Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Improved lung function and patient-reported outcomes with co-suspension delivery technology glycopyrrolate/formoterol fumarate metered dose inhaler in COPD: a randomized Phase III study conducted in Asia, Europe, and the USA
Authors Lipworth BJ ![]() , Collier DJ
, Collier DJ ![]() , Gon Y
, Gon Y ![]() , Zhong NS, Nishi K
, Zhong NS, Nishi K ![]() , Chen R
, Chen R ![]() , Arora S, Maes A, Siddiqui S
, Arora S, Maes A, Siddiqui S ![]() , Reisner C
, Reisner C ![]() , Martin UJ
, Martin UJ ![]()
Received 21 April 2018
Accepted for publication 23 July 2018
Published 26 September 2018 Volume 2018:13 Pages 2969—2984
DOI https://doi.org/10.2147/COPD.S171835
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Brian J Lipworth,1 David J Collier,2 Yasuhiro Gon,3 Nanshan Zhong,4 Koichi Nishi,5 Rongchang Chen,4 Samir Arora,6 Andrea Maes,7 Shahid Siddiqui,8 Colin Reisner,7,8 Ubaldo J Martin8
1Scottish Centre for Respiratory Research, Ninewells Hospital, University of Dundee, Dundee, Scotland, UK; 2William Harvey Research Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK; 3Itabashi Hospital, Nihon University School of Medicine, Itabashi, Tokyo, Japan; 4Guangzhou Institute of Respiratory Health, State Key Laboratory of Respiratory Diseases, National Clinical Research Center of Respiratory Diseases, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China; 5Ishikawa Prefectural Central Hospital, Kanazawa, Ishikawa, Japan; 6Aventiv Research Inc., Columbus, OH, USA; 7Pearl – a member of the AstraZeneca group, Morristown, NJ, USA; 8AstraZeneca, Gaithersburg, MD, USA
Background: COPD is a major global cause of mortality and morbidity. PINNACLE-4 evaluated the efficacy and safety of GFF MDI (glycopyrrolate/formoterol fumarate metered dose inhaler) in patients from Asia, Europe, and the USA with moderate-to-very severe COPD.
Methods: In this double-blind, placebo-controlled, Phase III study, patients were randomized to treatment with GFF MDI 18/9.6 µg, glycopyrrolate (GP) MDI 18 µg, formoterol fumarate (FF) MDI 9.6 µg, or placebo MDI (all twice daily) for 24 weeks. Lung function, patient-reported outcomes (symptoms and health-related quality of life), and safety were assessed.
Results: Of the 1,756 patients randomized, 1,740 patients were included in the intent-to-treat population (mean age 64.2 years, 74.1% male, and 40.2% Asian). GFF MDI significantly improved morning predose trough FEV1 at Week 24 (primary endpoint) vs placebo MDI, GP MDI, and FF MDI (least squares mean differences: 165, 59, and 72 mL, respectively; all P<0.0001). GFF MDI also significantly improved other lung function endpoints vs placebo MDI, GP MDI, and FF MDI and patient-reported outcomes vs placebo MDI and GP MDI. A larger proportion of patients treated with GFF MDI achieved the minimum clinically important difference in Transition Dyspnea Index score vs GP MDI and placebo MDI and in St George’s Respiratory Questionnaire score vs placebo MDI. Adverse event rates were similar across treatment groups.
Conclusion: These results demonstrated the efficacy of GFF MDI in patients with moderate-to-very severe COPD. GFF MDI was well tolerated, with a safety profile commensurate with long-acting bronchodilators.
Keywords: β2-agonist, bronchodilator, COPD, co-suspension delivery technology, muscarinic antagonist
Introduction
Bronchodilators are the cornerstone of maintenance therapy for COPD,1 one of the leading causes of mortality and morbidity worldwide.2 Combined treatment with a long-acting muscarinic antagonist (LAMA) and a long-acting β2-agonist (LABA) plays an important role in the stepwise management of COPD.3 The Global Initiative for Chronic Obstructive Lung Disease (GOLD) recommends combined LAMA/LABA treatment as a first-line therapy for patients with COPD in GOLD group D; and as a step-up treatment for patients in GOLD group C who experience frequent exacerbations despite LAMA or LABA monotherapy, and patients in GOLD group B who experience persistent symptoms despite bronchodilator monotherapy.1
Glycopyrrolate/formoterol fumarate metered dose inhaler (GFF MDI) 18/9.6 μg (Bevespi Aerosphere®; AstraZeneca, Wilmington, DE, USA) is a fixed-dose combination (FDC) of the LAMA glycopyrrolate (GP) and the LABA formoterol fumarate (FF), formulated using innovative co-suspension delivery technology. GFF MDI is approved in the USA for the long-term maintenance treatment of airflow obstruction in patients with COPD4 and, to date, is the first and only LAMA/LABA FDC available as an MDI.
The efficacy and safety of GFF MDI compared with respective monocomponents have been demonstrated over a period of up to 52 weeks in the pivotal Phase III studies PINNACLE-1, PINNACLE-2 (24 weeks; NCT01854645 and NCT01854658), and PINNACLE-3 (28-week safety extension study; NCT01970878), in patients from the USA, Australia, and New Zealand.5,6 Due to differences in COPD prevalence and burden between different countries and regions,7–10 as well as potential differences in the observed effects of pharmacological therapies,11 it was deemed important to evaluate the efficacy and safety of COPD maintenance treatments in other geographical patient populations. Here, we present the results of the PINNACLE-4 study (ClinicalTrials.gov: NCT02343458), which investigated the efficacy and safety of GFF MDI compared to its monocomponents (GP MDI and FF MDI) and placebo MDI in a population with moderate-to-very severe COPD, which included Asian and European patients.
Methods
Study design and treatment
PINNACLE-4 was a randomized, double-blind, parallel-group, placebo-controlled Phase III study conducted at multiple sites across Asia, Europe, and the USA. Patients were randomized 7:6:6:3 using an Interactive Web Response System (further details in the Supplementary materials) to receive treatment with GFF MDI 18/9.6 μg (equivalent to glycopyrronium/formoterol fumarate dihydrate 14.4/10 μg), GP MDI 18 μg, FF MDI 9.6 μg, or matched placebo MDI (all twice daily) for 24 weeks, with randomization stratified by reversibility to rescue albuterol sulfate and by COPD disease severity. Patients provided written informed consent prior to screening, and the study was conducted in accordance with Good Clinical Practice, including the Declaration of Helsinki and the International Council for Harmonisation. The protocol was approved by local institutional review boards (Table S1). Patients were required to discontinue prohibited COPD medications (including oral β2-agonists, LABAs, cromoglycate or nedocromil inhalers, leukotriene antagonists, ketotifen [except as eye drops], and LAMAs) following screening and were switched to sponsor-provided ipratropium bromide (administered four times daily) and albuterol sulfate (as needed) to control symptoms during the screening period. Patients using a maintenance FDC of an inhaled corticosteroid (ICS) and a LABA discontinued this, and were switched to the corresponding ICS monotherapy (fluticasone, mometasone, or budesonide) at an equivalent dose, as well as ipratropium bromide and albuterol sulfate (providing they had been maintained on a stable dose of the ICS component for ≥4 weeks prior to screening). Any patients taking a maintenance dose of an ICS not administered as an FDC with a LABA were allowed to continue using the ICS if they had been on a stable dose for ≥4 weeks prior to screening. Ipratropium bromide was discontinued after screening. Sponsor-provided albuterol sulfate was permitted, as needed, for the relief of symptoms throughout the study.
Study population
Patients were 40–80 years of age and had an established clinical history of COPD as defined by the American Thoracic Society/European Respiratory Society.12 Inclusion and exclusion criteria were the same as reported for PINNACLE-1 and PINNACLE-2.5 Briefly, eligible patients were current or former smokers (≥10 pack-years) with an FEV1/forced vital capacity ratio of <0.70 and an FEV1 of <80% predicted normal value at screening. Further details are provided in the Supplementary materials. Patients were required to demonstrate stable baseline FEV1, ie, mean predose FEV1 at randomization within ±20% or 200 mL of the mean of the predose FEV1 assessment obtained at the previous two screening visits. The ability of patients to use the MDI correctly was confirmed at screening, with additional training provided as necessary.
Assessments
The primary objective of the study was to compare the efficacy of GFF MDI with its monocomponents (GP MDI and FF MDI) and placebo MDI and also GP MDI and FF MDI with placebo MDI, in patients with moderate-to-very severe COPD. Study endpoints differed according to the regional regulatory registration requirements. This manuscript reports the approach that satisfies the filing requirements of the US and China regulatory authorities. Data for similar approaches and endpoints satisfying the filing requirements of other regions were also generated. The change from baseline in morning predose trough FEV1 at Week 24 was the primary endpoint. Secondary lung function endpoints included change from baseline in morning predose trough FEV1 over 24 weeks, peak change from baseline in FEV1 within 2 hours postdosing at Week 24, and time to onset of action on Day 1 (defined as the first time point at which the difference from placebo MDI was statistically significant).
Other secondary endpoints included Transition Dyspnea Index (TDI) focal score over 24 weeks, change from baseline in St George’s Respiratory Questionnaire (SGRQ) total score at Week 24 (intent-to-treat [ITT] population and symptomatic population), and change from baseline in mean daily rescue medication use over 24 weeks (rescue medication user population). Assessments of TDI focal score at Week 24 and SGRQ score over Weeks 12–24 were additional endpoints. Baseline Dyspnea Index (BDI) and TDI were assessed using the interviewer-administrated version of the BDI/TDI questionnaire.13,14 Other efficacy endpoints included responder analyses to determine the proportion of patients achieving an improvement of the minimal clinically important difference (MCID) threshold of ≥1 unit in TDI focal score15 over 24 weeks and ≥4 units in SGRQ score16 at Week 24.
Safety assessments included electrocardiograms (ECGs), clinical laboratory testing, and vital sign measurements. Adverse events (AEs) were monitored throughout the study.
Statistical analysis
Unless otherwise specified, results were based on analyses using the ITT population (all patients who were randomized and received any study treatment, even if <1 full dose). The safety population was the same as the ITT population, except patients who were analyzed according to treatment received rather than treatment assigned. The symptomatic population included all patients in the ITT population with a COPD assessment test (CAT) score of ≥15 at screening. The rescue medication user population included all patients in the ITT population with the mean baseline rescue medication use (albuterol sulfate) of ≥1 puff/day (calculated from the last 7 days of the 10–14 days screening period).
A sample size of 1,614 patients was estimated to provide 91% of power to detect differences for all primary comparisons (GFF MDI vs placebo MDI and each monocomponent and each monocomponent vs placebo MDI) in the primary endpoint (change from baseline in morning predose trough FEV1 at Week 24) with Type I error controlled at a two-sided α level of 0.05. The same sample size was estimated to provide 99% of power to detect differences for the same comparisons for change from baseline in morning predose trough FEV1 over 24 weeks.
The primary and secondary endpoints (with the exception of time to onset of action) were analyzed using repeated measures linear models (further details in the Supplementary materials). Strong control of Type I error (two-sided α=0.05) was implemented sequentially across the five key comparisons for the primary endpoint and then simultaneously across the secondary endpoints within a key comparison using the Hochberg procedure (two-sided α=0.05).
Results
Patient disposition
A total of 1,756 patients were randomized and received treatment (714 patients from Asia, 496 patients from the USA, and 546 patients from Europe [including Russia]), and 1,528 (87%) patients completed the study (Figure 1). The ITT and safety populations included 1,740 patients, of whom 841 patients were symptomatic (baseline CAT score ≥15). The rescue medication user population comprised 822 patients. Patient demographics and baseline characteristics are summarized in Table 1. The mean age of the patient was 64.2 years, 74.1% of them were male, and 40.2% of them were Asian (56.7% White).
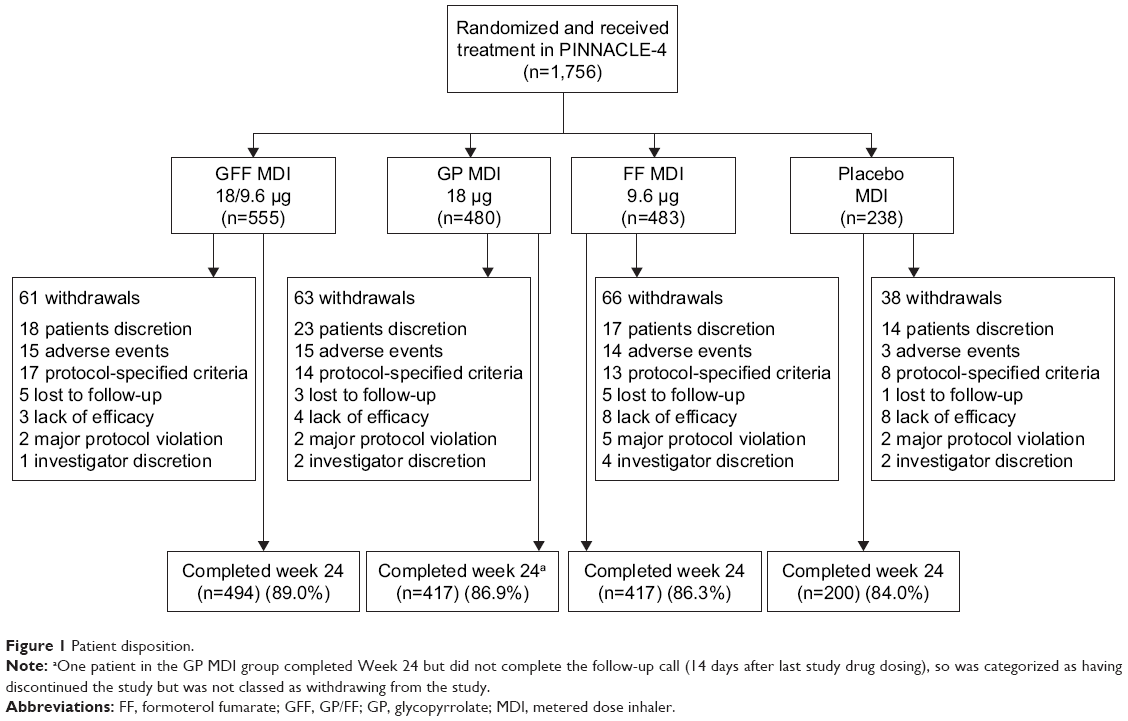 | Figure 1 Patient disposition. |
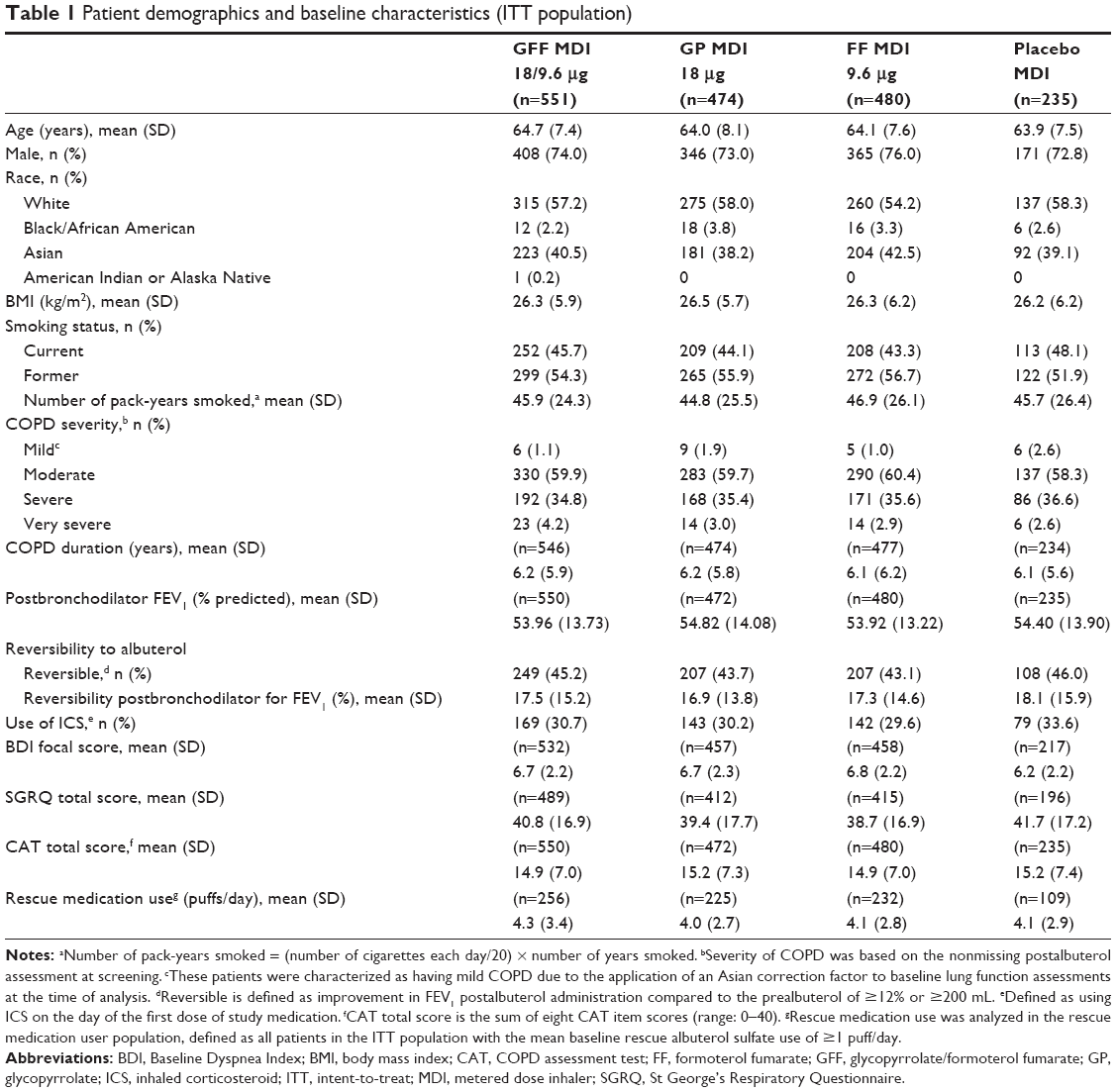 | Table 1 Patient demographics and baseline characteristics (ITT population) |
Efficacy
For the primary endpoint of change from baseline in morning predose trough FEV1 at Week 24, treatment with GFF MDI resulted in significantly greater improvements vs placebo MDI (least squares mean [LSM] difference: 165 mL; P<0.0001; Figure 2 and Table 2), GP MDI (LSM difference: 59 mL; P<0.0001), and FF MDI (LSM difference: 72 mL; P<0.0001). GP MDI and FF MDI treatments significantly increased morning predose trough FEV1 at Week 24 compared to placebo MDI (LSM difference 105 and 92 mL, respectively; both P<0.0001; Figure 2).
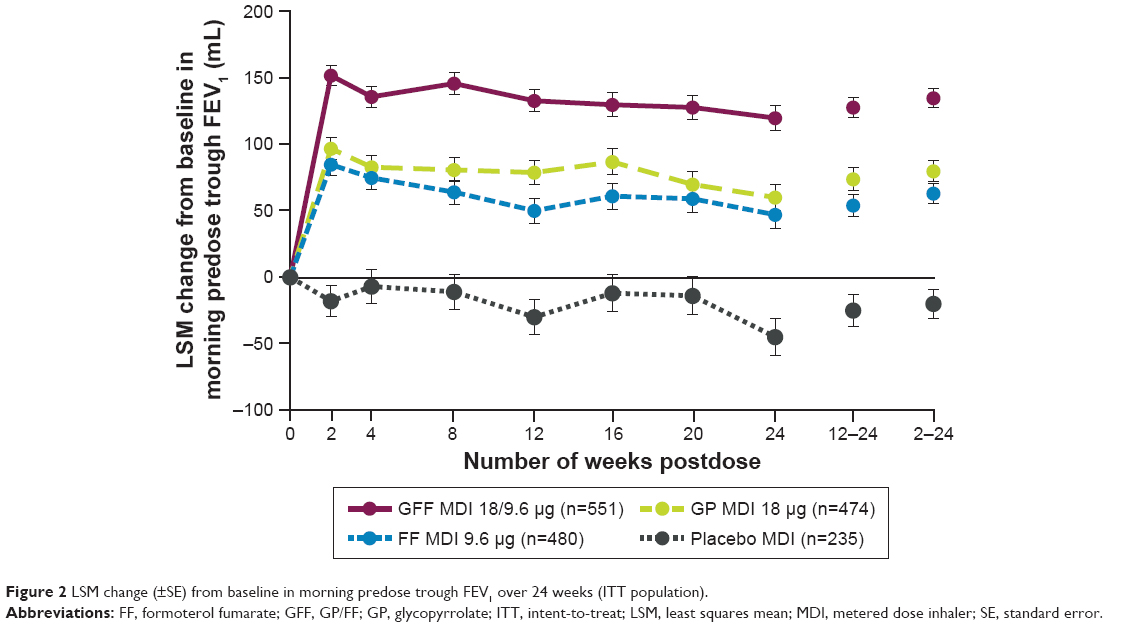 | Figure 2 LSM change (±SE) from baseline in morning predose trough FEV1 over 24 weeks (ITT population). |
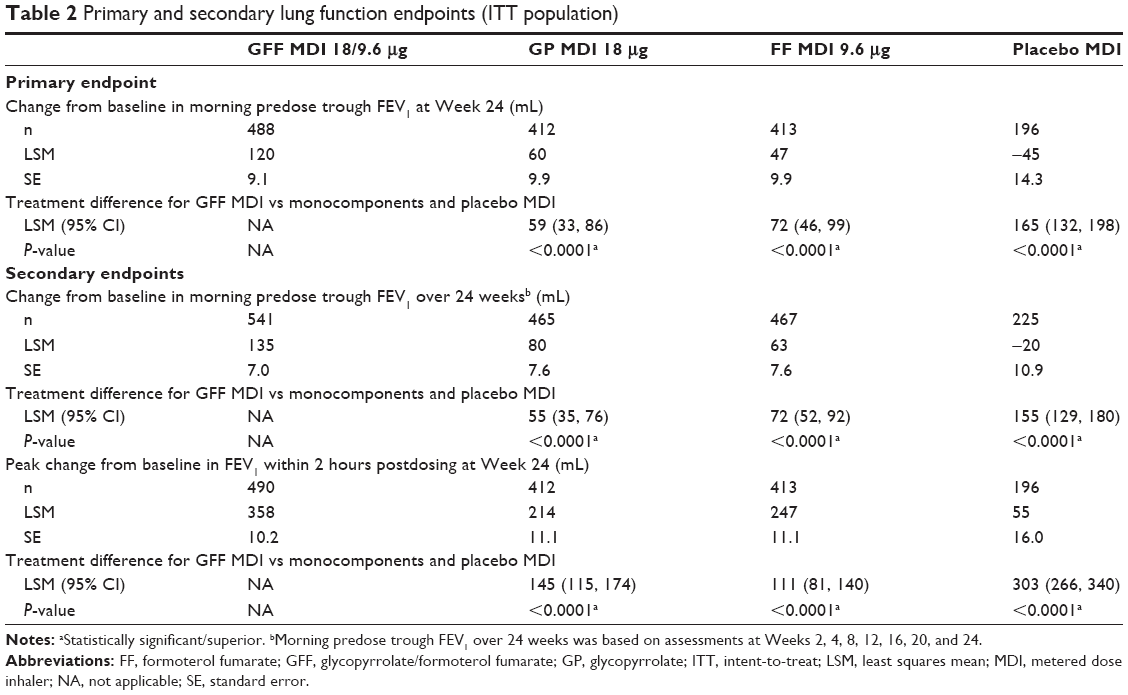 | Table 2 Primary and secondary lung function endpoints (ITT population) |
Similar improvements as for the primary endpoint were observed for change from baseline in morning predose trough FEV1 over 24 weeks (Figure 2 and Table 2). GFF MDI led to significant improvements in peak change from baseline in FEV1 within 2 hours postdose at Week 24 compared to GP MDI, FF MDI, and placebo MDI (Table 2). Onset of action for GFF MDI, GP MDI, and FF MDI occurred within 5 minutes postdose (LSM differences vs placebo MDI 179 mL [P<0.0001], 37 mL [P=0.0002], and 164 mL [P<0.0001], respectively).
Significant improvements in TDI focal score over 24 weeks and SGRQ score at Week 24 were observed in both the ITT population and the symptomatic population following treatment with GFF MDI compared with GP MDI and placebo MDI (P<0.05) but not with FF MDI (Table 3). Improvements in TDI score at Week 24 and SGRQ score over Weeks 12–24 were also greater following GFF MDI treatment compared to GP MDI and placebo MDI in both populations (Table S2). Patients treated with GFF MDI were more likely to achieve an improvement in at least the MCID for TDI score (≥1.0 unit) and SGRQ score (≥4.0 unit decrease) vs placebo MDI and versus GP MDI for TDI score (ITT population and symptomatic population; Table 4). Significant improvements in rescue medication use were observed for GFF MDI vs GP MDI (LSM difference: −0.77; P=0.0001) and placebo MDI in the rescue medication user population (LSM difference: −0.98; P<0.0001; Table 3).
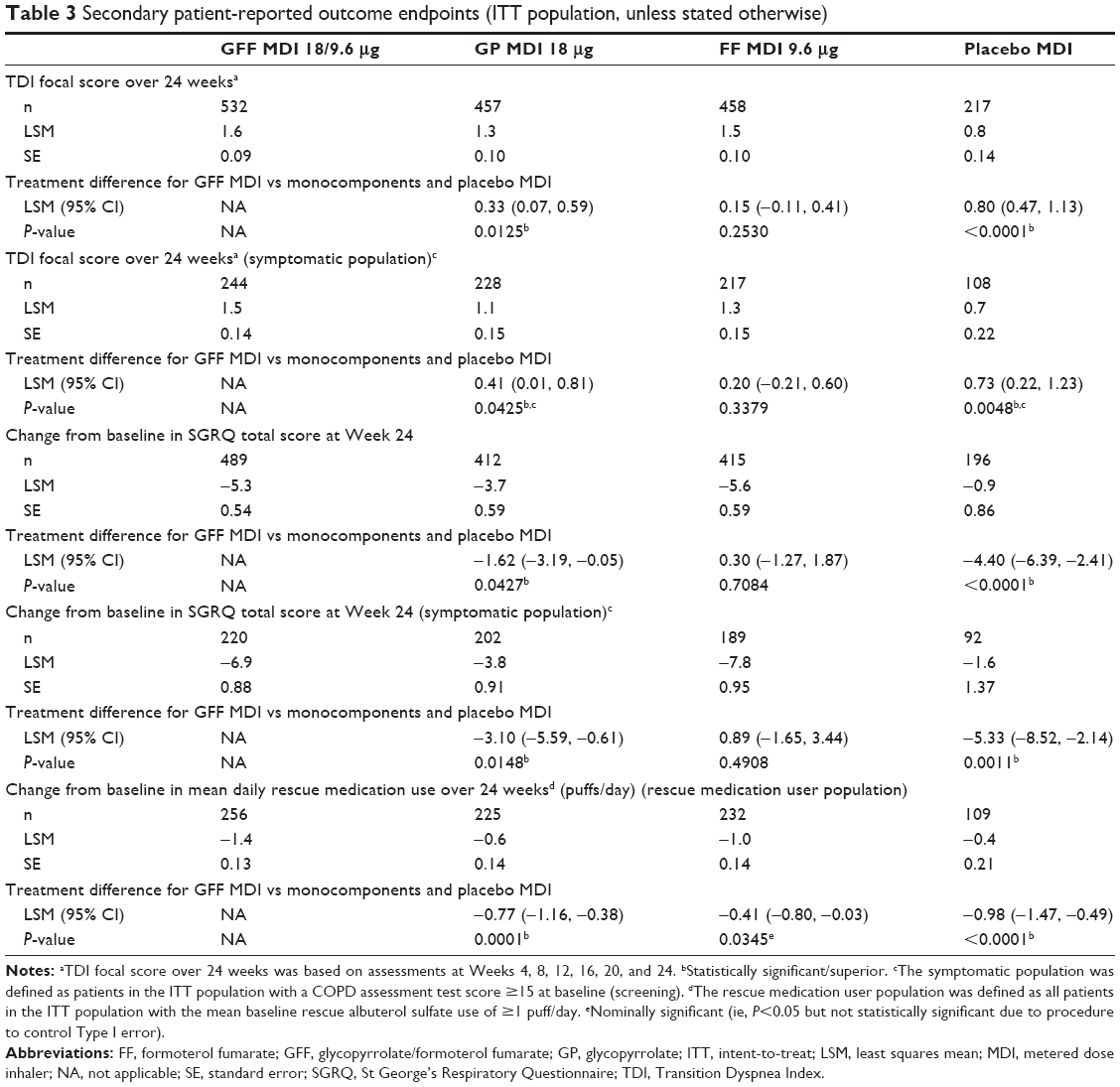 | Table 3 Secondary patient-reported outcome endpoints (ITT population, unless stated otherwise) |
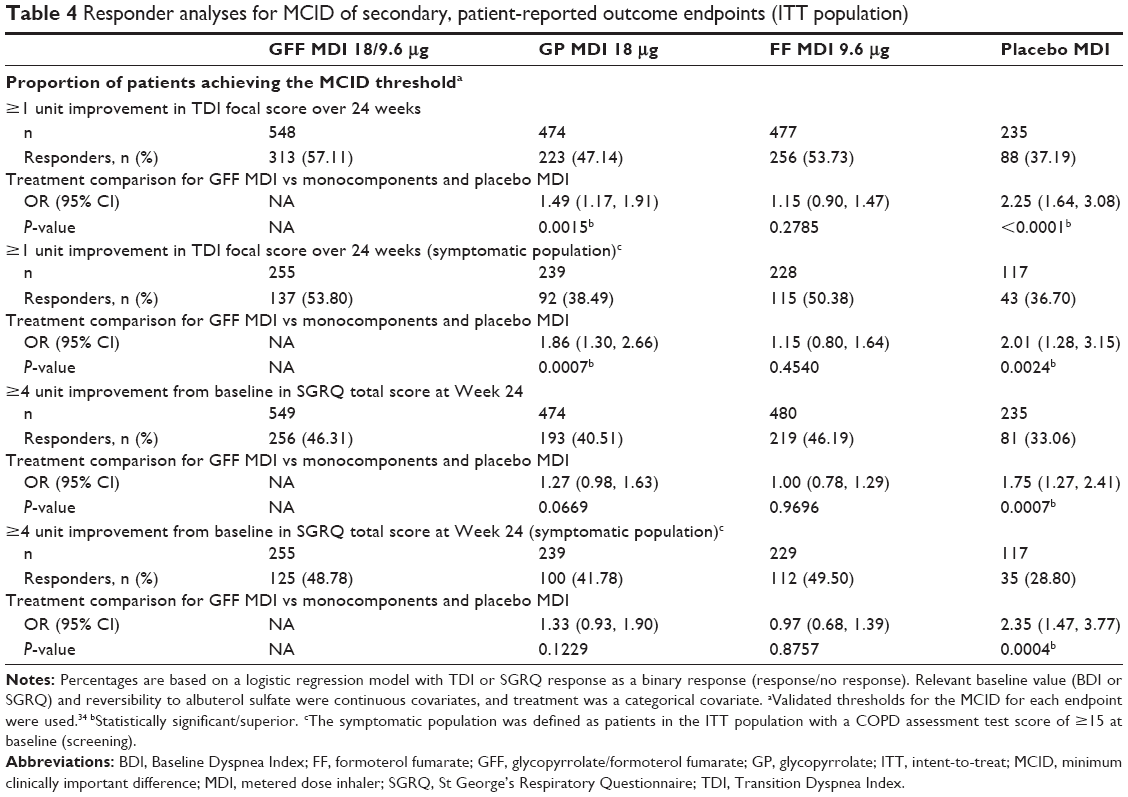 | Table 4 Responder analyses for MCID of secondary, patient-reported outcome endpoints (ITT population) |
Safety
The incidence of treatment-emergent AEs (TEAEs), treatment-related TEAEs, serious TEAEs, or TEAEs leading to discontinuation was similar across treatment groups (Table 5), with the majority of TEAEs being mild or moderate and not considered related to study treatment. A relatively low proportion of patients (ranging from 4.3% with placebo MDI to 5.3% with GP MDI) discontinued due to TEAEs. The most commonly reported TEAEs included upper respiratory tract infection, worsening of COPD, headache, and hypertension (Table 5).
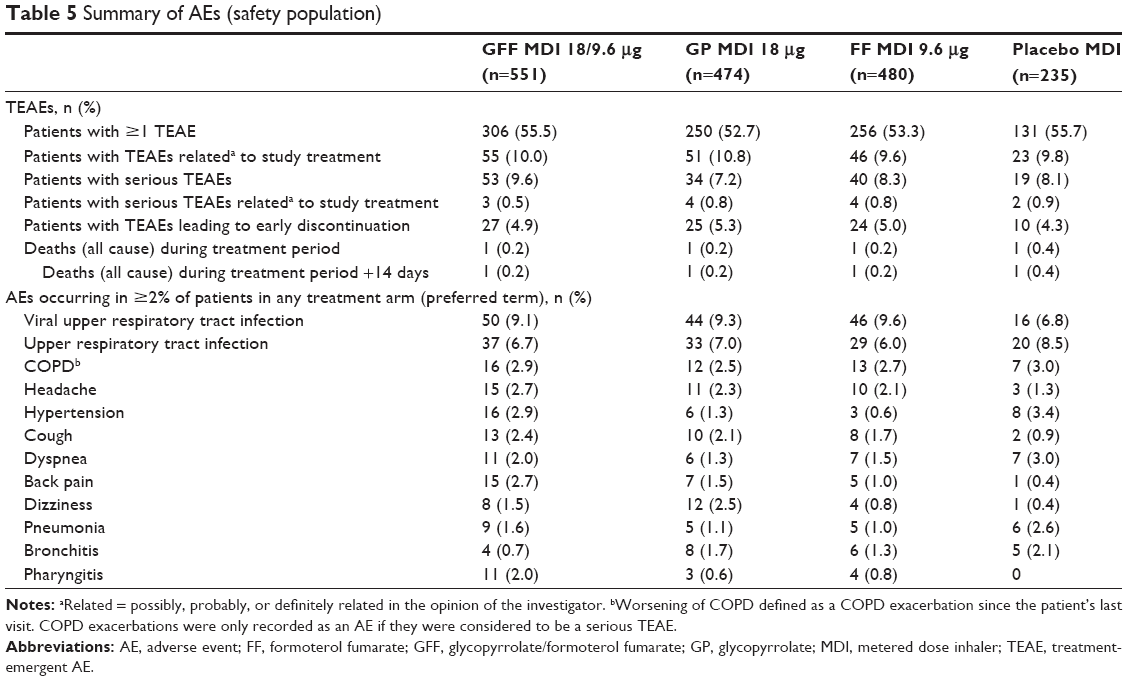 | Table 5 Summary of AEs (safety population) |
One death occurred in each of the treatment groups (lung cancer [metastatic; n=1 with both GFF MDI and placebo MDI], hemorrhagic stroke [GP MDI], and hypoglycemic coma [FF MDI]). None of these deaths were judged by the investigator to be related to study drug treatment.
Discussion
Treatment with the LAMA/LABA FDC, GFF MDI, improved lung function compared to placebo MDI and monocomponents and improved symptoms and patient-reported outcomes compared to placebo MDI and GP MDI in a population of patients with moderate-to-very severe COPD from Asia, Europe, and the USA. Improvements in the primary endpoint – change from baseline in morning predose trough FEV1 at Week 24 – exceeded the MCID of 100 mL17 for both GFF MDI and GP MDI vs placebo MDI and were significantly higher following treatment with GFF MDI vs monocomponents. Overall, results confirmed those from previous Phase III studies, which showed GFF MDI to be efficacious and well tolerated in a population that included patients from the USA, Australia, and New Zealand.5,6
Improvements in the secondary lung function endpoint (peak change from baseline in FEV1 within 2 hours postdose at Week 24) as well as rescue medication use were significantly larger in the GFF MDI treatment arm vs monocomponents and placebo MDI and similar to those observed in PINNACLE-1 and PINNACLE-2.5,18 TDI focal scores indicated greater reductions in breathlessness following GFF MDI treatment than comparators, although treatment differences were only significant compared with GP MDI and placebo MDI (ITT and symptomatic populations). The treatment difference for GFF MDI vs placebo MDI in TDI score over 24 weeks was larger than that observed in PINNACLE-1 and PINNACLE-2, which may be in part due to differences in the method of assessment of TDI score (the interviewer-administered version of the TDI was used in this study vs the self-administered, computerized version used in PINNACLE-1 and PINNACLE-2).19 A larger proportion of patients in the GFF MDI group achieved a clinically relevant improvement in TDI (total score ≥1 unit)15 vs GP MDI and placebo MDI in the symptomatic and ITT populations, demonstrating that GFF MDI was effective in reducing breathlessness in patients with COPD. The results of the SGRQ assessment in this study suggest that GFF MDI may improve health-related quality of life (HRQoL) compared with placebo MDI and GP MDI, which is consistent with the results of PINNACLE-1.5,18 Although the improvements seen with GFF MDI in patient-reported outcomes (TDI and SGRQ) were not statistically significant vs FF MDI, both treatments were effective in improving symptoms vs placebo. Differences between active treatments for patient-reported outcomes can be small and, therefore, these outcome measures may not be sensitive in indicating differences between active treatments. Ultimately, the superior effects of GFF MDI vs FF MDI on lung function may result in greater benefits for patients’ quality of life when sustained over a longer time period than the 24-week duration of the current study. In the long-term PINNACLE-3 safety study, treatment with GFF MDI resulted in statistically significant improvements in TDI score and numerical improvements in SGRQ, over 52 weeks compared with FF MDI.6
Although no head-to-head comparisons between GFF MDI and other LAMA/LABA FDCs have been reported, the magnitude of improvements in lung function, rescue medication use, and HRQoL vs monocomponents observed in this study followed a similar trend to those of pivotal studies with other LAMA/LABA FDCs.20–27 While several other efficacious and well-tolerated LAMA/LABA FDC combinations are available for the maintenance treatment of COPD, GFF MDI is notably the first to be delivered using an MDI. The co-suspension delivery technology used to formulate GFF MDI overcame formulation challenges encountered with MDIs,28 resulting in consistent in vitro aerosol performance, even in the presence of simulated patient-handling errors,29 providing reliable drug dose delivery to all regions of the lungs with high efficiency.30 As familiarity with an inhaler can result in more favorable clinical outcomes in respiratory disease31,32 and MDIs remain a commonly prescribed device type for rescue medication,33 the availability of a LAMA/LABA FDC delivered by MDI offers a useful option for the maintenance treatment of COPD. A Phase III study has shown that the addition of a spacer does not affect the lung function benefits and tolerability of GFF MDI,35 suggesting that this FDC could be a treatment option for patients who require a spacer to compensate for poor hand-to-breath coordination with an MDI.
A potential limitation of this study was the short duration (6 months) relative to expected use as prophylactic therapy. However, the long-term safety and efficacy of GFF MDI have been evaluated over a 1-year period during the PINNACLE-3 safety extension study.6 Additionally, patients could have potentially perceived benefit from participation in a study of the novel co-suspension delivery technology MDI. However, placebo was delivered by the same device as the active treatments to control for effects due to patient perception. The strength of this study was that patients were enrolled from sites across Asia, Europe, and the USA, allowing the efficacy and safety of GFF MDI to be evaluated in patients from a broad range of geographical locations and socioeconomic backgrounds. Patients were not required to be symptomatic at baseline for enrollment, though results were analyzed in a subgroup of patients with a CAT score of ≥15 (48% of patients randomized), which provided an insight into the efficacy of GFF MDI in symptomatic patients.
Conclusion
The results of PINNACLE-4 demonstrate that GFF MDI improves lung function, symptoms, and patient-reported outcomes in a study population including patients from Asia, Europe, and the USA. These results are consistent with previous Phase III studies with GFF MDI, which showed that this LAMA/LABA FDC was efficacious and well tolerated with no unexpected safety signals in patients with moderate-to-very severe COPD.
Abbreviations
AE, adverse event; BDI, Baseline Dyspnea Index; BMI, body mass index; CAT, COPD assessment test; ECG, electrocardiogram; FDC, fixed-dose combination; FF, formoterol fumarate; GFF, glycopyrrolate/formoterol fumarate; GP, glycopyrrolate; HRQoL, health-related quality of life; ICS, inhaled corticosteroid; ITT, intent-to-treat; LABA, long-acting β2-agonist; LAMA, long-acting muscarinic antagonist; LSM, least squares mean; MCID, minimum clinically important difference; MDI, metered dose inhaler; SE, standard error; SGRQ, St George’s Respiratory Questionnaire; TDI, Transition Dyspnea Index; TEAE, treatment-emergent adverse event.
Data sharing statement
All relevant data analyzed during this study are included in this article.
Acknowledgments
This study was supported by Pearl – a member of the AstraZeneca Group. Employees of the sponsor and employees of AstraZeneca were involved in various aspects of the conception and design of the study, acquisition of data and analysis and interpretation of data, and input into manuscript development. The sponsor did not place any restriction on authors about the statements made in the final article. The authors would like to thank all the patients and their families and the team of investigators, research nurses, and operations’ staff involved in these studies. Medical writing support, under the direction of the authors, was provided by Carol McNair, PhD, of CMC CONNECT, a division of Complete Medical Communications Ltd, Glasgow, UK, funded by AstraZeneca, Cambridge, UK, in accordance with Good Publication Practice (GPP3) guidelines.36 Data included in this manuscript have been presented in a poster at the American Thoracic Society International Conference 2018, San Diego, CA, USA. The PINNACLE studies were supported by Pearl – a member of the AstraZeneca Group.
Author contributions
CR is the guarantor and takes responsibility for the content of this manuscript, including the data and analysis. All authors participated in the analysis and interpretation of data reported. AM made significant contributions to the statistical analysis of the data. BJL, DJC, YG, NZ, KN, RC, and SA participated in the acquisition of reported data. AM, SS, CR, and UJM made substantial contributions to the conception or design of the study. All authors reviewed or critically revised the manuscript, provided final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
BJL is one of a number of co-investigators on an AstraZeneca-sponsored grant received by the University of Dundee to support genomic studies in COPD. He has also received speaker fees from AstraZeneca; payment for consulting and speaking from Boehringer Ingelheim and Chiesi; grant support from Boehringer Ingelheim, Chiesi, and Janssen; advisory board and speaker fees from Teva; and consulting fees from Sandoz, Cipla, Dr Reddys, and Lupin. DJC is supported by the National Institute for Health Research (NIHR) Barts Biomedical Research Center and received a grant from Pearl – a member of the AstraZeneca group, for normal recruitment of patients. YG received payment from AstraZeneca for medical institution costs for the clinical study and has received speaker fees from AstraZeneca, Boehringer Ingelheim Japan Co., Ltd, and Kyorin Pharmaceutical Co., Ltd. NZ has received consultancy and lecture fees from Boehringer Ingelheim and Novartis and was on the advisory board of the GOLD committee. KN and SA report no conflicts of interest in this work. RC is an advisory committee member and speaker for AstraZeneca. AM is an employee of Pearl – a member of the AstraZeneca Group. SS and UJM are employees of AstraZeneca, with stock options. CR is an employee of Pearl – a member of the AstraZeneca group and an employee of AstraZeneca.
References
Global Initiative for Chronic Obstructive Lung Disease [homepage on the Internet]. Global Strategy for the Diagnosis, Management and Prevention of COPD; 2018. Available from: http://www.goldcopd.org. Accessed February 5, 2018. | ||
GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. | ||
Currie GP, Lipworth BJ. Inhaled treatment for chronic obstructive pulmonary disease: what’s new and how does it fit? QJM. 2016;109(8):505–512. | ||
AstraZeneca Pharmaceuticals LP. Bevespi Aerosphere™ [prescribing information]; 2017. Available from: http://www.azpicentral.com/bevespi/bevespi_pi.pdf. Accessed February 5, 2018. | ||
Martinez FJ, Rabe KF, Ferguson GT, et al. Efficacy and safety of glycopyrrolate/formoterol metered dose inhaler formulated using co-suspension delivery technology in patients with COPD. Chest. 2017;151(2):340–357. | ||
Hanania NA, Tashkin DP, Kerwin EM, et al. Long-term safety and efficacy of glycopyrrolate/formoterol metered dose inhaler using novel Co-Suspension™ Delivery Technology in patients with chronic obstructive pulmonary disease. Respir Med. 2017;126:105–115. | ||
Tan WC, Seale P, Ip M, et al. Trends in COPD mortality and hospitalizations in countries and regions of Asia-Pacific. Respirology. 2009;14(1):90–97. | ||
Fang L, Gao P, Bao H, et al. Chronic obstructive pulmonary disease in China: a nationwide prevalence study. Lancet Respir Med. 2018;6(6):421–430. | ||
Fukuchi Y, Nishimura M, Ichinose M, et al. COPD in Japan: the Nippon COPD Epidemiology study. Respirology. 2004;9(4):458–465. | ||
Landis SH, Muellerova H, Mannino DM, et al. Continuing to Confront COPD International Patient Survey: methods, COPD prevalence, and disease burden in 2012–2013. Int J Chron Obstruct Pulmon Dis. 2014;9:597–611. | ||
Yasuda SU, Zhang L, Huang SM. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharmacol Ther. 2008;84(3):417–423. | ||
Celli BR, Macnee W. ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Mahler DA, Weinberg DH, Wells CK, Feinstein AR. The measurement of dyspnea. Contents, interobserver agreement, and physiologic correlates of two new clinical indexes. Chest. 1984;85(6):751–758. | ||
Mahler DA, Waterman LA, Ward J, Mccusker C, Zuwallack R, Baird JC. Validity and responsiveness of the self-administered computerized versions of the baseline and transition dyspnea indexes. Chest. 2007;132(4):1283–1290. | ||
Mahler DA, Witek TJ. The MCID of the transition dyspnea index is a total score of one unit. COPD. 2005;2(1):99–103. | ||
Jones PW. St. George’s Respiratory Questionnaire: MCID. COPD. 2005;2(1):75–79. | ||
Donohue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2(1):111–124. | ||
Martinez FJ, Fabbri LM, Ferguson GT, et al. Baseline symptom score impact on benefits of glycopyrrolate/formoterol metered dose inhaler in COPD. Chest. 2017;152(6):1169–1178. | ||
Rabe K, Martinez F, Rodriguez-Roisin R, et al. PT003, a novel co-suspension MDI glycopyrronium/formoterol fixed-dose combination is superior to monocomponents in patients with COPD. Eur Respir J. 2015;59(suppl):PA463. | ||
Bateman ED, Ferguson GT, Barnes N, et al. Dual bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE study. Eur Respir J. 2013;42(6):1484–1494. | ||
Celli B, Crater G, Kilbride S, et al. Once-daily umeclidinium/vilanterol 125/25 mcg in COPD: a randomized, controlled study. Chest. 2014;145(5):981–991. | ||
D’Urzo AD, Rennard SI, Kerwin EM, et al. Efficacy and safety of fixed-dose combinations of aclidinium bromide/formoterol fumarate: the 24-week, randomized, placebo-controlled AUGMENT COPD study. Respir Res. 2014;15:123. | ||
Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107(10):1538–1546. | ||
Singh D, Jones PW, Bateman ED, et al. Efficacy and safety of aclidinium bromide/formoterol fumarate fixed-dose combinations compared with individual components and placebo in patients with COPD (ACLIFORM-COPD): a multicentre, randomised study. BMC Pulm Med. 2014;14:178. | ||
Buhl R, Maltais F, Abrahams R, et al. Tiotropium and olodaterol fixed-dose combination versus mono-components in COPD (GOLD 2–4). Eur Respir J. 2015;45(4):969–979. | ||
Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2(6):472–486. | ||
Mahler DA, Kerwin E, Ayers T, et al. FLIGHT1 and FLIGHT2: efficacy and safety of QVA149 (indacaterol/glycopyrrolate) versus its monocomponents and placebo in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(9):1068–1079. | ||
Vehring R, Lechuga-Ballesteros D, Joshi V, Noga B, Dwivedi SK. Cosuspensions of microcrystals and engineered microparticles for uniform and efficient delivery of respiratory therapeutics from pressurized metered dose inhalers. Langmuir. 2012;28(42):15015–15023. | ||
Doty A, Schroeder J, Vang K, et al. Drug delivery from an innovative LAMA/LABA co-suspension delivery technology fixed-dose combination MDI: evidence of consistency, robustness, and reliability. AAPS PharmSciTech. 2018;19(2):837–844. | ||
Taylor G, Warren S, Dwivedi S, et al. Gamma scintigraphic pulmonary deposition study of glycopyrronium/formoterol metered dose inhaler formulated using co-suspension delivery technology. Eur J Pharm Sci. 2018;111:450–457. | ||
Bosnic-Anticevich S, Chrystyn H, Costello RW, et al. The use of multiple respiratory inhalers requiring different inhalation techniques has an adverse effect on COPD outcomes. Int J Chron Obstruct Pulmon Dis. 2017;12:59–71. | ||
Price D, Chrystyn H, Kaplan A, et al. Effectiveness of same versus mixed asthma inhaler devices: a retrospective observational study in primary care. Allergy Asthma Immunol Res. 2012;4(4):184–191. | ||
Lavorini F, Corrigan CJ, Barnes PJ, et al. Retail sales of inhalation devices in European countries: so much for a global policy. Respir Med. 2011;105(7):1099–1103. | ||
Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically important differences in pharmacological trials. Am J Respir Crit Care Med. 2014;189(3):250–255. | ||
Fakih F, Spangenthal S, Sigal B, et al. Randomized study of the effects of Aerochamber Plus® Flow-Vu® on the efficacy, pharmacokinetics and safety of glycopyrronium/formoterol fumarate dihydrate metered dose inhaler in patients with chronic obstructive pulmonary disease. Respir Med. 2018;138:74–80. | ||
Battisti WP, Wager E, Baltzer L, et al. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163(6):461–464. |
Supplementary materials
Study design and inclusion/exclusion criteria
A randomization ratio of 7:6:6:3 was used, as initial modeling suggesting that this is the most efficient for the sample size and treatments in this study. The appropriateness of this ratio was confirmed by the results of PINNACLE-1 and PINNACLE-2.1
Patients were 40–80 years of age, had an established clinical history of COPD as defined by the American Thoracic Society/European Respiratory Society,2 and were current or former smokers with a history of at least 10 pack-years. COPD had to be of at least moderate severity,3 defined as a FEV1/forced vital capacity ratio of <0.70 at screening, and FEV1 <80% predicted normal value (calculated using the Third National Health and Nutrition Examination Survey4 or local reference equations applicable to other regions) and ≥750 mL if FEV1 <30% predicted normal (ie, very severe COPD).
Exclusion criteria included diagnosis of a significant disease other than COPD (which, in the opinion of the investigator, could put the patient at risk or could influence either the study results or the patient’s ability to participate); poorly controlled COPD (defined as acute worsening of COPD that required treatment with oral corticosteroids or antibiotics within 6 weeks of, or during, screening); and hospitalization due to poorly controlled COPD within 3 months prior to, or during, screening. The need for long-term oxygen therapy (>12 hour/day), change in smoking status within 6 weeks of or during screening, and poor hand-to-breath coordination (requiring the use of a spacer device with an MDI) were also exclusion criteria.
Statistical analysis
The primary endpoint was analyzed using a repeated measures linear model with baseline FEV1 (the mean of evaluable 60- and 30-minute predose values on Day 1) and reversibility to albuterol sulfate as continuous covariates and visit, treatment, and treatment-by-visit interaction as categorical covariates. An unstructured variance–covariance matrix was applied, and two-sided P-values and point estimates with two-sided 95% CIs were produced for each treatment difference. Treatment group comparisons for the secondary endpoints were evaluated using a similar repeated measures linear model as for the primary endpoint but included the relevant baseline covariate for each endpoint. Time to onset of action on Day 1 was determined for each treatment using the 5 and 15-minute postdosing FEV1 assessments and analyzed using an analysis of covariance model, with baseline FEV1 and reversibility to albuterol sulfate as continuous covariates. For Transition Dyspnea Index and St George’s Respiratory Questionnaire responder analyses, logistic regression was used to compare treatment groups and P-values and odds ratios with 95% CIs were produced for each comparison. The procedure to control Type I error was applied to primary and secondary endpoints only and is described in the main body of the article.
  | Table S1 Institutional review boards and approval numbers |
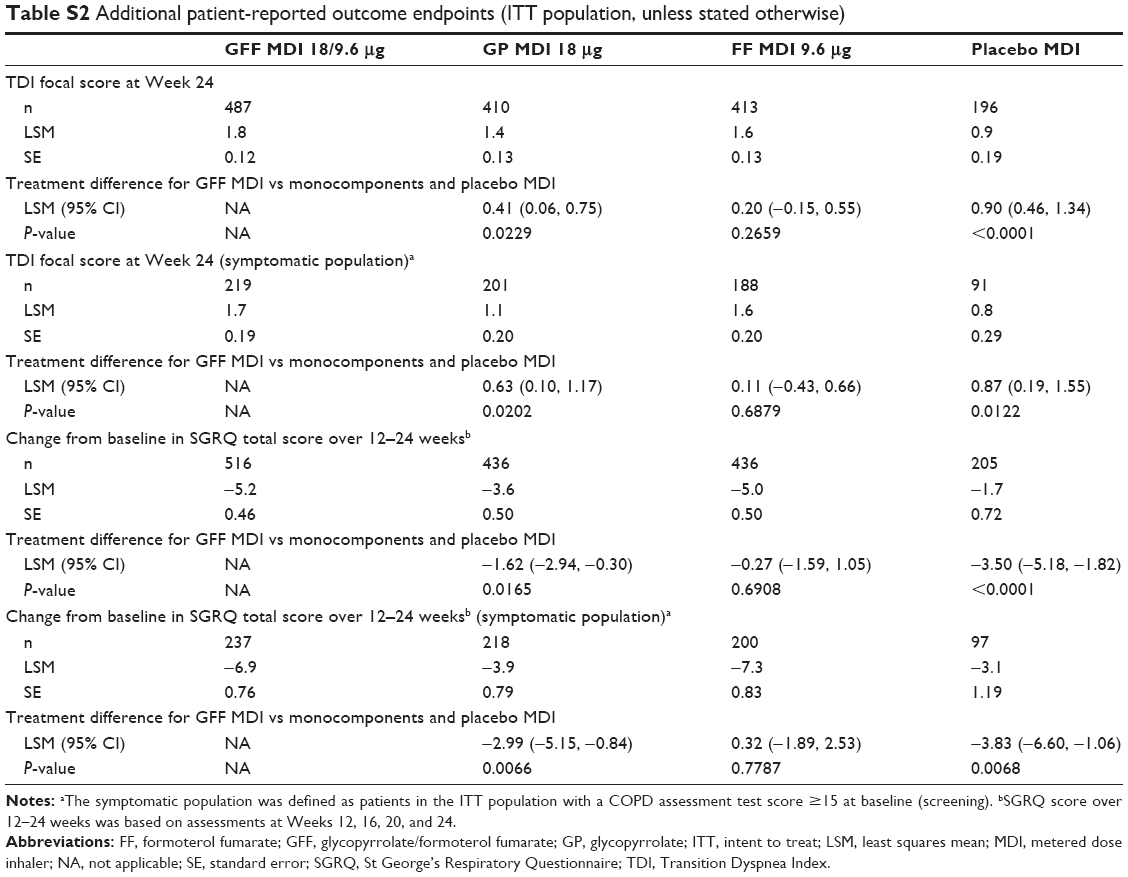 | Table S2 Additional patient-reported outcome endpoints (ITT population, unless stated otherwise) |
References
Martinez FJ, Rabe KF, Ferguson GT, et al. Efficacy and safety of glycopyrrolate/formoterol metered dose inhaler formulated using co-suspension delivery technology in patients with COPD. Chest. 2017;151(2):340–357. | ||
Celli BR, MacNee W. ATS/ERS Task Forcecommittee members. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. | ||
Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD. 2018. Available from: http://www.goldcopd.org. Accessed February 5, 2018. | ||
National Center for Health Statistics. Third National Health and Nutrition Examination Survey III Spirometry Procedure Manual. 1988. Available from: https://wwwn.cdc.gov/nchs/nhanes/nhanes3/manualsandreports.aspx. Accessed February 8, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.