 h; they were replaced with the same volume of fresh HBSS. In addition, the apical solution was collected after 5 h. Then, each collected sample was filtered through a polyvinylidene difluoride (PVDF) membrane filter (0.45 µm); the concentrations of OP or OP/DLM that permeated through the Caco-2 monolayer were determined using the HPLC system with a UV detector, as described above. The apparent permeability coefficient (Papp) of OP or OP/DLM was calculated using the following equation: Papp=dQ/dt×1/(A×C0), where dQ/dt indicates the linear appearance rate of mass in the basolateral compartment (μg/s), C0 is the initial concentration of OP or OP/DLM in the apical compartment (μg/mL), and A is the surface area of the monolayer membrane (cm2).
Intestinal Transport Mechanism of ODSF
Prior to establishment of the possible in vitro transcellular model and evaluation of the possible routes of ODSF transport, the differentiation and monolayer integrity of Caco-2 cells on the Transwell® membrane were measured using TEER (>350 Ω·cm2), as described above. To examine the mechanism involved in the transport of ODSF, 0.1 mL of HBSS containing inhibitors of specific cellular uptake pathways and 0.6 mL of free HBSS were added in the apical and basolateral compartment of each well, respectively, as described in our previous report (Table 1).39 After the plate had been incubated at 37°C for 30 min, the solution in the apical compartment was replaced with 0.1 mL of ODSF diluted in HBSS (equivalent to 100 µg/mL OP), along with corresponding inhibitors. The HBSS in the basolateral compartment was replaced with 0.6 mL of fresh HBSS; the plate was then incubated at 37°C. Aliquots of 200 µL of the sample solution were withdrawn from the basolateral compartment at predetermined times (ie, after 0.5, 1, 2, 3, 4, and 5 h) and replaced with the same volume of fresh HBSS.39–41
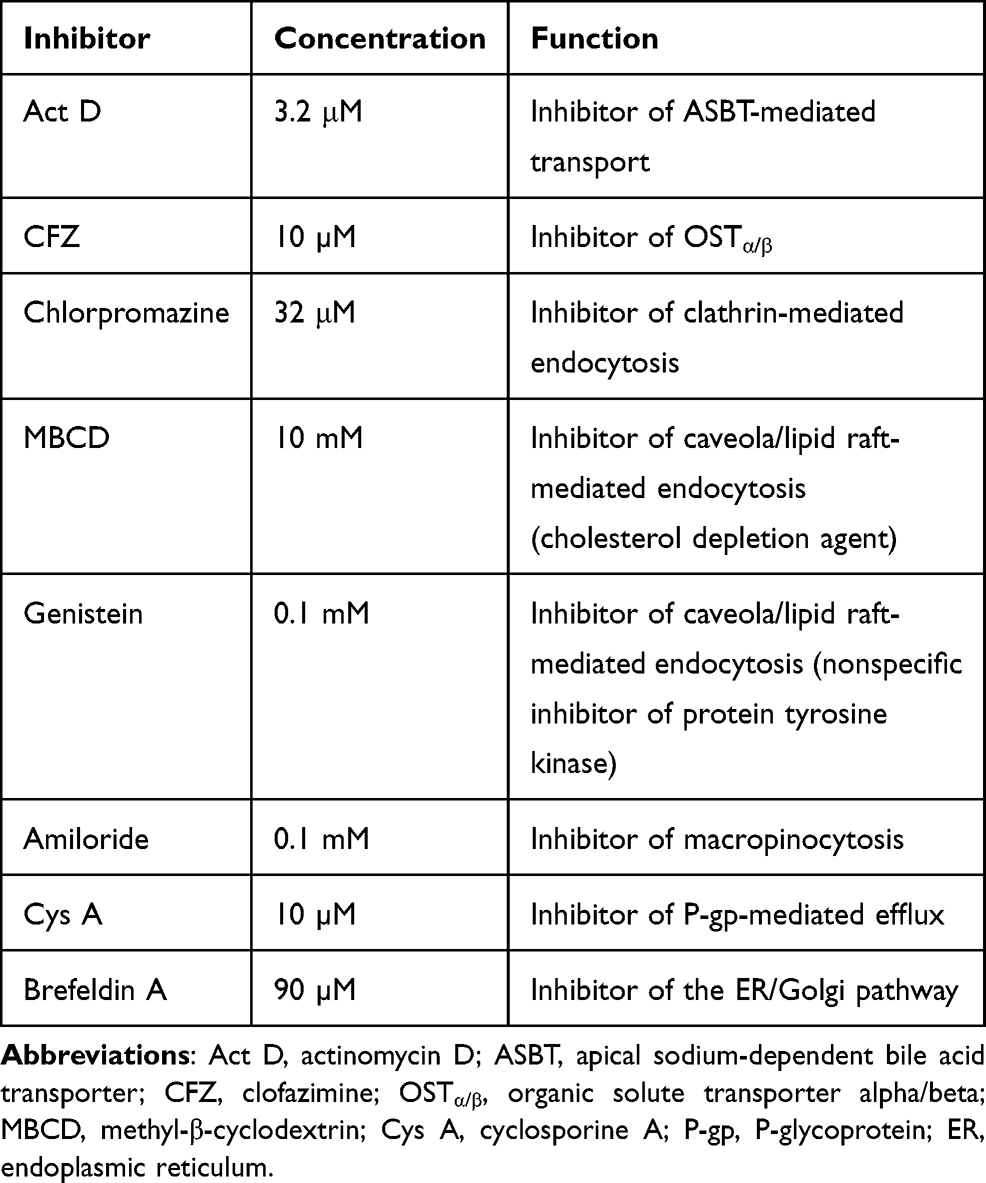 |
Table 1 Inhibitors Used in the Transport Study |
Moreover, Papp of OP from apical to basolateral (Papp, AB) and basolateral to apical (Papp, BA) in the presence of cyclosporine A (Cys A; specific inhibitor of P-gp) and all inhibitors except Cys A were examined to determine the role of P-gp-mediated efflux in the transport of ODSF. During apical to basolateral transport, 0.1 mL drug solution consisting of ODSF (equivalent to 100 µg/mL OP) was loaded into the apical compartment, along with Cys A (10 µM). Samples (100 µL) from the basolateral compartment were withdrawn at predetermined time points (ie, 0.5, 1, 2, 3, 4, and 5 h) and replaced with fresh HBSS. In addition, for the efflux study (basolateral to apical transport), 0.6 mL ODSF solution (equivalent to 100 µg/mL OP) containing Cys A or all inhibitors except Cys A was loaded into the basolateral compartment; 0.1 mL aliquots were withdrawn from the apical compartment, followed by replacement with the same volume of fresh HBSS. The Papp values in centimeters per second (cm/s) of apical to basolateral (Papp, AB) and basolateral to apical (Papp, BA) transport were calculated using the above equation.
Furthermore, ethylene glycol-bis(2-aminoethyl ether)-N,N,N,’N’-tetraacetic acid (EGTA), an extracellular calcium chelator, was used to reversibly open intracellular TJs in the Caco-2 cell monolayer and evaluate the effects of TJ opening on the transport of ODSF. In addition, the effects of EGTA in combination with all inhibitors except Cys A on the transport of ODSF were examined using a Caco-2 cell monolayer with TEER value >350 Ω·cm2. Then, the monolayer in the apical compartment was treated with 0.1 mL HBSS (Ca2+-free medium) containing 2.5 mM EGTA with or without all inhibitors except Cys A; 0.6 mL HBSS (Ca2+-free medium) was added to the basolateral compartment, followed by 45 min preincubation at 37°C. The opening of monolayer TJs was confirmed by measurement of cell monolayer integrity with the TEER value of ≤70 Ω·cm2. The medium in the apical compartment was replaced with 0.1 mL HBSS (with 1.8 mM Ca2+) containing ODSF (equivalent to 100 µg/mL OP), alone or with all inhibitors except Cys A; 0.6 mL HBSS (with 1.8 mM Ca2+) was added to each well in the basolateral compartment, followed by incubation at 37°C for an additional 5 h. Aliquots of 0.2 mL the solution from the basolateral compartment were withdrawn at predetermined time points, then replaced with the same volume of pre-warmed fresh HBSS. In addition, monolayer integrity at 2 and 5 h after drug loading was measured to identify the restoration of transmembrane resistance, which returned to ≥300 Ω·cm2.
Before analyses, the collected samples were filtered through PVDF membrane filters (0.45 µm) and the concentration of OP that permeated across the Caco-2 cell monolayer was determined by HPLC at 210 nm, as described above. Finally, the cumulative amount of OP that permeated through the Caco-2 membrane was plotted as a function of time and Papp was determined from the linear slope of the plot, using the above equation.
In vitro Cytotoxicity Study
To evaluate the cytotoxic effects of free OP, DLM, OP/DLM, DLM/P188, ODSF without OP (blank-ODSF), and ODSF on Caco-2 and Madin–Darby Canine Kidney (MDCK) cells, cell counting (CCK-8 kit; Dojindo Molecular Technologies, Rockville, MD, USA) assays were performed. Caco-2 or MDCK cells (ATCC® CCL-34™) were seeded at a density of 5×103 cells/well in 100 µL of DMEM containing 10% (v/v) fetal bovine serum and 1% penicillin/streptomycin. After cells had been incubated at 37°C for 24 h, they were treated with 100 µL of OP, OP/DLM, and ODSF in free DMEM (equivalent to 1, 10, 25, 50, 100, 250, and 500 µg/mL OP) along with the same volume and respective concentrations of DLM, DLM/P188, and blank-ODSF; the cells were then incubated for an additional 12 h. To determine cell viability, 10 μL of 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt (WST-8) solution diluted in 100 µL of DMEM was added to each well and cells were incubated for 2 h. The absorbance was then measured using a microplate reader (PerkinElmer multimode plate reader; PerkinElmer, Waltham, MA, USA) at 450 nm. The percentage of viable cells was calculated by comparing the numbers of treated and untreated cells.
In vitro Drug Release of ODSF
The in vitro release of OP from ODSF was measured in 900 mL amounts of 0.1 N HCl (pH 1.2) or phosphate buffer (pH 6.8) using the United States Pharmacopeia (USP) dissolution test apparatus I (LABINDIA DS 8000 basket, Maharashtra, India) with shaking at 100 rpm at 37±0.2°C. Ten milligrams of OP or ODSF (equivalent to 10 mg OP) was placed into hard gelatin capsules (size #00) that were subjected to dissolution testing with samples (0.5 mL) withdrawn at regular intervals (15, 30, 45, 60, 90, and 120 min). The samples were filtered and OP or OP/DLM contents were measured via HPLC using a UV detector, as described above.
Stability of ODSF
To determine the stability of the lyophilized OP formulation, ODSF (equivalent to 10 mg OP) was packaged in a hard gelatin capsule (size #00) and stored in sealed high-density polyethylene (HDPE) bottles under different storage conditions of temperature and RH (25±2°C with an RH of 65±5% and 40±2°C with an RH of 75±5%). At predetermined times (after 0, 1, 3, and 6 months), bottles were pulled from the stability chamber and the amount of drug in a capsule and cumulative drug release at 1 h in 900 mL of 0.1 N HCl (pH 1.2) were measured using the USP dissolution test apparatus I and HPLC system, as described above. In addition, particle size, PDI, and zeta potential were measured to evaluate the physical stability of the ODSF micelles, as previously described.
In vivo Pharmacokinetic Study of ODSF in Rats
To examine the distinction of pharmacokinetic parameters of OP after IV injection and oral administration of OP or various concentrations of ODSF, Sprague–Dawley rats were randomly divided into five groups of four animals each. Four groups were orally administered 400 µL OP-S (10 mg/kg OP dispersed in water), ODSF (2.5) (equivalent to 2.5 mg/kg OP), ODSF (10) (equivalent to 10 mg/kg OP), and ODSF (20) (equivalent to 20 mg/kg OP). To evaluate oral bioavailability, rats in one group underwent IV administration of 150 µL an aqueous solution of OP (5 mg/kg) via the tail vein. Blood samples (150 µL) were withdrawn at specified time points from the retroorbital plexus with the rats under mild anesthesia; then the blood samples were mixed with 50 µL sodium citrate (3.8% solution). Finally, plasma was separated through immediate centrifugation (2500×g, 15 min, 4°C) and stored at –70°C until analyses.
The plasma concentration of OP based on Pt was measured using inductively coupled plasma-mass spectroscopy. Blank plasma was spiked with 200 µL different concentrations of Pt, such that the final concentrations were 1, 10, 25, 50, and 100 ng/mL; this was followed by addition of iridium internal standard (200 µL, 100 ng/mL final concentration). The samples were digested with 3 mL concentrated nitric acid for 6 h at 100°C. After complete digestion, the samples were allowed to cool to room temperature and then diluted to 10 mL with deionized water. The concentrations of Pt in each standard and sample were determined by ICP-MS (SPECTRO Analytical Instruments GmbH, Kleve, Germany), which was conducted using a double-focusing magnetic sector field mass spectrometer and a semiconductor direct charge detector. The measurement conditions for Pt were as follows: radiofrequency power, 1450 W; coolant flow, 12 L/min; axillary flow, 2.3 L/min; and nebulizer flow, 0.89 L/min.
Absorption of Orally Administered ODSF in Monkeys
To explore oral absorption of ODSF in nonhuman primates, nonnaïve male cynomolgus monkeys (3.3–3.7 kg) were fasted for 16 h prior to drug administration and randomly divided into three groups of three animals each. Two groups received oral ODSF at 5 mL/kg (equivalent to 10 mg/kg OP) or OP (10 mg/kg) dispersed in normal saline (OP-S). Further three monkeys received 5 mL/kg OP (2 mg/kg) in normal saline (OP-IV) via the saphenous vein to determine oral bioavailability. After oral or IV administration, blood samples (1.8 mL) were collected from the femoral vein into BD Vacutainer® citrate tubes (Oakville, ON, Canada) at 0 (pre-dose), 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, and 48 h after oral administration, and at 0 (pre-dose), 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, and 48 h after IV administration. Plasma samples were obtained by centrifugation at 2500×g for 15 min and stored at –70°C until use. Plasma OP levels were determined based on Pt by ICP-MS using iridium as the internal standard, as described above.
In vivo Antitumor Efficacy of ODSF
The in vivo antitumor potential of orally administered ODSF was evaluated by inoculating CT26 (ATCC® CLR-2638™; 1×106 cells/100 µL phosphate-buffered saline, pH 7.4) or HCT116 (ATCC® CCL-247™; 1×107 cells/100 µL phosphate-buffered saline, pH 7.4) cells into the right dorsal flank of 6-week-old female BALB/c or BALB/c nude mice, respectively. When the tumor volume reached 70−100 mm3, the mice were randomly assigned to seven groups of 14 animals each:
Control (untreated),
OP-IV (3.3) (once-biweekly IV administration of 3.3 mg/kg OP),
OP-IV (10) (once-biweekly IV administration of 10 mg/kg OP),
OP-S (10) (once-daily oral administration of 10 mg/kg OP in aqueous solution),
ODSF (2.5) (once-daily oral administration of ODSF, equivalent to 2.5 mg/kg OP),
ODSF (10) (once-daily oral administration of ODSF, equivalent to 10 mg/kg OP), and
ODSF (20) (once-daily oral administration of ODSF, equivalent to 20 mg/kg OP).
During the 21-day treatment period, mice were fasted for 4 and 2 h before and after drug administration, respectively. Tumor volume and mouse body weight were measured at 3-day intervals; tumor volume was calculated as width2×length×0.52, using a Vernier caliper for the measurements. After 3 weeks of treatment, mice were sacrificed; tumor masses were measured and fixed in 10% formalin for histological evaluation. For immunohistochemical analyses, paraffin-embedded tissues were cut into 5-µm-thick sections for examination of proliferating cell nuclear antigen (PCNA) and CD31 expression patterns, as well as fluorescent terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) analyses, in accordance with the manufacturer’s protocol. In vivo proliferating cells were measured using immunostaining and quantification of PCNA-positive cells. In addition, the degrees of tumor angiogenesis and microvessel formation that indicated rapid tumor growth were evaluated by staining tumor sections with an antibody to the epithelial cell marker, CD31, which reveals endothelial cells or their clusters by coloring them brown. Analyses of apoptotic cells in the tumor tissue were performed using TUNEL staining, which causes dark green fluorescence.
Pharmacokinetic and Statistical Analyses
Pharmacokinetic parameters were determined using a noncompartmental method in WinNonlin® software (ver. 5.3; Pharsight Corporation, Mountain View, CA, USA). All data are expressed as means±standard deviations (SDs) or standard errors of the mean (SEMs). Student’s t-test was used for comparisons between two mean values for unpaired data; one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test, was used for comparisons among three or more mean values for unpaired data. In all analyses, P<0.05 was considered to indicate statistical significance.
Results and Discussion
Physical State Properties of ODSF
Construction of an ion-pairing complex of OP with DLM and further preparation of an oral powder formulation of OP/DLM were performed to improve the lipophilicity and intestinal membrane permeability of OP (Figure 1A). To confirm the ion-pairing complex formation between OP and DLM, the PXRD patterns of pure OP; DLM; P188; OP/DLM; and a physical mixture of OP, DLM, and P188 were compared to the PXRD pattern of ODSF (Figure 1B). Several characteristic peaks from PXRD of pure OP were observed at 2θ diffraction angles of 7.90°, 11.91°, 15.79°, 17.89°, 19.57°, 22.22°, 23.76°, 25.41°, 26.06°, 29.90°, 31.84°, 33.11°, and 41.14°; these peaks indicated that OP was present in crystalline form. Some crystalline drug signals were also detectable, with reduced intensity, in the physical mixture; these findings indicated that the drug remained present in crystalline form. The reduced intensities of peaks in the physical mixture may have been due to lower loading of the drug per unit weight of the physical mixture, compared to pure OP. These peaks were not observed in the PXRD pattern of OP/DLM, suggesting that OP was either molecularly dispersed with DLM or distributed in an amorphous form. Furthermore, ODSF did not exhibit any characteristic crystalline peaks from OP, suggesting that OP remained present in amorphous form after powder formulation with DLM, Labrasol, and P188. This suspicion was confirmed by DSC analyses (Figure 1C). The thermal curves for pure OP and P188 indicated different melting temperatures, with narrow endothermic peaks at 278°C and 55.8°C, respectively, characteristic of the crystalline forms. DLM did not exhibit an endothermic peak up to 250°C and melted above that temperature, indicating that it was amorphous. The thermograms of OP/DLM and ODSF revealed no clear endothermic OP peaks, confirming that OP was molecularly dispersed with DLM in an amorphous form after complex formation with DLM and formulation with P188 and Labrasol. However, a wide range of OP endothermic peaks was observed for the physical mixture of OP, DLM, and P188, suggesting that OP was molecularly dispersed into the molten states of DLM and P188, then partially converted into an amorphous state, presumably attributable to solid dispersion during heating.
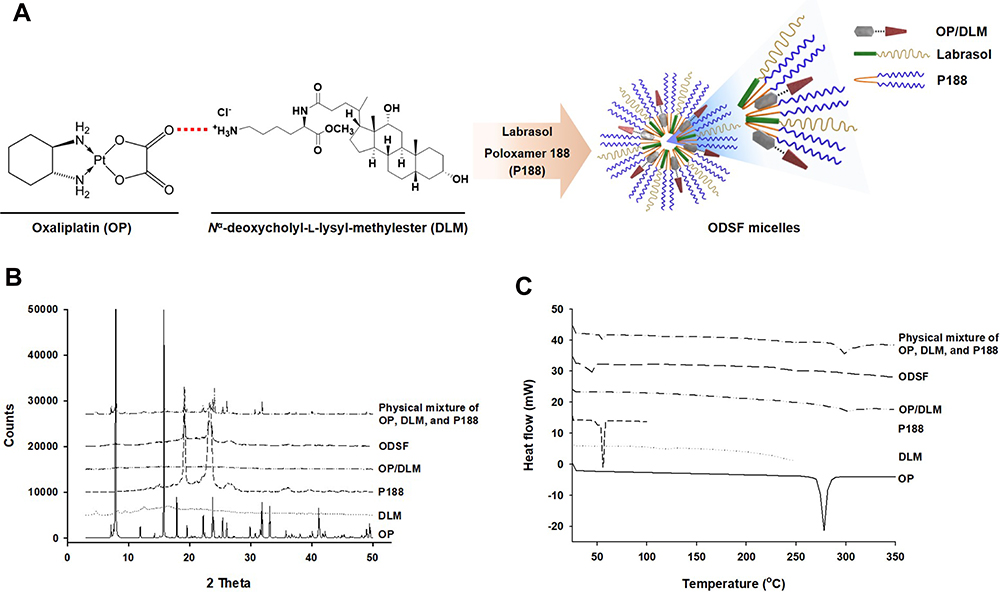 |
Figure 1 (A) Schematic representation of ion-pairing complex formation between OP and DLM, and the self-assembled micellar structure of ODSF in the aqueous phase. (B) Powder X-ray diffraction patterns and (C) differential scanning calorimetric thermograms of pure OP, DLM, P188, OP/DLM, ODSF, and a physical mixture of OP, DLM, and P188. |
Based on the above results, PXRD revealed the loss of distinct diffraction peaks and occurrence of an amorphous form of the solid, combined with the loss of the regular arrangement observed in ODSF. In addition, the DSC method is considered a “rule of thumb” technique to assess the crystallization properties and miscibility between drug and polymer. Moreover, the amorphous solid powder consists of a chemically homogenous phase in which all components were uniformly mixed at the molecular level.42 Several studies have suggested that the transformation of the crystalline form of drug to an amorphous form significantly increases its aqueous solubility, dissolution rate, and solid-state stability.42 In addition, polymers used in the formulation could act as crystallization inhibitors and reduce the particle size, thereby achieving better wettability and preventing recrystallization.43 Likewise, the use of the hydrophobic DC molecule in the ODSF formulation would increase the solubility, lipophilicity, and membrane permeability of OP without a trade-off between solubility and permeability. These changes are due to the amorphous form of the ODSF, which incorporates several solubilizing agents, resulting in supersaturation.44 Furthermore, inclusion of bile acids in the formulation inhibits crystallization and stabilizes the formulation.13,45 For these reasons, the amorphous form of ODSF and its reduced particle size, compared to OP/DLM, confirm the physical state of the formulation.
Characterization of ODSF Micelles
The excipients used in ODSF are P188, DLM, and Labrasol, diluted in an aqueous solution; their particles have sizes of 4.57±0.580, 191±9.87, and 21.0±0.32 nm, respectively, and PDIs of 0.293±0.063, 0.766±0.122, and 0.117±0.008, respectively. The particle size and PDI of OP/DLM were 420±135 nm and 0.564±0.17, respectively, whereas those of ODSF were 133±1.47 nm and 0.147±0.02, respectively (Figure 2A). The reduced particle size and PDI of ODSF, compared to OP/DLM, may have been due to the reduction in critical micellar concentration and the increase in micelle formation tendency caused by the combination of excipients.46 In addition, incorporation of Labrasol and P188 with OP/DLM in the formulation may have increased the number of surfactant molecules in each micelle, thereby assisting in the formation of well-arranged and compact self-assembled micelles, which would result in a reduced particle size.47,48 The zeta potential of ODSF was 50.3±3.02 mV, which was 1.56-fold greater than the potential of OP/DLM (Figure 2B). This increase in the zeta potential is related to the stability of ODSF micelles in aqueous conditions, which prevent the aggregation and flocculation caused by van der Waals attraction.49,50 The results of several studies have suggested that bile salts form mixed micelles or aggregates in aqueous solution, presumably due to the hydrophobic attractive interaction of the apolar regions.51,52 Furthermore, P188 and Labrasol, which are used as an amphiphilic block copolymer and a surfactant, respectively, self-assemble as micelles at concentrations above the critical micellar concentration.53,54 Thus, complex formation of OP with DLM, along with P188 and Labrasol as dispersants, results in the formation of stable micelles in the aqueous solution. In addition, transmission electron microscopy images of ODSF demonstrated the formation of spherical and homogenous nano-sized micelles, <200 nm in diameter (Figure 2C).
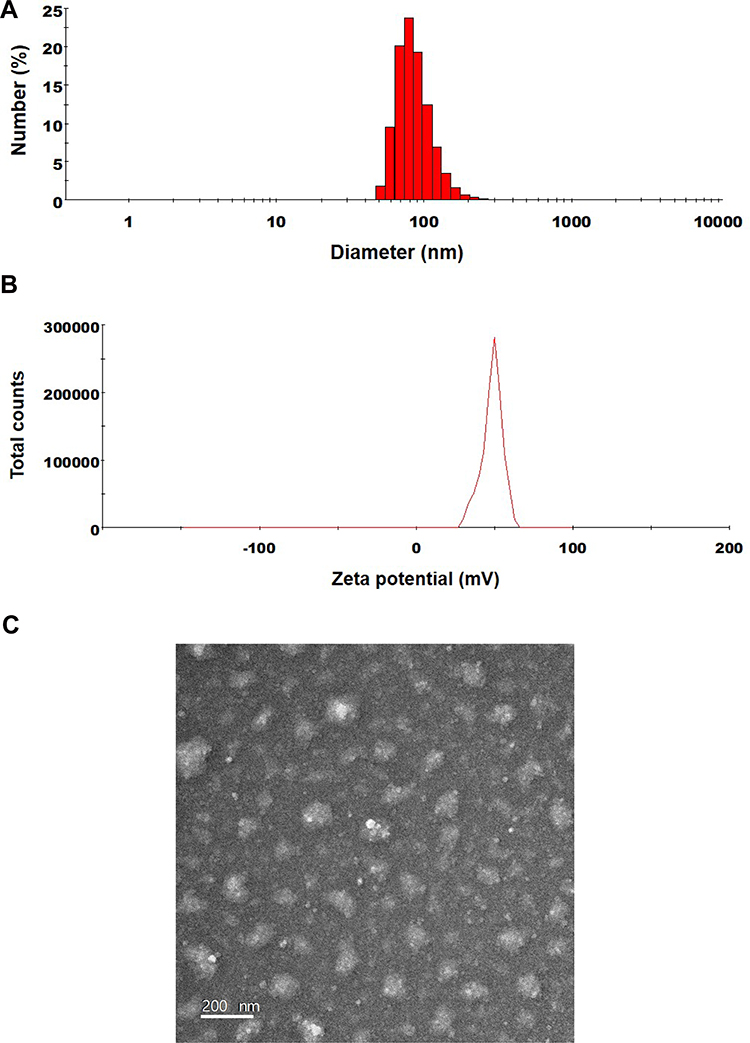 |
Figure 2 (A) Particle size, (B) zeta potential distribution, and (C) a transmission electron micrograph of ODSF micelles. Scale bar: 200 nm. |
In vitro Permeability of ODSF Through Parallel Artificial Intestinal Membrane and Caco-2 Cell Monolayer
To assess the passive diffusion of OP, OP/DLM, OP-SF, and ODSF through artificial intestinal membranes, permeability was determined using PAMPA. The Pe of OP after ion-pairing complex formation with DLM was significantly increased by 12.4-fold, compared to free OP (Table 2). This increase in artificial membrane permeability may have been due to the DLM-induced enhancement of OP hydrophobicity, which subsequently increased passive absorption through the phospholipid layer.26 Notably, bile acids act as crystallization inhibitors and potentially play roles in the formulation of supersaturated dosage forms or lipid-based drug delivery systems.45 Moreover, incorporation of OP/DLM into the oral powder formulation significantly increased the Pe of OP by 24.0-fold, 3.76-fold, and 1.94-fold, compared to free OP, OP-SF, and OP/DLM, respectively. The increased amphiphilicity and permeability of OP in this study may have been due to the synergistic activity of DLM, Labrasol, and P188 used in ODSF.
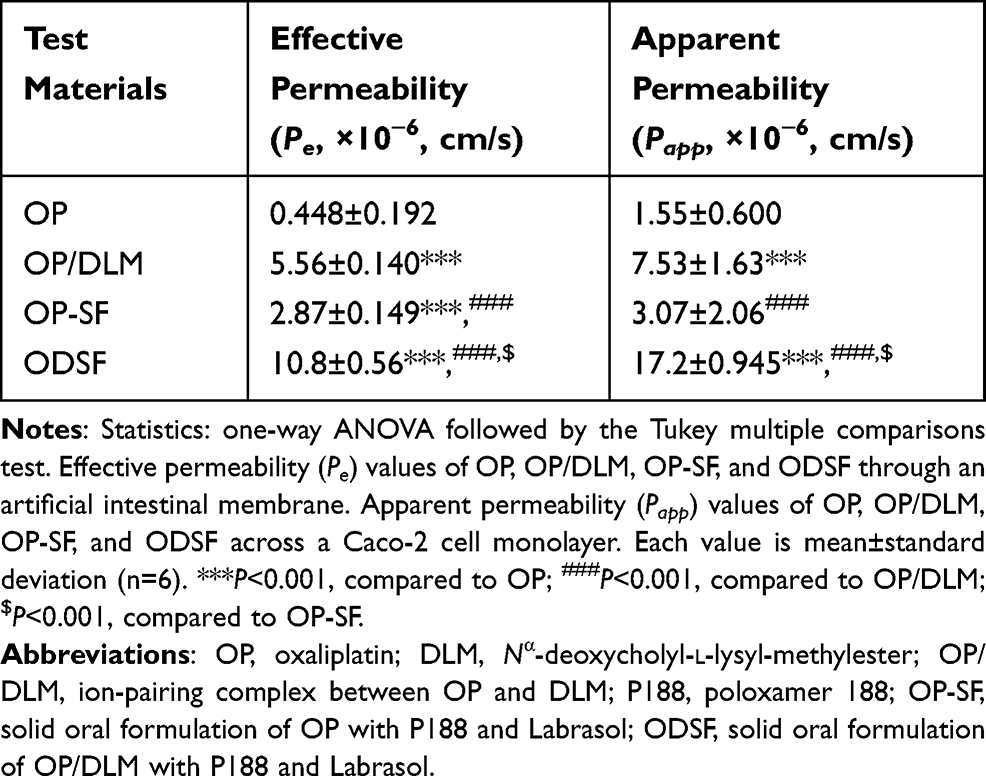 |
Table 2 Effective and Apparent Permeabilities of OP, OP/DLM, OP-SF, and ODSF |
The in vitro permeabilities of free OP, OP/DLM, OP-SF, and ODSF across a Caco-2 cell monolayer exhibited trends similar to those across the artificial intestinal membrane (Table 2). Addition of P188 and Labrasol to OP exhibited 98.1% higher Papp compared to free OP; this may have been due to the synergistic emulsifying activity of P188 and Labrasol, which resulted in formation of stable micelles on gentle agitation. The Papp of OP after ion-pairing complex formation with DLM was 386% and 145% higher, compared to free OP and OP-SF, respectively; this may have been due to the specific interaction of OP/DLM with apical sodium-dependent bile acid transporter (ASBT).55 Binding of bile acids to the calcium channels may have enhanced membrane flexibility and increased partitioning into the membrane, thereby causing transient opening of TJs and increasing the paracellular absorption of OP.30 In addition, the viscosity and elasticity of mucus adhering to the epithelial surface are reduced by bile acids, thereby enhancing intestinal membrane permeability.29,56 Intestinal membrane permeability is also reversibly modified by DC via autophosphorylation of epidermal growth factor receptor, as well as by dephosphorylation and rearrangement of occludin at the TJ level.57 The Papp of ODSF was further enhanced by 1010%, 460%, and 128%, compared to free OP, OP-SF, and OP/DLM, respectively. This significant enhancement in Papp of ODSF may have been due to the synergistic role of DLM, Labrasol, and P188 in the formation of stable micelles of OP/DLM, which might enhance drug transport through endocytosis and micropinocytosis.58 Moreover, Labrasol changes membrane permeability along with inhibition of the secretory systems in the intestinal membrane, thereby enhancing drug absorption.59 Surfactants (eg, Labrasol and P188) also act as absorption enhancers by reversibly opening TJs, causing redistribution of zonula occludens-1 (ZO-1); this enhances the absorption of hydrophilic drugs. Furthermore, administration of a formulation with positive charge binds to the epithelial cell membrane by electrostatic interaction, which could cause disruption of TJs.60 Therefore, the synergistic activity of DLM, Labrasol, and P188 may have contributed to the significant enhancement in Papp of ODSF. However, further studies using higher concentrations of these excipients alone or in combination are needed to confirm reversible changes in intestinal membrane properties.
Intestinal Transport Mechanism of ODSF
To study the mechanisms involved in the permeation, trafficking, and delivery of ODSF into the systemic circulation through enterocytes, different transport pathways were individually inhibited using specific pharmacological inhibitors (Table 1). As described above, the ion-pairing complex of OP with DLM enhanced the lipophilicity of OP, which may have increased both the paracellular and transcellular transport of the drug through the Caco-2 cell monolayer. In addition, ODSF in aqueous phase can exist as free OP/DLM or in micellar form, together with P188 and Labrasol. These self-assembled micelles of ODSF could also facilitate the uptake of OP through clathrin and caveola-mediated endocytosis, and macropinocytosis, respectively. Therefore, to confirm the presence and elucidate the predominant uptake mechanisms of these various pathways that may participate in the transport of ODSF, the Caco-2 monolayer was treated with specific inhibitors and their roles in the transport of OP were investigated. Notably, bile acids are mostly reclaimed through a combination of various processes, such as passive diffusion in the proximal intestine, active transport in the distal ileum, and passive absorption in the colon. Furthermore, bile acids are actively transported from the distal ileum via ASBT and shuttled across the basolateral membrane, thus entering the portal circulation by organic solute transporter alpha and beta (OSTα/β).61 To confirm involvement of ASBT in ODSF transport, actinomycin D (Act D) and clofazimine (CFZ) were used as inhibitors of ASBT and OSTα/β, respectively, alone or in combination.62,63 In the presence of Act D alone, the Papp of ODSF was reduced by 58.4%, compared to the control (without inhibitors) (Figure 3A). This significant reduction in Papp of ODSF after suppression of ASBT suggested that bile acid transporter plays an important role in increasing the permeability of ODSF. In addition, ODSF may be present as free OP/DLM or in micellar form; these micelles containing bile acids could form reverse micelles in aqueous solution, thereby exposing OP/DLM to the ASBT and enabling enhanced absorption through ASBT.64,65 OSTα/β primarily facilitates the transport of absorbed bile acids across the basolateral membrane of ileal enterocytes.66 Furthermore, the presence of an inhibitor of OSTα/β (CFZ) reduced the Papp of ODSF by 55.7%, compared to the control (without inhibitors) (Figure 3A). This result reaffirmed the active involvement of ASBT in the transport of ODSF across ileal enterocytes. When both CFZ and Act D were used in combination, the Papp of ODSF was reduced by 58.5%, compared to the control (without inhibitors); this finding was similar to the observations with Act D alone and CFZ alone (Figure 3A). These results confirmed the simultaneous involvement of ASBT-mediated transport, as well as the shuttling mechanism of OSTα/β across the basolateral membrane, during the absorption of ODSF.
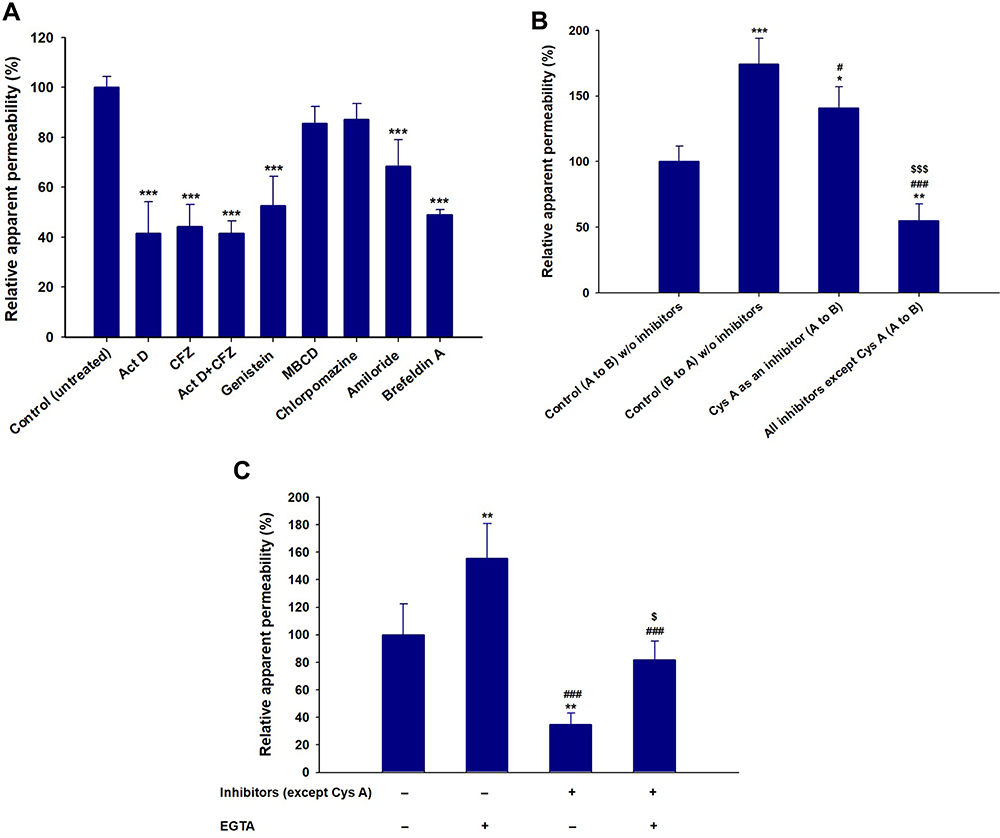 |
Figure 3 (A) Effects of various intestinal transport inhibitors on the relative apparent permeability of ODSF across a Caco-2 cell monolayer. ***P<0.001 compared to the Papp of ODSF in the absence of all inhibitors (untreated control). (B) Prediction of a role for P-gp in intestinal absorption of ODSF. *P<0.05, **P<0.01, ***P<0.001 compared to the Papp (A to B) of ODSF in the absence of all inhibitors (untreated control); #P<0.05, ###P<0.001 compared to the Papp (B to A) of ODSF in the absence of all inhibitors; $$$P<0.001 compared to the Papp (A to B) of ODSF in the absence of all inhibitors except Cys A. (C) Papp values with or without EGTA, in the presence or absence of all inhibitors except Cys A. **P<0.01, compared to the Papp without EGTA, in the absence of all inhibitors except Cys A; ###P<0.001 compared to the Papp after treatment with EGTA, in the absence of all inhibitors except Cys A; $P<0.05 compared to the Papp without EGTA, in the presence of all inhibitors except Cys A. |
In addition to ASBT-mediated transport, ODSF micelles can utilize endocytosis pathways to enter the cell. Furthermore, to understand the major endocytosis pathway and preferred trafficking routes involved in the transport of ODSF, specific endocytosis mechanisms were examined (ie, clathrin-mediated endocytosis, caveola-mediated endocytosis, and macropinocytosis) and their effects on Papp of ODSF were compared to the control condition (without inhibitors). First, to determine the role of clathrin-mediated endocytosis, the specific inhibitor chlorpromazine was used; this resulted in an only 12.8% reduction in the Papp of ODSF, compared to the control (without inhibitors) (Figure 3A). This result indicates that clathrin-mediated endocytosis does not play a substantial role in the uptake of ODSF micelles. Subsequently, to evaluate the role of the caveola-mediated pathway in the transport of ODSF, genistein was used as a specific inhibitor. Genistein is an important inhibitor of protein tyrosine kinase, which is involved in catalysis of tyrosine phosphorylation, activation of multiple signals for various transduction pathways, and regulation of endocytosis. In addition, genistein reportedly interrupts depolymerization of actin and recruitment of dynamin II, which is an important mechanism for the internalization and scission of caveola vesicles during caveola-mediated uptake from enterocytes.67,68 After treatment of Caco-2 cell monolayers with genistein, a 47.4% reduction was observed in Papp of ODSF, compared to the control (without inhibitors); this finding indicates an important role for caveola-mediated endocytosis in the uptake of ODSF (Figure 3A). By contrast, another inhibitor of caveola-mediated endocytosis, methyl-β-cyclodextrin (MBCD), slightly reduced the Papp of ODSF by 14.6%, compared to the control (without inhibitors) (Figure 3A); this result may have been due to its mode of action (ie, sequestration of cholesterol), which led to its depletion in the plasma membrane of enterocytes and caused inhibition of caveola-mediated endocytosis.69 The reduction of MBCD inhibitory activity in the presence of ODSF may be explained by the presence of a cholesterol derivative (DC) in DLM; this may neutralize the effects of MBCD.40 Importantly, macropinocytosis is another major pathway that may be involved in the internalization of ODSF. Initially in this process, the intestinal membrane forms a sac-like structure due to ruffling of the plasma membrane after contact with nanoparticles; this structure engulfs solutes or solvents present in the extracellular milieu of enterocytes to form cargo-loaded macropinosomes.70 To investigate the role-played by macropinocytosis in ODSF trafficking, we added the macropinocytosis inhibitor amiloride; this reduced the Papp of ODSF by 31.5% compared to the control (no inhibitor) (Figure 3A). Thus, macropinocytosis was involved in the absorption of ODSF micelles.
After internalization across the enterocyte membrane through endocytosis, ODSF is subjected to various pivotal stages of intracellular trafficking, which further determine its final destination. During this process, ODSF that entered through endocytosis or macropinocytosis can be internalized via formation of early or late endosomes, caveosomes, or macropinosomes; it can then be delivered to lysosomes or cytoplasm, or may exit enterocytes via the endoplasmic reticulum (ER) or ER/Golgi apparatus pathway.71–73 Moreover, effective lysosomal and cytoplasmic escape of ODSF is necessary to achieve optimum intracellular density and oral bioavailability to elicit an adequate pharmacological response. Therefore, to confirm exocytosis and involvement of the ER-to-Golgi apparatus pathway in the intracellular trafficking of ODSF micelles, the effects of brefeldin A, a specific inhibitor of the ER/Golgi secretory pathway, were examined.74 The results indicated a significant reduction in Papp of ODSF (51.1%), compared to the control (without inhibitors), after treatment of the cells with brefeldin A; this suggested active involvement of the ER/Golgi pathway in the exocytosis of ODSF (Figure 3A). In addition, involvement of the ER/Golgi apparatus in the intracellular transport of ODSF micelles clarified the roles of both endocytosis and macropinocytosis, and suggested substantial lysosomal escape of ODSF micelles.
P-gp is an ATP-mediated cassette that is highly expressed in various areas of the body, including the pancreas, liver, and villus of the intestinal epithelium. P-gp is reportedly mainly responsible for the generation of multidrug resistance to anticancer drugs by pumping them out of the cells through P-gp-mediated efflux.75 Therefore, to examine the role of P-gp in the transport of ODSF, the Papp of ODSF across the Caco-2 monolayer was examined bidirectionally: from apical to basolateral (A to B, absorptive function) (Papp, AB) and basolateral to apical (B to A, secretive function) (Papp, BA), in the presence of Cys A or all inhibitors other than Cys A; this was compared to the control (without inhibitors). The Papp, BA of ODSF across the Caco-2 monolayer (without inhibitors) was significantly enhanced by 74%, compared to Papp, AB (without inhibitors), which suggested the involvement of P-gp-mediated efflux in the transport of ODSF. In addition, inhibition of P-gp-mediated efflux by Cys A, as well as by a combination of inhibitors except Cys A, resulted in a 41% elevation and 45% reduction in Papp, AB, compared to control (without inhibitors), respectively (Figure 3B). Therefore, a distinctive feature of P-gp substrates is that they exhibit higher transport in the secretory direction (B to A), compared to the opposite direction (A to B).76 These results clearly demonstrated the involvement of P-gp-mediated efflux in the transport of ODSF. Our results were consistent with this notion; in the presence of Cys A, a significant enhancement in the Papp of ODSF was observed during A to B transport, whereas a marked reduction was observed in the opposite direction. Despite the presence of active P-gp-mediated efflux, the elevations in PAMPA and Caco-2 permeability (as described above) of ODSF, compared to free OP, suggested significant compensatory effects of other transport mechanisms (eg, ASBT-mediated facilitated transport, macropinocytosis, endocytosis, transcellular passive diffusion, and paracellular routes). This significantly enhanced permeation may have also been due to the surfactant-induced inhibition of transport proteins.59,77 Thus, intestinal transport of ODSF can be further improved by introduction of excipients with P-gp suppressant activity, such as
In addition to the abovementioned routes, transcellular passive diffusion and paracellular transport could also be major routes involved in enhancement of the permeability and bioavailability of orally administered drugs. To clarify the involvement of these routes in the transport of ODSF, the effects of EGTA on the uptake of ODSF were examined. EGTA is a selective calcium chelator, which is used to bind free extracellular Ca2+ ions and can reversibly open the intercellular TJs of Caco-2 monolayers.78 As shown in Figure 3C, when the Caco-2 monolayer was treated with all inhibitors other than Cys A, the Papp of ODSF decreased by 65.2%, compared to the control (without inhibitors); this finding indicates that passive diffusion and paracellular transport could have been responsible for 34.8% of ODSF permeation across the Caco-2 monolayer (Figure 3C). Moreover, this result was correlated with that of PAMPA as described above, whereby the passive diffusion of OP through the artificial intestinal membrane was markedly improved after ion-pairing complex formation with DLM, followed by adding P188 and Labrasol. Notably, the Papp of the EGTA-treated Caco-2 monolayer (with all inhibitors other than Cys A) was similar to that of the control (without inhibitors and EGTA); however, it was elevated by 47.0%, compared to the EGTA-untreated Caco-2 monolayer (with all inhibitors except Cys A) (Figure 3C). This observation clearly indicated the enhancement of Papp of ODSF after reversible opening of TJs using EGTA. In addition, this result indicates that reversible opening of TJs could increase the intestinal transport of ODSF; incorporation of this component with TJ-opening properties might favor intestinal permeation of OP. The modulation of P188 and Labrasol in ODSF micelles, which have proven TJ-opening properties, demonstrated the possibility of paracellular transport that may have favored the permeation of ODSF.79,80
In vitro Cytotoxicity of ODSF on Caco-2 and MDCK Cells
Cell viability assays were performed to evaluate the cytotoxic effects of a wide range of concentrations of free OP, DLM, OP/DLM, DLM/P188, blank-ODSF, and ODSF on Caco-2 and MDCK cells. As shown in Figure 4, DLM, a combination of DLM and P188, and blank-ODSF (without OP) had no cytotoxic effects on Caco-2 or MDCK cells; the cell viability was greater than 90% for a concentration range equivalent to 1−500 µg/mL of OP. Based on these results, neither DLM nor blank-ODSF showed toxicity at the experimental concentrations and duration, suggesting good tolerance and biocompatibility. In addition, a previous study demonstrated no cytotoxic activity for deoxycholic acid on Caco-2 cells.27 However, OP, OP/DLM, and ODSF demonstrated cytotoxicity at concentrations higher than 100 µg/mL. The cell viabilities of Caco-2 cells incubated with OP, OP/DLM, and ODSF at concentrations of 250 and 500 µg/mL were 82.5±1.11% and 66.6±6.99%, 59.5±5.39% and 47.6±6.65%, and 38.6±3.06% and 34.1±5.13%, respectively (Figure 4A). Similarly, MDCK cells treated with 250 µg/mL of OP, OP/DLM, and ODSF demonstrated reductions in cell viabilities to 74.8±6.82%, 67.8±6.25%, and 55.3±7.39%, respectively, compared to the control (Figure 4B). Increasing the concentration of OP, OP/DLM, and ODSF to 500 µg/mL significantly reduced the percentage cell viabilities to 55.3±7.79%, 47.5±6.79%, and 39.3±4.10%, respectively. The increased cell cytotoxicity of OP/DLM and ODSF in Caco-2 and MDCK cells at higher concentrations (250 and 500 µg/mL) might be attributable to increased cellular uptake, compared to pure OP. These results demonstrated the biocompatibility of the excipients used in the formulation; OP, OP/DLM, and ODSF had no cytotoxic effects on Caco-2 or MDCK cells at experimental concentrations. However, further comparative toxicity studies involving experimental animals are required for the individual excipients, blank formulation (without OP), and ODSF.
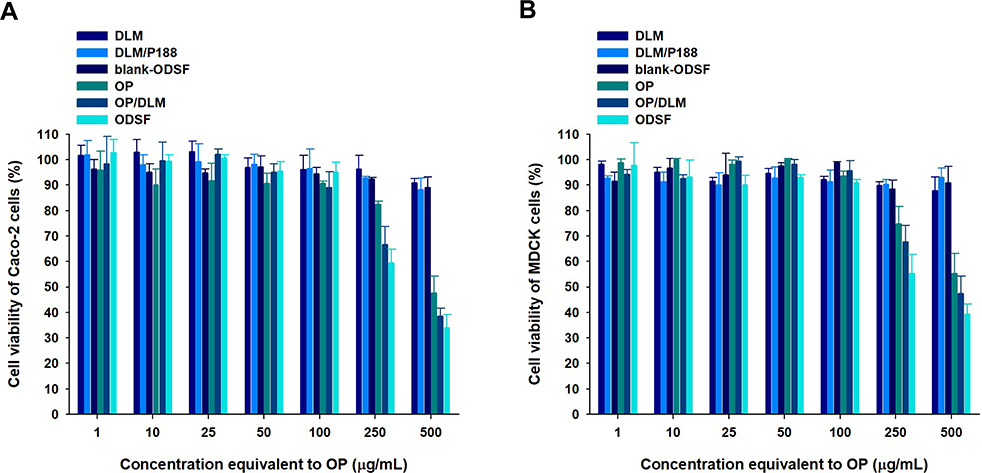 |
Figure 4 In vitro cytotoxic effects of OP, DLM, OP/DLM, DLM/P188, blank-ODSF, and ODSF on (A) Caco-2 and (B) Madin–Darby Canine Kidney (MDCK) cells. |
In vitro Dissolution of ODSF
The cumulative percentage release profiles of OP and ODSF in simulated gastric (pH 1.2) and intestinal (pH 6.8) media are shown in Figure 5. At pH 1.2, the drug release profiles of free OP and ODSF increased steadily over time and exceeded 90% at 60 min. The release of OP from ODSF was similar to that of free OP (>90% at 120 min) in the medium at pH 6.8. In addition, the similarity factors (f2) between OP and ODSF in pH 1.2 and 6.8 media were 50 and 76, respectively, indicating that the two dissolution profiles were similar. These similar release profiles suggested that ionic complex formation with DLM and incorporation into powder did not significantly affect the drug dissolution rate.
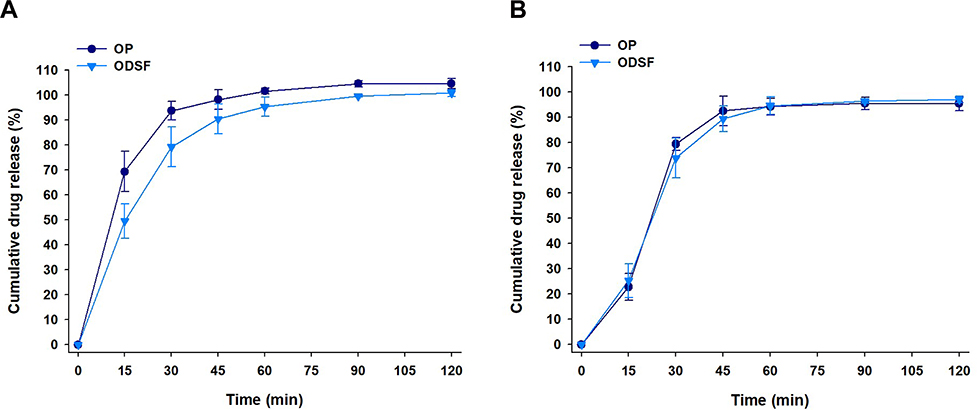 |
Figure 5 In vitro cumulative percentage release of OP and ODSF at (A) pH 1.2 and (B) pH 6.8. |
Stability of ODSF
The stability of a formulation is the extent to which that formulation retains similar properties and characteristics over a specified time, while under specified conditions. Evaluation of ODSF stability was performed at two different temperatures and RHs for 6 months (Table 3). The percentage drug contents at 25±2°C with 65±5% RH and at 40±2°C with 75±5% RH ranged from 97.2±2.21% to 101±2.02% over 6 months, suggesting that there was no change in the drug content in the capsules during storage. In addition, more than 90% drug release was observed at 1 h (in 0.1 N HCl, pH 1.2) under both sets of storage conditions. This result demonstrates that the storage of ODSF for 6 months does not affect the drug release rate. Moreover, after redispersion of the lyophilized formulation in water, the ODSF micelles maintained their former particle size, PDI, and zeta potential during 6 months of storage under both sets of conditions; these findings suggested that the overall storage stability of the formulation was good.
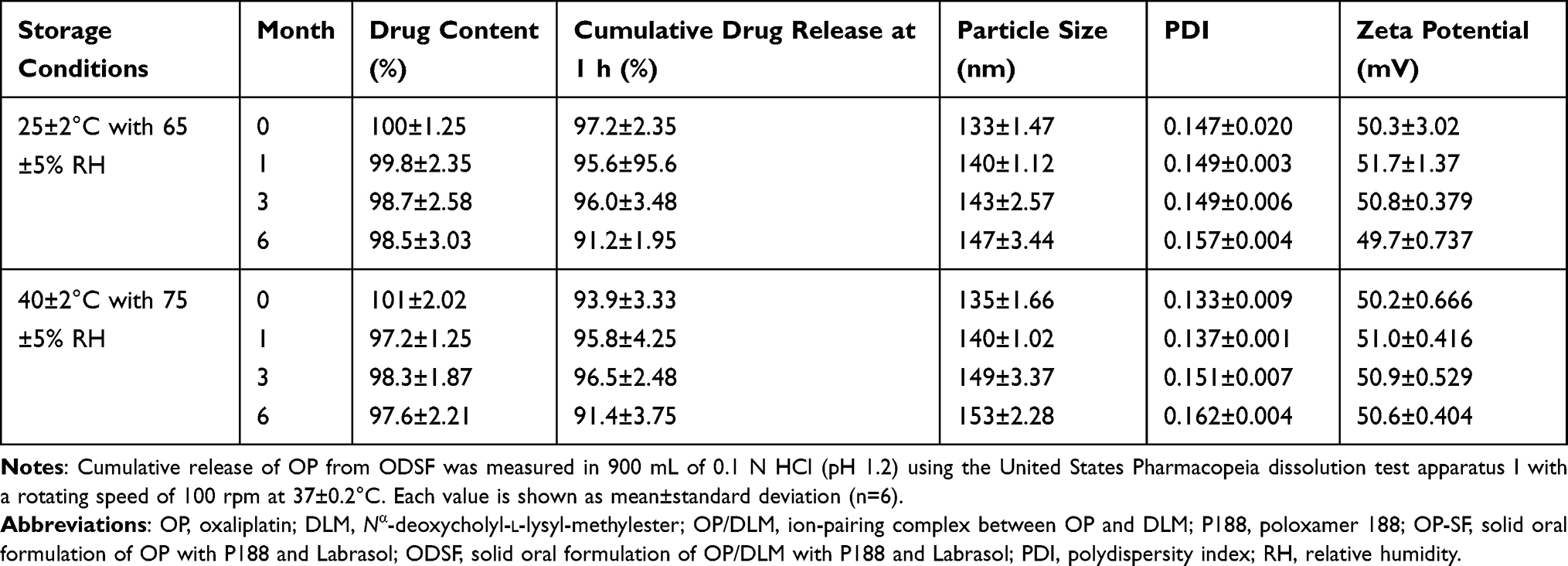 |
Table 3 Stability of ODSF |
In vivo Oral Absorption of ODSF in Rats
The comparative mean plasma concentration-time profiles after a single IV dose of OP (OP-IV, 5 mg/kg OP), oral administration of OP in aqueous solution (OP-S, 10 mg/kg), or various doses of ODSF (equivalent to 2.5, 10, and 20 mg/kg OP) are illustrated in Figure 6; the pharmacokinetic parameters are summarized in Table 4. The plasma concentration of OP increased rapidly after oral administration of OP-S (10 mg/kg) or the various doses of ODSF; it reached its maximum at 0.5 h after drug intake, indicating rapid absorption. After a single oral dose of ODSF (equivalent to 2.5 mg/kg OP), a significant increase was observed in the oral bioavailability of OP to 24.0±0.642%, which was 3.70-fold higher than the oral bioavailability of OP-S (10 mg/kg). In addition, the maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) after oral administration of ODSF (equivalent to 10 mg/kg OP) were 0.136±0.016 μg/mL and 1.62±0.125 μg·h/mL, respectively; these values were 1.92- and 1.68-fold higher, respectively, than those of OP-S (10 mg/kg), demonstrating 68.2% greater oral bioavailability. This increase in oral bioavailability may have been due to the increased solubility and lipophilicity of OP after incorporation into the nanoformulation. In addition, our findings regarding the intestinal membrane transport mechanism demonstrated significant reductions of Papp after inhibition of ASBT and OSTα/β-mediated transport by Act D and CFZ, respectively; these findings indicate an active role of the bile acid transporter, followed by OSTα/β-mediated transport, in increasing the oral bioavailability of OP via elevated uptake of OP/DLM. The DC in DLM may also enhance intestinal membrane flexibility via phosphorylation of the epidermal growth factor receptor and rearrangement of occludin at the TJ level.57,80 Transport of ODSF micelles via the caveola/lipid raft-mediated endocytosis pathway may explain their increased oral absorption, which was confirmed by the significant reduction in Papp after treatment of the Caco-2 cell monolayer with genistein. Notably, the presence of nonionic surfactants (eg, Labrasol and P188) in the formulation can reversibly modify intestinal membrane fluidity by dephosphorylation and rearrangement of occludin.79,81 Thus, the increased oral absorption of OP is supported by the entry of ODSF into enterocytes through macropinosome formation, paracellular transport, and passive transcellular diffusion (verified by intestinal transport mechanistic analyses).
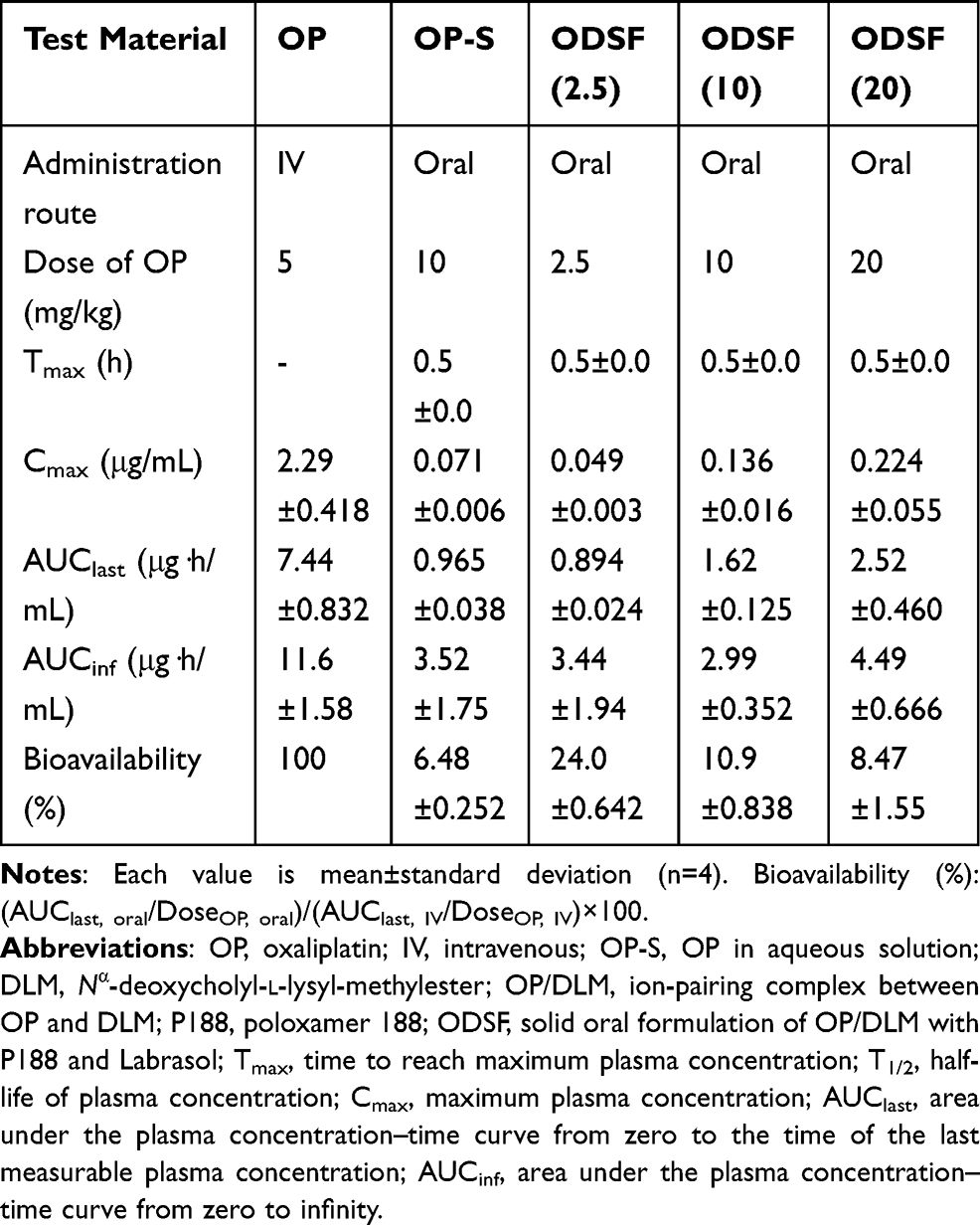 |
Table 4 Pharmacokinetic Parameters of OP in Rats After IV Injection of OP and Oral Administration of OP-S or ODSF with Various Levels of OP |
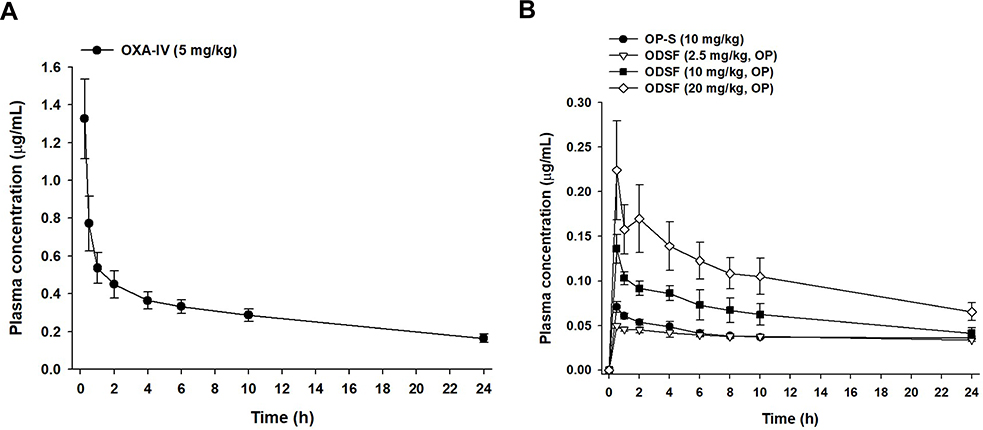 |
Figure 6 Venous plasma concentration–time profiles of OP after a single (A) IV dose (5 mg/kg) and (B) oral administration of OP in aqueous solution (OP-S, 10 mg/kg), ODSF equivalent to 2.5 mg/kg OP [ODSF (2.5)], ODSF equivalent to 10 mg/kg OP [ODSF (10)], and ODSF equivalent to 20 mg/kg OP [ODSF (20)] to rats. |
However, an increase in the ODSF dose from 10 to 20 mg/kg OP reduced the oral bioavailability from 10.9±0.838% to 8.47±1.55%. Furthermore, a single oral dose of ODSF (equivalent to 10 mg/kg OP) was associated with 2.78- and 1.81-fold increases in Cmax and AUC, respectively, compared to ODSF (equivalent to 2.5 mg/kg OP). Similarly, a 2-fold increase in the oral dose of ODSF (to 20 mg/kg OP) yielded Cmax and AUC values of 0.224±0.055 μg/mL and 2.52±0.460 μg·h/mL, respectively, which were 1.65- and 1.56-fold higher, respectively, than the corresponding values of ODSF (equivalent to 10 mg/kg OP). Therefore, the Cmax and AUC of ODSF increased in a dose-dependent manner between doses of 2.5 mg/kg OP and 20 mg/kg OP. This nonlinear relationship between dose and oral absorption of ODSF may reflect limited gut solubility due to the hydrophobic nature of the formulation, changes in intestinal membrane permeability, or saturation of intestinal transporters.82–84 However, a Phase I study to assess the dose proportionality of IV-administered OP revealed dose-dependent increases in the mean Cmax and AUC0–24, up to 180 mg/m2.85 Therefore, further studies are necessary to compare and correlate rat pharmacokinetic data for the dose dependency of oral ODSF with data for other animal species.
Pharmacokinetics of Oral ODSF in Monkeys
Given the enhanced oral absorption of ODSF in rats, we examined the mean plasma concentration–time profiles of orally administered OP (10 mg/kg) or ODSF (equivalent to 10 mg/kg OP), and IV-administered OP (2 mg/kg), in cynomolgus monkeys (Figure 7); the mean pharmacokinetic parameters are listed in Table 5. After IV administration of OP (2 mg/kg), the plasma levels of Pt decreased with time with an elimination T1/2 of 45.9±5.46 h. After oral administration of OP-S (10 mg/kg), OP was absorbed with a Cmax and Tmax of 0.043±0.006 µg/mL and 2.00±0.00 h, respectively. Oral ODSF significantly enhanced the intestinal absorption of OP with observed Cmax and AUClast values of 0.192±0.038 µg/mL and 3.97±0.659 μg·h/mL, respectively, thus 4.47- and 3.78-fold greater than those of OP-S, respectively. The oral bioavailability of ODSF was 377% that of OP-S. This significant increase likely reflects the fact that nanoformulation enhanced drug solubility and lipophilicity, as discussed above. The mean residence time (MRT) of ODSF was 18.4±0.049 h, slightly less than those of OP-S (19.7±0.124 h) and OP-IV (19.6±0.254 h). Therefore, ODSF was rapidly absorbed in both rats and monkeys.
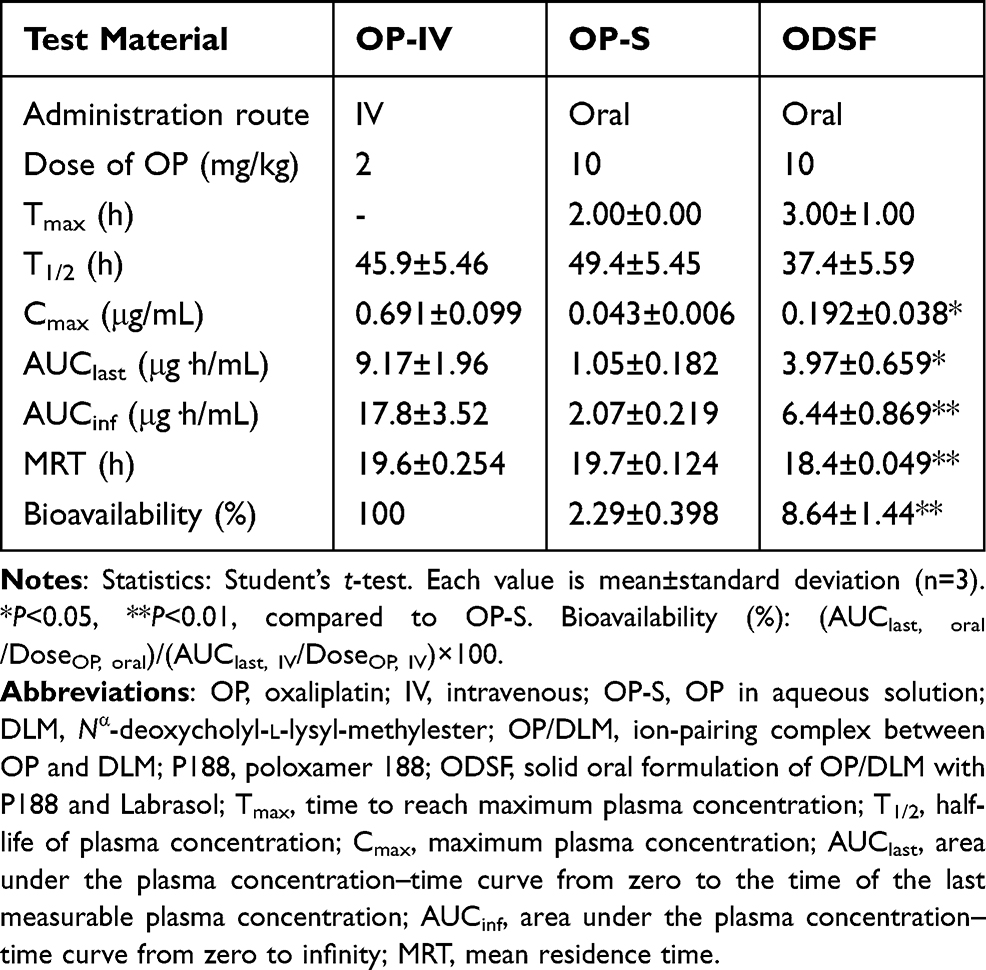 |
Table 5 Pharmacokinetic Parameters of OP in Monkeys After IV Injection of OP and Oral Administration of OP-S or ODSF |
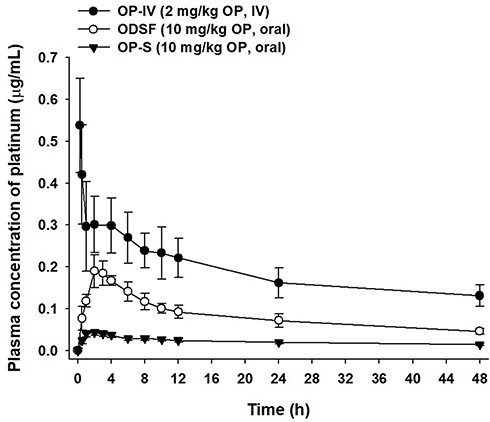 |
Figure 7 Venous plasma concentration–time profiles of OP after a single IV dose of OP (OP-IV, 2 mg/kg) and oral administration of OP in aqueous solution (OP-S, 10 mg/kg) or ODSF (equivalent to 10 mg/kg OP) to monkeys. |
In vivo Tumor Growth Inhibition of Orally Administered ODSF in a Murine Colon Cancer Cell Model
Next, the tumor growth inhibitory efficacy of orally administered ODSF was investigated in a murine colon cancer model (CT26 cells). As shown in Figure 8A, tumor growth in the control group occurred continuously and rapidly, reaching a mean of 3261±1651 mm3 by day 21. However, after once-daily oral administration of OP-S (10) and ODSF (2.5), respective tumor growth retardation rates of 34.8% and 32.4% were observed, compared to the control group; these similar rates may have been due to pharmacokinetic similarities. Administration of OP-IV (3.3) to tumor-bearing mice resulted in 40.0% inhibition of tumor growth rate, compared to the control group. Increasing the dose to OP-IV (10) resulted in significant enhancements of tumor suppression by 70.2% and 53.3%, compared to mice in the control and OP-S (10) groups, respectively. After oral administration for 21 days, ODSF (10) and ODSF (20) also significantly suppressed the tumor volume, by 76.9% and 87.1%, respectively, compared to the control group, and by 25.0% and 58.2% compared to OP-IV (10), respectively. Although the oral bioavailability of ODSF (10) in rats was 10.9%, its anticancer efficacy was higher than the efficacy of the maximum tolerated dose of OP-IV (10). This peculiar behavior may have been due to the oral metronomic scheduling of ODSF, which can activate innate and adaptive immune responses, reduce immune suppressive populations of regulatory T cells, and activate innate immune cells (eg, natural killer cells, dendritic cells, and macrophages).24,86,87 Metronomic chemotherapy is the protracted and regular administration of relatively low and minimally toxic doses of chemotherapy, with no drug-free breaks.88 Thus, a daily low dose of ODSF was implemented to maintain a low but sustained concentration of OP in the tumor cells and microenvironment, to minimize adverse drug reactions induced by the maximum tolerated dose, to target both endothelial and tumor cells at the proliferation stage, and to prolong therapeutic responses (rather than causing only short-term tumor regression). Moreover, metronomic chemotherapy prevents regrowth of resting tumor cells, thereby minimizing the possibility of chemoresistance.89,90 However, the mechanism of the anti-angiogenic property of ODSF requires further study. Although tumor growth in the ODSF (20) group was maximally inhibited, compared to the OP-IV (10) and control groups, the antitumor efficacy of that treatment was similar to treatment with ODSF (10); this finding corresponds well with the pharmacokinetics indicating the dose dependency of Cmax and AUC. Indeed, the maintenance of low, prolonged, and pharmacologically active plasma concentrations of OP during metronomic therapy with ODSF (10) may have direct and indirect effects on tumor cells and their microenvironment by inhibiting tumor angiogenesis through induction of apoptosis within activated endothelial cells and inhibition of endothelial cell migration.1,91–93 Moreover, recent studies have suggested an immunomodulatory effect of OP through T cell-mediated antitumor immunity and enhanced presentation of damage-associated molecular patterns in colorectal tumor cell lines, along with inherent cytotoxicity in tumor cells.22,94 Notably, tumor growth in mice treated with ODSF (10) and ODSF (20) began to decrease on day 15, whereas all other treatment groups and the control group exhibited continual increases in tumor volume. After 21 days of administration, the isolated tumor masses in mice treated with ODSF (20) were significantly reduced, by 76.9%, compared to the masses in the controls. Moreover, oral and IV administration of ODSF (10) and OP-IV (10) resulted in reductions in tumor mass of 50.1% and 71.7%, respectively, compared to the controls (Figure 8B and C). Treatment of mice with ODSF (20) resulted in body weight reduction at 6 days, compared to the control group and other treatment groups; this may have been due to drug-induced toxicity (Figure 8D). However, the reduced body weight was maintained until day 21. Thus, based on the dose of OP, further toxicity studies should be performed after repeated oral administration of ODSF for intervals longer than 3 weeks.
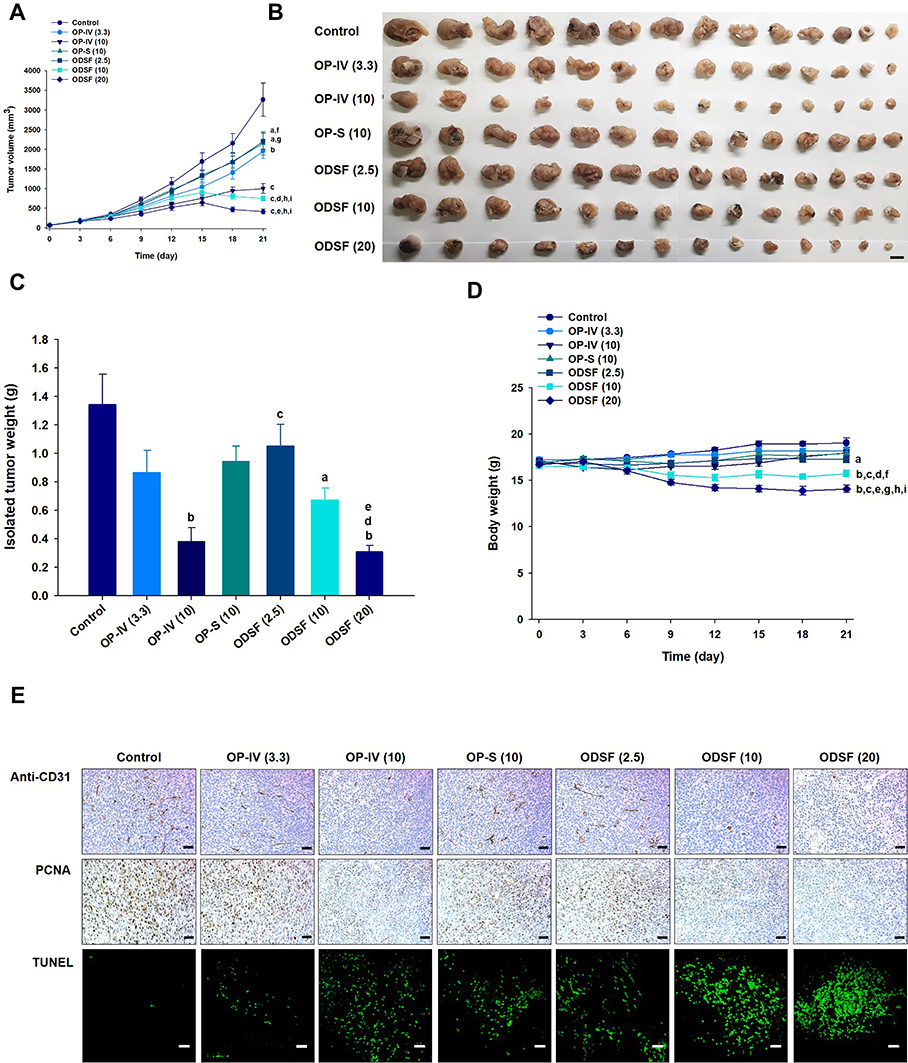 |
Figure 8 In vivo analyses of the inhibitory effects of the following on tumor growth in CT26 tumor-bearing mice: biweekly intravenous administration of 3.3 mg/kg OP [OP-IV (3.3)] and 10 mg/kg OP [OP-IV (10)]; once-daily oral administration of 10 mg/kg OP in aqueous solution (OP-S); once-daily oral administration of ODSF as 2.5 mg/kg OP [ODSF (2.5)]; once-daily oral administration of ODSF as 10 mg/kg OP [ODSF (10)]; and once-daily oral administration of ODSF as 20 mg/kg OP [ODSF (20)], for 21 days. (A) Tumor volumes in mice [aP< 0.05, bP<0.01, cP<0.001 compared to the control. dP<0.01, eP<0.001 compared to OP-IV (3.3). fP<0.05, gP<0.01 compared to OP-IV (10). hP<0.001 compared to OP-S (10). iP<0.001 compared to ODSF (2.5)]. (B) Photographs of tumors isolated from each group on day 21. Scale bar: 10 mm. (C) Tumor weights in CT26 tumor-bearing mice on day 21 [aP<0.05, bP<0.001 compared to the control. cP<0.05 compared to OP-IV (10). dP<0.05 compared to OP-S (10). eP<0.05 compared to ODSF (2.5)]. (D) Changes in mouse body weights during treatment [aP<0.05, bP<0.001 compared to the control. cP<0.001 compared to OP-IV (3.3). dP<0.01, eP<0.001 compared to OP-IV (10). fP<0.01, gP<0.001 compared to OP-S (10). hP<0.001 compared to ODSF (2.5). iP<0.05 compared to ODSF (10)]. (E) Representative cross-sectional images of isolated tumor tissues obtained 21 days after various treatments, stained as follows: With anti-CD31 antibody, indicating microvessels (brown); anti-proliferating cell nuclear antigen (PCNA) antibody, indicating proliferating cells (brown); and via fluorescent terminal deoxynucleotidyl transferase-mediated dUPT nick-end labeling (TUNEL), indicating apoptosis (green fluorescence). Scale bars: 50 μm for anti-CD31 antibody and anti-PCNA antibody staining; 20 μm for TUNEL staining. |
To investigate the biological effects of oral administration of ODSF, isolated tumors were subjected to immunohistochemical analyses for cell proliferation, apoptosis, and vessel density. Analyses of CD31 expression revealed the highest density of CD31-positive microvessels in the control group; however, the OP-IV (3.3) and OP-S (10) groups also exhibited greater numbers of microvessels, compared to other treatment groups (Figure 8E). In addition, labeling with anti-CD31 antibody confirmed that the ODSF (10) and ODSF (20) groups exhibited significant anti-angiogenic activity, compared to the controls. The numbers of PCNA-positive cells, an indicator of the rate of proliferation, were significantly higher in the control and OP-S treatment groups than in the OP-IV (10), ODSF (10), and ODSF (20) groups (Figure 8E). Moreover, the ODSF (10), ODSF (20), and OP-IV (10) treatment groups exhibited significant increases in the number of TUNEL-positive cells, indicating increased apoptosis after treatment, compared to the controls (Figure 8E). Thus, induction of apoptosis and inhibition of proliferation may be due to the anti-angiogenic mechanism of action of metronomic chemotherapy with OP, which upregulates the expression of endogenous angiogenesis inhibitors (eg, thrombospondin-1).95
In vivo Analyses of the Inhibitory Effects of Orally Administered ODSF on Tumor Growth in a Human Colon Cancer Cell Model
The antitumor efficacy of orally administered ODSF on human colon cancer cells (HCT116) was also assessed; findings regarding tumor growth inhibition were similar to those observed in the murine model. Treatment of tumor-bearing mice with OP-IV (3.3) resulted in a 33.5% reduction in tumor volume, compared to controls. Moreover, ODSF (10) resulted in 2.64- and 1.90-fold reductions in tumor volume, compared to the control and OP-IV (10) groups, respectively (Figure 9A). After oral administration of ODSF (20) for 21 days, the tumor growth rates and weights were significantly reduced with maximum suppression of 75.4% and 78.1%, respectively, compared to the control. Moreover, treatment of tumor-bearing mice with the same dose of OP resulted in significant reductions of the isolated tumor weights in the oral ODSF (10) group by 49.5% and 34.6%, respectively, compared to the OP-S (10) and OP-IV (10) groups (Figure 9B and C). None of the treatment groups or controls, except ODSF (20), revealed an effect on body weight of tumor-bearing mice. However, body weight was maintained after day 15 in the ODSF (20) group (Figure 9D). Thus, once-daily oral administration of ODSF (10) was well tolerated with maintenance of microenvironmental OP level sufficient to suppress tumor growth.
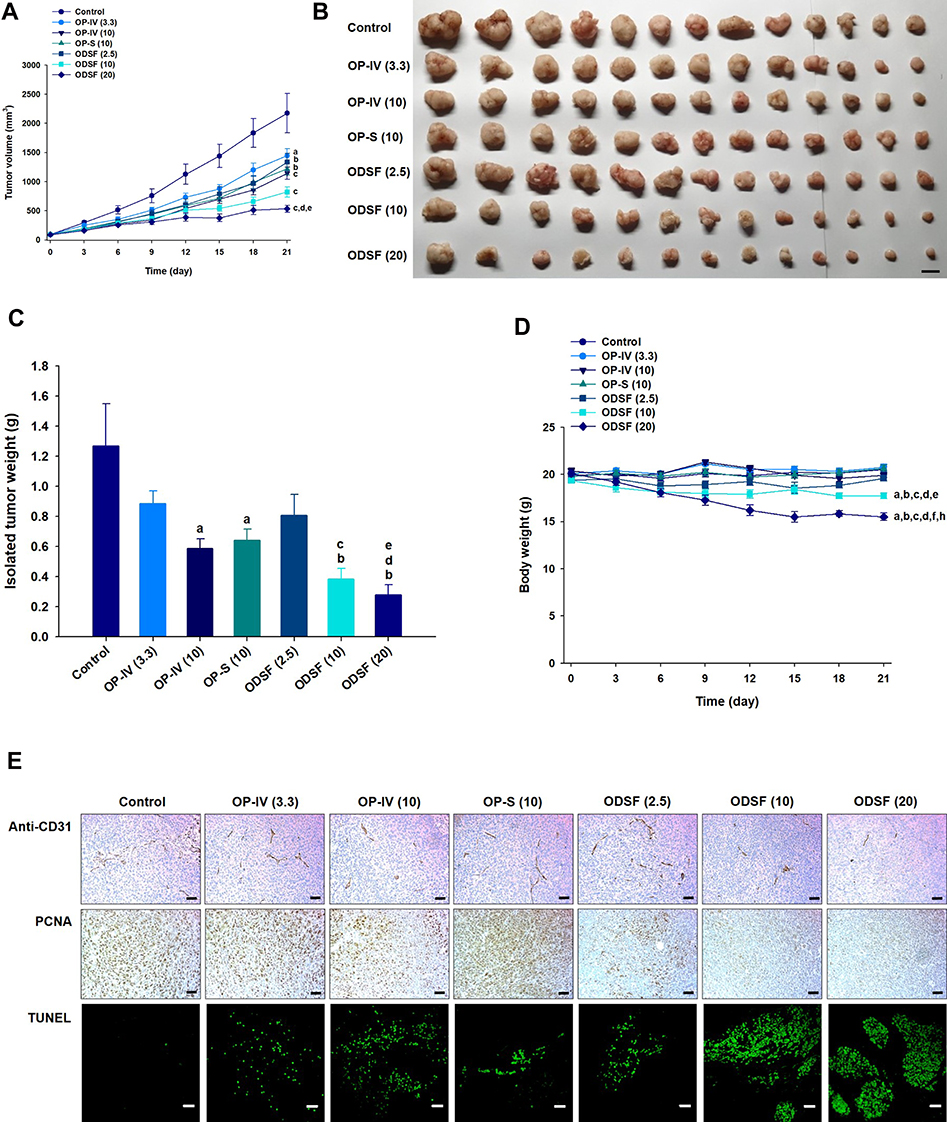 |
Figure 9 Antitumor effects of ODSF at various doses in HCT116 tumor-bearing mice after biweekly intravenous administration of 3.3 mg/kg OP [OP-IV (3.3)] and 10 mg/kg OP [OP-IV (10)]; once-daily oral administration of 10 mg/kg OP in aqueous solution (OP-S); once-daily oral administration of ODSF as 2.5 mg/kg OP [ODSF (2.5)]; once-daily oral administration of ODSF as 10 mg/kg OP [ODSF (10)]; and once-daily oral administration of ODSF as 20 mg/kg OP [ODSF (20)], for 21 days. (A) Tumor volumes in different groups [aP<0.05, bP<0.01, cP<0.001 compared to the control. dP<0.01 compared to OP-IV (3.3). eP<0.05 compared to ODSF (2.5)]. (B) Photographs of tumors isolated from each group on day 21. Scale bar: 10 mm. (C) Isolated tumor weights in HCT116 tumor-bearing mice on day 21 [aP<0.01, bP<0.001 compared to the control. cP<0.05, dP<0.01 compared to OP-IV (3.3). eP<0.05 compared to ODSF (2.5)]. (D) Changes in mouse body weight during treatment [aP<0.001 compared to the control. bP<0.001 compared to OP-IV (3.3). cP<0.001 compared to OP-IV (10). dP<0.001 compared to OP-S (10). eP<0.01, fP<0.001 compared to ODSF (2.5). hP<0.001 compared to ODSF (10)]. (E) Representative cross-sectional images of isolated tumor tissues obtained 21 days after various treatments, stained as follows: With anti-CD31 antibody, indicating microvessels (brown); with anti-proliferating cell nuclear antigen (PCNA) antibody, indicating proliferating cells (brown); and via fluorescent terminal deoxynucleotidyl transferase-mediated dUPT nick end labeling (TUNEL), indicating apoptosis (green fluorescence). Scale bars: 50 μm for anti-CD31 antibody and anti-PCNA antibody staining, and 20 μm for TUNEL staining. |
Similarly, after staining of histological sections of HCT116 tumor tissues with anti-CD31 antibody and anti-PCNA antibody, mice treated with OP-IV (10), ODSF (10), and ODSF (20) exhibited marked reductions in the numbers of CD31-positive microvessels and PCNA-positive proliferating cells, compared to the control and OP-S (10) groups (Figure 9E). Furthermore, TUNEL assay evaluation revealed significantly greater numbers of TUNEL-positive apoptotic cells in the OP-IV (10), ODSF (10), and ODSF (20) groups, compared to the control and OP-S (10) groups (Figure 9E). Therefore, considering the results of tumor efficacy and toxicity based on the administration dose, ODSF (10) was selected as the optimum formulation.
Taken together, the findings presented here revealed that formation of an ion-pairing complex of OP with DLM and further incorporation into a solid formulation enhanced the oral absorption and bioavailability of OP. In addition, daily oral administration of ODSF as metronomic chemotherapy may maintain a sufficient OP level in both tumor cells and their microenvironment, resulting in significant inhibition of tumor growth in both human and mouse tumor models. Therefore, delivery of ODSF as an oral metronomic chemotherapy may be a preferred alternative for use in patients with colorectal cancer.
Conclusion
We designed a solid oral formulation of OP by ion-pairing complex formation with DLM at a molar ratio of 1:2, along with P188 and Labrasol as dispersants (ODSF). The effective and apparent permeability of OP from ODSF was significantly improved by 24.0- and 11.1-fold, respectively, compared to free OP. Analyses of the intestinal transport mechanism indicated that inhibition of ASBT in the Caco-2 cell monolayer resulted in a significant reduction in its Papp, suggesting that ASBT-facilitated uptake represents the predominant pathway underlying enhanced oral absorption of OP from ODSF. In addition, caveola/lipid-raft mediated endocytosis, macropinocytosis, passive diffusion, and paracellular transport were actively involved in the transport of ODSF. The oral bioavailabilities of ODSF (equivalent to 10 mg/kg OP) in rats and monkeys were enhanced 1.68- and 3.77-fold compared to those of free OP. Moreover, enhanced oral absorption of the optimum formulation and maintenance of sustained plasma drug concentration during metronomic therapy with ODSF (10) resulted in maximal reductions in tumor volume by 25.0% and 34.6%, respectively, compared to the maximum tolerated dose of OP-IV (10) in CT26 and HCT116 tumor-bearing models. These observations suggest therapeutic potential for oral metronomic delivery of ODSF as a preferred alternative for the treatment of colorectal cancer.
Funding
This research was supported by the “Rediscovery of the Past R&D Result” program of the Ministry of Trade, Industry and Energy (MOTIE) and the Korea Institute for Advancement of Technology (KIAT) (grant no. P0010215), and the Basic Research Program through the National Research Foundation of Korea (NRF), funded by the Korean Government Ministry of Science and ICT (MSIT; Grant No. NRF-2020R1F1A1069889). We greatly appreciated it using the Convergence Research Laboratory (established by the MNU Innovation Support Project in 2020) to conduct this research.
Disclosure
No author has any possible conflict of interest for this work.
References
1. Pasquier E, Kavallaris M, Andre N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol. 2010;7(8):455–465. doi:10.1038/nrclinonc.2010.82
2. Aston WJ, Hope DE, Nowak AK, et al. A systematic investigation of the maximum tolerated dose of cytotoxic chemotherapy with and without supportive care in mice. BMC Cancer. 2017;17(1):684–693. doi:10.1186/s12885-017-3677-7
3. Andre N, Carre M, Pasquier E. Metronomics: towards personalized chemotherapy? Nat Rev Clin Oncol. 2014;11(7):413–431.
4. Opzoomer JW, Sosnowska D, Anstee JE, et al. Cytotoxic chemotherapy as an immune stimulus: a molecular perspective on turning up the immunological heat on cancer. Front Immunol. 2019;10(1654):1–9.
5. Thanki K, Gangwal RP, Sangamwar AT, et al. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. doi:10.1016/j.jconrel.2013.04.020
6. Mulkerin DL, Bergsbaken JJ, Fischer JA, et al. Multidisciplinary optimization of oral chemotherapy delivery at the university of wisconsin carbone cancer center. J Oncol Pract. 2016;12(10):e912–e923. doi:10.1200/JOP.2016.013748
7. Aisner J. Overview of the changing paradigm in cancer treatment: oral chemotherapy. Am J Health Syst Pharm. 2007;64(9 Supplement 5):S4–S7. doi:10.2146/ajhp070035
8. Chidambaram M, Manavalan R, Kathiresan K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Pharm Sci. 2011;14(1):67–77. doi:10.18433/J30C7D
9. Baek J-S, So J-W, Shin S-C, et al. Solid lipid nanoparticles of paclitaxel strengthened by hydroxypropyl-β-cyclodextrin as an oral delivery system. Int J Mol Med. 2012;30(4):953–959. doi:10.3892/ijmm.2012.1086
10. Zhang H, Wu F, Li Y, et al. Chitosan-based nanoparticles for improved anticancer efficacy and bioavailability of mifepristone. Beilstein J Nanotechnol. 2016;201:71861–71870.
11. Baidya D, Kushwaha J, Mahadik K, et al. Chrysin-loaded folate conjugated PF127-F68 mixed micelles with enhanced oral bioavailability and anticancer activity against human breast cancer cells. Drug Dev Ind Pharm. 2019;45(5):852–860. doi:10.1080/03639045.2019.1576726
12. Erdoğar N, Nemutlu E, İskit AB, et al. Improved oral bioavailability of anticancer drug tamoxifen through complexation with water soluble cyclodextrins: in vitro and in vivo evaluation. J Incl Phenom Macrocycl Chem. 2020;96(1):81–91.
13. Chen B, Wang X, Zhang Y, et al. Improved solubility, dissolution rate, and oral bioavailability of main biflavonoids from selaginella doederleinii extract by amorphous solid dispersion. Drug Deliv. 2020;27(1):309–322. doi:10.1080/10717544.2020.1716876
14. Arango D, Wilson AJ, Shi Q, et al. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br J Cancer. 2004;91(11):1931–1946. doi:10.1038/sj.bjc.6602215
15. Ficarra R, Calabrò ML, Cutroneo P, et al. Validation of a LC method for the analysis of oxaliplatin in a pharmaceutical formulation using an experimental design. J Pharm Biomed Anal. 2002;29(6):1097–1103. doi:10.1016/S0731-7085(02)00151-6
16. Oberoi HS, Nukolova NV, Kabanov AV, et al. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev. 2013;65(13–14):1667–1685. doi:10.1016/j.addr.2013.09.014
17. Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level. Oncol Rep. 2003;10(6):1663–1682.
18. Ibrahim A, Hirschfeld S, Cohen MH, et al. FDA drug approval summaries: oxaliplatin. Oncologist. 2004;9(1):8–12. doi:10.1634/theoncologist.9-1-8
19. Zeng C, Yu F, Yang Y, et al. Preparation and evaluation of oxaliplatin thermosensitive liposomes with rapid release and high stability. PLoS One. 2016;11(7):e0158517–e0158535. doi:10.1371/journal.pone.0158517
20. Kim YH, Lee SJ, Lee SH, et al. Preclinical efficacy and safety assessment of nano-oxaliplatin oral formulation prepared by novel fat employing supercritical nano system, the FESNS®. Pharm Dev Technol. 2012;17(6):677–686. doi:10.3109/10837450.2011.565349
21. Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482–491. doi:10.1038/onc.2009.356
22. Stojanovska V, Prakash M, McQuade R, et al. Oxaliplatin treatment alters systemic immune responses. Biochem Res Int. 2019;20194650695.
23. Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–970. doi:10.1126/science.1240527
24. Galluzzi L, Buque A, Kepp O, et al. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi:10.1016/j.ccell.2015.10.012
25. Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol. 2011;18(1):1–8. doi:10.3747/co.v18i1.708
26. Pangeni R, Choi SW, Jeon OC, et al. Multiple nanoemulsion system for an oral combinational delivery of oxaliplatin and 5-fluorouracil: preparation and in vivo evaluation. Int J Nanomed. 2016;201:116379–116399.
27. Xiao H, Li W, Qi R, et al. Co-delivery of daunomycin and oxaliplatin by biodegradable polymers for safer and more efficacious combination therapy. J Control Release. 2012;163(3):304–314. doi:10.1016/j.jconrel.2012.06.004
28. Urbanska AM, Karagiannis ED, Guajardo G, et al. Therapeutic effect of orally administered microencapsulated oxaliplatin for colorectal cancer. Biomaterials. 2012;33(18):4752–4761. doi:10.1016/j.biomaterials.2012.03.023
29. Pavlovic N, Golocorbin-Kon S, Ethanic M, et al. Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Front Pharmacol. 2018;9(1):1283–1306.
30. Moghimipour E, Ameri A, Handali S. Absorption-enhancing effects of bile salts. Molecules. 2015;20(8):14451–14473. doi:10.3390/molecules200814451
31. Kevresan S, Kuhajda K, Kandrac J, et al. Biosynthesis of bile acids in mammalian liver. Eur J Drug Metab Pharmacokinet. 2006;31(3):145–156. doi:10.1007/BF03190711
32. Sarenac TM, Mikov M. Bile acid synthesis: from nature to the chemical modification and synthesis and their applications as drugs and nutrients. Front Pharmacol. 2018;9939.
33. Jeon OC, Byun Y, Park JW. Preparation of oxaliplatin-deoxycholic acid derivative nanocomplexes and in vivo evaluation of their oral absorption and tumor growth suppression. J Nanosci Nanotechnol. 2016;16(2):2061–2064. doi:10.1166/jnn.2016.11914
34. Park JW, Byun Y. Ionic complex of risedronate with positively charged deoxycholic acid derivative: evaluation of physicochemical properties and enhancement of intestinal absorption in rats. Arch Pharm Res. 2014;37(12):1560–1569. doi:10.1007/s12272-013-0297-x
35. Jeon OC, Hwang SR, Al-Hilal TA, et al. Oral delivery of ionic complex of ceftriaxone with bile acid derivative in non-human primates. Pharm Res. 2013;30(4):959–967. doi:10.1007/s11095-012-0932-0
36. Sun S, Liang N, Kawashima Y, et al. Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin. Int J Nanomed. 2011;201:63049–63056.
37. Miller JM, Dahan A, Gupta D, et al. Enabling the intestinal absorption of highly polar antiviral agents: ion-pair facilitated membrane permeation of zanamivir heptyl ester and guanidino oseltamivir. Mol Pharm. 2010;7(4):1223–1234. doi:10.1021/mp100050d
38. Maharjan R, Pangeni R, Jha SK, et al. Anti-angiogenic effect of orally available pemetrexed for metronomic chemotherapy. Pharmaceutics. 2019;11(7):332–354. doi:10.3390/pharmaceutics11070332
39. Pangeni R, Jha SK, Maharjan R, et al. Intestinal transport mechanism and in vivo anticancer efficacy of a solid oral formulation incorporating an ion-pairing complex of pemetrexed with deoxycholic acid derivative. Int J Nanomed. 2019;14(1):6339–6356. doi:10.2147/IJN.S209722
40. He B, Lin P, Jia Z, et al. The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells. Biomaterials. 2013;34(25):6082–6098. doi:10.1016/j.biomaterials.2013.04.053
41. Mu H, Wang Y, Chu Y, et al. Multivesicular liposomes for sustained release of bevacizumab in treating laser-induced choroidal neovascularization. Drug Deliv. 2018;25(1):1372–1383. doi:10.1080/10717544.2018.1474967
42. Baghel S, Cathcart H, O’Reilly NJ. Investigation into the solid-state properties and dissolution profile of spray-dried ternary amorphous solid dispersions: a rational step toward the design and development of a multicomponent amorphous system. Mol Pharm. 2018;15(9):3796–3812. doi:10.1021/acs.molpharmaceut.8b00306
43. Ilevbare GA, Liu H, Edgar KJ, et al. Understanding polymer properties important for crystal growth inhibition—impact of chemically diverse polymers on solution crystal growth of ritonavir. Cryst Growth Des. 2012;12(6):3133–3143. doi:10.1021/cg300325p
44. Beig A, Miller JM, Lindley D, et al. Head-to-head comparison of different solubility-enabling formulations of etoposide and their consequent solubility-permeability interplay. J Pharm Sci. 2015;104(9):2941–2947. doi:10.1002/jps.24496
45. Li N, Mosquera-Giraldo LI, Borca CH, et al. A comparison of the crystallization inhibition properties of bile salts. Cryst Growth Des. 2016;16(12):7286–7300. doi:10.1021/acs.cgd.6b01470
46. Sharif Makhmal Zadeh B, Esfahani G, Salimi A. Permeability of ciprofloxacin-loaded polymeric micelles including ginsenoside as P-glycoprotein inhibitor through a Caco-2 cells monolayer as an intestinal absorption model. Molecules. 2018;23(8):1–15. doi:10.3390/molecules23081904
47. Funasaki N, Fukuba M, Hattori T, et al. Micelle formation of bile salts and zwitterionic derivative as studied by two-dimensional NMR spectroscopy. Chem Phys Lipids. 2006;142(1–2):43–57. doi:10.1016/j.chemphyslip.2006.02.025
48. Wang L, Peng M, Zhu Y, et al. Preparation of pluronic/bile salt/phospholipid mixed micelles as drug solubility enhancer and study the effect of the PPO block size on the solubility of pyrene. Iran J Pharm Res. 2014;13(4):1157–1163.
49. Lu GW, Gao P. CHAPTER 3 - emulsions and microemulsions for topical and transdermal drug delivery. In: Kulkarni VS, editor. Handbook of Non-Invasive Drug Delivery Systems. Boston: William Andrew Publishing; 2010:59–94.
50. Jang SB, Kim D, Kim SY, et al. Impact of micellar vehicles on in situ intestinal absorption properties of beta-lapachone in rats. Korean J Physiol Pharmacol. 2013;17(1):9–13. doi:10.4196/kjpp.2013.17.1.9
51. Cirin DM, Posa MM, Krstonosic VS. Interactions between selected bile salts and triton X-100 or sodium lauryl ether sulfate. Chem Cent J. 2011;5(1):589–597. doi:10.1186/1752-153X-5-89
52. George A, Vora S, Dogra A, et al. Mixed micelles of cationic surfactants and bile acid salts in aqueous media. J Surfactants Deterg. 1998;1(4):507–514. doi:10.1007/s11743-998-0050-2
53. Salimi A, Makhmal Zadeh B, Kazemi M. Preparation and optimization of polymeric micelles as an oral drug delivery system for deferoxamine mesylate: in vitro and ex vivo studies. Res Pharm Sci. 2019;14(4):293–307. doi:10.4103/1735-5362.263554
54. Koga K, Kusawake Y, Ito Y, et al. Enhancing mechanism of labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. Eur J Pharm Biopharm. 2006;64(1):82–91.
55. Dawson PA. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol. 2011;(201):169–203.
56. Nurunnabi M, Khatun Z, Revuri V, et al. Design and strategies for bile acid mediated therapy and imaging. RSC Adv. 2016;6(78):73986–74002.
57. Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G906–G913. doi:10.1152/ajpgi.00043.2007
58. Sahay G, Batrakova EV, Kabanov AV. Different internalization pathways of polymeric micelles and unimers and their effects on vesicular transport. Bioconjug Chem. 2008;19(10):2023–2040. doi:10.1021/bc8002315
59. Hu Z, Tawa R, Konishi T, et al. A novel emulsifier, labrasol, enhances gastrointestinal absorption of gentamicin. Life Sci. 2001;69(24):2899–2910. doi:10.1016/S0024-3205(01)01375-3
60. Sha X, Yan G, Wu Y, et al. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–486. doi:10.1016/j.ejps.2005.01.001
61. Sticova E, Jirsa M, Pawłowska J. New insights in genetic cholestasis: from molecular mechanisms to clinical implications. Can J Gastroenterol Hepatol. 2018;2018:1–12. doi:10.1155/2018/2313675
62. Kanda T, Foucand L, Nakamura Y, et al. Regulation of expression of human intestinal bile acid-binding protein in Caco-2 cells. Biochem J. 1998;330(Pt 1):261–265. doi:10.1042/bj3300261
63. Van de Wiel SMW, De Waart DR, Oude Elferink RPJ, et al. Intestinal farnesoid X receptor activation by pharmacologic inhibition of the organic solute transporter alpha-beta. Cell Mol Gastroenterol Hepatol. 2018;5(3):223–237.
64. Stojančević M, Pavlović N, Goločorbin-Kon S, et al. Application of bile acids in drug formulation and delivery. Front Life Sci. 2013;7(3–4):112–122. doi:10.1080/21553769.2013.879925
65. Posa M, Kevresan S, Mikov M, et al. Effect of cholic acid and its keto derivatives on the analgesic action of lidocaine and associated biochemical parameters in rats. Eur J Drug Metab Pharmacokinet. 2007;32(2):109–117. doi:10.1007/BF03190999
66. Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56(6):1085–1099. doi:10.1194/jlr.R054114
67. Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69(1):373–398. doi:10.1146/annurev.biochem.69.1.373
68. Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol. 1998;141(4):905–915.
69. Vercauteren D, Vandenbroucke RE, Jones AT, et al. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol Ther. 2010;18(3):561–569. doi:10.1038/mt.2009.281
70. Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10(4):364–371. doi:10.1111/j.1600-0854.2009.00878.x
71. Azevedo C, Macedo MH, Sarmento B. Strategies for the enhanced intracellular delivery of nanomaterials. Drug Discov Today. 2018;23(5):944–959. doi:10.1016/j.drudis.2017.08.011
72. Singh S, Kumar A, Karakoti A, et al. Unveiling the mechanism of uptake and sub-cellular distribution of cerium oxide nanoparticles. Mol Biosyst. 2010;6(10):1813–1820. doi:10.1039/c0mb00014k
73. Donahue ND, Acar H, Wilhelm S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv Drug Deliv Rev. 2019;143(1):68–96. doi:10.1016/j.addr.2019.04.008
74. Klausner RD, Donaldson JG, Lippincott-Schwartz J. A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116(5):1071–1080. doi:10.1083/jcb.116.5.1071
75. Lee G, Joung JY, Cho JH, et al. Overcoming P-glycoprotein-mediated multidrug resistance in colorectal cancer: potential reversal agents among herbal medicines. Evid Based Complement Alternat Med. 2018;2018(2018):3412074–3412083.
76. Manov I, Bashenko Y, Hirsh M, et al. Involvement of the multidrug resistance P-glycoprotein in acetaminophen-induced toxicity in hepatoma-derived HepG2 and Hep3B cells. Basic Clin Pharmacol Toxicol. 2006;99(3):213–224. doi:10.1111/j.1742-7843.2006.pto_443.x
77. Alqahtani S, Alayoubi A, Nazzal S, et al. Nonlinear absorption kinetics of self-emulsifying drug delivery systems (SEDDS) containing tocotrienols as lipophilic molecules: in vivo and in vitro studies. AAPS J. 2013;15(3):684–695. doi:10.1208/s12248-013-9481-7
78. Artursson P, Magnusson C. Epithelial transport of drugs in cell culture. II: effect of extracellular calcium concentration on the paracellular transport of drugs of different lipophilicities across monolayers of intestinal epithelial (Caco-2) cells. J Pharm Sci. 1990;79(7):595–600. doi:10.1002/jps.2600790710
79. DiMarco RL, Hunt DR, Dewi RE, et al. Improvement of paracellular transport in the Caco-2 drug screening model using protein-engineered substrates. Biomaterials. 2017;129152–129162.
80. Forsgard RA, Korpela R, Stenman LK, et al. Deoxycholic acid induced changes in electrophysiological parameters and macromolecular permeability in murine small intestine with and without functional enteric nervous system plexuses. Neurogastroenterol Motil. 2014;26(8):1179–1187. doi:10.1111/nmo.12383
81. Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013;1–16.
82. de Winter BC, Mathot RA, Sombogaard F, et al. Nonlinear relationship between mycophenolate mofetil dose and mycophenolic acid exposure: implications for therapeutic drug monitoring. Clin J Am Soc Nephrol. 2011;6(3):656–663. doi:10.2215/CJN.05440610
83. Hanlon N, Coldham N, Gielbert A, et al. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br J Nutr. 2008;99(3):559–564. doi:10.1017/S0007114507824093
84. Lin JH. Dose-dependent pharmacokinetics: experimental observations and theoretical considerations. Biopharm Drug Dispos. 1994;15(1):1–31. doi:10.1002/bdd.2510150102
85. Graham MA, Lockwood GF, Greenslade D, et al. Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res. 2000;6(4):1205–1218.
86. Zitvogel L, Apetoh L, Ghiringhelli F, et al. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73. doi:10.1038/nri2216
87. Ghiringhelli F, Menard C, Puig PE, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–648. doi:10.1007/s00262-006-0225-8
88. Maiti R. Metronomic chemotherapy. J Pharmacol Pharmacother. 2014;5(3):186–192. doi:10.4103/0976-500X.136098
89. Chan TS, Hsu CC, Pai VC, et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med. 2016;213(13):2967–2988. doi:10.1084/jem.20151665
90. Gasparini G. Metronomic scheduling: the future of chemotherapy? Lancet Oncol. 2001;2(12):733–740. doi:10.1016/S1470-2045(01)00587-3
91. Pasquier E, Andre N, Braguer D. Targeting microtubules to inhibit angiogenesis and disrupt tumour vasculature: implications for cancer treatment. Curr Cancer Drug Targets. 2007;7(6):566–581. doi:10.2174/156800907781662266
92. Bocci G, Nicolaou KC, Kerbel RS. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res. 2002;62(23):6938–6943.
93. Cai XJ, Wang Z, Cao JW, et al. Anti-angiogenic and anti-tumor effects of metronomic use of novel liposomal zoledronic acid depletes tumor-associated macrophages in triple negative breast cancer. Oncotarget. 2017;8(48):84248–84257. doi:10.18632/oncotarget.20539
94. Shimizu T, Abu Lila AS, Nishio M, et al. Modulation of antitumor immunity contributes to the enhanced therapeutic efficacy of liposomal oxaliplatin in mouse model. Cancer Sci. 2017;108(9):1864–1869. doi:10.1111/cas.13305
95. Scharovsky OG, Mainetti LE, Rozados VR. Metronomic chemotherapy: changing the paradigm that more is better. Curr Oncol. 2009;16(2):7–15.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.