Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Impact of acute exacerbations on platelet reactivity in chronic obstructive pulmonary disease patients
Authors Muñoz-Esquerre M, Ferreiro JL ![]() , Huertas D, Marcano AL, López-Sánchez M, Roura G, Gómez-Hospital JA, Dorca J, Cequier A, Santos S
, Huertas D, Marcano AL, López-Sánchez M, Roura G, Gómez-Hospital JA, Dorca J, Cequier A, Santos S
Received 27 September 2017
Accepted for publication 12 November 2017
Published 28 December 2017 Volume 2018:13 Pages 141—148
DOI https://doi.org/10.2147/COPD.S152660
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Mariana Muñoz-Esquerre,1 José Luis Ferreiro,2 Daniel Huertas,1,3 Ana Lucrecia Marcano,2 Marta López-Sánchez,1 Gerard Roura,2 Joan Antoni Gómez-Hospital,2 Jordi Dorca,1 Angel Cequier,2 Salud Santos1,3
1Department of Pulmonary Medicine, Bellvitge University Hospital, IDIBELL, University of Barcelona, L’Hospitalet de Llobregat, 2Heart Diseases Institute, Bellvitge University Hospital, IDIBELL, University of Barcelona, L’Hospitalet de Llobregat, 3Biomedical Research Networking Centre Consortium Respiratory Diseases, CIBERES, Barcelona, Spain
Background: A higher risk of atherothrombotic cardiovascular events, which are platelet-driven processes, has been described during acute exacerbations of chronic obstructive pulmonary disease (AECOPD). However, the relevance of platelet reactivity during AECOPD and whether this is affected by antiplatelet agents are not fully elucidated to date. This study aimed to evaluate whether platelet reactivity is augmented during an exacerbation in COPD patients with and without antiplatelet therapy and its association with systemic inflammatory parameters.
Materials and methods: Prospective, observational, ex vivo investigation was conducted in consecutive patients suffering an exacerbation of COPD. Platelet reactivity was assessed during AECOPD and at stable state. Platelet function assays included: 1) vasodilator-stimulated phosphoprotein assay expressed as P2Y12 reactivity index (PRI), 2) multiple electrode aggregometry and 3) optical aggregometry. Systemic inflammatory parameters such as leukocyte count, interleukin-6 and fibrinogen were also assessed.
Results: Higher platelet reactivity was observed during AECOPD compared to stability measured by vasodilator-stimulated phosphoprotein (PRI: 75.2%±1.9% vs 68.8%±2.4%, p=0.001). This augmented platelet aggregability was also observed in the subset of patients on antiplatelet therapy (PRI: 72.8%±3.1% vs 61.7%±7.5%, p=0.071). Consistent findings were observed with all other platelet function tests. Patients with greater enhancement of inflammatory markers during AECOPD were more likely to present a higher increase in platelet reactivity.
Conclusion: Platelet reactivity is increased during AECOPD, which may contribute to the augmented cardiovascular risk of these patients. Additionally, the increase in platelet reactivity might be associated with an increment in inflammatory markers during exacerbations.
Keywords: airflow limitation, inflammation, platelet aggregation, antiplatelets agents, thrombosis
Introduction
Cardiovascular disease is the most important comorbidity associated with chronic obstructive pulmonary disease (COPD) due to its impact on overall prognosis including mortality.1–3 Systemic inflammation has been suggested as one of the main factors that play a significant role in the pathogenesis of atherothrombosis in COPD,4,5 which is characterized by the presence of low-grade systemic inflammation in the stable state6 and, noteworthy, by an acute inflammatory response during exacerbations of the disease, as shown by Thomsen et al.7 The increased systemic inflammation associated with acute exacerbations is of particular relevance since it can be an underlying mechanism that may contribute to the impaired cardiovascular outcomes observed in COPD patients. In line with this, atherothrombotic complications such as myocardial infarction or stroke are particularly augmented during an acute exacerbation of COPD (AECOPD).8,9
Platelets are key elements in the development of atherosclerosis and its atherothrombotic complications.10 Several studies have observed that systemic inflammation may contribute to platelet activation and, thus, to an increased cardiovascular risk.11,12 In addition, an interesting mechanistic investigation by Maclay et al has shown increased platelet activation in COPD patients compared to non-COPD subjects, and during AECOPD compared to stability.13 However, the magnitude of the increment in platelet reactivity during an AECOPD is not fully elucidated to date, and if this is affected by antiplatelet agents has not been previously evaluated. The aim of this study was to evaluate whether platelet reactivity is augmented during an AECOPD compared with stability in COPD patients with and without antiplatelet therapy (APT) and its possible association with systemic inflammatory parameters.
Materials and methods
Population and study design
This was a prospective, ex vivo, observational investigation conducted in consecutive COPD patients attended during an exacerbation at the pulmonology day hospital from a tertiary centre. All patients were between 40 and 80 years of age and had been diagnosed of COPD following Global initiative for chronic Obstructive Lung Disease recommendations.1 An AECOPD was defined as an acute event characterized by worsening of the patient’s respiratory symptoms that is beyond normal day-to-day variations, which leads to a change in medication.1 Exacerbations were classified as moderate (outpatient treatment requiring antibiotics and/or oral corticosteroids) and severe (requiring hospital admission).14 Exclusion criteria were the presence of other pulmonary disease, current therapy with anticoagulant agents or glycoprotein IIb/IIIa inhibitors, any active malignancy or hematologic disease, platelet count <100×106/μL, severe chronic kidney disease (creatinine clearance <30 mL/min) and pregnant females. Patients could receive APT with aspirin or clopidogrel for concomitant chronic cardiovascular conditions.
The study had a prospective design with paired data, in which assessments were performed at two time points: the first one during AECOPD and the second one after the patient had recovered stability (4–6 weeks after the first visit and no symptoms of exacerbation or administration of antibiotics or oral corticosteroids for at least 2 weeks). In patients receiving APT, blood samples were collected for platelet function testing before the daily maintenance dose of aspirin or clopidogrel was administered. The study was approved by the local ethics committee “Comitè Ètic d’ Investigació Clínica Del Hospital de Bellvitge”, No PR077/13, and performed in accordance with the Declaration of Helsinki. All subjects provided written informed consent to the study.
Platelet function assays
Blood samples were collected from antecubital vein, discarding the first 2–4 mL of blood to avoid spontaneous platelet activation. Samples were processed by trained laboratory personnel blinded to the patient status and medication within 2 hours of blood drawing. Several agonists were used in platelet function assessments in order to explore different platelet activation signaling pathways. In patients receiving APT, aspirin-mediated effects were specifically evaluated by using arachidonic acid and collagen as agonists, while clopidogrel-mediated effects were specifically assessed by using adenosine diphosphate (ADP) as an agonist. Platelet function assays included flow cytometric analysis of the phosphorylation status of vasodilator-stimulated phosphoprotein (VASP), light transmission aggregometry (LTA) and multiple electrode aggregometry.
Analysis of the phosphorylation status of VASP
The P2Y12 reactivity index (PRI) measures the functional status of the platelet P2Y12 signaling pathway and was determined according to standard protocols.15 In brief, VASP phosphorylation (VASP-P) was measured by quantitative flow cytometry using commercially available labeled monoclonal antibodies (Biocytex Inc., Marseille, France). The PRI was calculated after measuring the mean fluorescence intensity (MFI) of VASP-P levels following challenge with prostaglandin E1 (PGE1) and PGE1 + ADP. PGE1 increases the VASP-P levels through stimulation of adenylate cyclase, and ADP binding to purinergic receptors leads to inhibition of adenylate cyclase; thus, the addition of ADP to PGE1-stimulated platelets reduces the levels of PGE1-induced VASP-P. The PRI was calculated as follows: ([MFI PGE1]−[MFI PGE1 + ADP]/[MFI PGE1]) ×100%. An increased PRI is indicative of higher platelet reactivity.
LTA
LTA was performed according to standard protocols as previously described.16 Briefly, blood-citrate tubes were centrifuged at 100× g for 10 min to recover platelet-rich plasma (PRP) and further centrifuged at 2,400× g for 15 min to recover platelet-poor plasma (PPP). Platelet aggregation was assessed using PRP and PPP by the turbidometric method in a two-channel aggregometer (Chrono-Log 490 Model; Chrono-Log Corp., Havertown, PA, USA). Light transmission was adjusted to 0% for PRP and 100% for PPP for each measurement. Maximal platelet aggregation is reported as a percentage and was measured following stimuli with several agonists: ADP (5 and 20 μmol/L), arachidonic acid (1 mmol/L) and collagen (2 μg/mL).
Multiple electrode aggregometry
Blood was collected in hirudin-treated tubes. Multiple electrode aggregometry was performed in whole blood with the Multiplate™ analyzer (Hoffman-La Roche Ltd., Basel, Switzerland) as previously described.17 This instrument assesses the change in impedance caused by the adhesion of platelets onto sensor units formed by silver-covered electrodes. In this investigation, the following agonists were used: ADP (6.4 μmol/L), arachidonic acid (0.5 mmol/L), collagen (3.2 μg/mL) and thrombin receptor activating peptide (32 μmol/L). Aggregation curves were recorded for 6 min and platelet aggregation was determined as the area under the curve of arbitrary aggregation units (AU × min).
Systemic inflammatory parameters
Three inflammatory parameters involved in COPD exacerbations were assessed: leukocyte count, interleukin-6 (IL-6) and fibrinogen.7 Leukocytes were measured as part of complete cell blood counts performed at the time points described above. To quantify the IL-6 (expressed in pg/mL) and fibrinogen (expressed in g/L) serum levels, individual enzyme-linked immunosorbent assay tests were used following the manufacturer’s instructions. The lower limit of detection for IL-6 was 0.156 pg/mL (HS600B; R&D Systems, Inc., Minneapolis, MN, USA) and for fibrinogen was 1.56 ng/mL (ab208036; Abcam, Cambridge, UK).
Study endpoints and sample size
The primary endpoint of this study was the difference in platelet reactivity measured as PRI of COPD patients during the AECOPD compared to stable state. Assuming an SD of 20%, a sample size of 36 subjects was needed to detect a minimal difference between time points of 10%, with 85% power and a two-tailed p-value <0.05 for a paired data comparison. Considering an approximate 10% dropout rate (eg, inadequate samples or loss in the follow-up), the inclusion of 40 subjects was allowed to ensure that data from 36 patients were available for analysis. The secondary endpoints included: 1) differences in platelet reactivity measured with the other platelet function assays during the AECOPD compared to stable state and 2) changes in inflammatory parameters during the AECOPD compared to stable state and their association with platelet reactivity.
Statistical analysis
Conformity to the normal distribution was evaluated for continuous variables with the Kolmogorov–Smirnov and the Shapiro–Wilk tests. For baseline characteristics, categorical variables were expressed as frequencies and percentages, whereas continuous variables were summarized by mean ± SD or by median (interquartile range) if a normal distribution could be assumed or not, respectively. A repeated-measures analysis of variance (ANOVA) model was used to evaluate the primary endpoint and to make all other intragroup comparisons between the AECOPD compared to the stable state. In order to evaluate the association between inflammation and platelet aggregability, exploratory analyses comparing the change in platelet reactivity measured by VASP (ΔPRI: PRI during exacerbation–PRI during stability) according to tertiles of the difference (Δ) of each inflammatory marker (levels during exacerbation–levels during stability) were performed using an ANOVA method with a general linear model. Results are reported as least squares mean ± standard error of the mean for the above-detailed ANOVA analyses. A p-value <0.05 was considered statistically significant for all comparisons. Statistical analysis was performed using SPSSv18.0 software (SPSS Inc., Chicago, IL, USA).
Results
Forty consecutive severe COPD patients consulting with an AECOPD were prospectively included. Two patients were excluded because they were lost in the follow-up, and one patient was excluded because anticoagulant therapy for atrial fibrillation was prescribed before visit 2 occurred. Therefore, a total of 37 patients were included in this analysis: 29 (78.4%) with a moderate AECOPD and 8 (21.6%) with a severe AECOPD, thus requiring admission. In this cohort, 10 patients receiving APT were included: 8 on aspirin and 2 on clopidogrel. Demographic and clinical characteristics of the overall population are summarized in Tables 1 and 2. Characteristics of the AECOPD as well as treatment approach are listed in Table 3.
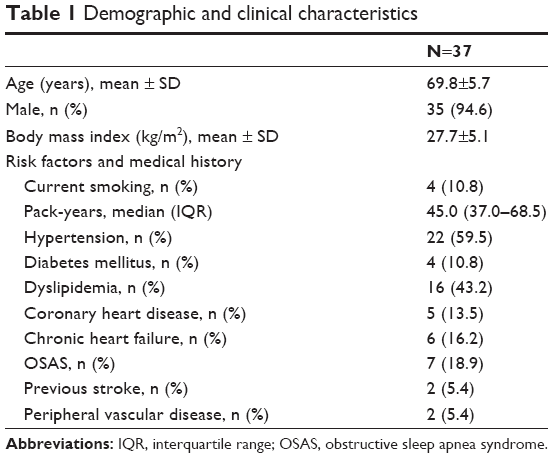 | Table 1 Demographic and clinical characteristics |
 | Table 2 Baseline pulmonary function parameters, laboratory data and medical therapy |
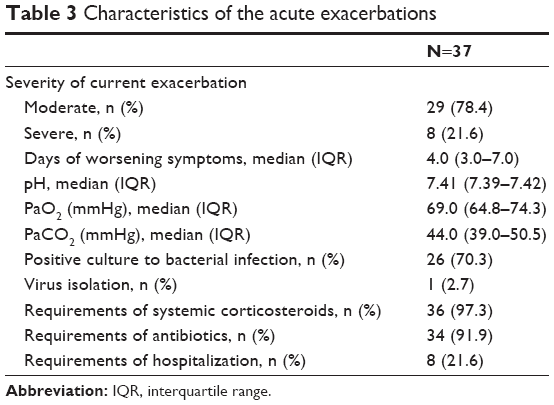 | Table 3 Characteristics of the acute exacerbations |
Platelet reactivity during an AECOPD
Higher platelet reactivity was observed during AECOPD compared to stability, as measured by VASP (PRI: 75.2%±1.9% vs 68.8%±2.4%, p=0.001) in the overall sample (Figure 1). Consistent results were obtained with the other platelet function assays performed and all agonists used, which showed a numerically greater platelet reactivity during exacerbations, although statistical significance was not reached in all of them (Table 4).
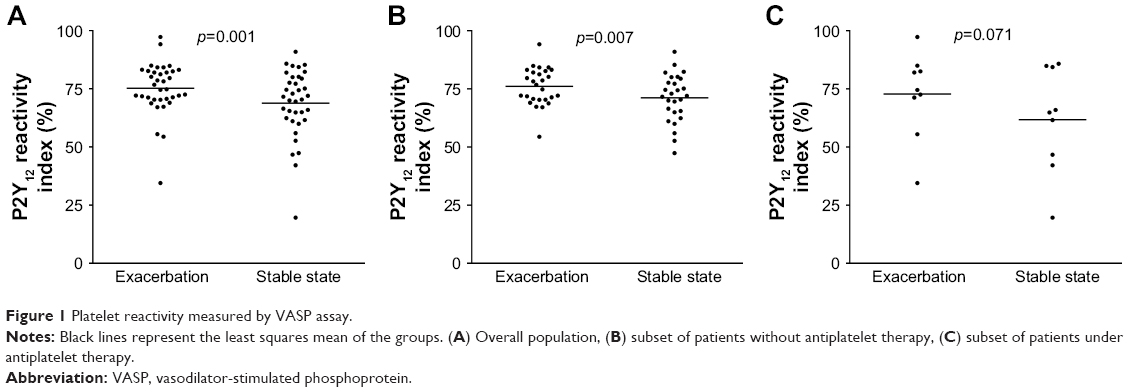 | Figure 1 Platelet reactivity measured by VASP assay. |
 | Table 4 Platelet reactivity during the acute exacerbation and stable state |
The increase in platelet reactivity during exacerbations was consistent in patients with or without concomitant APT. In patients not receiving APT, a statistically significant greater PRI was observed during AECOPDs compared to stability (76.0%±1.5% vs 71.2%±2.0%, p=0.007; Figure 1), with parallel findings obtained with the other platelet function tests used (Table 4).
A numerically higher platelet reactivity was seen during exacerbations in the subset of patients with APT, although not achieving statistical significance (PRI: 72.8%±3.1% vs 61.7%±7.5%, p=0.071; Figure 1). Among those receiving aspirin (n=8), a higher collagen-induced platelet aggregation measured with LTA was observed during AECOPD (56.2%±6.1% vs 43.8%±6.4%, p=0.022), whereas the numerical differences did not reach statistical significance with the other assays sensitive to aspirin therapy (data not shown). In the two patients on clopidogrel, no differences in platelet reactivity were observed during exacerbations compared to stability (data not shown).
Systemic inflammatory parameters
The levels of all inflammatory parameters assessed were augmented during AECOPD compared to the stable state (Figure 2): leukocytes (8.8±0.5 vs 7.3±0.4×109/L, p=0.001), IL-6 (5.7±0.8 vs 3.0±0.4 pg/mL, p=0.002) and fibrinogen (5.5±0.7 vs 4.7±0.8 g/L, p=0.021).
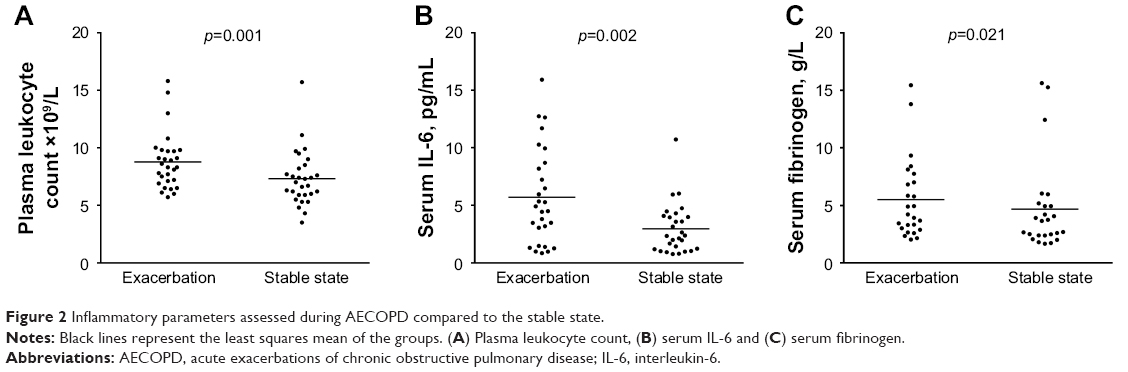 | Figure 2 Inflammatory parameters assessed during AECOPD compared to the stable state. |
The change in platelet reactivity measured by VASP was numerically higher in those patients in whom a greater difference in the levels of inflammatory markers between exacerbation and stability was observed. The most evident and almost statistically significant association was found between ΔPRI and Δleukocyte count split into tertiles (lower tertile: −0.5%±3.4%, mid tertile: 7.2%±3.2%, upper tertile: 9.0%±3.2%; p for trend 0.051; Figure 3), while the association was less marked for IL-6 and fibrinogen (p for trend 0.072 and 0.227, respectively; Figure 3).
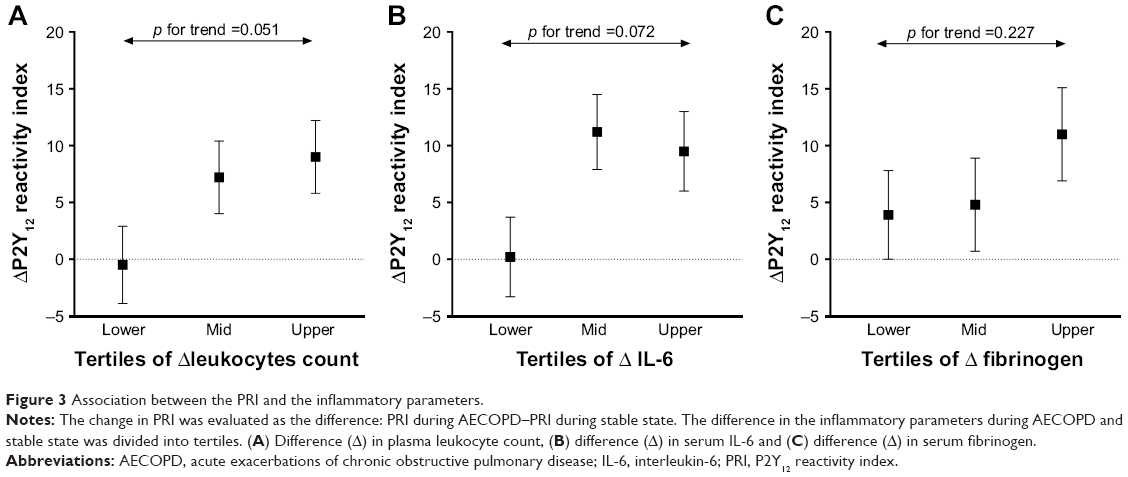 | Figure 3 Association between the PRI and the inflammatory parameters. |
Discussion
In this investigation, we evaluated the impact of an acute exacerbation on platelet reactivity in COPD patients with a comprehensive panel of assays in order to explore different platelet signaling pathways. Noteworthy, patients receiving APT were not excluded, expanding upon prior studies by evaluating the impact of these agents on the results. The main findings of this study can be summarized as follows: 1) platelet reactivity is increased during AECOPD; 2) this augmented platelet aggregability is also observed in patients on APT and 3) those patients with greater enhancement of inflammatory markers during an AECOPD are more likely to present a higher increase in platelet reactivity.
Cardiovascular disease is the leading cause of mortality in COPD patients when respiratory failure is excluded.18 In fact, prior investigations have observed that COPD subjects have an increased risk of ischemic heart disease irrespective of age, gender or smoking status.19–22 Remarkably, a 2–3-fold increased risk of myocardial infarction has been described during the first 5 days following an AECOPD,8 which underscores the need for a better understanding of the mechanisms linking AECOPD, inflammation and cardiovascular atherothrombotic complications. Few studies have previously evaluated and observed augmented platelet activation in COPD patients compared to control subjects.23–26 Further, the impact of an AECOPD in platelet reactivity has been only evaluated in a single experience by Maclay et al who demonstrated increased circulating platelet–monocyte aggregates and, thus, higher platelet reactivity during exacerbations compared to stable COPD.13 In line with this, our investigation confirmed that exacerbations of COPD lead to a hyperreactive platelet phenotype, showing upregulation of multiple signaling pathways. Noteworthy, this is the first study evaluating the impact of AECOPD on platelet reactivity with platelet function assays with proven ability to predict clinical outcomes in patients with ischemic heart disease.27
It has been reported that COPD patients suffering a myocardial infarction have worse prognosis than non-COPD patients.28 This can be partially explained by the fact that COPD patients are less likely to receive guideline-recommended secondary preventive medications with proven benefit, such as antiplatelet agents.28 Another important feature of our investigation is that patients receiving APT were also included in the study, whereas they were specifically excluded in prior experiences. The finding that patients receiving APT with aspirin or clopidogrel also displayed increased platelet reactivity during an AECOPD raises questions about the efficacy of these two agents in this setting and suggests that a more potent antiplatelet drug could be of help for COPD patients in certain scenarios. Moreover, the results of a recent post hoc analysis of a randomized large-scale trial showed that ticagrelor, a very potent platelet P2Y12 inhibitor, was more effective in terms of reducing the risk of ischemic events than clopidogrel (both in addition to aspirin) in COPD patients with an acute coronary syndrome.29 However, further clinical studies are required to consider if potent antiplatelet treatment must be used during exacerbations in COPD patients with concomitant ischemic cardiovascular diseases.
The exact underlying mechanisms of platelet activation in COPD patients have not been identified, although several factors have been suggested to contribute, such as increased systemic inflammation, hypoxemia and oxidative stress.30 In particular, COPD is characterized by the presence of low-grade systemic inflammation and an acute inflammatory response during exacerbations of the disease.7,31 Moreover, the role of inflammation in the formation of atherosclerotic lesions and the occurrence of atherothrombotic complications, where platelets also play a key role, is undisputed.11,12 In line with this idea, an association between leukocyte count and platelet reactivity has been previously reported.32 In our investigation, those patients with greater increment of inflammatory markers such as leukocyte count had a numerical trend towards larger increases in platelet reactivity without reaching statistical significance. This finding might support the idea of an association between inflammation and platelet activation in COPD patients.
This study has several inherent limitations due to its observational design and relatively small sample size. However, the consistent findings obtained using a variety of assays and agonists are supportive of our study conclusions. The subset of patients receiving APT is particularly small, making the study underpowered to detect differences in this subgroup; therefore, results obtained in this subgroup should be considered exploratory and hypothesis generating. In addition, all patients included in our study had severe and very severe airway limitation, representing an advanced stage of the disease. Therefore, whether the findings of our study can be extrapolated to mild or moderate COPD patients cannot be ascertained.
Conclusion
Platelet reactivity is augmented during acute exacerbations of COPD, irrespective of treatment with antiplatelet agents. This platelet hyperreactivity may contribute to the augmented cardiovascular risk of these patients. In addition, the increase in platelet reactivity could be directly associated with an increment in systemic inflammatory markers during exacerbations.
Acknowledgments
The authors would like to thank E Aliagas and M Gracida for their contributions to acquisition and analysis of data. Similarly, the authors would like to thank MJ Manuel, M Navarro and G Sosa for their assistance in sample collection and performing laboratory techniques. The study was funded by a grant from The Spanish Society of Pulmonology and Thoracic Surgery (SEPAR-116/2013) awarded to Dr Muñoz-Esquerre and by a grant from the Bellvitge Biomedical Research Institute (IDIBELL) as part of a Pre-doctoral Research Fellowship awarded to Dr Muñoz-Esquerre. The Catalan Foundation of Pulmonology (FUCAP 2013) supported this study with a grant awarded to Dr Santos. JL Ferreiro reports receiving honoraria for lectures from Eli Lilly Co, Daiichi Sankyo Inc., Astra Zeneca, Pfizer; consulting fees from AstraZeneca, Eli Lilly Co, Daiichy Sankyo Inc.; and research grants from AstraZeneca. This work is solely the responsibility of the authors.
Author contributions
MME is the guarantor of the paper, taking responsibility for the integrity of the work as a whole, from inception to the published article. MME and JLF contributed substantially to the study concept and design, data collection, data analysis and interpretation, and the writing of the manuscript. DH, ALM, MLS, GR, JAGH, JD, ÁC and SS contributed substantially to the study design, data analysis and interpretation, and the writing of the manuscript. All authors have read and approved the final version of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Anthonisen NR, Connett JE, Enright PL, Manfreda J; Lung Health Study Research Group. Hospitalizations and mortality in the Lung Health Study. Am J Respir Crit Care Med. 2002;166(3):333–339. | ||
Soriano JB, Rigo F, Guerrero D, et al. High prevalence of undiagnosed airflow limitation in patients with cardiovascular disease. Chest. 2010;137(2):333–340. | ||
Engström G, Hedblad B, Janzon L, Valind S. Respiratory decline in smokers and ex-smokers an independent risk factor for cardiovascular disease and death. J Cardiovasc Risk. 2000;7(4):267–272. | ||
Fimognari FL, Scarlata S, Conte ME, Incalzi RA. Mechanisms of atherothrombosis in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(1):89–96. | ||
Walter RE, Wilk JB, Larson MG, et al. Systemic inflammation and COPD: the Framingham Heart Study. Chest. 2008;133(1):19–25. | ||
Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA. 2013;309(22):2353–2361. | ||
Donaldson GC, Hurst JR, Smith CJ, Hubbard RB, Wedzicha JA. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest. 2010;137(5):1091–1097. | ||
Rothnie KJ, Yan R, Smeeth L, Quint JK. Risk of myocardial infarction (MI) and death following MI in people with chronic obstructive pulmonary disease (COPD): a systematic review and meta-analysis. BMJ Open. 2015;5(9):e007824. | ||
Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482–2494. | ||
Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–1695. | ||
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. | ||
Maclay JD, McAllister DA, Johnston S, et al. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax. 2011;66(9):769–774. | ||
Rodriguez-Roisin R. Toward a consensus definition for COPD exacerbations. Chest. 2000;117(5 Suppl 2):S398–S401. | ||
Aleil B, Ravanat C, Cazenave JP, Rochoux G, Heitz A, Gachet C. Flow cytometric analysis of intraplatelet VASP phosphorylation for the detection of clopidogrel resistance in patients with ischemic cardiovascular diseases. J Thromb Haemost. 2005;3(1):85–92. | ||
Angiolillo DJ, Shoemaker SB, Desai B, et al. Randomized comparison of a high clopidogrel maintenance dose in patients with diabetes mellitus and coronary artery disease: results of the Optimizing Antiplatelet Therapy in Diabetes Mellitus (OPTIMUS) study. Circulation. 2007;115(6):708–716. | ||
Sibbing D, Braun S, Jawansky S, et al. Assessment of ADP-induced platelet aggregation with light transmission aggregometry and multiple electrode platelet aggregometry before and after clopidogrel treatment. Thromb Haemost. 2008;99(1):121–126. | ||
Sin DD, Anthonisen NR, Soriano JB, Agusti AG. Mortality in COPD: role of comorbidities. Eur Respir J. 2006;28(6):1245–1257. | ||
Hole DJ, Watt GC, Davey-Smith G, Hart CL, Gillis CR, Hawthorne VM. Impaired lung function and mortality risk in men and women: findings from the Renfrew and Paisley prospective population study. BMJ. 1996;313(7059):711–715. | ||
Sorlie PD, Kannel WB, O’Connor G. Mortality associated with respiratory function and symptoms in advanced age. The Framingham Study. Am Rev Respir Dis. 1989;140(2):379–384. | ||
Bhatt SP, Dransfield MT. Chronic obstructive pulmonary disease and cardiovascular disease. Transl Res. 2013;162(4):237–251. | ||
Campo G, Pavasini R, Malagù M, et al. Chronic obstructive pulmonary disease and ischemic heart disease comorbidity: overview of mechanisms and clinical management. Cardiovasc Drugs Ther. 2015;29(2):147–157. | ||
Wedzicha JA, Syndercombe-Court D, Tan KC. Increased platelet aggregate formation in patients with chronic airflow obstruction and hypoxaemia. Thorax. 1991;46(7):504–507. | ||
Cordova C, Musca A, Violi F, Alessandri C, Perrone A, Balsano F. Platelet hyperfunction in patients with chronic airways obstruction. Eur J Respir Dis. 1985;66(1):9–12. | ||
Davì G, Basili S, Vieri M, et al. Enhanced thromboxane biosynthesis in patients with chronic obstructive pulmonary disease. The Chronic Obstructive Bronchitis and Haemostasis Study Group. Am J Respir Crit Care Med. 1997;156(6):1794–1799. | ||
Malerba M, Olivini A, Radaeli A, Ricciardolo FL, Clini E. Platelet activation and cardiovascular comorbidities in patients with chronic obstructive pulmonary disease. Curr Med Res Opin. 2016;32(5):885–891. | ||
Ferreiro JL, Angiolillo DJ. Clopidogrel response variability: current status and future directions. Thromb Haemost. 2009;102(1):7–14. | ||
Andell P, Koul S, Martinsson A, et al. Impact of chronic obstructive pulmonary disease on morbidity and mortality after myocardial infarction. Open Heart. 2014;1(1):e000002. | ||
Andell P, James SK, Cannon CP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes and chronic obstructive pulmonary disease: an analysis from the platelet inhibition and patient outcomes (PLATO) trial. J Am Heart Assoc. 2015;4(10):e002490. | ||
Malerba M, Clini E, Malagola M, Avanzi GC. Platelet activation as a novel mechanism of atherothrombotic risk in chronic obstructive pulmonary disease. Expert Rev Hematol. 2013;6(4):475–483. | ||
Wedzicha JA, Seemungal TA, MacCallum PK, et al. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost. 2000;84(2):210–215. | ||
Faraday N, Yanek LR, Vaidya D, et al. Leukocyte count is associated with increased platelet reactivity and diminished response to aspirin in healthy individuals with a family history of coronary artery disease. Thromb Res. 2009;124(3):311–317. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.