")
Back to Journals » International Journal of General Medicine » Volume 16
Impact of Sickle Cell Disease on Affected Individuals in Nigeria: A Critical Review
Authors Adigwe OP , Onavbavba G , Onoja SO
Received 24 February 2023
Accepted for publication 8 June 2023
Published 14 August 2023 Volume 2023:16 Pages 3503—3515
DOI https://doi.org/10.2147/IJGM.S410015
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Obi Peter Adigwe,1 Godspower Onavbavba,1 Solomon Oloche Onoja2
1National Institute for Pharmaceutical Research and Development, Abuja, Federal Capital Territory, Nigeria; 2Department of Medical Laboratory Sciences, University of Nigeria, Enugu, Nigeria
Correspondence: Obi Peter Adigwe; Godspower Onavbavba, National Institute for Pharmaceutical Research and Development, Abuja, Federal Capital Territory, Nigeria, Email [email protected]; [email protected]
Abstract: Sickle cell disease is an autosomal recessive disorder of the beta-globin gene, with resultant deformation of the red blood cells and variable clinical outcomes. Nigeria is recognised as the country with the highest burden of sickle cell disease globally. This study aimed at critically reviewing available literature on impact of sickle cell disease in Nigeria. A literature search was carried out on four databases, and a total of 116 articles that met the inclusion criteria were included in the critical review. It was observed that majority of the studies were carried out in South-Western part of Nigeria (47.4%), whilst the North-East had the least number of studies undertaken in this area, more than a quarter of the studies (27.6%) were related to hematologic and serologic screening. Major themes that emerged from this review were morbidity and mortality; prevalence of sickle cell disease; issues relating to blood transfusion; psychosocial impact; and anatomical dysfunction in sickle cell disease. Intervention programs from both government and non-governmental organizations aimed at reducing the burden of sickle cell disease and its socio-economic impact were identified as key to strategies aimed at overcoming challenges associated with the disease. Findings from this study also revealed that education and awareness interventions were central to reducing the prevalence of sickle cell disease in this setting.
Keywords: sickle cell anemia, public health, hematology, genotype, hemoglogin, gene, red blood cell, vaso-occlusive crises
Introduction
Sickle cell disease is an autosomal recessive genetic disorder which occurs as a result of mutated beta-globin gene inherited from both parents.1,2 Hemoglobin SS genotype (Hb SS) is the most common type of sickle cell disease.3 Other familiar sickle cell genotypes include sickle hemoglobin C disease (Hb SC) and the beta thalassemia (Hb Sb° and Sb+). Rare variants such as hemoglobin SE, hemoglobin SD Los Angeles, and hemoglobin SG-Philadelphia have also been identified.4,5 Fully functional beta-globin arises from the normal wild-type allele of the β- globin gene (HbA). Over 400 mutant alleles have been identified and are known to give rise to variant hemoglobin or thalassemia.6 Out of these, the allele for sickle cell disease has attracted the greatest attention because of its frequency and the severity of sickle cell disease.7
The basic pathophysiology of sickle cell disease involves the de-oxygenation and polymerization of red blood cells, leading to deformation and hemolysis. This therefore gives rise to varieties of clinical manifestations such as acute pain episodes and anemia.8 The hallmark of sickle cell disease is vaso-occlusive crises, and it is the most common reason for frequent hospital visits for persons affected with the disease.9
Sickle cell disease has been recognized by the United Nations General Assembly as a disease of global public health concern. It is noteworthy that 20 to 25 million people are affected with sickle cell disease globally, out of which between 12 to 15 million reside in Africa, whilst developed countries only account for 10% cases.10 Sub-Saharan Africa accounts for the highest percentages of various indices associated with sickle cell disorder. It accommodates 75% of all patients with sickle cell disease and 70% of all sickle cell disease births globally, with some of the affected children dying before the age of 5 years.11 Nigeria is believed to be the most sickle cell endemic country in sub-Saharan Africa with between 2% and 3% of the total population affected.12 This study aimed at critically reviewing the impact of sickle cell disease in Nigeria, alongside relevant emergent issues. The critical review strategy can help assess the impact of sickle cell disease holistically and provide policy direction for government in improving the quality of care for individuals with the condition.
Methods
Data Sources and Search Strategy
A detailed literature search was undertaken to identify publications that focused on sickle cell disease in Nigeria. Relevant articles were searched, screened, and included in this study accordingly. Articles were searched through Web of Science, PubMed, Google Scholar, and African Journal Online using different combinations of keywords which includes “sickle cell disease”, “sickle cell anemia”, “Nigeria” “burden” and “impact”. All the four databases were carefully searched for only English-language full-text original articles published. Titles and abstracts of the retrieved documents were examined to identify those that were within the area of focus.
Review Selection
After the exclusion of duplicate literature, titles and abstracts of selected articles were carefully reviewed. All relevant full-text studies obtained were assessed for eligibility. Original articles from peer-reviewed scientific journals with primary data on various areas that focused on the impact of sickle cell disease in Nigeria were considered potentially eligible for inclusion in this review. All studies outside Nigeria were excluded.
Data Extraction and Analysis
Data were extracted from the selected studies into an Excel spreadsheet (Microsoft Office Excel 2013). The variables registered for each article were author, year of publication, name of the journal, and article title. Others include area of focus, geopolitical zone, method of data collection and age range of subjects. Data were then imported into Statistical Package for Social Sciences software version 25 for descriptive analysis, and results were summarized as figures.
Results
The literature search undertaken across various databases yielded 4812 articles that were related to sickle cell disease in Nigeria, and this covers between 1985 to 2021. After the elimination of duplicates, a total of 621 were retained. Following titles and abstracts screening, up to 178 full-text articles were eligible for consideration. Finally, a total number of 116 full-text articles (See Supplementary Table 1) which met the inclusion criteria were selected for critical review.1,3–9,11,13–118 Further details on the search strategy are presented in Figure 1.
 |
Figure 1 Review flow chart. |
Bibliometry
Sickle Cell Disease Studies by Geopolitical Zone
Figure 2 shows the various geopolitical zones with their respective percentage of studies on sickle cell disease in Nigeria. It was observed that a considerable proportion of the studies (55; 47.4%) were carried out in the South-West zone. A total of ten studies were carried out each in North-Central, North-West, and South-South, whilst 20 studies were recorded in South-East.
 |
Figure 2 Articles according to geopolitical zone. Abbreviations: NC, North Central; NW, North West; NE, North East; SS, South South; SW, South West; SE, South East. |
Findings in Figure 2 show that studies encompassing all the zones were relatively few as only two (1.7%) of the articles in this review captured all six geopolitical zones in their data collection process.
Sickle Cell Disease Studies by the Method of Data Collection
Figure 3 gives a graphical illustration of sickle cell disease studies by the method of data collection. It was observed that data from laboratory tests dominated under this category (49; 42.2%).
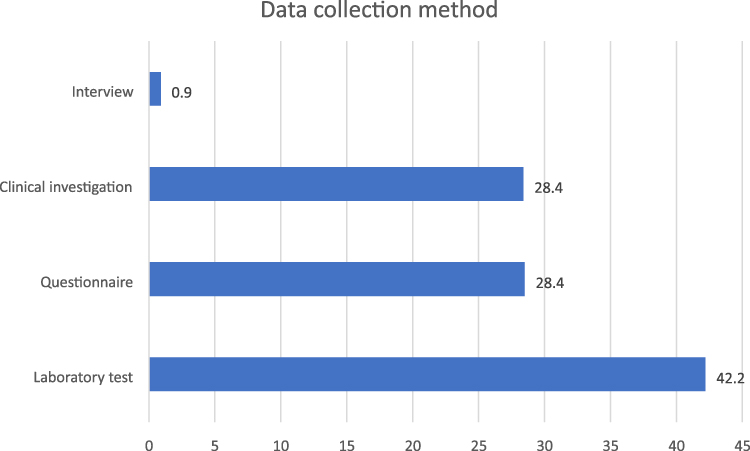 |
Figure 3 Sickle cell disease studies according to method of data collection. |
Findings in Figure 3 indicate that only one of the articles’ data collection process was underpinned by the use of interview. Studies involving questionnaires were of similar proportion with those that employed clinical investigation as their data collection strategy.
Sickle Cell Disease According to Age Group
All articles evaluated were classified according to three age groups which include adults, children, and all age groups. The studies in the category of “all age group” were those that targeted both adults and children, and findings in Figure 4 show that majority of the articles reviewed (62; 53.4%) fell under this category.
 |
Figure 4 Classification according to age group. |
Child mortality from sickle cell disease is high in Nigeria, and this has contributed to a substantial portion of the Country’s under 5 years mortality over the years. Findings in Figure 4, however, show that slightly above a quarter of the studies (32; 27.6%) were undertaken among children.
Data Driven Thematic Explorations of Sickle Cell Disease
Cardiovascular Effect of Sickle Cell Disease
Various articles in this review focused on the cardiovascular effect of sickle cell anemia. Oguanobi et al reported a marked increase in pulse rate and pulse pressure with a decrease in diastolic blood pressure and mean arterial blood pressure among patients with sickle cell disease.13 Lower diastolic blood pressure and increased pulse pressure have been attributed to hemodynamic circulation in chronic anemia.14 Sokunbi et al recommended cardiovascular examination for signs of pulmonary hypertension for children with sickle cell anemia due to increased risk of pulmonary hypertension.15 Furthermore, pulmonary dysfunction has been observed among adolescents and adults with sickle cell disease in Nigeria. This was also attributed to raised white blood cells, leading to a cascade of inflammatory events.16 VanderJagt et al suggested early nutritional intervention for children with sickle cell disease owing to reduced pulmonary function highlighted in their study.17
Heart rate variability and electrocardiogram reports had also been investigated among sickle cell disease patients. Adebiyi et al reported a reduction in heart rate variability among patients with vaso-occlusive crises.18 Significant electrocardiographic abnormalities were also reported among patients with sickle cell disease.19,20
Morbidity and Mortality in Sickle Cell Disease
Findings by Akingbola et al revealed varying degree of abdominal pain among sickle cell disease patients with multifactorial etiology.21 Some identified factors responsible for abdominal pain include retroperitoneal lymph node enlargement, bone marrow hyperplasia, hepatobiliary disease, splenic disorders, and mesenteric arterial thrombosis.22,119
Mortality in sickle cell anemia patients was linked to bacterial infection, necessitating the need for proper treatment with appropriate antibiotics.23 Some bacteria commonly associated with mortality in sickle cell anemia include Streptococcus pneumonia, Salmonella species, and Hemophilus influenza.24,120
Effect of sickle cell disease on pregnant women had also been explored; Afolabi et al reported high maternal mortality in women with HbSS.25 Ugboma and George highlighted malaria and vaso-occlusive crisis as the most common complications encountered in pregnancy.26 Other relevant issues and complications documented include emergency cesarean section, low birth weight, and fetal mortality.27
Pattern of Sickle Cell Disease
Several studies explored the prevalence and pattern of sickle cell disease in Nigeria. Taiwo et al reported a prevalence of 2.4% for HbSS genotype in South-Western Nigeria.7 Similarly, Stephen et al reported a gradual increase in the prevalence rate of sickle cell disease in Jos, Nigeria, in 2013 and 2014.11 On the contrary, the case fatality rate for sickle cell disease gradually decreased from 15.4% in 2012 to 11.1% in 2013 and 10.3% in 2014. Other researchers that studied children in North Central Nigeria identified 5% prevalence rate of sickle cell disease among the study population.28 However, Kingsley et al revealed a decline in prevalence of sickle cell disease from 2011 with the least prevalence reported in the years 2016 and 2017.29
Hematological and Serological Screening
Some studies reported a marked decrease in hemoglobin concentration and packed cell volume among patients with sickle cell disease.30–32 Decreased mean cell hemoglobin and mean cell volume had also been reported among people with this condition.33 Low serum zinc levels as well as increased copper levels were prominent among patients with sickle cell disease.34
A study among children affected with sickle cell disease revealed a decrease in Myeloperoxidase and Malondialdehyde.35 In a study undertaken by Akinbami et al, it was observed that serum ferritin was higher in males than females.36 Similarly, Odunlade et al reported increased serum ferritin levels among patients with sickle cell disease and this was attributed to iron overload.37
Studies on analysis of electrolyte activities and kidney function among sickle cell disease patients also revealed a marked decrease in sodium, potassium, creatinine, and urea, and this was indicative of chronic kidney disease.8,38 Additionally, increased levels of alkaline phosphatase had been noted among people with sickle cell, and this was believed to be associated with vaso-occlusive crises involving the bones.90
Sickle Cell Disease and Blood Transfusion
The chances of acquiring infections are higher when blood is not properly screened prior to transfusion. Also, evidence suggests that blood transfusion services in some public hospitals had fallen short of best practices.39 There were reported cases of human immunodeficiency virus infections following blood transfusion among patients with sickle cell disease.40 Blood transfusion was identified as a significant risk factor for the development of post-transfusion hepatitis in sickle cell anemia.41–45 Furthermore, complications of red cell alloimmunization in multi‑transfused patients with sickle cell anemia had also been highlighted as major challenges of blood transfusion in patients with sickle cell disease.46,47,121
Psychosocial Impact of Sickle Cell Disease
Interaction between patients, their disease condition, and social environment can lead to maladjustment which could provoke psychosocial dysfunctions.48 Previous studies had reported cases of psychosocial problems in children suffering from sickle cell anemia, as well as their caregivers, and it was suggested that psychosocial management should focus more on families rather than affected individuals.49,50 Available evidence also suggests that family members of patients with sickle cell disease suffer considerable financial, interpersonal, and psychological challenges due to the high cost of living up with sickle cell disease.48 Previous findings, however, indicate that such challenges can significantly be reduced by external financial support from both government and non-governmental organizations.51,52
Anatomical Dysfunction in Sickle Cell Disease
Varying degrees of anatomical dysfunctions are reportedly associated with sickle cell disease. Nwadiaro et al identified Staphylococcus aureus as the leading cause of chronic osteomyelitis in patients with sickle cell disease.53 The first decade of life was reported as the peak age for incidence of chronic osteomyelitis.54 Osteonecrosis also constituted anatomic dysfunctions in sickle cell disease with multifactorial etiology.55 Factors associated with increased predisposition to osteonecrosis of the femoral head are euglobulin clot lysis time, hemoglobin level, and co-existence of other variants of abnormal hemoglobin like alpha-thalassemia with hemoglobin SS genotype.122 Iwegbu and Fleming (1985) revealed the most susceptible age for avascular necrosis, as between 6 and 15 years, and they also indicated that females were more prone to avascular necrosis, compared to males.56
Musculoskeletal complications of sickle cell disease have also been explored in the extant literature. Onyemaechi et al and Balogun et al studied the spectrum of musculoskeletal disorders in sickle cell disease.1,57 They revealed musculoskeletal complications as major root causes of morbidity and disability in people affected with the disease. They also highlighted bacterial infections of bones and joints as a precipitating factor for osteomyelitis and septic arthritis.
Discussion
Interesting findings have emerged from this critical review based on the methodological approach adopted. Majority of the studies were undertaken in the South-Western part of Nigeria, and this was followed by the South-East zone. North-East had the least proportion of articles, and this may be attributed to the current unrest caused by insurgency which has been ongoing for over a decade in the region.123,124 The ongoing issues relating to the terrorism and insecurity in the North-East may have constituted access and implementation challenges for scientists interested in working in that part of the country. This consequently would reduce research output.
Findings from this review revealed that data utilized for majority of the studies were collected via laboratory tests. Only one out of the reviewed studies adopted a qualitative approach through the use of interviews. This suggests that knowledge gaps may exist, especially in areas where interpretivist epistemologies are best suited to improve practice.125 Qualitative data collection can encourage communication on critical issues which may be difficult to express in quantitative form. Interpersonal interview has been found to increase confidence as well as clinical outcomes for patients.49 It is, therefore, critical for more qualitative research be considered in undertaking studies that aim at better understanding sickle cell disease, with a view to improving its management.
A considerable proportion of the studies focused on hematological and serological screening, and this could be due to the fact that sickle cell disease is primarily a blood disorder.31,58 Similarly, significant problems associated with white blood cells, platelets, and coagulation in sickle cell anemia condition could be responsible for the extensive research in this area.30,59 Another factor that could have contributed to the preponderance of research in this area could be the routine serologic evaluations of sickle cell patients’ body electrolytes, liver enzymes, as well as micro and macro nutrients.4,34,60–62,90
Sickle cell disease poses a serious burden to Nigeria’s health system and the society at large.63 In this study, various pathophysiologic effects associated with this condition were identified. Findings revealed relatively lower arterial blood pressure in patients with sickle cell anemia.13,18 Furthermore, indicators such as hematocrit, frequency of crisis, body mass index, and body surface area emerged as significant determinants of blood pressure issues for people affected with the disease.16,64 Additionally, peripheral vascular resistance was identified as a determinant of blood pressure in patients with severe anemia.14,16
Findings from this study suggest the presence of electrocardiographic abnormalities among patients with sickle cell disease.19,20 Electrocardiographic abnormalities observed among these individuals were attributed largely to chronic anemia and vaso-occlusion.18 Chronic anemia was believed to be responsible for increased cardiac output with a minimal increase in heart rate.14 Progressive vasculopathy occurring as a result of inflammatory cytokines and oxidative stress was reported to be associated with sickling and intravascular hemolysis, and this also contributed to progressive cardiac lesions with subsequent abnormal electrocardiographic readings.16
There were indications from this review that sickle cell disease had significant effect on pregnancy, as severe complications were reported among pregnant women which were accompanied by increased fetal and maternal mortality.27 Importantly, pain crisis, urinary tract infection, low birth weight, retained placenta and pre-eclampsia were identified as some of the common complications during pregnancy.26 It is interesting to note that prophylactic transfusion had been shown to reduce the frequency of painful crises in pregnant women.25
Morbidity and mortality patterns in hospitalized patients with sickle cell disease indicated vaso-occlusive crisis, abdominal pain, hyper-hemolytic crisis, and acute splenic sequestration, as common clinical presentations. Other parameters identified, include septicemia, acute osteomyelitis, pneumonia, priapism, urinary tract infection, and septic arthritis.23,59,126 These findings imply the need for development of a contextual framework that emphasises early diagnosis, intensive counseling, and appropriate antibiotic prophylaxis for affected individuals.65,127
Evidence from this study also suggests a high level of sexual dysfunction among male patients with sickle cell disease. Priapism was identified as a common sexual organ dysfunction seen among male patients with sickle cell disease between 18 and 20 years of age.62,66 Risk factors predisposing patients to priapism were unclear,66 suggesting the need for further research in this area.
This study further revealed a musculoskeletal crisis in sickle cell disease, acute osteomyelitis was seen in children with sickle cell disease, and the tibia bone had been identified as the most common site.127 This may also be one of the contributing factors to low life expectancy in children with sickle cell disease. Furthermore, leg ulcer was observed to be common among persons suffering from sickle cell anemia.67,68 Ulceration was believed to occur due to thrombi formation in small capillaries, which consequently results to ischemia. This phenomenon is further promoted by up-regulation of integrins by micro-thrombin which promotes platelets aggregation and adherence to the endothelium.4,60 Also, the release of injurious cytokines, as well as conditions such as thrombocytosis and antithrombin deficiency, were identified as other factors that may promote ulceration.69
Therapeutic red blood cell transfusion remains a cardinal intervention in the management of sickle cell disease. However, blood transfusion could also introduce immune alloantibodies in transfused individuals, with resultant clinical consequences such as hemolytic transfusion reactions and incompatibility crisis.47 The prevalence of alloantibodies in transfused patients with sickle cell disease is high in Nigeria.46,121 Likewise, a high prevalence of transfusion transmissible infections had been reported.40–44 This high prevalence can be attributed to poor screening modalities for blood and blood products in some healthcare facilities. Proper screening of blood before transfusion is therefore critical in reducing this occurrence. Additionally, awareness and educational interventions that improve knowledge regarding voluntary blood donation and screening for blood transmissible infections need to be encouraged.42,45
Findings from this study revealed some psychosocial impacts of sickle cell disease.70 Societal attitudes and perceptions were identified as triggers for major psychosocial issues among sickle cell disease patients. Impaired psychosocial health-related quality of life had been associated with a number of negative effects, including low self-esteem; anxiety and depression; a loss of interest in basic life activities.71,72 The need for active involvement of social workers in the overarching healthcare for persons with sickle cell disease cannot therefore be over emphasized. Government, non-governmental organizations, and other stakeholders involved in sickle cell care must also synergize efforts in improving psychosocial and socioeconomic support for individuals with sickle cell disease. This is especially fundamental in improving access to health and quality of life for persons with sickle cell disease.73,74 Several of the clinical issues observed in this review can be invaluable in contributing to the revision of the 2014 national guideline for the control and management of sickle cell disease.128
Current interventions for sickle cell disease in Nigeria were reported to be suboptimal, and this was mainly attributed to a lack of relevant infrastructure and equipment in this area.129,130 In order to reduce the burden of sickle cell disease in Nigerian setting, there is a need to improve quality of care alongside intensifying relevant campaigns for prevention of the disease.131,132
Limitation and Strength
The possible limitation of this review is associated with the search strategy that was limited to the title, keywords and abstract of each article. More in-depth search could have perhaps resulted in identification of more studies. Despite this potential weakness, the review adopted a robust method of analysis through the use of a combination of both descriptive statistical analysis and narrative analysis in the synthesis of various outcomes, and presentation of relevant findings. Further research to determine relevant interventions to cushion the relevant impacts identified in this review can be invaluable in reducing the burden of sickle cell disease in Nigeria.
Conclusion
This study adopted a novel approach to critically review issues relating to sickle cell disease and its health impact on affected individuals in Nigeria. Majority of the articles in this review were undertaken in the South-Western part of the Country, and a considerable proportion of them involved laboratory investigations. Findings from the study revealed that sickle cell disease had both health and social consequences. The disease was identified as a major healthcare issue in Nigeria, given that the country had highest prevalence of sickle cell disease globally, while also contributing to significantly high mortality rates of children under 5 years of age. Key aspects of disease reduction and elimination strategies for sickle cell disorder in Nigeria include continuous enlightenment programmes; social awareness campaigns; and premarital genetic counselling about the condition.
It is also critical to establish more sickle cell clinics with adequate state of the art equipment and well-trained personnel for prompt diagnosis and treatment of various clinical manifestations of sickle cell disease. Further health system-wide measures that can help control and eliminate the disease include free routine genetic screening as well as psychosocial and financial support for persons with sickle cell disease. It is important for sickle cell patients to have access to critical services like stroke risk screening with transcranial doppler ultrasound and pulmonary hypertension risk with echocardiography. Also, it is important to prioritise access to relevant affordable therapies such as hydroxyurea, niprisan, prophylactic antibiotics, antimalarials and other medications. A synergistic implementation of these interventions identified in the study will not only help reduce sickle cell morbidity and mortality for Nigerians with sickle cell disease, they would also make a significant contribution towards controlling and elimination of the disease. The innovative study design adopted by the study enabled the identification of thematic areas, geopolitical zones, methodological approaches and disease specifics neglected by the extant literature. Further robust research in these critical areas can yield strong contextual evidence that will underpin comprehensive strategies for sustainable and impactful policy and practice reforms.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Onyemaechi NO, Enweani UN, Maduka CO. Musculoskeletal complications of sickle cell disease in Enugu, Nigeria. Niger J Med. 2011;20(4):456–461.
2. Onoja SO, Eluke BC, Dangana A, Musa S, Abdullahi IN. Evaluation of von Willebrand factor and other coagulation homeostasis profile of patients with sickle cell anaemia attending a tertiary hospital at Enugu, Nigeria. Med J Zambia. 2020;47(4):269–275. doi:10.55320/mjz.47.4.715
3. Abdullahi SU, DeBaun MR, Jordan LC, Galadanci NA. Increased prevalence of stroke recurrence and stroke related mortality in children with sickle cell disease in Nigeria: evidence for a secondary stroke prevention trial. Pediatr Neurol. 2019;95:73–78. doi:10.1016/j.pediatrneurol.2019.01.008
4. Smith OS, Ajose OA, Adegoke SA, Adegoke OA, Adedeji TA, Oderinu KA. Plasma level of antioxidants is related to frequency of vaso-occlusive crises in children with sickle cell anaemia in steady state in Nigeria. Pediatr Hematol Oncol J. 2019;4(1):17–22. doi:10.1016/j.phoj.2019.03.003
5. Oladipo OO, Temiye EO, Ezeaka VC, Obomanu P. Serum magnesium, phosphate and calcium in Nigerian children with sickle cell disease. West Afr J Med. 2005;24(2):120–123. doi:10.4314/wajm.v24i2.28180
6. Maigatari DM, Abdulrasheed I, Lawal DI, Hassan A, Augustine B, Muhammad PK. Preoperative blood transfusion in adults with sickle cell disease undergoing elective surgical procedures: a survey of practice and outcome in Zaria, Nigeria. Arch Int Surg. 2017;7:17–21. doi:10.4103/ais.ais_45_17
7. Taiwo IA, Oloyede OA, Dosumu AO. Frequency of sickle cell genotype among the Yorubas in Lagos: implications for the level of awareness and genetic counseling for sickle cell disease in Nigeria. J Community Genet. 2011;2:13–18. doi:10.1007/s12687-010-0033-x
8. Bukar AA, Sulaiman MM, Ladu AI, et al. Chronic kidney disease amongst sickle cell anaemia patients at the university of Maiduguri teaching hospital, northeastern Nigeria: a study of prevalence and risk factors. Mediterr J Hematol Infect Dis. 2019;11(1):e2019010. doi:10.4084/MJHID.2019.010
9. Nwabuko OC, Okoh DA, Iyalla C, Omunakwe H. Prevalence of sickle cell disease among pregnant women in a tertiary health center in south-south Nigeria. Sub Saharan Afr J Med. 2016;3:132–136. doi:10.4103/2384-5147.190843
10. Aygun B, Odame I. A global perspective on sickle cell disease. Pediatr Blood Cancer. 2012;59:386–390. doi:10.1002/pbc.24175
11. Stephen N, Nden N, Gusen NJ, Kumzhi PR, Gaknung B, Auta DA. Prevalence of sickle cell disease among children attending plateau specialist hospital, Jos, Nigeria. Acta Med Int. 2018;5:20–23. doi:10.4103/ami.ami_60_17
12. Nwongoh B, Adewoyin AS, Iheanacho OE, Bazuaye GN. Prevalence of haemoglobin variants in Benin City, Nigeria. Ann Biomed Sci. 2012;2(2):60–64.
13. Oguanobi NI, Onwubere BJ, Ibegbulam OG, et al. Arterial blood pressure in adult Nigerians with sickle cell anemia. J Cardiol. 2010;56:326–331. doi:10.1016/j.jjcc.2010.07.001
14. Juwah AI, Nlemadim EU, Kaine W. Types of anaemic crises in paediatric patients with sickle cell anaemia seen in Enugu, Nigeria. Arch Dis Child. 2014;89:572–576. doi:10.1136/adc.2003.037374
15. Sokunbi OJ, Ekure EN, Temiye EO, Anyanwu R, Okoromah CAN. Pulmonary hypertension among 5 to 18 year old children with sickle cell anaemia in Nigeria. PLoS One. 2017;12(9):e0184287. doi:10.1371/journal.pone.0184287
16. Ozoh OB, Kalejaiye OO, Eromesele OE, Adelabu YA, Dede SK, Ogunlesi FO. Pulmonary dysfunction among adolescents and adults with sickle cell disease in Nigeria: implications for monitoring. Ann Thorac Med. 2019;14(4):269–277. doi:10.4103/atm.ATM_58_19
17. VanderJagt DJ, Trujillo MR, Jalo I, Bode-Thomas F, Glew RH, Agabac P. Pulmonary function correlates with body composition in Nigerian children and young adults with sickle cell disease. J Pediatr. 2007;54(2):87–93. doi:10.1093/tropej/fmm070
18. Adebiyi AA, Oyebowale OM, Olaniyi AJ, Falase AO. Heart rate variability study in adult Nigerian subjects with sickle cell disease during vaso-occlusive crisis. Niger Postgrad Med J. 2019;6:8–12. doi:10.4103/npmj.npmj_186_18
19. Oguanobi NI, Onwubere BJ, Ike SO, Anisiuba BC, Ejim EC, Ibegbulam OG. Electocardiographic findings in adult Nigerians with sickle cell anaemia. Afr Health Sci. 2010;10(3):235–241.
20. Dosunmu A, Akinbami A, Uche E, Adediran A, John-Olabode S. Electrocardiographic Study in adult homozygous sickle cell disease patients in Lagos, Nigeria. J Trop Med. 2016;2016:1–5. doi:10.1155/2016/4214387
21. Akingbola TS, Kolude B, Aneni EC, et al. Abdominal pain in adult sickle cell disease patients: a Nigerian Experience. Ann Ibd Pg Med. 2011;9(2):100–104.
22. Jebbin NJ, Adotey JM. Acute abdominal conditions in people with sickle cell disease: a 10-year experience in PortHarcourt, Nigeria. Ann Afr Med. 2011;10:165–170. doi:10.4103/1596-3519.82072
23. Brown BJ, Jacob NE, Lagunju IA, Jarrett OO. Morbidity and mortality pattern in hospitalized children with sickle cell disorders at the University College Hospital, Ibadan, Nigeria. Niger J Paed. 2013;40(1):34–39. doi:10.4314/njp.v40i1.6
24. Bello N, Dawakin Kudu AT, Adetokun AB, et al. Characterization and antimicrobial susceptibility profile of bacteraemia causing pathogens isolated from febrile children with and without sickle cell disease in Kano, Nigeria. Mediterr J Hematol Infect Dis. 2018;10(1):e2018016. doi:10.4084/MJHID.2018.016
25. Afolabi BB, Iwuala NC, Iwuala IC, Ogedengbe OK. Morbidity and mortality in sickle cell pregnancies in Lagos, Nigeria: a case control study. J Obstet Gynaecol. 2009;29(2):104–106. doi:10.1080/01443610802667112
26. Ugboma HA, George IO. Sickle cell disease in pregnancy: maternal and fetal outcome in Port Harcourt, Nigeria. Br J Med Med Res. 2015;7(1):40–44. doi:10.9734/BJMMR/2015/11602
27. Ocheni S, Onah HE, Ibegbulam OG, Eze MI. Pregnancy outcomes in patients with sickle cell disease in Enugu, Nigeria. Niger J Med. 2007;16(3):227–230.
28. Diwe K, Iwu AC, Uwakwe K, et al. Prevalence and patterns of sickle cell disease among children attending tertiary and non-tertiary health care institutions in a South Eastern State, Nigeria: a 10 year survey. J Res Med Dent Sci. 2016;4(3):75–81. doi:10.5455/jrmds.2016432
29. Kingsley A, Enang O, Essien O, Legogie A, Cletus O, Oshatuy O. Prevalence of sickle cell disease and other haemoglobin variants in Calabar, Cross River State, Nigeria. Annu Res Rev Biol. 2019;13:1–6. doi:10.9734/ARRB/2019/v33i530132
30. Salawu L, Orimolade EA, Durosinmi MA. Immuno-haematological characteristics of Nigerian sickle cell disease patients in asymptomatic steady state. Eur J Gen Med. 2009;6(3):170–174.
31. Akinbami A, Dosunmu A, Adediran A, Oshinaike O, Adebola P, Arogundadeet O. Haematological values in homozygous sickle cell disease in steady state and haemoglobin phenotypes AA controls in Lagos, Nigeria. BMC Res Notes. 2012;5:396. doi:10.1186/1756-0500-5-396
32. Akinbami A, Dosunmu A, Adediran A, et al. Steady state hemoglobin concentration and packed cell volume in homozygous sickle cell disease patients in Lagos, Nigeria. Caspian J Intern Med. 2012;3(2):405–409.
33. Omoti CE. Haematological values in sickle cell anaemia in steady state and during vaso-occlusive crisis in Benin City, Nigeria. Ann Afr Med. 2005;4:62–67.
34. Bot YS, Benjamin A, Nyango DY, et al. Analyses of Cu and Zn in serum of sickle cell disease patients in Jos. Int J Med Sci. 2015;3(3):207–209.
35. Adelakun A, Ajani O, Ogunleye T, Disu E, Kosoko A, Arinola G. Respiratory burst enzymes and oxidant antioxidant status in Nigerian Children with Sickle Cell Disease. Br Biotechnol J. 2014;4(3):270–278. doi:10.9734/BBJ/2014/7411
36. Akinbami AA, Dosunmu AO, Adediran AA, et al. Serum ferritin levels in adults with sickle cell disease in Lagos, Nigeria. J Blood Med. 2013;4:59–63. doi:10.2147/JBM.S42212
37. Odunlade OC, Adeodu OO, Owa JA, Obuotor EM. Iron overload in steady state, non-chronically transfused children with sickle cell anaemia in Ile-Ife, Nigeria. Pediatr Hematol Oncol J. 2017;2(2):35–38. doi:10.1016/j.phoj.2017.08.001
38. Ibe EO, Ezeoke AC, Emeodi I, Akubugwo EI, Elekwa E, Ugonabo MC. Electrolyte profile and prevalent causes of sickle cell crisis in Enugu, Nigeria. Afr J Biochem. 2009;3(11):370–374.
39. Diaku-Akinwumi IN, Abubakar SB, Adegoke SA, et al. Blood transfusion services for patients with sickle cell disease in Nigeria. Int Health. 2016;8(5):330–335. doi:10.1093/inthealth/ihw014
40. Ubesie AC, Emodi IJ, Ikefuna AN, Ilechukwu GC, Ilechukwu G. Prevalence of human immunodeficiency virus transmission among transfused children with sickle cell anemia in Enugu Nigeria. Ann Med Health Sci Res. 2012;2:109–113. doi:10.4103/2141-9248.105655
41. Fasola FA, Otegbayo IA. Post transfusion viral hepatitis in sickle cell anemia: retrospective-prospective analysis. Niger J Clin Pract. 2002;5(1):16–19.
42. Ejiofor OS, Ibe BC, Emodi IJ, et al. The role of blood transfusion on the prevalence of hepatitis c virus antibodies in children with sickle cell anemia in Enugu, south east Nigeria. Niger J Clin Pract. 2009;12(4):355–358.
43. Nwannadi IA, Alao OO, Bazuaye GN, Omoti CE, Hali NK. Seroprevalence of hepatitis c virus antibody in sickle cell anaemia patients in Benin-city, Nigeria. GJMS. 2012;10(1):2.
44. Babatola AO, Olatunya OS, Faboya AO, et al. Hepatitis B and C infections among pediatric patients with sickle cell disease at a tertiary hospital in Nigeria. Arch Pediatr Infect Dis. 2020;8(4):e101632. doi:10.5812/pedinfect.101632
45. Bolarinwa RA, Aneke JC, Olowookere SA, Salawu L. Seroprevalence of transfusion transmissible viral markers in sickle cell disease patients and healthy controls in Ile-Ife, SouthWestern Nigeria: a case–control study. J Appl Hematol. 2015;6(4):162–167. doi:10.4103/1658-5127.171985
46. Ugwu NI, Awodu OA, Bazuaye GN, Okoye AE. Red cell alloimmunization in multi- transfused patients with sickle cell anemia in Benin City, Nigeria. Niger J Clin Pract. 2015;18:522–526. doi:10.4103/1119-3077.154204
47. Kangiwa U, Ibegbulam O, Ocheni S, Madu A, Mohammed N. Pattern and prevelence of alloimmunization in multiply transfused patients with sickle cell disease in Nigeria. J Blood Med. 2015;3(1):59–63. doi:10.2147/JBM.S42212
48. Ojelabi AO, Bamgboye AE, Ling J. Preference-based measure of health-related quality of life and its determinants in sickle cell disease in Nigeria. PLoS One. 2019;4(11):e0223043. doi:10.1371/journal.pone.0223043
49. Tunde-Ayinmode MF. Children with sickle cell disease who are experiencing psychosocial problems concurrently with their mothers: a Nigerian study. Afr J Psychiatry. 2011;14:392–401. doi:10.4314/ajpsy.v14i5.8
50. Aloba O, Eyiolawi D. Correlates of hopelessness in clinically stable Nigerian adults with sickle cell disease. Niger J Clin Pract. 2020;23:219–225. doi:10.4103/njcp.njc_119_19
51. Ohaeri JU, Shokunbi WA. Psychosocial burden of sickle cell disease on caregivers in a Nigerian setting. J Natl Med Assoc. 2002;94(12):1058–1070.
52. Adegoke SA, Kuteyi EA. Psychosocial burden of sickle cell disease on the family, Nigeria. Afr J Prm Health Care Fam Med. 2012;4(1). doi:10.4102/phcfm.v4i1.380
53. Nwadiaro HC, Ugwu BT, Legbo JN. Chronic osteomyelitis in patients with sickle cell disease. East Afr Med J. 2000;77(1):23–26. doi:10.4314/eamj.v77i1.46370
54. Ebong WA. Acute osteomyelitis in Nigerians with sickle cell disease. Ann Rheum Dis. 1986;45:911–915. doi:10.1136/ard.45.11.911
55. Akinyoola AL, Adediran IA, Asaleye CM, Bolarinw AR. Risk factors for osteonecrosis of the femoral head in patients with sickle cell disease. Int Orthop. 2009;33:923–926. doi:10.1007/s00264-008-0584-1
56. Iwegbu CG, Fleming AF. Avascular necrosis of the femoral head in sickle cell disease. A series from the Guinea Savannah of Nigeria. J Bone Joint Surg. 1985;67(1):29–33. doi:10.1302/0301-620X.67B1.3968138
57. Balogun RA, Obalum DC, Giwa SO, Adekoya-Cole TO, Ogo CN, Enweluzo GO. Spectrum of musculo-skeletal disorders in sickle cell disease in Lagos, Nigeria. J Orthop Surg Res. 2010;5:1–6. doi:10.1186/1749-799X-5-2
58. Buseri FI, Jeremiah ZA, Shokunbi WA. Plasma levels ofsome blood coagulationparameters in Nigerian homozygous sickle cell patients (HbSS) in steady state. Hematology. 2006;11(5–6):375–379. doi:10.1080/10245330600841287
59. Kotila R, Okesola A, Makanjuola O. Asymptomatic malaria parasitaemia in sickle-cell disease patients: how effective is chemoprophylaxis? J Vect Borne Dis. 2007;44:52–55.
60. Okocha CE, Manafa PO, Ozomba JO, Ulasi TO, Chukwuma GO, Aneke JC. C‑reactive protein and disease outcome in Nigerian sickle cell disease patients. Ann Med Health Sci Res. 2014;4(5):701–705. doi:10.4103/2141-9248.141523
61. Fakunle EE, Eteng KI, Shokunb WA. D-D dimer levels in patients with sickle cell disease during bone pain crises and in the steady state. PLMI. 2012;4:21–25. doi:10.2147/PLMI.S29393
62. Abudu EK, Akanmu SA, Soriyan OO, et al. Serum testosterone levels of HbSS (sickle cell disease) male subjects in Lagos, Nigeria. BMC Res Notes. 2011;4:298. doi:10.1186/1756-0500-4-298
63. Akinbami AA, Ajibola S, Bode-Shojobi I, et al. Prevalence of significant bacteriuria among symptomatic and asymptomatic homozygous sickle cell disease patients in a tertiary hospital in Lagos, Nigeria. Niger J Clin Pract. 2014;17:163–167. doi:10.4103/1119-3077.127441
64. Adeniyi AF, Saminu KS. Non communicable diseases local incentive spirometry improves peak expiratory flow rate in teenage sickle cell anaemia patients: a randomized pilot trial. Afr Health Sci. 2011;11(3):303–308.
65. Oniyangi O, Ahmed P, Otuneye OT, et al. Strokes in children with sickle cell disease at the National Hospital Abuja Nigeria. Niger J Paed. 2013;40(2):158–164. doi:10.4314/njp.v40i2.10
66. Adediran A, Wright K, Akinbami A, Dosunmu A, Oshinaike O, Osikomaiya B. Prevalence of priapism and its awareness amongst male homozygous sickle cell patients in Lagos, Nigeria. Adv Urol. 2013;2013:1–4. doi:10.1155/2013/890328
67. Hassan A, Gayus DL, Abdulrasheed I, Umar MA, Ismail DL, Babadoko AA. Chronic leg ulcers in sickle cell disease patients in Zaria, Nigeria. Arch Int Surg. 2014;4:141–145. doi:10.4103/2278-9596.146405
68. Bazuaye GN, Nwannadi AI, Olayemi EE. Leg ulcers in adult sickle cell disease patients in Benin City Nigeria. GJMS. 2010;8(2):190–194.
69. Adegoke SA, Smith OS, Akinlosotu MA. Total oxidant status of children with sicklecell anaemia: correlation with rate of pain episodes and haematological indices. Pediatr Hematol Oncol J. 2018;3(3):70–73. doi:10.1016/j.phoj.2018.10.002
70. Iliyasu Z, Borodo AM, Jibir BW, Nass NS, Aliyu MH. “A child with sickle cell disease can’t live with just anyone.” A mixed methods study of socio-behavioral influences and severity of sickle cell disease in northern Nigeria. Health Sci Rep. 2020;4:e222. doi:10.1002/hsr2.222
71. Uchendu UO, Ikefuna AN, Nwokocha AR, Emodi IJ. Impact of socioeconomic status on sexual maturation of Nigerian boys living with sickle cell anaemia. Hematolog. 2010;15(6):414–421. doi:10.1179/102453310X12647083621209
72. Olatunya OS, Babatola AO, Adeniyi AT, et al. Determinants of care-seeking practices for children with sickle cell disease in Ekiti, Southwest Nigeria. J Blood Med. 2021;2:123–132. doi:10.2147/JBM.S294952
73. Adegboyega LO. Psycho-social problems of adolescents with sickle-cell anaemia in Ekiti State, Nigeria. Afr Health Sci. 2021;21(2):775–781. doi:10.4314/ahs.v21i2.37
74. Nwagha T, Omotowo BI. Determinants of psychosocial health‑related quality of life of adults with sickle cell disease in a Nigerian setting. Niger Med J. 2020;61:114–119. doi:10.4103/nmj.NMJ_122_19
75. Ayoola OO, Bolarinwa RA, Onakpoya UU, Adedeji TA, Onwuka CC, Idowu BM. Intima-media thickness of the common femoral artery as a marker of leg ulceration in sickle cell disease patients. Blood Adv. 2018;2(22):3112–3117. doi:10.1182/bloodadvances.2018023267
76. Okafor LA, Nonnoo DC, Ojehanon PI, Aikhionbare O. Oral and dental complications of sickle cell disease in Nigerians. Angiology. 1986;37(9):672–675. doi:10.1177/000331978603700909
77. Alabi S, Ernest K, Eletta P, Owolabi A, Afolabi A, Suleiman O. Otological findings among Nigerian children with sickle cell anaemia. Int J Pediatr Otorhinolaryngol. 2008;72(5):659–663. doi:10.1016/j.ijporl.2008.01.024
78. Agholor CA, Akhigbe AO, Atalabi OM. The prevalence of cholelithiasis in Nigerians with sickle cell disease as diagnosed by ultrasound. Br J Med Med Res. 2014;4(15):2866. doi:10.9734/BJMMR/2014/8645
79. Olaniyi JA, Abjah UM. Frequency of hepatomegaly and splenomegaly in Nigerian patients with sickle cell disease. West Afr J Med. 2007;26(4):274–277. doi:10.4314/wajm.v26i4.28326
80. Brown BJ, Fatunde OJ, Sodeinde O. Correlates of steady-state haematocrit and hepatosplenomegaly in children with sickle cell disease in Western Nigeria. West Afr J Med. 2012;31(2):86–91.
81. Odunvbun ME, Adeyekun AA. Ultrasonic assessment of the prevalence of gall stones in sickle cell disease children seen at the University of Benin Teaching Hospital, Benin City, Nigeria. Niger J Paediatr. 2014;41(4):370–374. doi:10.4314/njp.v41i4.16
82. Eze CU, Offordile GC, Agwuna KK, Ocheni S, Nwadike IU, Chukwu BF. Sonographic evaluation of the spleen among sickle cell disease patients in a teaching hospital in Nigeria. Afr Health Sci. 2015;15(3):949–958. doi:10.4314/ahs.v15i3.32
83. VanderJagt DJ, Okolo SN, Rabasa AI, Glew RH. Bioelectrical impedance analysis of the body composition of Nigerian children with sickle cell disease. J Trop Pediatr. 2000;46(2):67–72. doi:10.1093/tropej/46.2.67
84. Dosunmu AO, Akinola RA, Onakoya JA, et al. Pattern of chronic lung lesions in adults with sickle cell disease in Lagos, Nigeria. Caspian J Intern Med. 2013;4(4):754.
85. Luntsi G, Eze CU, Ahmadu MS, Bukar AA, Ochie K. Sonographic evaluation of some abdominal organs in sickle cell disease patients in a tertiary health institution in Northeastern Nigeria. J Med Ultrasound. 2018;26(1):31. doi:10.4103/JMU.JMU_5_17
86. Madu AJ, Okoye AE, Ajuba IC, Madu KA, Anigbo C, Agu K. Prevalence and associations of symptomatic renal papillary necrosis in sickle cell anemia patients in South‑Eastern Nigeria. Niger J Clin Pract. 2016;19(4):471–474. doi:10.4103/1119-3077.183299
87. Babadoko AA, Ibinaye PO, Hassan A, et al. Autosplenectomy of sickle cell disease in Zaria, Nigeria: an ultrasonographic assessment. Oman Med J. 2012;27(2):121. doi:10.5001/omj.2012.25
88. Oredugba FA, Savage KO. Anthropometric findings in Nigerian children with sickle cell disease. Pediatr Dent. 2002;24(4):321–325.
89. Arinola OG, Olaniyi JA, Akiibinu MO. Evaluation of antioxidant levels and trace element status in Nigerian sickle cell disease patients with Plasmodium parasitaemia. Pak J Nutr. 2008;7(6):766–769. doi:10.3923/pjn.2008.766.769
90. Nnodin JK, Opara AU, Nwanjo HU, Ibeaja OA. Plasma lipid profile in sickle cell disease patients in Owerri, Nigeria. Pak J Nutr. 2012;11(1):64–65. doi:10.3923/pjn.2012.64.65
91. Iheanacho O. Haematological parameters of adult and paediatric subjects with sickle cell disease in steady state, in Benin City, Nigeria. Int Blood Res Rev. 2015;3(4):171–177. doi:10.9734/IBRR/2015/18339
92. Olaniyi JA, Arinola OG, Odetunde AB. Foetal haemoglobin (HbF) status in adult sickle cell anaemia patients in Ibadan, Nigeria. Ann Ib Postgrad Med. 2010;8(1):30–33. doi:10.4314/aipm.v8i1.63955
93. Olorunshola KV, Audu L. ABO (H) secretor status of sickle cell disease patients in Zaria, Kaduna State, Nigeria. Niger J Physiol Sci. 2013;28(1):29–34.
94. Brown BJ, Okereke JO, Lagunju IA, Orimadegun AE, Ohaeri JU, Akinyinka OO. Burden of health‐care of carers of children with sickle cell disease in Nigeria. Health Soc Care Community. 2010;18(3):289–295.
95. Aneke JC, Adegoke AO, Oyekunle AA, et al. Degrees of kidney disease in Nigerian adults with sickle-cell disease. Med Princ Pract. 2014;23(3):271–274. doi:10.1159/000361029
96. VanderJagt DJ, Shores J, Okorodudu A, Okolo SN, Glew RH. Hypocholesterolemia in Nigerian children with sickle cell disease. J Trop Pediatr. 2002;48(3):156–161. doi:10.1093/tropej/48.3.156
97. Kotila T, Adedapo K, Adedapo A, Oluwasola O, Fakunle E, Brown B. Liver dysfunction in steady state sickle cell disease. Ann Hepatol. 2005;4(4):261–263. doi:10.1016/S1665-2681(19)32049-6
98. Isah IZ, Udomah FP, Erhabor O, et al. Foetal haemoglobin levels in sickle cell disease (SCD) patients in Sokoto, Nigeria. Br J Med Health Sci. 2013;1(6):36–47.
99. Ukoha OM, Emodi IJ, Ikefuna AN, Obidike EO, Izuka MO, Eke CB. Comparative study of nutritional status of children and adolescents with sickle cell anemia in Enugu, Southeast Nigeria. Niger J Clin Pract. 2020;23(8):1079–1086. doi:10.4103/njcp.njcp_476_19
100. VanderJagt DJ, Trujillo MR, Bode-Thomas F, Huang YS, Chuang LT, Glew RH. Phase angle correlates with n-3 fatty acids and cholesterol in red cells of Nigerian children with sickle cell disease. Lipids Health Dis. 2003;2:1–8. doi:10.1186/1476-511X-2-2
101. Idris IM, Abba A, Galadanci JA, et al. Men with sickle cell disease experience greater sexual dysfunction when compared with men without sickle cell disease. Blood Adv. 2020;4(14):3277–3283. doi:10.1182/bloodadvances.2020002062
102. Ikefuna AN, Emodi IJ, Ocheni S. Clinical profile and home management of sickle cell-related pain: the Enugu (Nigeria) experience. Pediatr Hematol Oncol. 2009;26(5):309–312. doi:10.1080/08880010902754818
103. Madu AJ, Galadanci NA, Nalado AM, et al. Stroke prevalence amongst sickle cell disease patients in Nigeria: a multi-centre study. Afr Health Sci. 2014;14(2):446–452. doi:10.4314/ahs.v14i2.22
104. Dosunmu AO, Balogun TM, Adeyeye OO, et al. Prevalence of pulmonary hypertension in sickle cell anaemia patients of a tertiary hospital in Nigeria. Niger J Med. 2014;55(2):161. doi:10.4103/0300-1652.129661
105. Adeyemo T, Ojewunmi O, Oyetunji A. Evaluation of high performance liquid chromatography (HPLC) pattern and prevalence of beta-thalassaemia trait among sickle cell disease patients in Lagos, Nigeria. Pan Afr Med J. 2014;18. doi:10.11604/pamj.2014.18.71.4239
106. Akubuilo UC, Ayuk A, Ezenwosu OU, Okafor UH, Emodi IJ. Persistent hematuria among children with sickle cell anemia in steady state. Hematol Transfus Cell Ther. 2020;42:255–260. doi:10.1016/j.htct.2019.07.007
107. Lagunju I, Sodeinde O, Telfer P. Prevalence of transcranial Doppler abnormalities in Nigerian children with sickle cell disease. Am J Hematol. 2012;87(5):544–547. doi:10.1002/ajh.23152
108. Bolarinwa RA, Akinlade KS, Kuti MA, Olawale OO, Akinola NO. Renal disease in adult Nigerians with sickle cell anemia: a report of prevalence, clinical features and risk factors. Saudi J Kidney Dis Transpl. 2012;23(1):171–175.
109. Nnaji GA, Ezeagwuna DA, Nnaji IJ, Osakwe JO, Nwigwe AC, Onwurah OW. Prevalence and pattern of sickle cell disease in premarital couples in Southeastern Nigeria. Niger J Clin Pract. 2013;16(3):309–314. doi:10.4103/1119-3077.113452
110. Adegoke SA, Adeodu OO, Adekile AD. Sickle cell disease clinical phenotypes in children from South. Western, Nigeria. Niger J Clin Pract. 2015;18(1):95–101. doi:10.4103/1119-3077.146987
111. Duru A, Madu AJ, Okoye H, et al. Variations and characteristics of the various clinical phenotypes in a cohort of Nigerian sickle cell patients. Hematology. 2021;26(1):684–690. doi:10.1080/16078454.2021.1972242
112. Saganuwan SA. The pattern of sickle cell disease in sickle cell patients from Northwestern Nigeria. Therapeutics. 2016;8:CMT–S38164.
113. Isa H, Adegoke S, Madu A, et al. Sickle cell disease clinical phenotypes in Nigeria: a preliminary analysis of the Sickle Pan Africa Research Consortium Nigeria database. Blood Cells Mol Dis. 2020;84:102438. doi:10.1016/j.bcmd.2020.102438
114. Anie KA, Egunjobi FE, Akinyanju OO. Psychosocial impact of sickle cell disorder: perspectives from a Nigerian setting. Global Health. 2010;6:1–6. doi:10.1186/1744-8603-6-2
115. Ola B, Coker R, Ani C. Stigmatising attitudes towards peers with sickle cell disease among secondary school students in Nigeria. Int J Child Fam Stud. 2013;4(4):391–402. doi:10.18357/ijcyfs44201312693
116. Adeyemo TA, Ojewunmi OO, Diaku‐Akinwumi IN, Ayinde OC, Akanmu AS. Health related quality of life and perception of stigmatisation in adolescents living with sickle cell disease in Nigeria: a cross sectional study. Pediatr Blood Cancer. 2015;62(7):1245–1251. doi:10.1002/pbc.25503
117. Adewoyin S. Erythrocyte transfusion and alloimmunisation patterns among sickle cell disease patients, Benin City, Nigeria. Br J Med Med Res. 2016;11(10):1–8. doi:10.9734/BJMMR/2016/21755
118. Emokpae A, Kuliya-Gwarzo A. The association of transfusion status with antioxidant enzymes and malondialdehyde level in Nigerians with sickle cell disease. Asian J Transfus Sci. 2014;8(1):47. doi:10.4103/0973-6247.126692
119. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X
120. Bebe T, Odetoyin B, Bolarinwa R. Occurrence of multidrug-resistant uropathogens implicated in asymptomatic bacteriuria in adults with sickle cell disease in ile-ife, Southwest Nigeria. Oman Med J. 2020;35(2):e109. doi:10.5001/omj.2020.27
121. Adewoyin AS, Oyewale OA. Complications of allogeneic blood transfusion: current approach to diagnosis and management. Int Blood Res Rev. 2015;3(4):135–151. doi:10.9734/IBRR/2015/17874
122. Adekile AD, Gupta R, Yacoub F, Sinan T, Al-Bloushi M, Haider MZ. Avascular necrosis of the Hip in children with sickle cell disease and the high Hb F: magnetic resonance imaging findings and influence of alpha-thalassemia trait. Acta Haematol. 2001;105(1):27–31. doi:10.1159/000046529
123. Weeraratne S. Theorizing the expansion of the Boko Haram insurgency in Nigeria. Terror Polit Violenc. 2017;29(4):610–634. doi:10.1080/09546553.2015.1005742
124. Dunn G. The impact of the Boko Haram insurgency in Northeast Nigeria on childhood wasting: a double-difference study. Confl Health. 2018;12(1):1–2. doi:10.1186/s13031-018-0136-2
125. Williams R. The epistemology of knowledge and the knowledge process cycle: beyond the “objectivist” vs “interpretivist”. J Knowl Manag. 2008;12(4):72–85. doi:10.1108/13673270810884264
126. Ikemefuna AN, Emodi IJ. Hospital admission of patients with sickle cell anemia pattern and outcome in Enugu area of Nigeria. Niger J Clin Pract. 2007;10(1):24–29.
127. Ambe JP, Mava Y, Chama R, Farouq G, Machoko Y. Clinical features of sickle cell Anaemia in Northern Nigerian children. WAJM. 2012;31(2):81–85.
128. Federal Ministry of Health. National guideline for the control and management of sickle cell disease; 2014. Available from: http://scsn.com.ng/wp-content/uploads/2014/11/National-Guideline-for-The-Control-and-Management-of-Sickle-Cell-Disease.pdf.
129. Adigwe OP. Access to healthcare for people with sickle cell disease: views of healthcare professionals on policies and practices. Mol Genet Genomic Med. 2023;11(2):e2142. doi:10.1002/mgg3.2142
130. Galadanci N, Wudil BJ, Balogun TM, et al. Current sickle cell disease management practices in Nigeria. Int Health. 2014;6(1):23–28. doi:10.1093/inthealth/iht022
131. Adigwe OP, Onavbavba G, Onoja SO. Attitudes and practices of unmarried adults towards sickle cell disease: emergent factors from a cross sectional study in Nigeria’s capital. Hematology. 2022;27(1):488–493. doi:10.1080/16078454.2022.2059629
132. Adigwe OP. Knowledge and awareness of sickle cell disease: a cross sectional study amongst unmarried adults in Nigeria’s capital city. J Community Genet. 2022;13(6):579–585. doi:10.1007/s12687-022-00607-x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.