")
Back to Journals » OncoTargets and Therapy » Volume 13
Impact of Mutational Profile on the Management of Myeloproliferative Neoplasms: A Short Review of the Emerging Data
Authors Loscocco GG , Guglielmelli P, Vannucchi AM
Received 21 October 2020
Accepted for publication 18 November 2020
Published 1 December 2020 Volume 2020:13 Pages 12367—12382
DOI https://doi.org/10.2147/OTT.S287944
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Giuseppe G Loscocco, Paola Guglielmelli, Alessandro M Vannucchi
CRIMM, Centro di Ricerca e Innovazione per le Malattie Mieloproliferative, Azienda Ospedaliero-Universitaria Careggi, Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy
Correspondence: Alessandro M Vannucchi
CRIMM, Centro di Ricerca e Innovazione per le Malattie Mieloproliferative, Azienda Ospedaliero-Universitaria Careggi, Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy
Tel +390557948902
Fax +390557947688
Email [email protected]
Abstract: Philadelphia-chromosome negative myeloproliferative neoplasms (MPN) are a heterogeneous group of clonal hematopoietic stem cell disorders characterized by an increased risk of thrombosis and progression to acute myeloid leukemia. MPN are associated with driver mutations in JAK2, CALR and MPL which are crucial for the diagnosis and lead to a constitutive activation of the JAK-STAT signaling, independent of cytokine regulation. Moreover, most patients have concomitant mutations in genes involved in DNA methylation, chromatin modification, messenger RNA splicing, transcription regulation and signal transduction. These additional mutations may arise before, in the context of clonal hematopoiesis of indeterminate potential (CHIP), or after the acquisition of the driver mutation. The clinical phenotype of MPN results from complex interactions between mutations and host factors. The increased application of next-generation sequencing (NGS) techniques to a large series of patients with MPN has expanded the knowledge of mutational landscape and contributed to define the clinical significance of mutations. This molecular information is being increasingly used to refine diagnosis, risk stratification, monitoring of residual disease and response to treatment. ASXL1, SRSF2, EZH2, IDH1/IDH2 and U2AF1 mutations are associated with a more advanced disease and reduced overall survival in primary myelofibrosis (PMF), whereas spliceosome mutations in Polycythemia vera (PV) and essential thrombocythemia (ET) adversely affect both overall (SF3B1, SRSF2 in ET and SRSF2 in PV) and myelofibrosis-free (U2AF1, SF3B1 in ET) survival. This review discusses current knowledge of the molecular landscape of MPN, and how the availability of those molecular information may impact patient management.
Keywords: myeloproliferative neoplasms, polycythemia vera, essential thrombocythemia, myelofibrosis, JAK-STAT pathway, gene mutations
Introduction
The classic Philadelphia-chromosome negative myeloproliferative neoplasms (MPN) are a heterogeneous group of clonal hematopoietic stem cell diseases characterized by overproduction of one or more types of cells of the myeloid lineage.1 According to the 2016 World Health Organization (WHO) criteria, MPN include Polycythemia Vera (PV), Essential Thrombocythemia (ET) and myelofibrosis, which may be classified as primary (PMF) or secondary to PV and ET (post-PV-MF and post-ET-MF).2 This classification has been updated with the introduction of pre-fibrotic/early PMF, distinguishable from ET on the basis of bone marrow (BM) morphology, that has a higher tendency to develop overt MF and is characterized by a reduced overall survival compared to true ET3 (Table 1). The diagnostic criteria of PPV-MF and PET-MF were developed by the International Working Group for MPN Research and Treatment – IWG-MRT4,5 (Table 2). All MPN can transform into secondary acute myeloid leukemia, referred to as MPN-blast phase (BP), which is typically refractory to conventional chemotherapy and has a poor prognosis.6 The 15-year risk of leukemia is estimated at 2.1% to 5.3% for ET, 5.5% to 18.7% for PV and more than 20% for PMF whereas fibrotic progression rates of ET and PV, during a similar time interval, are estimated at 4% to 11% and 6% to 14%, respectively.7
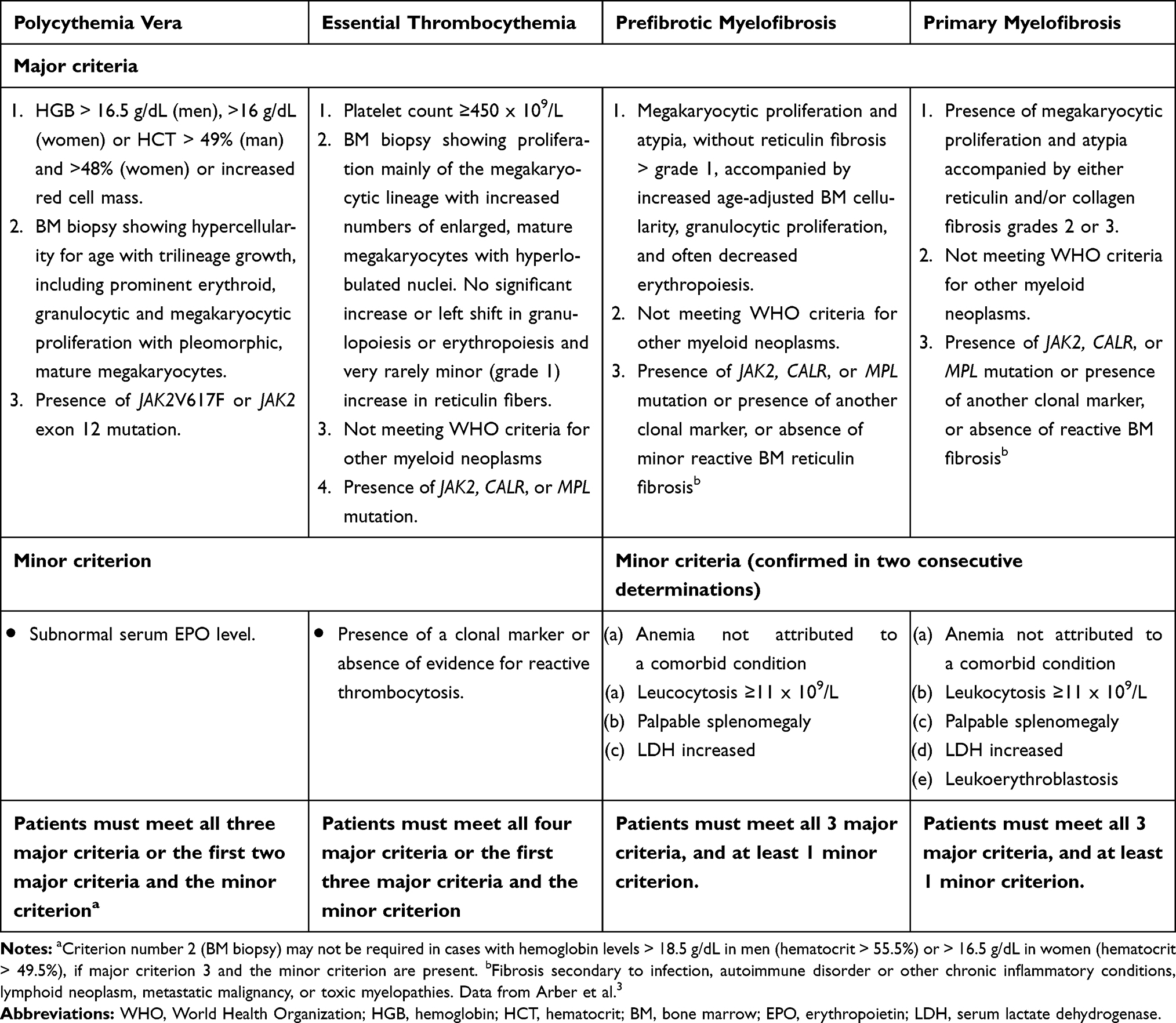 |
Table 1 Summary of WHO Criteria for MPN |
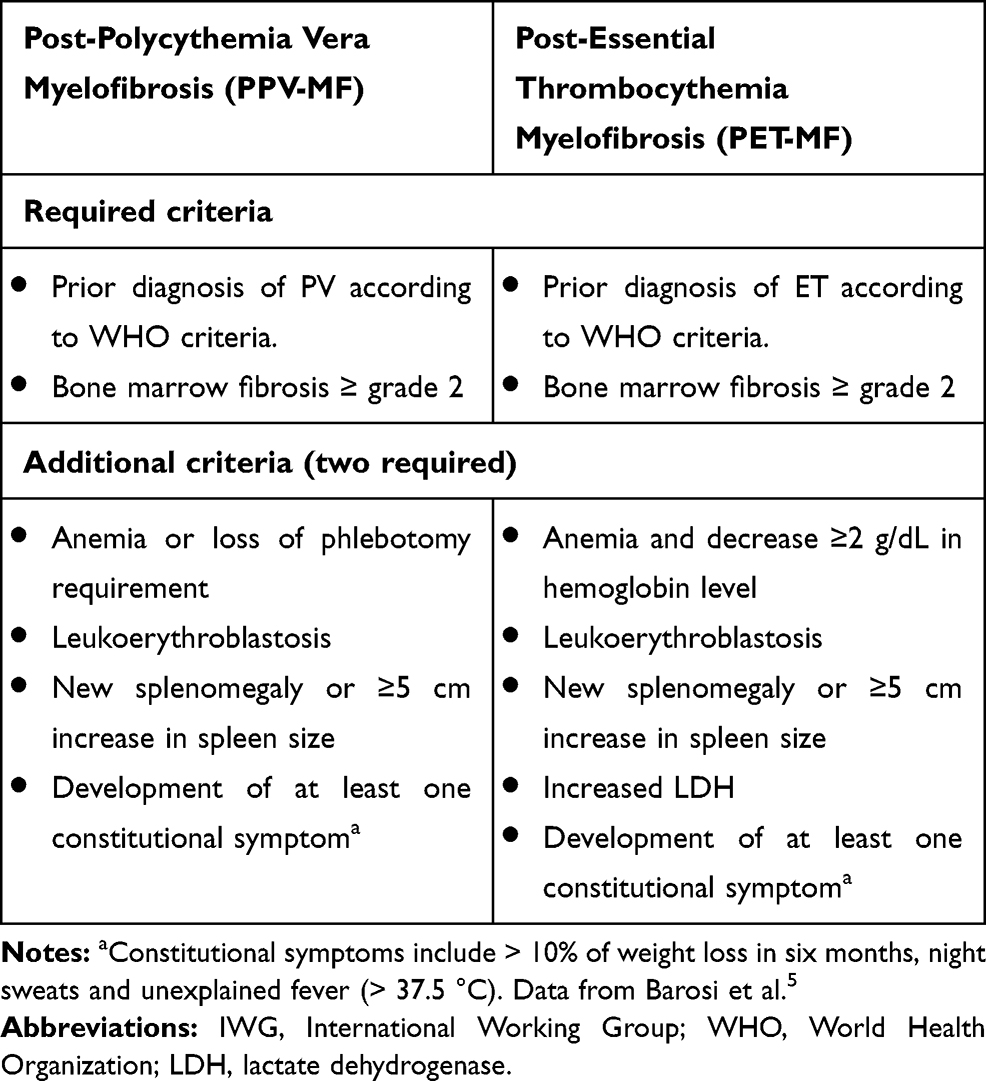 |
Table 2 Summary of IWG Criteria for PPV-MF and PET-MF |
In Europe, the incidence of MPN varies from 0.4 to 2.8/100.000 in PV, 0.38 to 1.7/100.000 in ET, and 0.1 to 1/100.000 in PMF, while prevalence remains difficult to determine.8 MPN have clinical heterogenicity, with some patients having normal lifespan and others developing disease progression or life-threatening complications. Median survival is around 20 years for ET, 14 years for PV and 6 years for PMF; the corresponding values for patients younger than 60 years are 33, 24 and 15 years.9 PV and ET are characterized by cardiovascular events, mainly thrombosis and less frequently hemorrhage, a varying burden of symptoms, and an intrinsic risk of evolution to MF or BP. Treatment is mainly focused on the reduction of thrombosis risk, control of myeloproliferation, improvement of symptoms, and management of related complications. MF is a protean disease with variable levels of cytopenias, constitutional symptoms, often as a result of a hypercytokinaemia, extramedullary hematopoiesis and marrow fibrosis. Splenomegaly is present at the varying extent and can cause abdominal pain, early satiety, splenic infarction, and portal hypertension. Progressive MF can be associated with marrow failure and evolution to BP. Treatment, both standard and experimental, is variably effective in reducing myeloproliferation, splenomegaly, constitutional symptoms and improving the quality of life. However, some patients do not respond to treatment, and others become resistant. Nowadays, the only curative approach is allogeneic hematopoietic stem cell transplant (allo-HSCT) which has a high mortality risk and is often not considered for older patients and those with comorbidities.
Mutational Landscape of MPN
Driver Mutations: JAK2, CALR and MPL
Constitutive activation of the JAK-STAT pathway is the hallmark of MPN. Mutations in JAK2, CALR and MPL are referred as “driver mutations”, based on their role in driving the MPN phenotype and are crucial for the diagnosis along with clinical and histopathological features. These three mutations are mutually exclusive, but cases of co-occurrence have been reported.10,11 Patients with features of MPN without any one of these mutations are classified as “triple negative”. Two types of JAK2 mutations are described in MPN. The first, discovered in 2005, is a valine to phenylalanine substitution at amino acid position 617 (V617F) in exon 14;12–15 the second, described in 2007, comprises many different mutations, particularly in-frame insertions or deletions, in exon 12 of JAK2.16,17 JAK2V617F is detected in approximately 95% of PV cases and 50–60% of ET and PMF. Conversely, JAK2 exon 12 mutations are extremely rare and are found exclusively in 2–3% of all PV patients, that are negative for JAK2V617F. JAK2V617F can drive all the different MPN phenotypes through ligand-independent activation of receptors for erythropoietin (EPO), thrombopoietin (TPO) and granulocyte-colony stimulating factor (G-CSF), resulting in the expansion of erythroid, megakaryocytic and granulocytic lineages, respectively.18–20 In comparison to V617F mutation, patients with exon-12 mutations predominantly activate EPO receptor and are characterized by erythroid‐dominant myeloproliferation with lower leukocytosis and thrombocytosis, and younger age at diagnosis.21,22 The different phenotypes of JAK2V617F mutated MPN may reflect different host characteristics, different development stages at which the mutation arises, the presence and order of other molecular variants and characteristics of bone marrow microenvironment. The timing of mutation acquisition may affect the clinical phenotype with the “JAK2-first” more commonly occurring in PV, and “TET2-first” more commonly in ET.23 Similar to TET2, patients are more likely to present with PV when JAK2V617F is acquired before DNMT3A mutation, compared with patients who first acquired DNMT3A mutation and more currently have an ET phenotype.24 Moreover, allele burden has been associated with different phenotypes; homozygous mutation and higher mutant allele burden (>50%) have been described in most PV patients, and associated with increased risk of thrombosis and fibrotic evolution, whereas ET patients tend to have a lower burden.25
CALR encodes for calreticulin, a 46-kDa chaperone protein located in endoplasmic reticulum (ER) lumen, which has a key role in the maintenance of calcium homeostasis and protein folding.26 To date, more than 50 different CALR mutations, all in exon 9, have been described in 20–25% of ET and 25–30% of PMF cases, but not in PV patients.27,28 Two CALR mutations account for approximately 80% of all the subtypes; type-1 is a 52-bp deletion (c.1092_1143del p.L367fs*46) and type-2 is a 5-bp insertion (c.1154_1155insTTGTC p.K385fs*47), resulting in mutant proteins that loss the ER-retention motif (KDEL) at the C-terminus. All the other CALR mutations are grouped as type 1-like and type 2-like in relation to their corresponding structural similarities and effect on C-terminal. CALR subtypes are associated with many clinical phenotypes and outcomes in MPN. In PMF, type-1 CALR mutations are more prevalent than type-2 (70% vs 13%), while in ET they are more balanced (51% vs 39%).29 Moreover, type-1 mutations have been associated with a significantly higher risk of myelofibrotic transformation in ET.30 In a large series of ET patients both CALR variants, compared to mutant JAK2, were associated with higher platelet and lower hemoglobin and leukocyte counts.31 The role of mutated CALR in driving the clinical phenotype of MPN has yet to be fully elucidated. Recently, some studies demonstrated that mutant CALR binds to MPL within the ER, resulting in the constitutive activation of MPL, and consequently of the JAK-STAT signaling.32–35 Moreover, MPL-CALR complex has been detected on the cell surface and this translocation appears essential for the oncogenic activity,36,37 raising the prospect for future target therapies.
MPL is the cell surface receptor for TPO, which regulates megakaryopoiesis and platelet production through the JAK-STAT signaling and is also expressed on the hematopoietic stem cells. MPL mutants largely account for the marked thrombocytosis in patients with ET and PMF.38 MPL gain of function mutations of tryptophan 515 (W515) in exon 10, located in the transmembrane domain of the protein, are described in 3–8% of ET and PMF cases.39 The most common mutations are W515L and W515K. Other rare mutations at the same position, as W515R, W515A and W515G have been reported.40 Although the MPLS505N allele was identified initially as an inherited mutation in familial thrombocythemia,41 it can also be acquired as a somatic event in ET patients. Moreover, in a recent report, several second-site mutations that enhance S505N-driven activation were described.42
Triple-Negative MPN
Mutations of JAK2, CALR, and MPL, account for over 90% of MPN cases and are usually mutually exclusive. However, in approximately 15% of patients with essential thrombocythemia (ET) and 8–10% of primary myelofibrosis (PMF), driver mutations are absent and these patients are referred as triple‐negative (TN).43 A small number of TN MPN patients acquired JAK2 mutations after some time from diagnosis; this acquisition may reflect the clonal expansion of a very-low burden mutation or false negative in initial testing. Approximately 10% of TN ET and PMF patients may have mutations outside of MPL exon 10 and JAK2 exon 14. These “non-canonical” MPL mutations include T119I, S204F/P and E230G in the extracellular domain and Y591D/N in the intracellular domain.44 Non-canonical JAK2 mutations include V625F, F556V, R683G and E627A.45 As demonstrated in functional studies, most of these rare variants led to constitutive activation of the JAK-STAT signaling.44,45 Some are somatic mutations, while others represent germline variants, with the possibility that many patients may have a form of non-clonal erythrocytosis or thrombocytosis with variable family penetrance.
Additional Mutations in Myeloid Genes
High-throughput next-generation sequencing (NGS) analyses have discovered a remarkable number of additional somatic mutations with prognostic and therapeutic relevance, particularly in PMF (Figure 1). More than 50% of MPN patients harbor additional mutations.46 These mutations are not restricted to MPN but are shared by other myeloid malignancies including acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). They are more frequent with increasing age, not only in patients with hematologic neoplasms but also in healthy individuals in the context of clonal hematopoiesis of indeterminate potential (CHIP), which is defined as a cell population associated with a recognized hematological neoplasm driver mutation at a variant allele frequency (VAF) ≥2%, in the absence of severe cytopenias or a WHO-defined disorder. Some studies demonstrated that CHIP predisposes to the development of a hematological malignancy and cardiovascular death, the latter probably resulting from proinflammatory interactions between endothelium and macrophages derived from clonal monocytes.47 Affected genes are involved in DNA methylation (TET2, DNMT3A, IDH1 and IDH2), histone modification (ASXL1, EZH2), mRNA splicing (SF3B1, SRSF2, U2AF1 and ZRSR2), signaling pathways (LNK/SH2B3, CBL, NRAS, KRAS and PTPN11), transcription factors (RUNX1, NFE2, PPM1D and TP53). In Table 3 main clinical features associated with recurrent additional mutations in MPN are summarized.
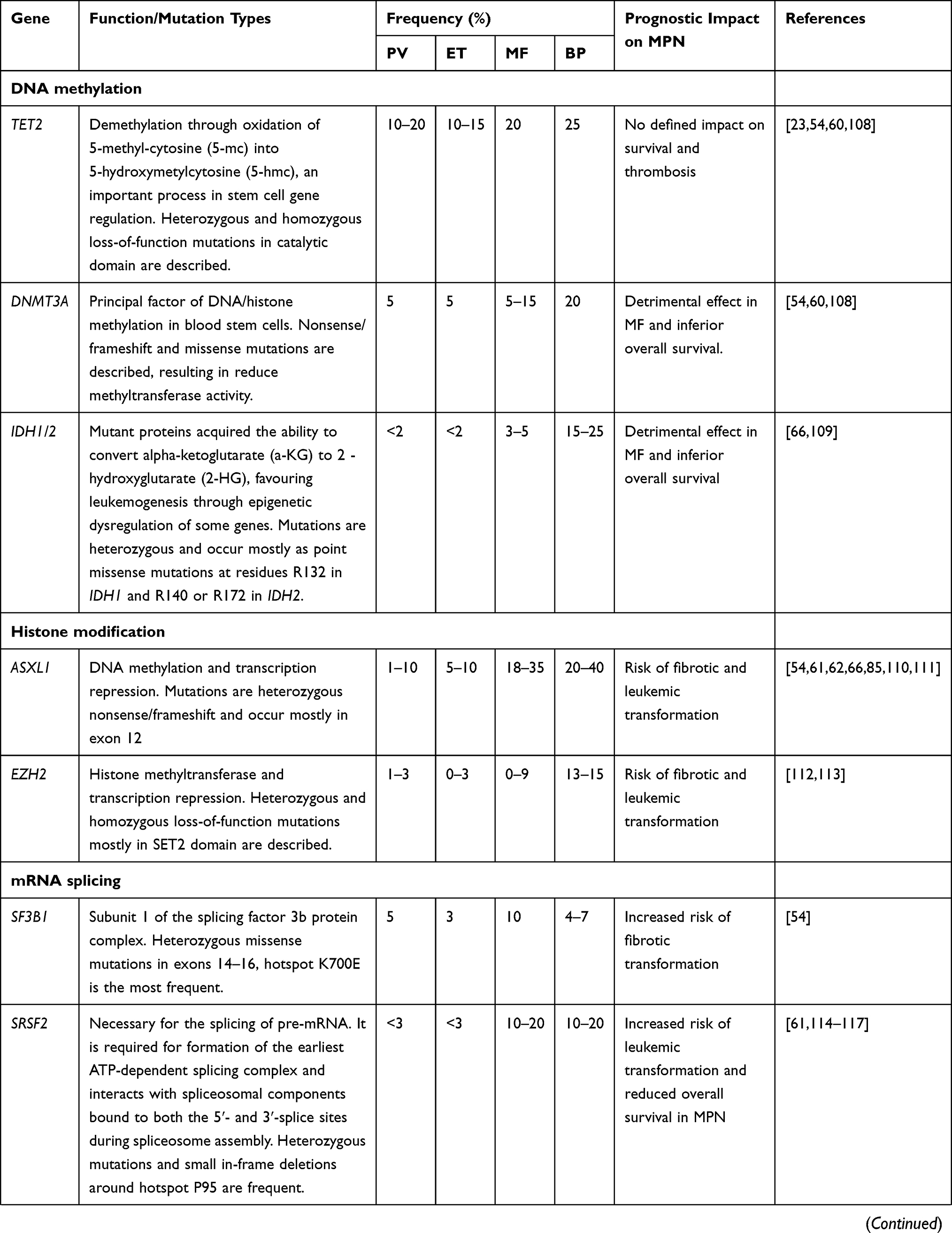 | 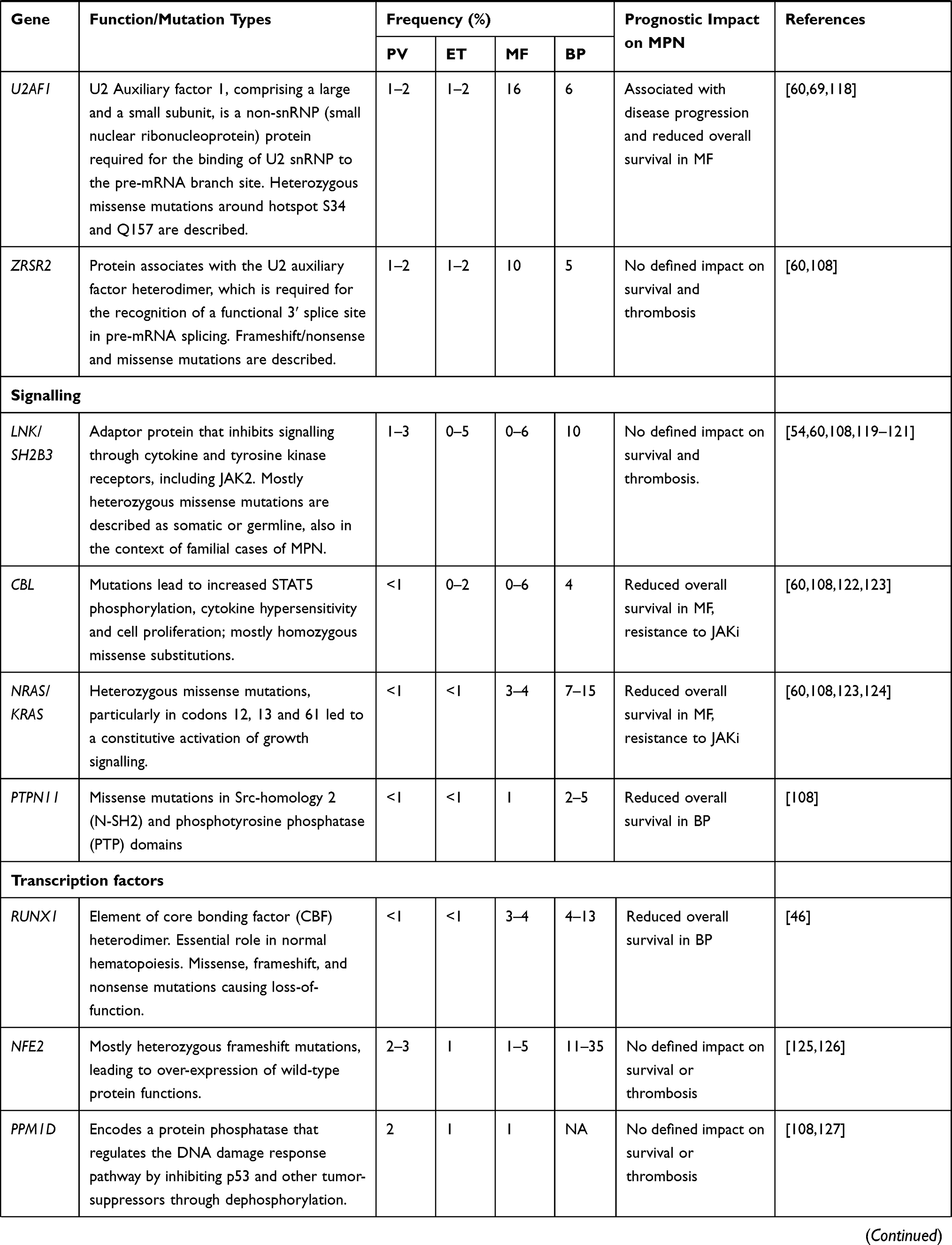 |  |
Table 3 Recurrent Additional Mutations in MPN |
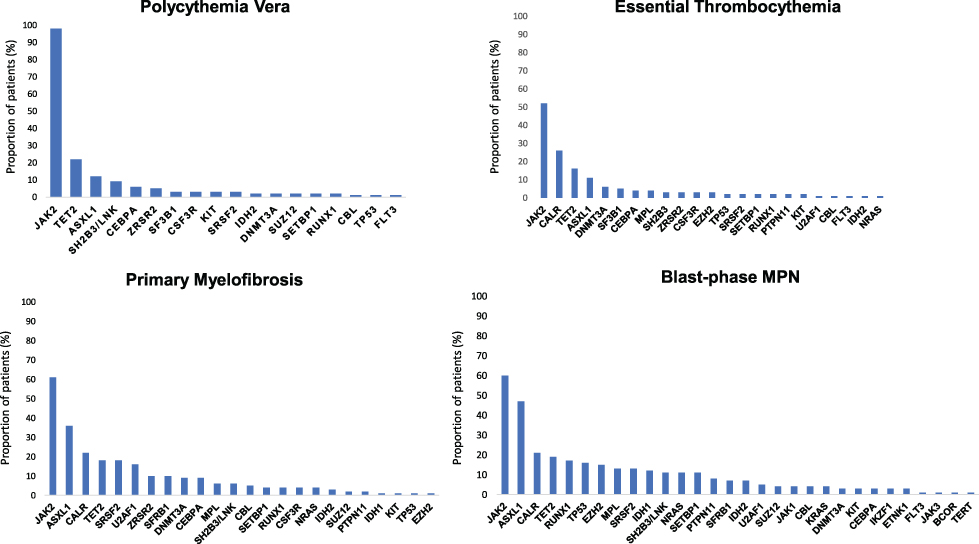 |
Figure 1 Mutational landscape of MPN. Results from a target next-generation sequencing analyses in patients with polycythemia vera, essential thrombocythemia, primary myelofibrosis and blast-phase MPN. The graphs show the proportion of patients for each corresponding mutation (both driver JAK2, CALR and MPL mutations and additional somatic mutations). Data from Tefferi et al54,60 and Lasho et al.108 |
Clinical and Molecular-Integrated Prognostic Scores in MPN
Polycythemia Vera and Essential Thrombocythemia
For many years, thrombosis prediction in PV and ET relied on clinical variables only (age >60 years and prior history of thrombosis). Recent findings, however, have highlighted the contribution of genetic information. While patients with PV almost exclusively carry mutations in JAK2, the effect of mutated JAK2 in patients with ET was compared with JAK2 wild-type ones, highlighting that JAK2 wild-type patients showed a lower risk of thrombosis.48,49 In 2012, these studies led to the development of a three-tiered International Prognostic Scoring of thrombosis in ET (IPSET-thrombosis) with the addition of JAK2 status and cardiovascular risk factors (2 and 1 point, respectively) to age and prior thrombosis (1 point each).50 The score was re-analyzed leading to a refined 4-tiered version, which excluded cardiovascular risk factors evaluation,51 (Table 4) and was validated in a large independent cohort of patients.52 Because of their very low-risk of thrombosis, ET CALR mutated young patients without a prior history of thrombosis and cardiovascular risk factors may not require aspirin. In these patients, as reported, aspirin might rather increase the risk of bleeding without improving the intrinsically low thrombotic risk.51,53
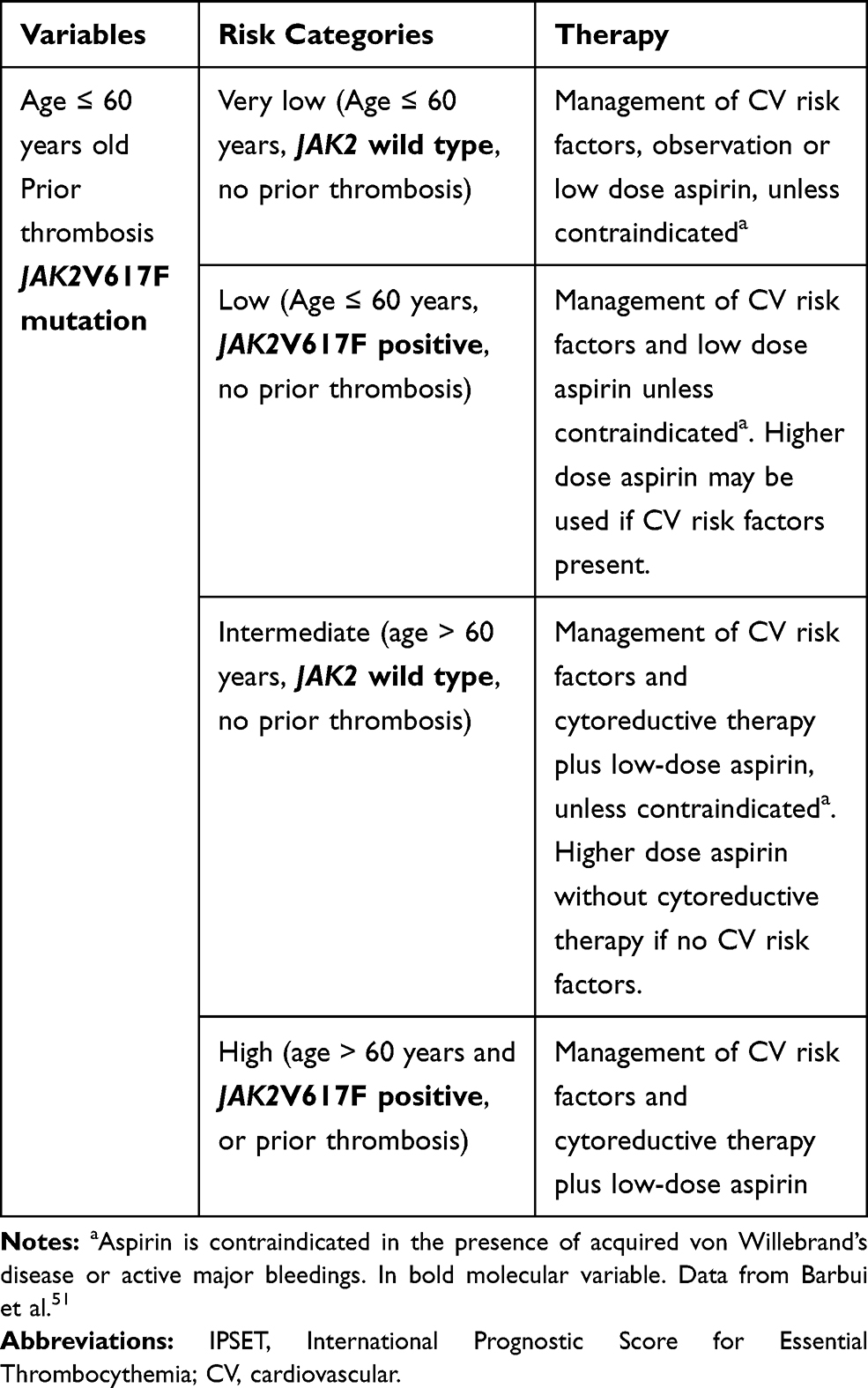 |
Table 4 Revised IPSET-Thrombosis Model for Essential Thrombocythemia |
Recently, the prognostic relevance of somatic mutations was investigated in large PV and ET cohorts of patients.54 Adverse molecular variants in PV included ASXL1, SRSF2, and IDH2, and in ET SH2B3/LNK, SF3B1, U2AF1, TP53, IDH2, and EZH2, based on age-adjusted multivariable analysis of the impact on overall, leukemia-free and myelofibrosis-free survival. Their presence was associated with inferior survival in both PV (median, 7.7 vs 16.9 years) and ET (median, 9 vs 22 years). The authors incorporated mutational information into a new prognostic model, the Mutation-Enhanced International Prognostic Scoring System (MIPSS) specific for PV and ET (Table 5). Further studies are necessary to validate these models and to determine their role in clinical decision-making.55
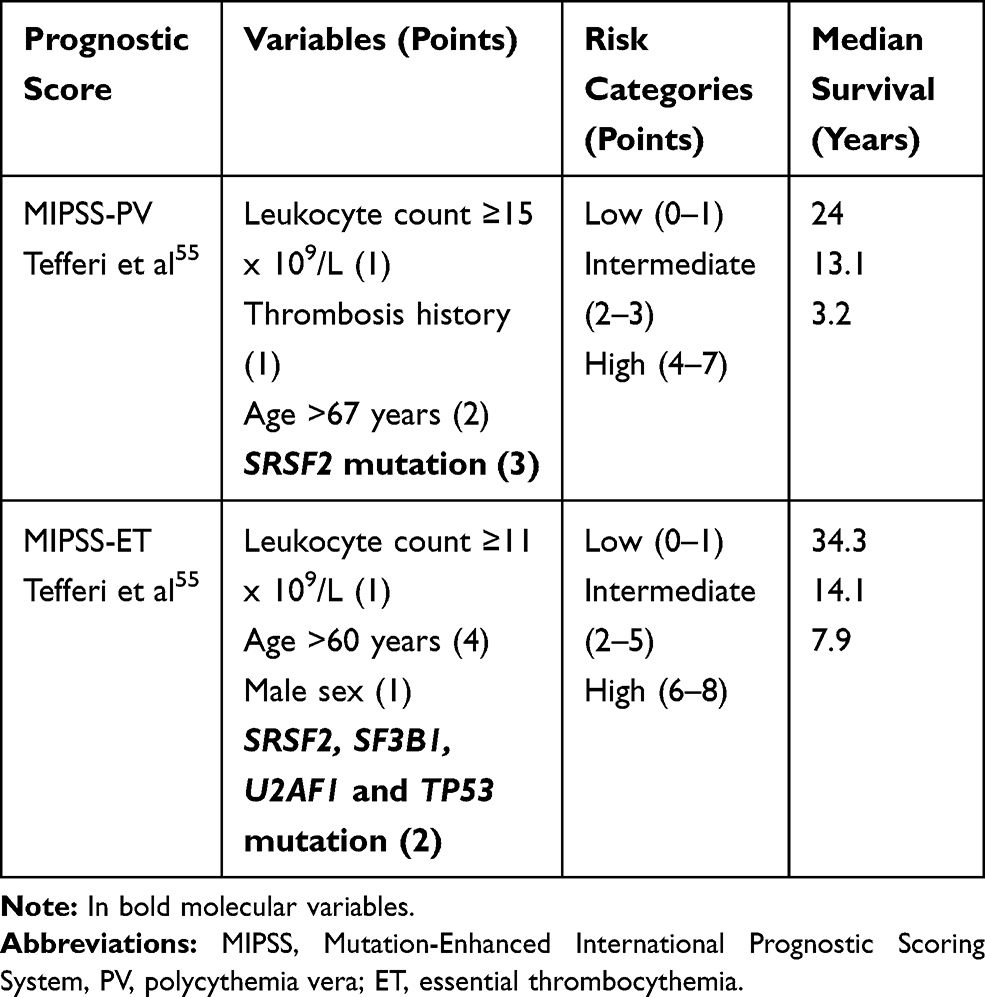 |
Table 5 Clinical-Molecular Prognostic Scores in Polycythemia Vera and Essential Thrombocythemia |
Primary and Post-PV/ET Myelofibrosis
Traditionally, two prognostic models, which include exclusively clinical parameters, are used to stratify PMF patients into risk categories with significant differences in overall survival: International Prognostic Scoring System (IPSS),56 that is used at the time of diagnosis, and Dynamic IPSS (DIPSS),57 applicable at any time during the clinical course. These models use five variables that independently predict inferior survival: age >65 years, hemoglobin <10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1%, and constitutional symptoms. Subsequently, DIPSS was revised to DIPSS plus,58 including red blood cells transfusion need, platelet count <100 × 109/L, and unfavorable karyotypes (complex karyotype or sole or 2 abnormalities that included +8, −7/7q-, i(17q), inv(3), −5/5q-, 12p- or 11q23 rearrangement).59 In addition to the driver mutations, more than 80% of patients with PMF harbor other DNA variants in myeloid genes, as discussed above, often in multiple combinations.60 Beyond their presumed pathogenetic relevance, driver and other mutations in PMF were shown to influence overall survival and leukemia-free survival, independent of IPSS and DIPSS-plus.61–63 The effect of driver mutations on outcome supports prognostic distinction based on the presence or absence of CALR type-1 mutation,64,65 whereas additional ASXL1, SRSF2, EZH2, and IDH1/IDH2 mutations were defined as high–molecular risk (HMR), with a prognostic relevance amplified by the number of such mutations in an individual patient.60,61,66 Building upon the complementary nature of molecular information, the Molecular enhanced International Prognostic Score Systems (three-tiered MIPSS70 and four-tiered MIPSS70-plus) were developed using a cohort of patients younger than 70 years, potentially eligible for allo-HSCT, recruited from multiple Italian centers and the Mayo Clinic.67 The MIPSS70-plus score additionally included unfavorable karyotype defined by any abnormal karyotype other than normal karyotype or sole abnormalities of 20q-, 13q-, +9, chromosome 1 translocation/duplication, -Y, or sex chromosome abnormality other than -Y. Leukocytosis and BM fibrosis grade ≥2 were included in MIPSS70 but not retained in MIPSS70-plus. Both models predict LFS and OS, but MIPSS70-plus seemed to have the best performance in identifying a very high-risk category of patients, 23% of whom developed acute leukemia, due to additional cytogenetic abnormalities. Further revision, the MIPSS70 v2.0,68 was published incorporating a new HMR mutation U2AF1Q157.69 Additional refinement of risk categories was provided with different thresholds for anemia (defined severe by hemoglobin levels of <8 g/dL in women and <9 g/dL in men, and moderate by hemoglobin levels of 8 to 9.9 g/dL in women and 9 to 10.9 g/dL in men)70 and a new three-tiered cytogenetic risk distribution with the introduction of very high risk (VHR) group for those with single/multiple abnormalities of −7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23, or other autosomal trisomies not including + 8/+ 9.71
These updates were also used for the development of a Genetically Inspired Prognostic Scoring System (GIPSS) that is exclusively based on genetic (ASXL1, SRSF2, U2AF1Q157, absence of type-1 CALR mutations) and cytogenetic markers.72 The authors demonstrated the non-inferiority of GIPSS in discrimination ability and prediction accuracy, compared to MIPSS70-plus and DIPSS. These data were confirmed in an external validation cohort showing also that GIPSS score performs equally well for both primary and secondary myelofibrosis.73 Primarily, all these scores were developed exclusively for patients with a diagnosis of PMF and their performance in patients with secondary MF was suboptimal. Therefore, a specific Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) was developed. This four-tiered score includes the following variables: hemoglobin level, circulating blasts, platelet count, age, presence of constitutional symptoms and CALR mutational status.74 Finally, to overcome the shortcoming of clinical scores for MF in the setting of HSCT and to accurately predict outcome following transplantation, the Myelofibrosis Transplant Scoring System (MTSS) was recently formulated for patients with PMF and post-PV-ET MF. The score variables incorporated were age (≥57 years), Karnofsky performance status, platelet and leukocyte count prior to transplantation (<150x109/L and >25x109/L, respectively), HLA-mismatched unrelated donor, CALR/MPL mutation and ASXL1 mutational status.75 The last to be developed, based on sequencing effort of 69 genes in more than 2000 patients with MPN, including 309 with myelofibrosis, identified a prognostic role for CBL, NRAS, RUNX1, TET2 and P53 and a contribution from GNAS, IDH2 and U2AF1 in both overall survival and leukemia-free survival. This study integrated a remarkable number of demographics, clinical and genomic variables and through the random effects modelling, led to the creation of a personalized predictive individual model for disease transformation and death at each phase into a single multi-stage model.46 A prognostic calculator of individualized patient outcome is available online (https://jg738.shinyapps.io/mpn_app/). An interactive web application is similarly available for MYSEC-PM (http://www.mysec-pm.eu/) and MIPSS70 (http://mipss70score.it). Table 6 provides a comprehensive overview of the clinical-molecular prognostic scoring systems described.
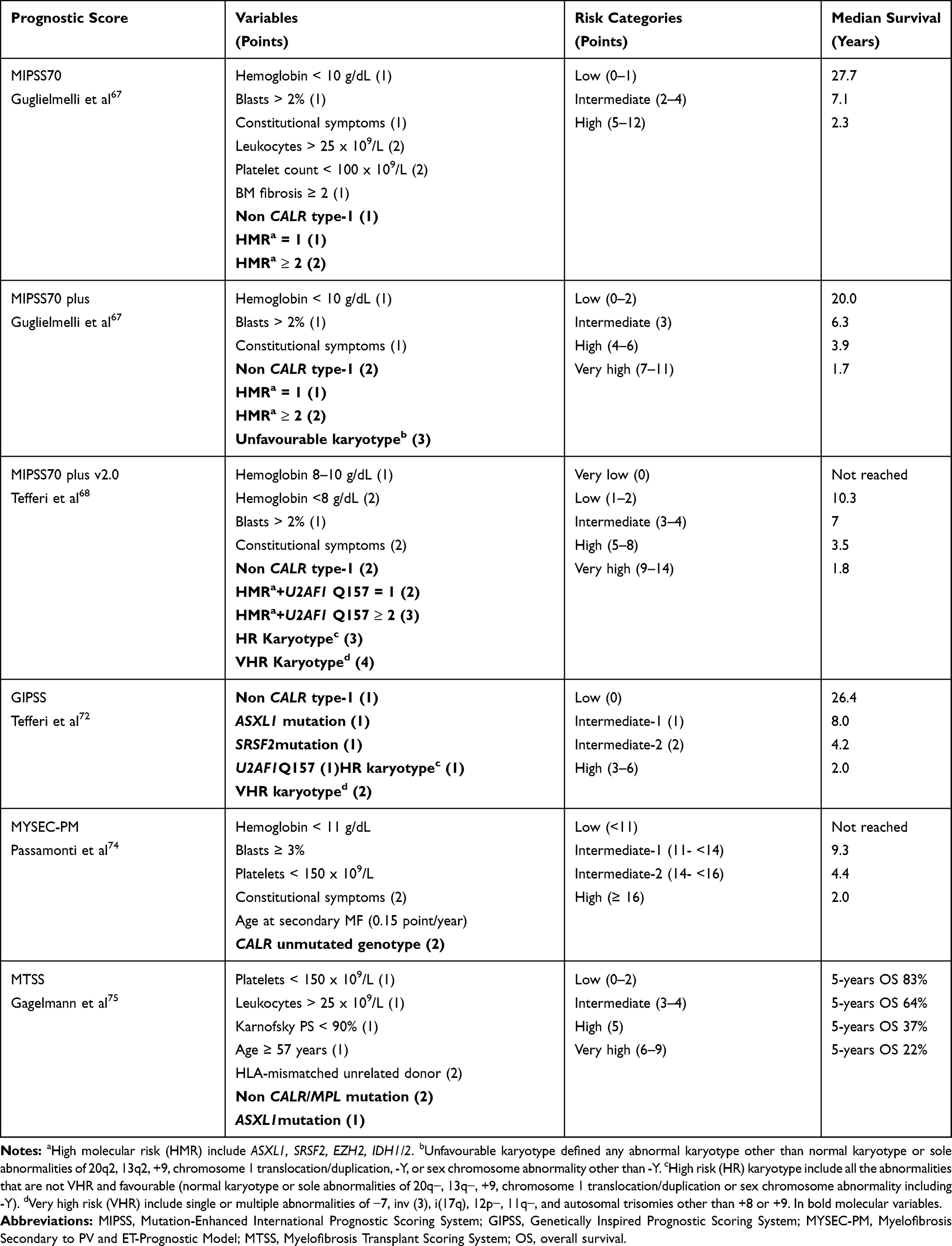 |
Table 6 Clinical-Molecular Prognostic Scores in Myelofibrosis |
Clinical and Therapeutic Implications
Since JAK2V617, MPL and CALR mutations are driver abnormalities of MPN through activation of the JAK-STAT signaling, developing JAK inhibitors (JAKi) has raised a great interest. Ruxolitinib was the first JAK1/2 inhibitor that received approval in myelofibrosis based on COMFORT I/II clinical trials.76,77 Ruxolitinib was effective in reducing splenomegaly and alleviating constitutional symptoms, with possible effects on survival. Long term follow-up studies suggested a reduction in allele burden, with rare cases of molecular remission.78,79 Subsequently, ruxolitinib was tested in PV patients resistant or intolerant to hydroxyurea, according to European Leukemia-Net (ELN) criteria,80 in RESPONSE81 and RESPONSE-282 studies in patients with and without splenomegaly, respectively. A progressive decline in JAK2 mutant burden in patients treated with ruxolitinib was seen in RESPONSE trial, although without definite clinical correlation.83 Therefore, this provides a challenge to the use of JAK2 allele burden reduction as a marker of treatment efficacy. Moreover, one small study highlighted that MF patients starting with a higher allele burden may benefit the most from ruxolitinib, showing a higher probability of spleen response if allele burden was greater than 50% at entry.84 It is important to underline that ruxolitinib targets the kinase domain of JAK1/2 kinases, without a greater affinity against mutant JAK2; this explains the clinical efficacy also in JAK2 negative patients, with similar response rates. Unfortunately, most patients with myelofibrosis on ruxolitinib will become resistant to therapy with the progression of symptoms and splenomegaly, worsening cytopenias, or evolution to BP; in the COMFORT-II study, responding patients had a < 50% chance of maintaining response at 5 years.78 Intriguingly, as reported by Newberry et al,85 clonal evolution after ruxolitinib discontinuation, defined by the acquisition of at least one additional mutation, was reported in 35% of patients. Mutations mostly occurred in ASXL1, followed by TET2, EZH2 and TP53. Moreover, overall survival after ruxolitinib discontinuation was shorter for patients with clonal evolution compared with the others (6 vs 16 months). One drawback of this study was the lack of a control cohort of patients.86 A subsequent study which included 25 MF patients treated with hydroxyurea as a control group confirmed these data but also demonstrated that clonal progression is independent of the treatment.87 However, acquisition of new mutations under ruxolitinib has important clinical correlates, since it is associated with a higher rate of discontinuation due to resistance to treatment and death.
Allo-HSCT remains the only therapeutic approach that can modify the natural history of MF, but it is associated with a relevant morbidity and mortality and only a minority of patients is eligible for such an intensive procedure. As a consequence, research aimed at the discovery of more effective drugs, also in the context of novel JAKi, is extremely active. In recent years, Interferon-α (IFNα), especially better tolerate pegylated forms (Peg-IFNα), has emerged as a promising approach in MPN, particularly in PV and ET, with high rates of both hematological and molecular remissions.88–92 In this respect, monopegylated Ropeginterferon alpha-2b (Ropeg) has received approval in Europe as a first-line therapy for PV patients following the demonstration of its superior efficacy over hydroxyurea in the PROUD/CONTINUATION-PV trial.92 The treatment appears to selectively target the mutant JAK2 clone, as suggested by the high rate of reduction of JAK2V617F allele burden compared to CALR positive ones.93 As recently reported, enhanced sensitivity of JAK2V617F mutated cells to IFNα was related to high expression and phosphorylated levels of STAT1.94 Interestingly, the presence of concomitant mutations is associated with smaller mean decreases in JAK2V617F allele burden under Peg-IFNα treatment.89 IFNα may have decreased ability to eradicate TET2 clones even in cases where JAK2V617F-mutant clone is markedly reduced, indirectly suggesting that the genomic landscape of MPN patients can predict responses to treatment.95 However, Peg-IFNα used at a higher dose in a cohort of 31 CALR positive ET patients, induced hematologic responses in all patients and median CALR allele burden decreased from 41% to 26%, with two patients achieving complete molecular remission.96 In the latter study, decreases in CALR variant allele frequency correlated with laboratory parameters of disease burden including platelets and white blood cell counts, hemoglobin, and lactate dehydrogenase. Similar to findings in JAK2 mutated patients, the presence of additional mutations such as TET2, ASXL1, IDH2, and TP53 was associated with poorer molecular responses. These responses seem to be also durable, which is an appealing aspect of IFNα treatment. In a long-term 83-month follow up of ET and PV patients treated with Peg-IFNα, median duration of hematologic and molecular response was 66 and 53 months, respectively.97 In addition, three patients had a complete molecular remission even after discontinuation of therapy, although in most patients, JAK2V617F allele burden increased after the first 2 years of treatment stop.
Imetelstat, a 13-mer lipid-conjugated oligonucleotide that targets the RNA template of human telomerase reverse transcriptase, was tested in MF and ET. A pilot study evaluated the efficacy and safety in 33 patients with intermediate-2 and high-risk MF according to DIPPS-plus score. A complete or partial response was observed in seven patients and responses correlated with the presence of JAK2V617F, SF3B1 or U2AF1 mutations and the absence of ASXL1 mutations.98
Conversely, in 18 ET patients treated with imetelstat, a partial molecular response was detected in seven of eight JAK2V617F mutated patients. The median JAK2V617F mutant allele burden was reduced by 71% at 3 months after the initiation of treatment. MPL and CALR mutant allele burdens were also reduced, by 15% to 66%.99 Additional mutations significantly reduced the depth of response and had an impact on the duration of response. Of acquired mutations with known adverse prognosis, ASXL1, EZH2 and U2AF1 mutations were responsive to imetelstat, while SF3B1 and TP53 mutations persisted.100
Mutations in IDH1/2 and TP53 are enriched in BP. These molecular findings may have clinical implications given the role of ivosidenib and enasidenib (anti-IDH1 and IDH2, respectively) in patients with relapsed/refractory IDH-mutated AML101,102 and high response rate of TP53-mutated AML to 10-day decitabine.103 Another promising option is venetoclax, a BCL2 inhibitor that can be used in combination with low-dose cytarabine or hypometilating agents. In a recent trial for patients with AML ineligible for standard induction chemotherapy, azacytidine plus venetoclax was superior to azacytidine alone, also in the subset of IDH1/2 and TP53 mutated patients.104 Although these therapies are promising, patients with MPN BP have a dismal outcome without allo-HSCT. Further studies are needed, but these target therapies may represent a valid therapeutic approach also as a bridge to transplantation, given the frequent refractoriness to conventional chemotherapies of MPN BP patients.
The presence of acquired mutations in the driver and/or myeloid genes in the most part of MF patients offers the opportunity to use these markers as indicators of minimal residual disease (MRD) after allo-HSCT. In some studies, the persistence of JAK2V617F mutation after allo-HSCT was associated with a higher incidence of relapse and a shorter overall survival.105,106 More recently, in a series of 136 patients, Wolschke et al demonstrated that patients with detectable JAK2, MPL or CALR mutation at either day +100 or day +180 after allo-HSCT had a significantly higher risk of relapse at 5 years compared to those in molecular remission (62% vs 10%, and 70% vs 10%, respectively).107 Based on these studies, monitoring of molecular MRD in MF patients after allo-HSCT is strongly recommended.
Conclusions
The discovery of JAK2, CALR and MPL driver mutations elucidated the genetic basis of MPN although some patients, so-called triple negative, while having clinical and histological characteristics of MPN, remain molecularly mute. The introduction of high-throughput NGS techniques expanded the mutational landscape and further raised pathophysiological knowledge. Furthermore, these molecular information let to refine the diagnosis, attribute better prognosis score, and monitor the response to treatments. The development of JAKi offered new hopes to patients with MPN allowing them to achieve significant advances in the control of symptoms and quality of life but are largely insufficient to cure the diseases and prolong survival. The last unmet needs regard the understanding of molecular mechanisms at the basis of loss of response to JAKi and the identification of new molecular abnormalities suitable for target therapy.
Acknowledgments
This work was supported by AIRC 5x1000 called “Metastatic disease: the key unmet need in oncology” to MYNERVA project, #21267 (MYeloid NEoplasms Research Venture AIRC) and by PRIN (Progetti di rilevante interesse nazionale)-MIUR called “Myeloid neoplasms: an integrated clinical, molecular and therapeutic approach” Prot. 2017WXR72T. The funding had no role in manuscript preparation.
Disclosure
AMV received personal fees for the advisory board from Novartis, Incyte, Bristol-Myers Squibb and AOP and speaker bureau from Novartis, CTI and Celgene. PG received personal fees for the advisory board from Novartis. The authors report no other potential conflicts of interest for this work.
References
1. Vannucchi AM, Guglielmelli P, Tefferi A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin. 2009;59(3):171–191. doi:10.3322/caac.20009
2. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi:10.1182/blood-2009-03-209262
3. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
4. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the international working group for myelofibrosis research and. Leuk Res. 2007;31(6):737–740. doi:10.1016/j.leukres.2006.12.002
5. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the international working group for myelofibrosis research and treatment. Leukemia. 2008;22(2):437–438. doi:10.1038/sj.leu.2404914
6. Tefferi A, Mudireddy M, Mannelli F, et al. Blast phase myeloproliferative neoplasm: mayo-AGIMM study of 410 patients from two separate cohorts. Leukemia. 2018;32(5):1200–1210. doi:10.1038/s41375-018-0019-y
7. Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5(11):e366–e366. doi:10.1038/bcj.2015.95
8. Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol. 2014;92(4):289–297. doi:10.1111/ejh.12256
9. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):
10. Boddu P, Chihara D, Masarova L, Pemmaraju N, Patel KP, Verstovsek S. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann Hematol. 2018;97(11):2071–2080. doi:10.1007/s00277-018-3402-x
11. Zamora L, Xicoy B, Cabezón M, et al. Co-existence of JAK2 V617F and CALR mutations in primary myelofibrosis. Leuk Lymphoma. 2015;56(10):2973–2974. doi:10.3109/10428194.2015.1015124
12. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi:10.1016/j.ccr.2005.03.023
13. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi:10.1056/NEJMoa051113
14. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi:10.1016/S0140-6736(05)71142-9
15. James C, Ugo V, Le Couédic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi:10.1038/nature03546
16. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. doi:10.1056/NEJMoa065202
17. Scott LM. The JAK2 exon 12 mutations: a comprehensive review. Am J Hematol. 2011;86(8):668–676. doi:10.1002/ajh.22063
18. Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci. 2005;102(52):18962–18967. doi:10.1073/pnas.0509714102
19. Lu X, Huang LJ-S, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem. 2008;283(9):5258–5266. doi:10.1074/jbc.M707125200
20. Sangkhae V, Etheridge SL, Kaushansky K, Hitchcock IS. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood. 2014;124(26):3956–3963. doi:10.1182/blood-2014-07-587238
21. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 2011;117(10):2813–2816. doi:10.1182/blood-2010-11-316810
22. Tefferi A, Lavu S, Mudireddy M, et al. JAK2 exon 12 mutated polycythemia vera: mayo-Careggi MPN alliance study of 33 consecutive cases and comparison with JAK2V617F mutated disease. Am J Hematol. 2018;93(4):E93–E96. doi:10.1002/ajh.25017
23. Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372(7):601–612. doi:10.1056/NEJMoa1412098
24. Nangalia J, Nice FL, Wedge DC, et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica. 2015;100(11):e438–e442. doi:10.3324/haematol.2015.129510
25. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21(9):1952–1959. doi:10.1038/sj.leu.2404854
26. Gold LI, Eggleton P, Sweetwyne MT, et al. Calreticulin: non‐endoplasmic reticulum functions in physiology and disease. FASEB J. 2010;24(3):665–683. doi:10.1096/fj.09-145482
27. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. doi:10.1056/NEJMoa1312542
28. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi:10.1056/NEJMoa1311347
29. Cabagnols X, Defour JP, Ugo V, et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocythemia: relevance for disease evolution. Leukemia. 2015;29(1):249–252. doi:10.1038/leu.2014.270
30. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. 2016;30(2):431–438. doi:10.1038/leu.2015.277
31. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol. 2014;89(8):E121–124. doi:10.1002/ajh.23743
32. Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016;127(10):1325–1335. doi:10.1182/blood-2015-11-681932
33. Balligand T, Achouri Y, Pecquet C, et al. Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia. 2016;30(8):1775–1778. doi:10.1038/leu.2016.47
34. Nivarthi H, Chen D, Cleary C, et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia. 2016;30(8):1759–1763. doi:10.1038/leu.2016.32
35. Elf S, Abdelfattah NS, Chen E, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016;6(4):368–381. doi:10.1158/2159-8290.CD-15-1434
36. Pecquet C, Chachoua I, Roy A, et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood. 2019;133(25):2669–2681. doi:10.1182/blood-2018-09-874578
37. Masubuchi N, Araki M, Yang Y, et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia. 2020;34(2):499–509. doi:10.1038/s41375-019-0564-z
38. Staerk J, Lacout C, Sato T, Smith SO, Vainchenker W, Constantinescu SN. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood. 2006;107(5):1864–1871. doi:10.1182/blood-2005-06-2600
39. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi:10.1371/journal.pmed.0030270
40. Defour J-P, Chachoua I, Pecquet C, Constantinescu SN. Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: implications for myeloproliferative neoplasms. Leukemia. 2016;30(5):1214–1216. doi:10.1038/leu.2015.271
41. Ding J, Komatsu H, Wakita A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103(11):4198–4200. doi:10.1182/blood-2003-10-3471
42. Bridgford JL, Lee SM, Lee CMM, et al. Novel drivers and modifiers of MPL-dependent oncogenic transformation identified by deep mutational scanning. Blood. 2020;135(4):287–292. doi:10.1182/blood.2019002561
43. Tefferi A, Vannucchi AM. Genetic risk assessment in myeloproliferative neoplasms. Mayo Clin Proc. 2017;92(8):1283–1290. doi:10.1016/j.mayocp.2017.06.002
44. Cabagnols X, Favale F, Pasquier F, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. 2016;127(3):333–342. doi:10.1182/blood-2015-07-661983
45. Milosevic Feenstra JD, Nivarthi H, Gisslinger H, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127(3):325–332. doi:10.1182/blood-2015-07-661835
46. Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416–1430. doi:10.1056/NEJMoa1716614
47. Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018;2(22):3404–3410. doi:10.1182/bloodadvances.2018020222
48. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–1551. doi:10.1182/blood-2013-11-539098
49. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123(10):1552–1555. doi:10.1182/blood-2013-11-538983
50. Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an international prognostic score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood. 2012;120(26):
51. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5(11):e369. doi:10.1038/bcj.2015.94
52. Haider M, Gangat N, Lasho T, et al. Validation of the revised international prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis) in 585 Mayo clinic patients. Am J Hematol. 2016;91(4):390–394. doi:10.1002/ajh.24293
53. Alvarez-Larrán A, Pereira A, Guglielmelli P, et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica. 2016;101(8):926–931. doi:10.3324/haematol.2016.146654
54. Tefferi A, Lasho TL, Guglielmelli P, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30. doi:10.1182/bloodadvances.2016000216
55. Tefferi A, Guglielmelli P, Lasho TL, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291–302. doi:10.1111/bjh.16380
56. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood. 2009;113(13):2895–2901. doi:10.1182/blood-2008-07-170449
57. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. doi:10.1182/blood-2009-09-245837
58. Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. doi:10.1200/JCO.2010.32.2446
59. Caramazza D, Begna KH, Gangat N, et al. Refined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patients. Leukemia. 2011;25(1):82–88. doi:10.1038/leu.2010.234
60. Tefferi A, Lasho TL, Finke CM, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1(2):105–111. doi:10.1182/bloodadvances.2016000208
61. Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. doi:10.1038/leu.2013.119
62. Tefferi A, Guglielmelli P, Lasho TL, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. 2014;28(7):1494–1500. doi:10.1038/leu.2014.57
63. Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28(7):1472–1477. doi:10.1038/leu.2014.3
64. Tefferi A, Lasho TL, Tischer A, et al. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood. 2014;124(15):2465–2466. doi:10.1182/blood-2014-07-588426
65. Guglielmelli P, Rotunno G, Fanelli T, et al. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015;5(10):e360–e360. doi:10.1038/bcj.2015.90
66. Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–1810. doi:10.1038/leu.2014.76
67. Guglielmelli P, Lasho TL, Rotunno G, et al. MIPSS70: mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J Clin Oncol. 2018;36(4):310–318. doi:10.1200/JCO.2017.76.4886
68. Tefferi A, Guglielmelli P, Lasho TL, et al. MIPSS70+ version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J Clin Oncol. 2018;36(17):1769–1770. doi:10.1200/JCO.2018.78.9867
69. Tefferi A, Finke CM, Lasho TL, et al. U2AF1 mutation types in primary myelofibrosis: phenotypic and prognostic distinctions. Leukemia. 2018;32(10):2274–2278. doi:10.1038/s41375-018-0078-0
70. Nicolosi M, Mudireddy M, Lasho TL, et al. Sex and degree of severity influence the prognostic impact of anemia in primary myelofibrosis: analysis based on 1109 consecutive patients. Leukemia. 2018;32(5):1254–1258. doi:10.1038/s41375-018-0028-x
71. Tefferi A, Nicolosi M, Mudireddy M, et al. Revised cytogenetic risk stratification in primary myelofibrosis: analysis based on 1002 informative patients. Leukemia. 2018;32(5):1189–1199. doi:10.1038/s41375-018-0018-z
72. Tefferi A, Guglielmelli P, Nicolosi M, et al. GIPSS: genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia. 2018;32(7):1631–1642. doi:10.1038/s41375-018-0107-z
73. Kuykendall AT, Talati C, Padron E, et al. Genetically inspired prognostic scoring system (GIPSS) outperforms dynamic international prognostic scoring system (DIPSS) in myelofibrosis patients. Am J Hematol. 2019;94(1):87–92. doi:10.1002/ajh.25335
74. Passamonti F, Giorgino T, Mora B, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia. 2017;31(12):2726–2731. doi:10.1038/leu.2017.169
75. Gagelmann N, Ditschkowski M, Bogdanov R, et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood. 2019;133(20):2233–2242. doi:10.1182/blood-2018-12-890889
76. Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi:10.1056/NEJMoa1110557
77. Harrison C, Kiladjian -J-J, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi:10.1056/NEJMoa1110556
78. Harrison CN, Vannucchi AM, Kiladjian -J-J, et al. Long-term findings from COMFORT-II, a Phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701–1707. doi:10.1038/leu.2016.148
79. Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10(1):55. doi:10.1186/s13045-017-0417-z
80. Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057–1069. doi:10.1038/s41375-018-0077-1
81. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. doi:10.1056/NEJMoa1409002
82. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017;18(1):88–99. doi:10.1016/S1470-2045(16)30558-7
83. Vannucchi AM, Verstovsek S, Guglielmelli P, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol. 2017;96(7):1113–1120. doi:10.1007/s00277-017-2994-x
84. Barosi G, Klersy C, Villani L, et al. JAK2V617F allele burden < 50% is associated with response to ruxolitinib in persons with MPN-associated myelofibrosis and splenomegaly requiring therapy. Leukemia. 2016;30(8):1772–1775. doi:10.1038/leu.2016.45
85. Newberry KJ, Patel K, Masarova L, et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood. 2017;130(9):1125–1131. doi:10.1182/blood-2017-05-783225
86. Vannucchi AM, Guglielmelli P. Traffic lights for ruxolitinib. Blood. 2017;130(9):1075–1077. doi:10.1182/blood-2017-07-795880
87. Pacilli A, Rotunno G, Mannarelli C, et al. Mutation landscape in patients with myelofibrosis receiving ruxolitinib or hydroxyurea. Blood Cancer J. 2018;8(12). doi:10.1038/s41408-018-0152-x
88. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27(32):5418–5424. doi:10.1200/JCO.2009.23.6075
89. Quintás-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon α-2a. Blood. 2013;122(6):893–901. doi:10.1182/blood-2012-07-442012
90. Them NCC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol. 2015;90(4):288–294. doi:10.1002/ajh.23928
91. Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, et al. Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015;126(15):1762–1769. doi:10.1182/blood-2015-04-637280
92. Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–e208. doi:10.1016/S2352-3026(19)30236-4
93. Kjær L, Cordua S, Holmström MO, et al. Differential dynamics of CALR mutant allele burden in myeloproliferative neoplasms during interferon alfa treatment. PLoS One. 2016;11(10):e0165336. doi:10.1371/journal.pone.0165336
94. Czech J, Cordua S, Weinbergerova B, et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia. 2019;33(4):995–1010. doi:10.1038/s41375-018-0295-6
95. Kiladjian -J-J, Massé A, Cassinat B, et al. Clonal analysis of erythroid progenitors suggests that pegylated interferon α-2a treatment targets JAK2V617F clones without affecting TET2 mutant cells. Leukemia. 2010;24(8):1519–1523. doi:10.1038/leu.2010.120
96. Verger E, Cassinat B, Chauveau A, et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood. 2015;126(24):2585–2591. doi:10.1182/blood-2015-07-659060
97. Masarova L, Patel KP, Newberry KJ, et al. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, Phase 2 trial. Lancet Haematol. 2017;4(4):e165–e175. doi:10.1016/S2352-3026(17)30030-3
98. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373(10):908–919. doi:10.1056/NEJMoa1310523
99. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373(10):920–928. doi:10.1056/NEJMoa1503479
100. Oppliger Leibundgut E, Haubitz M, Burington B, et al. Dynamics of mutations in patients with essential thrombocythemia treated with imetelstat. Haematologica. 2020;
101. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–2398. doi:10.1056/NEJMoa1716984
102. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi:10.1182/blood-2017-04-779405
103. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med. 2016;375(21):2023–2036. doi:10.1056/NEJMoa1605949
104. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi:10.1056/NEJMoa2012971
105. Alchalby H, Badbaran A, Zabelina T, et al. Impact of JAK2V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood. 2010;116(18):3572–3581. doi:10.1182/blood-2009-12-260588
106. Lange T, Edelmann A, Siebolts U, et al. JAK2 p.V617F allele burden in myeloproliferative neoplasms one month after allogeneic stem cell transplantation significantly predicts outcome and risk of relapse. Haematologica. 2013;98(5):722–728. doi:10.3324/haematol.2012.076901
107. Wolschke C, Badbaran A, Zabelina T, et al. Impact of molecular residual disease post allografting in myelofibrosis patients. Bone Marrow Transplant. 2017;52(11):1526–1529. doi:10.1038/bmt.2017.157
108. Lasho TL, Mudireddy M, Finke CM, et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018;2(4):370–380. doi:10.1182/bloodadvances.2018015875
109. Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302–1309. doi:10.1038/leu.2010.113
110. Tefferi A, Lasho TL, Finke C, et al. Prognostic significance of ASXL1 mutation types and allele burden in myelofibrosis. Leukemia. 2018;32(3):837–839. doi:10.1038/leu.2017.318
111. Kröger N, Panagiota V, Badbaran A, et al. Impact of molecular genetics on outcome in myelofibrosis patients after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23(7):1095–1101. doi:10.1016/j.bbmt.2017.03.034
112. Shimizu T, Kubovcakova L, Nienhold R, et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med. 2016;213(8):1479–1496. doi:10.1084/jem.20151136
113. Guglielmelli P, Biamonte F, Score J, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011;118(19):5227–5234. doi:10.1182/blood-2011-06-363424
114. Lasho TL, Jimma T, Finke CM, et al. SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood. 2012;120(20):4168–4171. doi:10.1182/blood-2012-05-429696
115. Zhang S-J, Rampal R, Manshouri T, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119(19):4480–4485. doi:10.1182/blood-2011-11-390252
116. Tefferi A, Nicolosi M, Mudireddy M, et al. Driver mutations and prognosis in primary myelofibrosis: mayo-Careggi MPN alliance study of 1095 patients. Am J Hematol. 2018;93(3):348–355. doi:10.1002/ajh.24978
117. Tefferi A, Lasho TL, Hanson CA, Ketterling RP, Gangat N, Pardanani A. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br J Haematol. 2018;183(4):678–681. doi:10.1111/bjh.15010
118. Chowdhury O, O’Sullivan J, Barkas N, et al. Spliceosome mutations are common in persons with myeloproliferative neoplasm-associated myelofibrosis with RBC-transfusion-dependence and correlate with response to pomalidomide. Leukemia. 2020. doi:10.1038/s41375-020-0979-6
119. Maslah N, Cassinat B, Verger E, Kiladjian -J-J, Velazquez L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia. 2017;31(8):1661–1670. doi:10.1038/leu.2017.139
120. Loscocco GG, Mannarelli C, Pacilli A, et al. Germline transmission of LNKE208Q variant in a family with myeloproliferative neoplasms. Am J Hematol. 2016;91(9):E356–E356. doi:10.1002/ajh.24437
121. Rumi E, Harutyunyan AS, Pietra D, et al. LNK mutations in familial myeloproliferative neoplasms. Blood. 2016;128(1):144–145. doi:10.1182/blood-2016-04-711150
122. Sanada M, Suzuki T, Shih L-Y, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460(7257):904–908. doi:10.1038/nature08240
123. Coltro G, Rotunno G, Mannelli L, et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020;4(15):3677–3687. doi:10.1182/bloodadvances.2020002175
124. Braun BS, Shannon K. Targeting Ras in myeloid leukemias. Clin Cancer Res. 2008;14(8):2249–2252. doi:10.1158/1078-0432.CCR-07-1005
125. Jutzi JS, Bogeska R, Nikoloski G, et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J Exp Med. 2013;210(5):1003–1019. doi:10.1084/jem.20120521
126. Guglielmelli P, Pacilli A, Coltro G, et al. Characteristics and clinical correlates of NFE2 mutations in chronic Myeloproliferative neoplasms. Am J Hematol. 2020;95(1). doi:10.1002/ajh.25668
127. Kahn JD, Miller PG, Silver AJ, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood. 2018;132(11):1095–1105. doi:10.1182/blood-2018-05-850339
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.