")
Back to Journals » International Journal of General Medicine » Volume 15
Impact of Geographical Location on the Gut Microbiota Profile in Egyptian Children with Type 1 Diabetes Mellitus: A Pilot Study
Authors Elsherbiny NM , Ramadan M, Abu Faddan NH, Hassan EA , Ali ME, Abd El-Rehim ASE , Abbas WA , Abozaid MAA, Hassanin E, Mohamed GA, Hetta HF , Salah M
Received 6 February 2022
Accepted for publication 10 June 2022
Published 15 July 2022 Volume 2022:15 Pages 6173—6187
DOI https://doi.org/10.2147/IJGM.S361169
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Scott Fraser
Nahla M Elsherbiny,1 Mohammed Ramadan,2 Nagla H Abu Faddan,3 Elham Ahmed Hassan,4 Mohamed E Ali,2 Abeer Sharaf El-Din Abd El-Rehim,4 Wael A Abbas,5 Mohamed AA Abozaid,5 Ebtisam Hassanin,6 Ghada A Mohamed,7 Helal F Hetta,1 Mohammed Salah8
1Department of Medical Microbiology and Immunology, Faculty of Medicine, Assiut University, Assiut, Egypt; 2Department of Microbiology and Immunology, Faculty of Pharmacy, Al-Azhar University, Assiut Branch, Assiut, Egypt; 3Department of Pediatrics, Faculty of Medicine, Assiut University, Assiut, Egypt; 4Department of Gastroenterology and Tropical Medicine, Faculty of Medicine, Assiut University, Assiut, Egypt; 5Department of Internal Medicine, Gastroenterology and Hepatology Unit, Faculty of Medicine, Assiut University, Assiut, Egypt; 6Clinical Pathology, Ministry of Health, Assiut, Egypt; 7Department of Internal Medicine, Endocrine Unit, Faculty of Medicine, Assiut University, Assiut, Egypt; 8Department of Microbiology and Immunology, Faculty of pharmacy, Port Said University, Port Said, Egypt
Correspondence: Elham Ahmed Hassan, Department of Gastroenterology and Tropical Medicine, Faculty of Medicine, Assiut University, Assiut, 71515, Egypt, Tel +20 882410285, Fax +20 882333327, Email [email protected]; [email protected]
Purpose: To investigate the compositional and functional characteristics of T1DM-associated gut microbiota in two Egyptian cities and to study the geographical locality effects.
Patients and Methods: This case-control study included 32 children with controlled T1DM and 16 controls, selected from two different regions of Egypt. The gut microbiota of both diabetic and control children was analyzed through 16S rRNA gene sequencing; this was done using the Illumina MiSeq platform.
Results: Consistent findings among the diabetic children included significantly lower alpha diversity than the control children, as well as a lower mean Firmicutes/Bacteroidetes (F/B) ratio, and reduced proportions of Firmicutes and the genera Prevotella and Ruminococcus. In the diabetic children, there were also significantly enriched representations of Actinobacteria, Bacteroidetes, and Proteobacteria and the genera Lactobacilli, Bacteroides, and Faecalibacterium. When comparing the two diabetic groups, the Ismailia group (IsDM) was found to have a significantly higher F/B ratio and diversity indices, with resultant differences at the functional level.
Conclusion: There are a number of consistent changes in the microbiota profile characterizing the diabetic groups irrespective of the geographical location including significantly lower alpha diversity, mean Firmicutes/ Bacteroidetes (F/B) ratio, and reduced proportions of Firmicutes and genera Prevotella and Ruminococcus. There are also significantly enriched representations of Actinobacteria, Bacteroidetes, and Proteobacteria and genera Lactobacilli, Bacteroides, and Faecalibacterium pointing to the greater driving power of the disease.
Keywords: gut microbiota, dysbiosis, type 1 diabetes mellitus, children
Introduction
Type 1 diabetes mellitus (T1DM) is an autoimmune disease characterized by insufficient insulin production due to partial or complete destruction of pancreatic islet beta cells in genetically susceptible individuals; the condition is induced by innate and adaptive immunity, especially by autoreactive T-cells.1
A number of environmental factors may act as triggers for T1DM, including pregnancy-related and perinatal factors, viruses, ingestion of cereals and cow’s milk, and the use of antibiotics and probiotics. These factors also affect the microbiota, which explains the hypothesis that the gut microbiome has a role in linking environmental influences with the development of T1DM.2
Each individual differs not only in his/her own genetic material but also in his/her gut microbiota which form a complex microbial community of ~4X1013 cells.3 These organisms form symbiotic relationships with their host and have many beneficial functions, including the maintenance of intestinal epithelial integrity and the regulation of immunological activities.4 What this means is that dysbiosis can lead to the loss of immune regulatory effects on the gut mucosa, a loss which is associated with a number of inflammatory and immune-mediated diseases, including T1DM; this occurs especially in childhood.5
The role of the gut microbiota in the pathogenesis of T1DM has become an important subject of investigation.6 It is, however, difficult to determine the alterations in the microbiota that are consistently associated with T1DM during the early childhood period, when the gut microbiota is unstable, and the seroconversion is occurring early in those children who will develop diabetes. At this point it has not yet been determined whether these microbial changes are a cause of diabetes, a response to it, or both7 and whether these changes are consistent irrespective of the geographical locality which is a known important factor shaping the microbiota composition.8
When trying to assess the complexity of the thousands of bacterial species in the gut, one option is a categorization method that assigns an individual’s microbial profile to certain clusters, known as enterotypes, which are suggested to be biomarkers of the gut ecology and might reduce the large interindividual microbial variation and are geographically stratified.9 These are named depending on the abundance of signature taxa, such as Bacteroides and Prevotella, the main identified enterotypes, followed by Ruminococcaceae.10,11
The key methodology to study these bacterial populations and to analyze microbiome data is 16S ribosomal RNA sequencing, which focuses on the relative abundances and diversity of the various taxa found in microbiome samples.12 However, these analyses do not show relationships between the taxa in a community, which are represented using networks.13
Although several studies have linked dysbiosis to T1DM by tracking changes in microbial composition, very few have studied the enterotypes, the relationships between the taxa in a community and whether locality has any effect on these findings. This study therefore aimed to bridge this gap by studying the microbiota of a sample of healthy and controlled diabetic Egyptian children in two Egyptian cities. The locations selected were Assiut in Upper, or Southern, Egypt and Ismailia in Lower, or Northern, Egypt. Northern Egypt is more industrialized and has among the highest socioeconomic and educational levels in the country, while Assiut is one of the poorest towns in the country, and lies in the southern region, where the economy relies upon agriculture and the population is often exposed to chemical fertilizers.
Materials and Methods
Study Participants and Design
This was a case-control study that was conducted over a period of one year from January to December 2017. Representative participants were recruited from two geographical regions with different socioeconomic settings. The first group was from Assiut in Upper Egypt and was represented by seventeen diabetic children (AsDM) and eight healthy subjects as controls (AsC). The second group was from Ismailia in Lower Egypt and was composed of fifteen diabetic children (IsDM) and eight healthy subjects as controls (IsC). The study was carried out according to the principles of the Declaration of Helsinki and was approved by the Ethics Committee of the Faculty of Medicine, Assiut University (Institutional Review Board (IRB) reference number 17300404/2016). The parents of the studied children were informed about the purpose of the study and consents were taken from them for the enrollment of their children in the study.
The diagnosis of T1DM was made according to the criteria of the American Diabetes Association.14 The treatment and follow-up of the diabetic children were done according to a standard medical protocol, in which patients were undergoing treatment with multiple doses of insulin. The inclusion criteria included controlled diabetic status in terms of normal blood glucose and glycosylated hemoglobin (HbA1C), the absence of any allergic, inflammatory disease, or diarrhea. Also included were patients who had not had any antibiotics or probiotics or any other medical treatment that may affect the intestinal microbiota two months before sampling. The controls were healthy children who were matched according to gender, age, mode of delivery and duration of breastfeeding. All participants had normal body mass indices (BMI).
Sample Collection and Microscopic Parasitological Examination
Stool samples were obtained from the diabetic children during their regular follow-up in the outpatient clinic of the Assiut University Pediatric Hospital, Assiut, Egypt and from the Health Insurance Organization Hospital, Ismailia, Egypt. Samples were also collected from matched healthy subjects who fulfilled the inclusion criteria. In Assiut, the samples were sent for processing to the Medical Research Centre of the Faculty of Medicine at Assiut University; in Ismailia, they were sent to the Microbiology Department in the Faculty of Pharmacy at Port Said University. A parasitological examination was done on all the stool samples when they reached the laboratories.
DNA Extraction and PCR Amplification and Sequencing of 16S rRNA Gene
This was performed using the PureLinkTM Microbiome DNA Purification Kit from Thermofisher Scientific, USA, according to the manufacturer’s instructions. The DNA concentration was estimated by Nanodrop spectrophotometer.
The PCR cycling conditions followed those reported by Ramadan et al.15 The V3-V4 regions of the 16S rRNA gene were amplified using the following primers with included Illumina adaptor, as follows:
Forward Primer.
5'TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG3'.
Reverse Primer.
5'GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC'.
Validation of the size and quality of the amplified products was done using 1% agarose gel. Amplicons were purified by the Agencourt XP Ampure Beads (Beckman Coulter, USA). Finally, PCR amplicons of negative controls and fecal samples were sent to IGA Technology Services in Udine, Italy, where they were sequenced using the Illumina MiSeq platform (Illumina, USA).
Bioinformatic Analyses
Preprocessing of the Raw 16S rRNA Datasets
The QIIME software package was used for sequence analysis.16 In brief, merging of the paired-end sequences generated by the Illumina MiSeq was done using the join_paired_ends.py script with default parameters. After that, merged fastq sequences were quality filtered from low-quality sequences (Phred score value ≤28 and ≤465 bp, chimeric sequences and ambiguous reads). The generated sequences were classified as Operational Taxonomic Units (OTUs) at 97% similarity by UCLUST.17 Finally, OTUs were assigned to the corresponding taxonomic levels against the Greengenes database v13_8.18
OTU Based Data Analyses and Statistical Analysis
Three alpha diversity-based methods (Observed species, Chao1 and Shannon index) were used to calculate the alpha diversity of the datasets. The microbial relations among the gut microbiomes were elucidated using beta diversity analysis by Principal Coordinate Analysis (PCoA) based on weighted and unweighted uniFrac distances. Statistical significance of differences among the studied groups was estimated using the unpaired Wilcoxon rank sum test and the Kruskal Wallis rank sum test. Also, to identify the statistically significant genera that differed between AsDM and IsDM, Welch’s t-tests (confidence interval method, P < 0.05) was measured. For multigroup comparisons, false discovery rate method (FDR) was performed to adjust all P-values.19 Furthermore, for inferring the correlations of bacterial community members to participants’ clinical metadata, Spearman correlation coefficient was applied to genera that were detected with mean relative proportion ≥ 0.23% using R package, Hamsic.20
To determine the Enterotypes of the gut microbiota, the relative abundance of genera in all datasets were used for clustering using Partitioning around medoids (PAM) algorithm and Jensen–Shannon divergence (JSD), then the optimum number of clusters was identified using the Calinski-Harabasz (CH) index.21
For defining the core microbiome in our datasets, the compute_core_microbiome.py script was used. The OTU was considered as a core taxon of the whole dataset if it was present in at least 80% of all samples, while it was considered as a core taxon of a given dataset if it presented in 100% of samples of that study group.
Biomarker Determination and Function Prediction of Gut Microbiota
This was done by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) where Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) Database pathways were predicted based on the taxa ID in Greengenes.22,23 Moreover, LEfSe (ver. 1.0) analysis tool was used for determining the potential biomarkers (Taxa or KEGG metabolic pathways with logarithmic LDA scores > 3.0 and α= 0.05 at level 2 level 3).24
Availability of Data
The 16S rRNA raw sequences have been deposited in SRA at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA647206 under accession number PRJNA647206.
Results
Population Characteristics
The study population consisted of a total of 32 children with controlled T1DM (17 from Assiut and 15 from Ismailia with mean age of 7.3 ± 2.8 and 7.5 ± 2.5 years respectively) and 16 healthy controls aged 7.3 ± 1.8 years (eight from each city). The clinical characteristics are listed in Table 1. Giardia cyst was detected in four stool samples from the AsDM group.
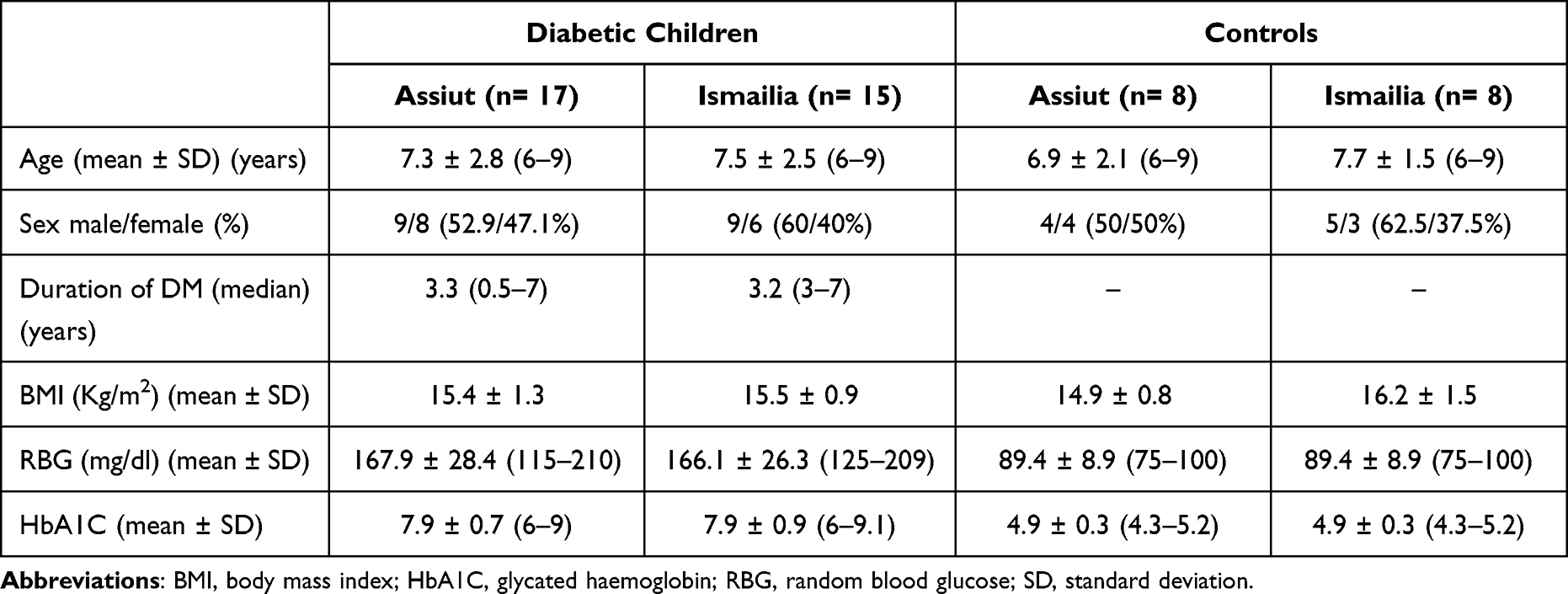 |
Table 1 Characteristics of the Study Population |
Distinct Structure of Microbial Communities in Harmonious Association to State of Health and Locality
A total number of 3,754,432 raw sequences was obtained from the study groups. After the quality- filtering and removal of potentially chimeric sequences, a total number of 2,965,462 high-quality reads were available, and were used for downstream analyses.
Taxonomic assignments of 16S rRNA reads resulted in 5269 OTUs, 21 phyla, 58 classes, 113 orders, 209 families, and 518 genera across all samples. Specifically, analysis of bacterial phyla showed Firmicutes and Bacteroides as the major phyla amongst all the groups (Figure 1). Their relative abundance was 94.8% in the healthy control groups, while for the diabetic groups, the proportions were 86.9% in AsDM and 95.4% in IsDM.
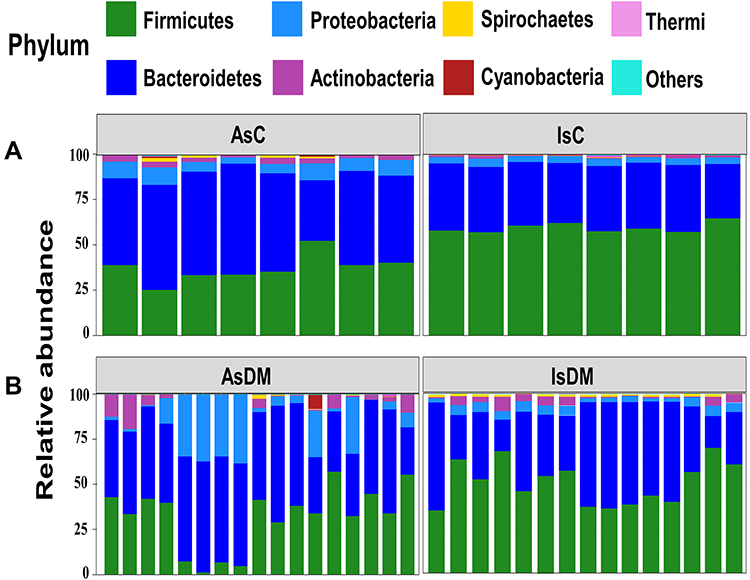 |
Figure 1 A bar plot of the relative abundance of different phyla in the gut microbiota of the study groups; (A) Control groups (Assiut and Ismailia) and (B) Diabetic groups (Assiut and Ismailia). |
Analysis of the mean Firmicutes and Bacteroides ratio (F/B) showed that it was generally higher in the control groups than in the corresponding diabetic groups from the same locality. However, IsC showed a significantly higher mean ratio (1.7) than AsC (0.75) (Kruskal–Wallis, P= 2.96×10−4). For the Ismailia region, the mean F/B ratio was 1.7 in the control group and 1.5 in the diabetic group. Notably, the F/B ratios in the Assiut groups were much lower than those in the Ismailia groups, with 0.75 for the control group and 0.6 for the diabetic group. The phyla Proteobacteria and Actinobacteria were next in order, though at much lower percentages, as shown in Figure 1. There was a distinct subset of samples consisting of the four children of the AsDM group with giardia. Their taxonomic profiles were different from those of the other diabetic samples, with Bacteroides (~59.4%) and Proteobacteria (~32.47%,) being the major phyla, with a relatively diminished representation of Firmicutes.
The overall alpha diversity indices showed statistically significant differences in terms of the evenness and richness within the datasets (Wilcoxon test, P= 0.001). The control groups in both localities showed statistically higher diversity than their corresponding diabetic groups (Wilcoxon test, P= 0.001). The IsC manifested significantly more diverse communities than did AsC (Wilcoxon test, P= 0.001) (Figure 2A).
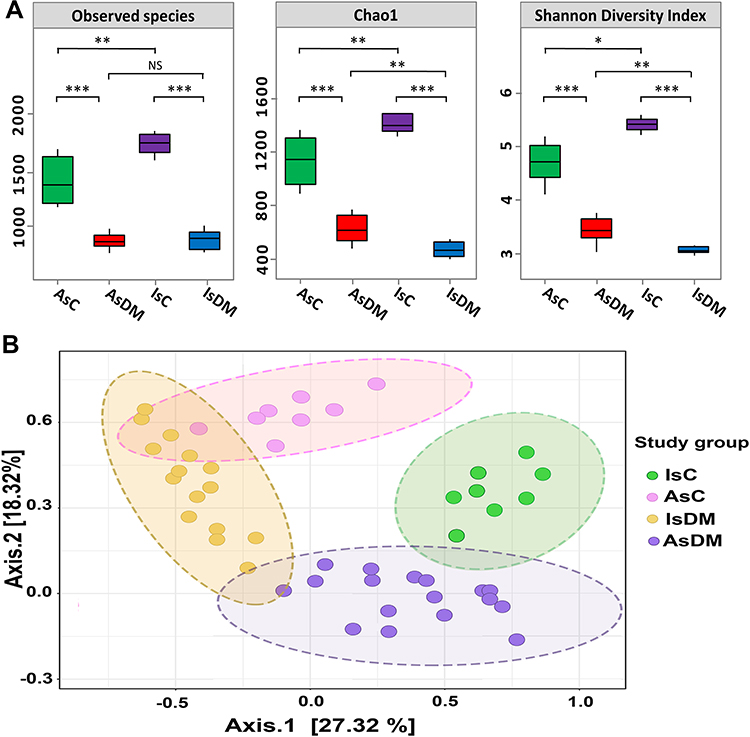 |
Figure 2 Diversity analyses of gut microbiomes. (A) Bacterial alpha diversity indices were represented by box plots of the following indices: the number of observed species Chao1 index and Shannon diversity index. X-axis represented the study groups, while alpha indices were plotted on the Y-axis. The median was represented by the line in each box, the interquartile range (IQR) between the 25th and 75th percentile was delimited by the box, and the range was outlined by the whisker. The nonparametric Wilcoxon rank-sum test was used to define the statistical significance of pairwise comparisons. Significant differences were displayed with either NS (P> 0.05), * (P< 0.05), ** (P< 0.01) or *** (P< 0.001). (B) Principal coordinates analysis (PCoA) of gut microbiomes based on weighted UniFrac distance matrices. PCoA plot represented the similarity distances between gut microbiota of healthy and diseased groups. The significant clustering was denoted by ellipses (Adonis: r2= 0.047, P= 0.00092). |
Moreover, visualization of weighted UniFrac distance by PCoA plot showed significant distinction in bacterial communities between all study groups (PERMANOVA; R2= 0.169, P= 0.001) (Figure 2B). Surprisingly, although there was a distinct clustering for each study group, the bacterial communities of the diseased groups were relatively close to each other, in contrast to the bacterial communities of the controls.
At the genus level, there were 21 genera exhibiting varied patterns in each study group, as shown in Figure 3. Prevotella, Faecalibacterium, Bacteroides and Lactobacilli were among the genera representing the core microbiome of the entire dataset, but they were in relatively different proportions, especially in the diabetic groups. For instance, Prevotella and Faecalibacterium were significantly enriched in AsDM and IsDM groups, respectively. Meanwhile, many genera played dual roles as core genera for all the datasets and also as potential biomarkers for a given study group; for example, Collinsella for the AsDM group, and Succinivibrio, Blautia, Catenibacterium and Bifidobacterium for the IsDM group. See Table 2 and Supplementary Tables S1 and S2.
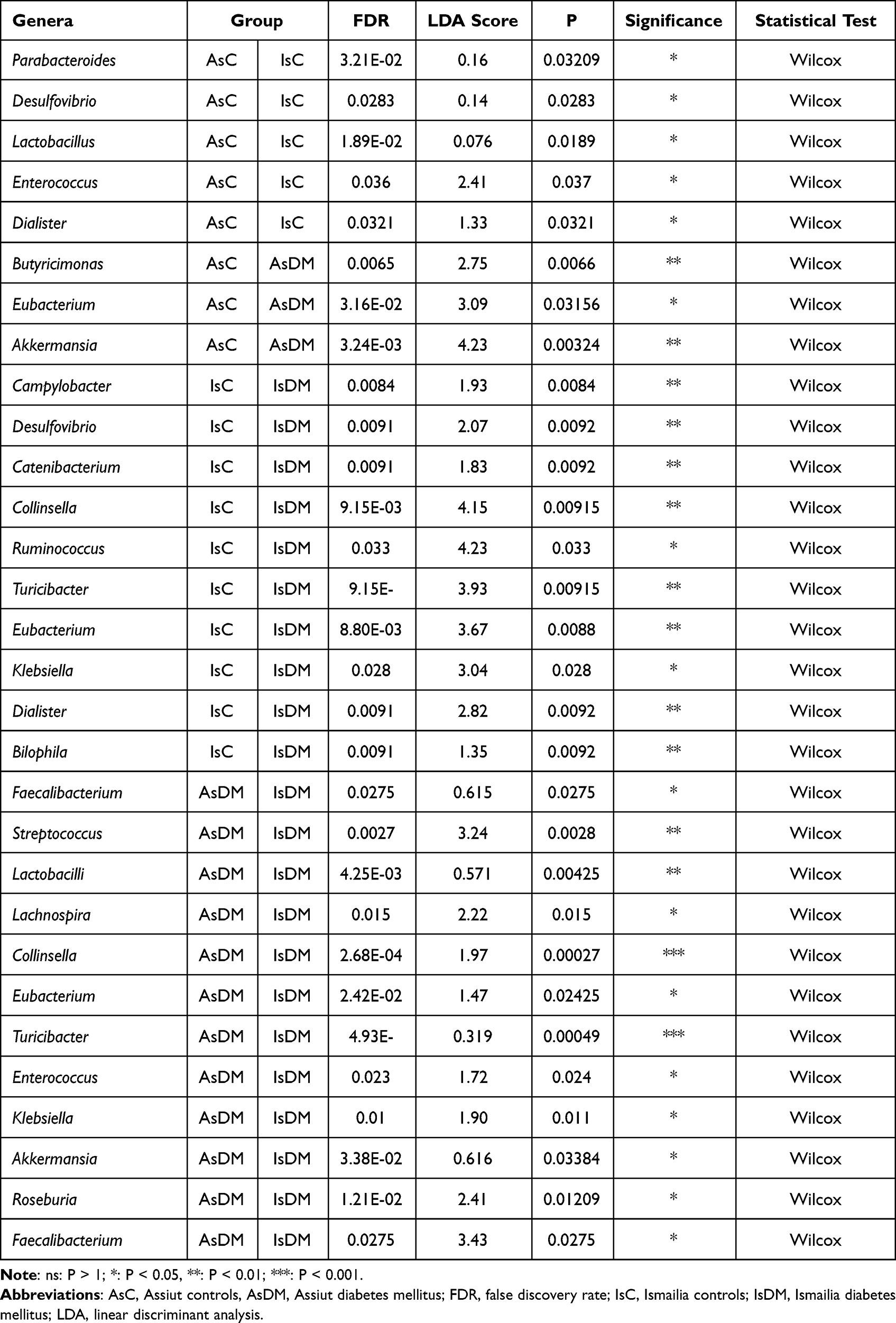 |
Table 2 Significant Genera Amongst the Studied Groups |
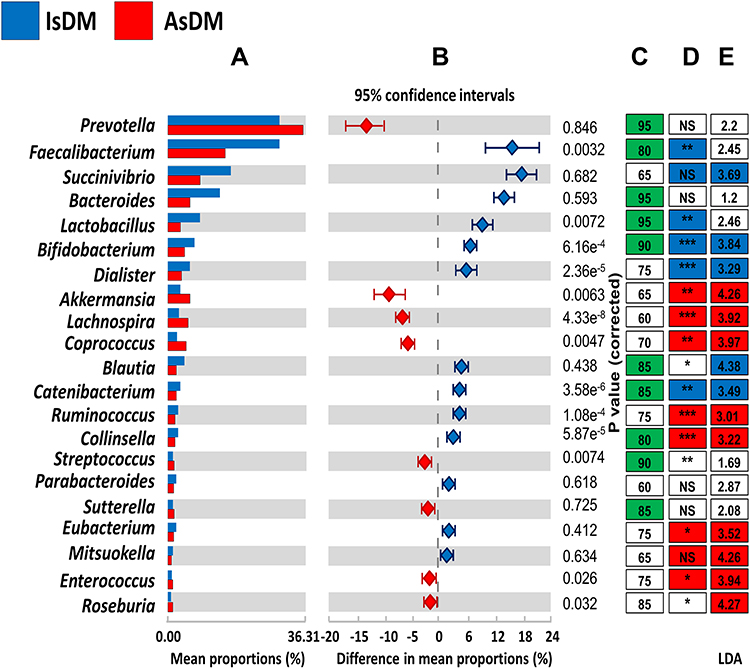 |
Figure 3 Genus level analysis of gut microbiomes. (A) Relative abundance of most predominant genera in diabetic groups. (B) Difference in mean proportions between diabetic groups measured by confidence of intervals. (C) Green boxes represented core genera in all samples and the numbers defined the percentage of the presence of genera in all data set. (D) Colored boxes indicated the core microbiome of each diabetic group and strikes represented the significance difference of genera between diabetic groups [NS (P< 0.05), * (P< 0.05), ** (P< 0.01) or *** (P< 0.001)]. (E) LEfSe inferred potential biomarkers for each group and numbers indicated LDA scores. |
Gut Inhabitants Notably Shape the Compositional Characteristics of Enterotypes
The samples were assigned to five enterotypes (E 1–5), and these were mainly dependent on the health status and locality. Enterotype E1 (AsC) had 18 genera, E2 (IsDM) had 17 genera, E3 (IsC) had 19 genera, E4 (the four samples of giardia in AsDM) presented a cluster distinct from the other AsDM samples and E5 (AsDM) had 17 genera. The leading genera in the five enterotypes were Prevotella in E1, E3 and E5, Faecalibacterium in E2 and Bacteroides in E4, as shown in Figure 4 and Supplementary Tables S3 and S4.
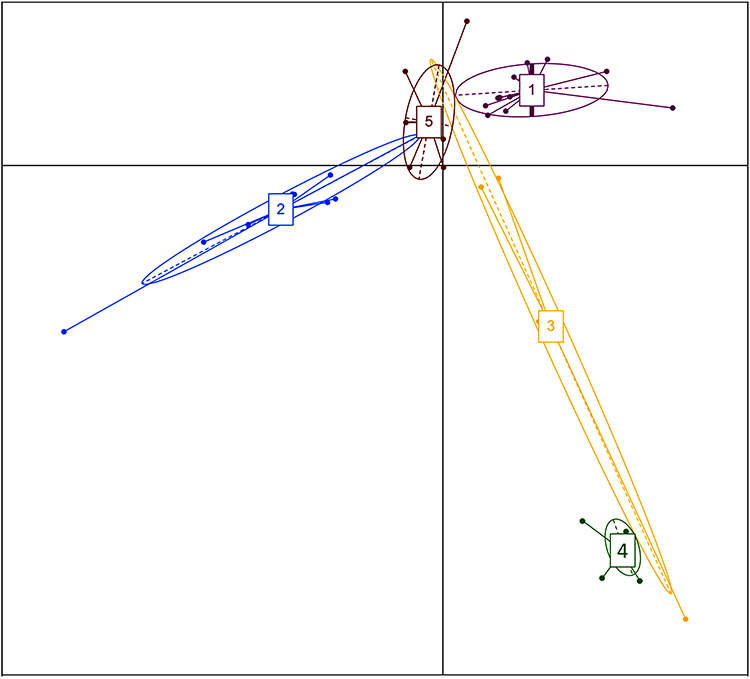 |
Figure 4 Enterotype clusters identified using PCoA analysis in the gut microbiota. Enterotype cluster 1 (Assiut controls), enterotype cluster 2 (Ismailia DM), enterotype cluster 3 (Ismailia controls), enterotype cluster 4 (Assiut DM with giardiasis) and enterotype cluster 5 (AsDM). |
Network Correlation Analysis
The strength of the correlation between the top 29 of the most abundant genera was estimated using the Spearman correlation coefficient (r ≥ ± 0.63, P ≤ 0.01). The over-representation of Prevotella had a negative impact on the abundance of the Faecalibacterium, Blautia, Lachnospira and Lactococcus genera. The Mitsoukella, Catenibacterium, Slackia, Eubacterium, Dialister and Succinivibrio genera were positively correlated to each other. The genus Bacteroides was positively correlated with Parabacteroides and negatively correlated with Sarcina and Collinsella genera, as shown in Figure 5.
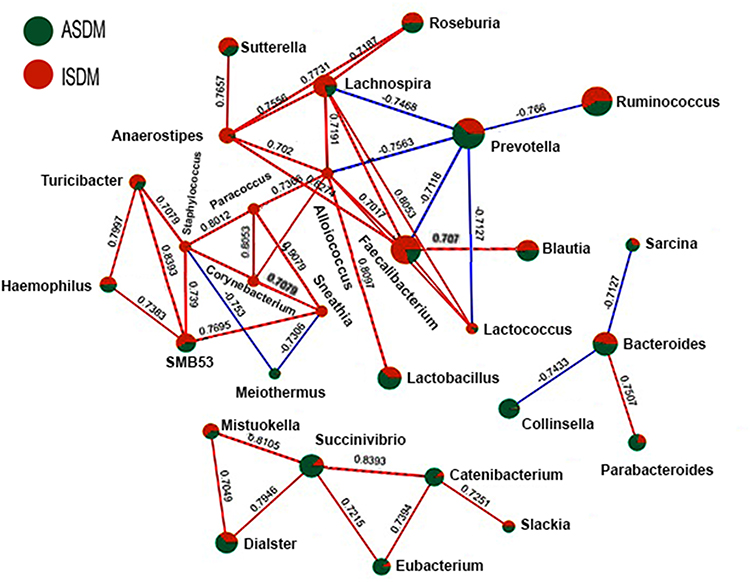 |
Figure 5 Estimated co-occurrence at the genus level among the DM samples. Assiut samples were presented by the green color, while Ismailia samples were presented by the red color. The size of a node represented its relative abundance. The blue lines represented negative association, and the red lines represented positive associations. |
Function Prediction of the Abundant KEGG
In the KEGG modules of the AsDM group, we observed gene functions for neurodegenerative diseases (LDA= 2.9, P= 0.003), carbohydrate metabolism (amino sugar and nucleotide sugar metabolism, LDA= 4.1, P= 0.009), as well as for xenobiotic biodegradation and metabolism (toluene degradation, LDA= 2.9, P= 0.03), while for the IsDM group, we observed gene function of carbohydrate metabolism (starch and sucrose metabolism, LDA= 4, P= 0.04).
In the AsC control groups, a number of gene functions were observed: of carbohydrate metabolism (fructose and mannose metabolism, LDA= 3.9, P= 0.001 and ascorbate and aldarate metabolism, LDA= 2.9, P= 0.0001). In the IsC group, the gene functions found were replication, recombination and repair proteins (LDA= 3.9, P= 0.02), carbohydrate metabolism (propanoate metabolism, LDA= 3.7, P= 0.02) and amino acid metabolism (LDA= 3.2, P= 0.02).
Discussion
Children are considered a unique category because in early childhood their gut microbiota is unstable, before evolving into its adult configuration.25 Generally, differences in gut microbiota have been reported normally not only across wide international geographical areas with different socio-economic settings but also among people with similar cultural and genetic backgrounds.26 So, this raised the research question, whether the geographical locality influenced the compositional and functional characteristics of T1DM-associated gut microbiota in controlled diabetic children from two Egyptian cities. Unfortunately, worldwide there are only limited studies discussing this point; for the situation in Egypt, studies are almost non-existent.
In our study, the alpha diversity of the samples was significantly higher among the controls than the diabetic groups of the same locality. The effect of the geographical difference was clearly apparent among the same groups in both areas.
Similar results were reported in previous studies,27,28 which also detected significant variations in the gut microbiota of healthy subjects than the diabetic groups. The lack of bacterial diversity could mean that their ability to digest a diverse diet would be limited and could lead to a number of gut microbial fermentation products, which might eventually cause metabolic diseases, including diabetes.29
When examining the phylum level among the diabetic groups, the increased prevalence of Actinobacteria and Proteobacteria was generally accompanied with an under-representation of Firmicutes. IsDM exhibited a higher frequency of Bacteroidetes than that found in their corresponding controls. Other studies have reported similar findings of a reduced abundance of Firmicutes in children with T1DM, lower than that found in healthy children.28,30 The special group of four children in the Assiut diabetes group who had giardia cysts in their stool samples surprisingly showed enriched abundance of Bacteroidetes and Proteobacteria and markedly diminished Firmicutes, in line with findings previously reported by Barash et al.31 Giardia infection increases bacterial load and invasiveness as a sequela to intestinal barrier disruption,32 which highlights its possible role in diabetes. It is also a reflection of the poor socioeconomic status. Other findings are in line with results of previous studies. For example, the control groups in this study generally showed a higher mean F/B ratio than the DM groups from the same locality; similar results were previously presented in studies by Murri et al28 and De Goffau et al.33 There were also differences in the mean F/B ratios of the two control groups, with IsC exhibiting a significantly higher F/B ratio than that found in AsC, a result highlighting the influence of geographical location with its resultant difference in the socioeconomic status, poverty, education, hygiene, and nutritional habits. Members of the Bacteroidetes decrease the mucosal integrity of the gut, activate inflammatory pathways and block T-reg differentiation by producing acetate, succinate and propionate,30,34 while Firmicutes, by the production of butyrate, enhance the gut barriers by reverse mechanisms, as explained in the findings of Louis and Flint.35
In this study, Bifidobacterium, a core genus in all the samples, was higher in the IsC group than in the IsDM group, while the reverse was true in the Assiut samples. The IsDM group reported a significantly higher percentage of Bifidobacterium than did the AsDM group; it was also a biomarker for IsDM. Most other studies have reported that Bifidobacterium was found in higher abundance in control groups than in diabetic groups.28,30,36,37 Another core genus in all samples was Lactobacilli, but here, in both localities, the percentage was unusually higher among the diabetic groups than in their corresponding controls, with the abundance being significantly higher in the IsDM group than in the AsDM group. Other studies have produced contradictory results regarding the abundance of Lactobacilli in people with diabetes mellitus.28,38 It appears that the protective effects of these two genera may depend on the presence of other gut bacteria to work together in a complementary way.39 They produce lactate and acetate, but not butyrate and/or propionate,40 thus contributing to the health effects indirectly mediated by SCFA.
An interesting finding is that the diabetic groups in both localities showed greater enrichment of Bacteroides than did their corresponding control groups. This finding - that Bacteroides are the prominent genus leading to T1DM-associated dysbiosis, regardless of the age, ethnicity or geography of the sample - has also been reported by other studies.6,28,30,36 The abundance of this genus is proportional to the number of T1DM autoantibodies.33,41 It has also been reported that this genus is positively correlated with levels of zonulin, which is a physiological modulator of intercellular tight junctions; it increases gut permeability and disrupts the intestinal barrier function, thus leading to the genesis of metabolic disorders.30 Bacteroides also produce Glutamic acid decarboxylase (GAD) which can stimulate GAD autoimmunity by molecular mimicry.34
There is a controversy regarding the association of the genus Prevotella with T1DM, and researchers differ as to whether they see it as beneficial or harmful. Some studies have associated Prevotella spp. with gut inflammation, autoimmune diseases, insulin resistance and diabetes.42 The present study, in contrast, found Prevotella to be lower in the diabetic groups than in their corresponding control groups in the same locality, a finding in line with that of many other studies. A number of other researchers have argued that Prevotella species are beneficial microbes, and see them as able to improve glucose metabolism stimulated by the intake of prebiotics and to produce more SCFA from carbohydrates in the diet, thus maintaining the integrity of the intestinal wall;43,44 this argument is supported by the results of our study.
The Ruminococcus genus was found at a higher abundance in the control groups than in the corresponding diabetic groups in both cities; this was similar to data reported in previous studies.6 A negative correlation was found between the Ruminococcus genus and the levels of HbA1C and zonulin, which could affect gut permeability in T1DM.30 Many members of this genus have a strong gastrointestinal mucin-degrading ability and release lactate, acetate and propionate, some of which can be used by other bacteria to produce such end products as butyrate.34
There is contradictory evidence regarding the genus Faecalibacterium, which is a member of the Ruminococcaceae. In this study, it was found to be a core genus in all samples and was found in higher abundance amongst the diabetic groups in both cities than in their corresponding control groups; this was similar to the findings of a previous study.36 It was also significantly higher in IsDM than AsDM. F. prausnitzii was found to be a biomarker for IsC; it has a predominant butyrate-producing capacity and a known anti-inflammatory molecule (MAM), a metabolite needed for gut homeostasis.45
Many genera in this study were strongly dependent on geographical location and thus were not consistent with T1DM. These included Dialister, lachnospira, Blautia, Dorea and Veillonella, and their role suggests that geographical location may have a greater driving effect than the disease itself.
In the current study, the overall structure of the gut microbiota is categorized into five distinct and discrete patterns called enterotypes with interactions among community members. The leading genera in the five enterotypes were Prevotella in E1, E3 and E5, Faecalibacterium in E2 and Bacteroides in E4. It was previously reported that the Egyptian gut enterotype belongs to the Prevotella enterotype,46 an enterotype which has been associated with carbohydrate consumption, a fact which would explain its abundance in many groups in this study. Bacteroides was not significantly abundant in all the enterotypes, a situation arising from the lack of meat in the children’s diet, an indication of their low socioeconomic level.47 The enterotypes in children are affected by breast-feeding duration in early life and pre-school dietary lifestyle. Three enterotypes were identified in 281 Dutch school-age children (6–9 years) dominated by the genera Bacteroides, Prevotella, and Bifidobacterium.48 In that study, the children, which were in the same age as this study, were found to have an overall adult-like gut microbiota.48
It was also found that species tend to co-occur most frequently either with species with which they most strongly compete, or with genetically similar taxa, or with those which respond to environmental stimuli in the same manner, that is, with similar functionality.49
In this study, Prevotella (a Bacteroidetes member) had a negative impact on the abundance of many of the Firmicutes members, such as the Faecalibacterium, Blautia, Lachnospira and Lactococcus genera, which include many SCFA producers. Also, many of the polysaccharide-degrader genera that previously characterized the Egyptian gut, such as Eubacterium, Mitsuokella and Catenibacterium, were shown in this study to be positively related to each other. The Succinivibrio genus, which was linked to pathogenicity and infections and was previously reported to be enriched in the Egyptian gut, was also found in an earlier study to have a positive correlation with the polysaccharide degraders.46
The assembly of functional microbial communities can be driven by an analysis of microbial co-occurrence patterns.50 Several microbial functions were significantly over- or under-represented in the four groups in the study, which was due to significant differences in their gut composition. The relative abundance of genes associated with a given pathway may indicate an increased metabolic activity of the gut microbiota with regard to this pathway.30 As Egyptians consume a large quantity of cereals in their diet, it has not surprisingly been found previously that many carbohydrate utilization pathways tend to be abundant in Egyptian stool samples.46 The increase in the abundance of Prevotella in the control groups may be one of the factors behind the increased expression of amino acid and carbohydrate metabolism pathways, because Prevotella actively interacts with a variety of microbes to accelerate the metabolism of carbohydrates in the body.51
There was a significant expression of the gene functions for neurodegenerative diseases (NDs) in the AsDM group. This might be attributable to a significant increase in several genera such as Akkermansia and Lachnospira, as well as to the fact that there was significantly less Faecalibacterium and Bifidobacterium in AsDM than in IsDM that was in line with previous studies.52,53
Like several recent studies, we observed the enrichment of many modules in the gut microbiota of the diabetic groups, including the amino sugar and nucleotide sugar metabolism pathways, as well as the biodegradation and metabolism of xenobiotics in the AsDM group.54 The starch and sucrose metabolism found in the IsDM group was also in line with the results of several recent studies.55 The latter pathway was also reported in a previous study to be strongly associated with the development of T2D.56 An interesting finding was the enhanced expression of the toluene degradation pathway in the AsDM group, which could be attributed to the increased exposure to toluene found in pesticides and other pollutants, as agriculture is common in Assiut. This may also explain the enhanced expression of gene functions for neurodegenerative diseases in the AsDM group, as exposure to toluene has previously been linked to central nervous tissue diseases.57 These findings highlight the influence of locality on the gut functions.
It was clear in this study that in the diabetic groups there were many compensatory mechanisms attempting to compensate for increased glucose levels. For example, the significant increase in the amino sugar metabolism was an attempt to stimulate insulin secretion by combining amino acids with glucose; this would lead to protein synthesis and amino acid transport in the target tissues. The degrading of complex carbohydrates such as cellulose and starch from the diet leads to the production of glucose,58 which explains the increased expression of starch metabolism in the IsDM group.
While many studies in this field indicate that there are generally great variations in the patterns of relative abundance of gut microbiota, the group-specific differentiation between cases and controls in this study does pose a challenge. A recent systematic review has reported that twenty-four scientific articles have confirmed the association between gut microbiota dysbiosis and T1DM,59 with several mechanisms being suggested as regards the role of the gut microbiota in the pathogenesis of T1DM. One of these mechanisms is that some bacteria increase mucin degradation and thus reduce the integrity and increase the permeability of intestinal mucosa, which leads to bacterial penetration and stimulation of the immune system as well as to the production of antibodies against these bacteria. These antibodies then cross-react with the surface antigens of the pancreatic beta cells; this, along with T cell cross-reactivity, results in the destruction of beta cells and the formation of T1DM.60
This study highlights the way that exposure to different environments acts as a selective pressure in shaping gut microbial ecology.61 Northern or Lower Egypt, the region surrounding Ismailia, is more industrialized than the south (also known as Upper Egypt) where Assiut is located. The North generally is characterized by higher socioeconomic and educational levels than the South, while Southern Egypt (especially its rural areas) has higher levels of poverty and Illiteracy Rate. The economy of the south relies upon agriculture, so that the population may suffer from exposure to agricultural chemicals like fertilizers. Assiut, in fact, has the highest incidence of poverty in all of Egypt (~65), a figure much higher than that in Ismailia (~ 15),62 in addition to the high Illiteracy Rate (Ismailia 17.7 and Assiut 28.4).63 This definitely affects the dietary habits of the people and results in a different pattern of gut microbiota.
The main limitation of this study is the limited number of subjects in the sample, which means that it can only be considered as a pilot study. Other limitations include the fact that the genetic status of the control group subjects is unknown, especially the presence or absence of T1DM susceptibility genes. The study also lacked the dietary history of the children enrolled, especially as any deficiency of iron, vitamin A or zinc could affect the gut microbiota. In addition, it was impossible to prove the cause-and-effect relationships in the study, as the design was a cross-sectional one. The strengths of our study, however, lie in its being the first to be done in Egypt, as well as in its careful design, its well-matched groups, and in the next-generation sequencing of the microbiome.
Conclusion
Irrespective of their geographical location, the diabetic groups were characterized by several consistent microbiota profile including significantly lower alpha diversity than the controls as well as a lower mean Firmicutes/ Bacteroidetes (F/B) ratio, and reduced proportions of Firmicutes and the genera Prevotella and Ruminococcus. There were also significantly enriched representations of Actinobacteria, Bacteroidetes and Proteobacteria and the genera Lactobacilli, Bacteroides and Faecalibacterium. On the other hand, many genera were strongly dependent on geographical location and thus were not consistent with T1DM. These included Dialister, lachnospira, Blautia, Dorea and Veillonella, suggesting that the geographical location may have a greater driving effect than the disease itself. Lowered bacterial diversity associated with T1DM clearly stratified microbiomes into separate clusters that were based on the health status and the geographical locality of the groups. It was also clear that Giardia shaped the compositional characteristics of enterotypes, especially a diminished abundance of Firmicutes. The overall functional potential of microbiomes associated with T1DM was characterized by the over-representation of many genes, and included some mechanisms compensating for increased glucose levels, such as the metabolism of amino sugar to stimulate insulin secretion.
Acknowledgments
We would like to thank Professor Enas Daef, the director of the Medical Research Laboratory, Faculty of Medicine, Assiut University, and the team for the technical support.
Funding
This research was funded by the Grant Office of the Faculty of Medicine, Assiut University, grant number 2016/12/29-004.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Abdellatif AM, Sarvetnick NE. Current understanding of the role of gut dysbiosis in type 1 diabetes. J Diabetes. 2019;11(8):632–644. doi:10.1111/1753-0407.12915
2. Durazzo M, Ferro A, Gruden G. Gastrointestinal microbiota and type 1 diabetes mellitus: the state of art. J Clin Med. 2019;8(11):1843. doi:10.3390/jcm8111843
3. Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164(3):337–340. doi:10.1016/j.cell.2016.01.013
4. Mcdermott AJ, Huffnagle GB. The microbiome and regulation of mucosal immunity. Immunology. 2014;142(1):24–31. doi:10.1111/imm.12231
5. Skonieczna-żydecka K, Marlicz W, Misera A, Koulaouzidis A, Łoniewski I. Microbiome—The missing link in the gut-brain axis: focus on its role in gastrointestinal and mental health. J Clin Med. 2018;7(12):521. doi:10.3390/jcm7120521
6. Gavin PG, Mullaney JA, Loo D, et al. Intestinal metaproteomics reveals host-microbiota interactions in subjects at risk for type 1 diabetes. Diabetes Care. 2018;41(10):2178–2186. doi:10.2337/dc18-0777
7. Zhou H, Sun L, Zhang S, Zhao X, Gang X, Wang G. Evaluating the causal role of gut microbiota in type 1 diabetes and its possible pathogenic mechanisms. Front Endocrinol (Lausanne). 2020;11. doi:10.3389/fendo.2020.00125.
8. Kaplan RC, Wang Z, Usyk M, et al. Gut microbiome composition in the Hispanic Community Health Study/Study of Latinos is shaped by geographic relocation, environmental factors, and obesity. Genome Biol. 2019;20(1). doi:10.1186/S13059-019-1831-Z
9. Xiao L, Wang J, Zheng J, Li X, Zhao F. Deterministic transition of enterotypes shapes the infant gut microbiome at an early age. Genome Biol. 2021;22(1):1–21. doi:10.1186/s13059-021-02463-3
10. Costea PI, Hildebrand F, Manimozhiyan A, et al. Enterotypes in the landscape of gut microbial community composition. Nat Microbiol. 2018;3(1):8–16. doi:10.1038/S41564-017-0072-8
11. Martinez-Medina JN, Flores-Lopez R, López-Contreras BE, et al. Effect of gut microbial enterotypes on the association between habitual dietary fiber intake and insulin resistance markers in Mexican children and adults. Nutrients. 2021;13(11):1–16. doi:10.3390/nu13113892
12. Fuks G, Elgart M, Amir A, et al. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome. 2018;6(1). doi:10.1186/s40168-017-0396-x
13. McGregor K, Lie Labbe A, Greenwood CMT, et al. MDiNE: a model to estimate differential co-occurrence networks in microbiome studies. Bioinformatics. 2020;36(6):1840–1847. doi:10.1093/bioinformatics/btz824
14. Marathe PH, Gao HX, Close KL. American Diabetes Association Standards of Medical Care in Diabetes 2017. J Diabetes. 2017;9(4):320–324. doi:10.1111/1753-0407.12524
15. Ramadan M, Solyman S, Taha M, Hanora A. Preliminary characterization of human skin microbiome in healthy Egyptian individuals. Cell Mol Biol. 2016;62(8):21–27. doi:10.14715/cmb/2016.62.8.4
16. Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi:10.1038/nmeth.f.303
17. Edgar RC, Bateman A. Search and clustering orders of magnitude faster than BLAST. Bioinforma Appl NOTE. 2010;26(19):2460–2461. doi:10.1093/bioinformatics/btq461
18. McDonald D, Price MN, Goodrich J, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6(3):610–618. doi:10.1038/ismej.2011.139
19. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300.
20. Harrell FE, Dupont CJR Harrell: Hmisc: Harrell miscellaneous - Google Scholar. R Packag version; 2008. Available from: https://scholar.google.com/scholar?cluster=1135968011042410304&hl=en&oi=scholarr.
21. Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi:10.1038/nature09944
22. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(D1):D199–D205. doi:10.1093/NAR/GKT1076
23. Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi:10.1038/nbt.2676
24. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
25. Bäckhed F, Roswall J, Peng Y, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703. doi:10.1016/j.chom.2015.04.004
26. Fontana A, Panebianco C, Picchianti-Diamanti A, et al. Gut microbiota profiles differ among individuals depending on their region of origin: an Italian pilot study. Int J Environ Res Public Health. 2019;16(21):4065. doi:10.3390/ijerph16214065
27. Manfredo Vieira S, Hiltensperger M, Kumar V, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359(6380):1156–1161. doi:10.1126/science.aar7201
28. Murri M, Leiva I, Gomez-Zumaquero JM, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11(1). doi:10.1186/1741-7015-11-46
29. Han H, Li Y, Fang J, et al. Gut microbiota and type 1 diabetes. Int J Mol Sci. 2018;19(4):1–11. doi:10.3390/ijms19040995
30. Leiva-Gea I, Sánchez-Alcoholado L, Martín-Tejedor B, et al. Gut microbiota differs in composition and functionality between children with type 1 diabetes and MODY2 and healthy control subjects: a case-control study. Diabetes Care. 2018;41(11):2385–2395. doi:10.2337/dc18-0253
31. Barash NR, Maloney JG, Singer SM, Dawson SC. Giardia alters commensal microbial diversity throughout the murine gut. Infect Immun. 2017;85(6). doi:10.1128/IAI.00948-16
32. Chen TL, Chen S, Wu HW, et al. Persistent gut barrier damage and commensal bacterial influx following eradication of Giardia infection in mice. Gut Pathog. 2013;5(1). doi:10.1186/1757-4749-5-26
33. De Goffau MC, Luopajärvi K, Knip M, et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes. 2013;62(4):1238–1244. doi:10.2337/db12-0526
34. Huang Y, Li SC, Hu J, et al. Gut microbiota profiling in Han Chinese with type 1 diabetes. Diabetes Res Clin Pract. 2018;141:256–263. doi:10.1016/j.diabres.2018.04.032
35. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. doi:10.1111/1462-2920.13589
36. Pinto E, Anselmo M, Calha M, et al. The intestinal proteome of diabetic and control children is enriched with different microbial and host proteins. Microbiology. 2017;163(2):161–174. doi:10.1099/mic.0.000412
37. Salamon D, Sroka-Oleksiak A, Kapusta P, et al. Characteristics of gut microbiota in adult patients with type 1 and type 2 diabetes based on next-generation sequencing of the 16S rRNA gene fragment. Polish Arch Intern Med. 2018;128(6):336–343. doi:10.20452/pamw.4246
38. Alkanani AK, Hara N, Gottlieb PA, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes. 2015;64(10):3510–3520. doi:10.2337/db14-1847
39. Rivière A, Gagnon M, Weckx S, Roy D, De Vuyst L. Mutual cross-feeding interactions between Bifidobacterium longum subsp. longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan oligosaccharides. Appl Environ Microbiol. 2015;81(22):7767–7781. doi:10.1128/AEM.02089-15
40. Bindels LB, Delzenne NM, Cani PD, Walter J. Opinion: towards a more comprehensive concept for prebiotics. Nat Rev Gastroenterol Hepatol. 2015;12(5):303–310. doi:10.1038/nrgastro.2015.47
41. Kostic AD, Gevers D, Siljander H, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17(2):260–273. doi:10.1016/j.chom.2015.01.001
42. Pedersen HK, Gudmundsdottir V, Nielsen HB, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535(7612):376–381. doi:10.1038/nature18646
43. Kovatcheva-Datchary P, Nilsson A, Akrami R, et al. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of prevotella. Cell Metab. 2015;22(6):971–982. doi:10.1016/j.cmet.2015.10.001
44. Wang J, Li W, Wang C, et al. Enterotype bacteroides is associated with a high risk in patients with diabetes: a pilot study. J Diabetes Res. 2020;2020:1–11. doi:10.1155/2020/6047145
45. Xu J, Liang R, Zhang W, et al. Faecalibacterium prausnitzii-derived microbial anti-inflammatory molecule regulates intestinal integrity in diabetes mellitus mice via modulating tight junction protein expression. J Diabetes. 2020;12(3):224–236. doi:10.1111/1753-0407.12986
46. Shankar V, Gouda M, Moncivaiz J, et al. Differences in gut metabolites and microbial composition and functions between Egyptian and U.S. children are consistent with their diets. mSystems. 2017;2(1). doi:10.1128/mSystems.00169-16.
47. Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (80-). 2011;334(6052):105–108. doi:10.1126/science.1208344
48. Zhong H, Penders J, Shi Z, et al. Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in young school-age children. Microbiome. 2019;7(1):1–14. doi:10.1186/s40168-018-0608-z
49. Chaffron S, Rehrauer H, Pernthaler J, Von Mering C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010;20(7):947–959. doi:10.1101/gr.104521.109
50. Endesfelder D, Engel M, Davis-Richardson AG, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome. 2016;4(1). doi:10.1186/s40168-016-0163-4
51. Petersen LM, Bautista EJ, Nguyen H, et al. Community characteristics of the gut microbiomes of competitive cyclists. Microbiome. 2017;5(1):98. doi:10.1186/s40168-017-0320-4
52. Sasmita AO. Modification of the gut microbiome to combat neurodegeneration. Rev Neurosci. 2019;30(8):795–805. doi:10.1515/revneuro-2019-0005
53. De Angelis M, Piccolo M, Vannini L, et al. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One. 2013;8(10):10. doi:10.1371/journal.pone.0076993
54. Chávez-Carbajal A, Pizano-Zárate ML, Hernández-Quiroz F, et al. Characterization of the gut microbiota of individuals at different T2D stages reveals a complex relationship with the host. Microorganisms. 2020;8(1):94. doi:10.3390/microorganisms8010094
55. Ma S, You Y, Huang L, et al. Alterations in gut microbiota of gestational diabetes patients during the first trimester of pregnancy. Front Cell Infect Microbiol. 2020;10:58. doi:10.3389/fcimb.2020.00058
56. Sun H, Zhang S, Zhang A, et al. Metabolomic analysis of diet-induced type 2 diabetes using UPLC/MS integrated with pattern recognition approach. PLoS One. 2014;9(3):e93384. doi:10.1371/journal.pone.0093384
57. Sumathy J. A study on toluene degrading bacteria; 2018:414–425.
58. Dedysh SN, Yilmaz P. Refining the taxonomic structure of the phylum Acidobacteria. Int J Syst Evol Microbiol. 2018;68(12):3796–3806. doi:10.1099/ijsem.0.003062
59. Jamshidi P, Hasanzadeh S, Tahvildari A, et al. Is there any association between gut microbiota and type 1 diabetes? A systematic review. Gut Pathog. 2019;11(1):1–10. doi:10.1186/s13099-019-0332-7
60. Cole DK, Bulek AM, Dolton G, et al. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J Clin Invest. 2016;126(6):2191–2204. doi:10.1172/JCI85679
61. Das B, Ghosh TS, Kedia S, et al. Analysis of the gut microbiome of rural and urban healthy Indians living in sea level and high altitude areas. Sci Rep. 2018;8(1). doi:10.1038/s41598-018-28550-3
62. El-Laithy H The ADCR 2011: poverty in Egypt (2009); 2011. Available from: https://scholar.google.com/scholar?rlz=1C5CHFA_enEG903EG903¨1¡UTF-8&lr&q=related:xhW7QSQU8uaqpM:scholar.google.com/.
63. Baseera undefined. Localizing the targets of the sustainable development goals on governorate level study; 2018. Available from: https://www.mendeley.com/catalogue/047cf53b-8668-38e5-bc57-b03fe49a08b9/?utm_source=desktop&utm_medium=1.19.8&utm_campaign=open_catalog&userDocumentId=%7B3952b62f-2157-47f9-94ea-5a08c57a2afb%7D.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.