")
Back to Journals » ImmunoTargets and Therapy » Volume 3
Immunotherapy for the treatment of Alzheimer's disease: amyloid-β or tau, which is the right target?
Authors Castillo-Carranza D, Guerrero-Muñoz M, Kayed R
Received 25 September 2013
Accepted for publication 26 November 2013
Published 27 December 2013 Volume 2014:3 Pages 19—28
DOI https://doi.org/10.2147/ITT.S40131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Diana L Castillo-Carranza,1,2 Marcos J Guerrero-Muñoz,1,2 Rakez Kayed1–3
1Mitchell Center for Neurodegenerative Diseases, 2Departments of Neurology, Neuroscience, and Cell Biology, 3Sealy Center for Vaccine Development, University of Texas Medical Branch, Galveston, TX, USA
Abstract: Alzheimer's disease (AD) is characterized by the presence of amyloid plaques composed mainly of amyloid-β (Aβ) protein. Overproduction or slow clearance of Aβ initiates a cascade of pathologic events that may lead to formation of neurofibrillary tangles, neuronal cell death, and dementia. Although immunotherapy in animal models has been demonstrated to be successful at removing plaques or prefibrillar forms of Aβ, clinical trials have yielded disappointing results. The lack of substantial cognitive improvement obtained by targeting Aβ raises the question of whether or not this is the correct target. Another important pathologic process in the AD brain is tau aggregation, which seems to become independent once initiated. Recent studies targeting tau in AD mouse models have displayed evidence of cognitive improvement, providing a novel therapeutic approach for the treatment of AD. In this review, we describe new advances in immunotherapy targeting Aβ peptide and tau protein, as well as future directions.
Keywords: immunotherapy, Alzheimer's disease, β-amyloid, tau
Introduction
Alzheimer’s disease (AD) is a complex, debilitating disorder and the most common cause of dementia affecting those over the age of 65 years. Currently available drugs for the treatment of AD only provide relief of symptoms with no effect on the course of the disease. As the longevity of the worldwide population increases, the amount of people susceptible to AD will continue to rise. Therefore, there is an urgent need to develop new therapeutic strategies to modify or prevent the progression of AD. The cause or causes of sporadic AD are unknown. Histopathologic features of the disease are accumulation of extracellular amyloid plaques, mainly composed of amyloid β (Aβ) peptide and intracellular neurofibrillary tangles (NFTs) composed of tau protein. Aβ peptide is derived from amyloid precursor protein (APP) and while missense mutations in APP or in presenilin (PS) genes, PS1 and PS21 can cause familial forms of AD, the sporadic form is the most prevalent, representing 95% of all cases. Regardless of the cause of AD, the consequence is accumulation of Aβ monomers, which eventually aggregate, forming oligomers, fibrils, and finally large metastable plaques.
For over two decades, the Aβ peptide has been considered the main culprit of AD. According to the amyloid cascade hypothesis, Aβ peptide accumulates into plaques, triggering a series of events leading to formation of NFTs, neuronal cell death, and dementia.2 However, this theory fails to explain the discrepancy between amyloid plaque deposition and cognitive impairment in AD. The lack of correlation comes from post-mortem tissue of patients included in the first clinical trial. Although immunization against Aβ induced a reduction of amyloid plaques, the patients had severe dementia before death.3 Furthermore, it seems likely that neurodegeneration may begin prior to amyloid deposition. Moreover, the presence of amyloid plaques in the brains of cognitively normal adults indicates that another entity is involved in cognitive decline.4 Prefibrillar forms of Aβ or oligomers have been identified in AD brains. Recent evidence suggests that Aβ oligomers and not fibrils are the primary toxic species in AD.5,6 Toxicity of oligomers was demonstrated in vitro and in vivo.7–10
Importantly, observations from post-mortem tissue revealed that tau pathology correlates better with the severity of dementia.11,12 Moreover, it has been reported that soluble forms of Aβ correlate with the onset of the disease only in the presence of tau.13–15 Additionally, tau pathology occurs in brain areas lacking amyloid plaques. These findings suggest that tau mediates Aβ toxicity. However, mechanistic relationships between Aβ deposition and tau pathology remain contentious. Despite the evidence that tau pathology contributes to disease progression, therapeutic approaches have been concentrated almost entirely on modulating Aβ production or accumulation. These interventions include: reduction of APP processing by blocking the β or γ secretases;16–19 prevention of Aβ aggregation;20,21 and promoting Aβ clearance by immunotherapy.22 However, γ secretase inhibitors have been found to have toxic effects,19 while β secretase inhibitors are high molecular weight compounds with limited ability to cross the blood–brain barrier.17 On the other hand, immunotherapeutic approaches targeting Aβ for the treatment of AD failed to slow cognitive decline. The disappointing results obtained by lowering Aβ questioned whether or not Aβ is the correct target. This review provides a description of new advances in Aβ and tau immunotherapy as well as future directions.
Targeting Aβ assemblies by immunotherapy
In AD, Aβ converts from its soluble form to β-sheet-rich, toxic oligomers and highly organized fibrils, which eventually assemble into plaques. However, the toxic role of Aβ aggregates found in AD brains remains unclear. For a long time, senile plaques were considered to be the toxic species in AD. In the brain cortex of aged rhesus monkeys, Aβ fibrils induced toxicity of neurons in the vicinity of plaques.23 Toxicity of plaques was also observed previously using longitudinal in vivo multiphoton imaging in mice. Plaque formation was fast and followed by neuritic changes.24 However, recent evidence from animal models suggests that plaque accumulation has a protective rather than toxic effect. Animal models exposed to an enriched environment showed cognitive improvement associated with an increase in plaque load,25 confirming the lack of correlation between brain amyloid plaque deposition and cognition in AD.
Amyloid plaques
Immunotherapeutic approaches to treat AD have been well executed in various mouse models. These approaches can be broadly classified as either active or passive immunization strategies. In 1999, Schenk et al published the first active immunization study in the PDAPP transgenic mouse model.26 Initial findings revealed a reduction of plaque deposition in aged mice after administration of Aβ-42. Treatment in young mice prevented Aβ plaque formation. No signs of damage were observed in the brains of treated animals. Later, two other groups reported the immunization of different AD mouse models using aggregated Aβ-42.27,28 Immunization of TgCRND8 mice resulted in a reduction in amyloid plaque load. No changes in total Aβ levels were observed. Furthermore, improvement in cognition was observed by evaluation with the Morris water maze.27 In a separate study, immunization of Tg2576 and Tg2576-PS1 mice also recapitulated the effect of the Aβ-42 treatment described previously. In these cases, active immunotherapy had beneficial effects on cognition, which correlated with reduction of amyloid plaque burden.28 Not only were full Aβ1–42 immunizations effective, but fragments of Aβ peptide including tandems of Aβ(1–15) also induced reduction of plaque load29 and lowered levels of Aβ-40 and Aβ-42 in the brain, which correlated with an efflux of Aβ to the blood.30 Moreover, combination of Aβ fragments with either diphtheria toxoid or tetanus toxoid was used to stimulate the immune system, while avoiding an Aβ-specific T-cell response.31,32 Afterwards, several active immunization studies reproduced the same results using Aβ peptide in different AD models33 including rats, nonhuman primates,34–36 and mice. Contrary to other active immunizations, Austin et al described no improvement in memory after several months of treatment with Aβ-42. This work suggests that immunization could be more beneficial in the early stages of the disease.37 Adverse effects were also reported in old lemurs after Aβ immunization, including microbleeds and iron deposits in the choroid plexus.38
Passive immunizations have also been performed in animal models of AD. Although the use of antibodies represents a safer alternative to active immunotherapy, treatment may need to be administered frequently in order to reach the necessary concentration in serum to be effective. It has been estimated that only 0.1% of the antibody in blood crosses the blood–brain barrier.39 Studies in mice immunized with mouse monoclonal antibodies against Aβ peptide indicated that the antibody can cross the blood–brain barrier and reduce amyloid plaques,40,41 as well as levels of soluble Aβ-42.40 However, removal of plaques in the PDAPP mouse does not necessarily require the antibody to enter the brain. Peripherally administered m266 antibody, that recognizes Aβ13–28, clears Aβ deposits from the brain with no evidence of antibody-plaque interaction. These findings suggest that removal of amyloid pathology from the brain alters the Aβ dynamics inducing protein efflux to plasma.42 Immunization of Tg2576 mice with BAM-10 antibody that recognizes Aβ1–12 fully reversed memory deficits, as demonstrated by the Morris water maze task. However, treatment with BAM-10 had an insignificant effect on reduction of soluble Aβ levels in the mouse brain. Researchers propose that treatment causes BAM-10 to enter the central nervous system, reversing deleterious effects of small Aβ assemblies that interfere with cognitive function, thus restoring normal memory in Tg2576 mice.43 Subsequently, others failed to find a correlation between performance in behavioral tests and amyloid pathology.44–47 Although passive immunization reverses cognitive deficits in AD mouse models, some antibodies induce vascular pathology,48 including intracerebral hemorrhage. When PDAPP mice were immunized with an anti-Aβ1–5 antibody known as 3D6, it interacted with soluble and insoluble Aβ, as well as induced removal of Aβ deposits from the vasculature. This treatment increased microhemorrhage and Aβ deposits in capillaries, but side effects were eventually resolved.49,50 Antibodies against amyloid plaques have also been developed. Chronic treatment of PDAPP mice with an antibody that recognizes Aβ-42 lowered pre-existing plaques without microhemorrhage. However, treatment was less effective at preventing plaque deposition in immunized mice.51 This phenomenon is associated with those antibodies that bind Aβ in plaques.52–54 However, the mechanism by which immunization reduced plaques is not understood. It has been suggested that neoangiogenesis, a key event underlying plaque formation in AD, can be modulated by removal of plaques in mice immunized with Aβ peptide by inducing reversion of hypervascularization.55 Additionally, two possible mechanisms of antibody-mediated removal of amyloid plaque have been proposed. These are microglia activation40 and the peripheral sink hypothesis.42 Microglia activation implies that antibody enters the brain to stimulate phagocytosis of the antibody-Aβ complex. On the other hand, the sink mechanism does not require the antibody to enter the brain, in that a peripheral sink is created when the antibodies in serum alter the equilibrium of Aβ across the blood–brain barrier, inducing an efflux of proteins from brain to blood. Both mechanisms have been demonstrated in preclinical studies.
Clinical trials
Although the first clinical trial using immunotherapy for AD was stopped because of adverse effects, some are currently in progress (Table 1). The first active immunization in humans consisted of a mixture of Aβ-42 peptide and adjuvant QS-21, known as AN1792. Treatment with this preparation did not find significant differences between the antibody and placebo groups in a variety of behavioral tests, including Alzheimer’s Disease Assessment Scale-cognitive (ADAS-Cog), Disability Assessment for Dementia, Clinical Dementia Rating, Mini-Mental State Examination, or Clinical Global Impression of Change.56 Although the Phase I trial showed good tolerability, the Phase IIa trial was interrupted because 6% of immunized patients developed acute meningoencephalitis.57 A follow-up of patients treated with AN1792 in a Phase I trial showed clearance of amyloid plaques without preventing progression of the disease, and after 5 years there was no evidence of reduction of neurodegeneration.3 Major adverse events observed with AN1792 include meningoencephalitis and cerebral microhemorrhage. Post-mortem analysis of encephalitis cases revealed that brain infection was associated with T-cell activation. This seems to be related to the sequence of the protein, given that the Aβ-carboxyl terminus contains epitopes for T-cells.58,59 Therefore, different N-terminal Aβ peptides like AD0, AD02, ACC-001, CAD106, and ACI-24 were developed and are now in Phase II clinical trials. Affitopes (AD01 and AD02) contain a six amino acid peptide that mimics part of the native Aβ N-terminus60 while ACC-001 is a vaccine composed of a seven amino acid fragment of the Aβ N-terminal conjugated to a mutated diphtheria toxin protein known as CRM197.61,62 Patients immunized with the N-terminal of the Aβ peptide called CAD106 had some adverse effects, including nasopharyngitis and injection site erythema. Nine patients experienced serious adverse events, none of which were related to the peptide. This treatment increased total Aβ levels and reduced free Aβ in plasma. However, no significant changes in Aβ or tau levels were observed in cerebrospinal fluid.63 Another active immunization is the ACI-24 vaccine, consisting of a liposome-based vaccine with a tetra-palmitoylated Aβ1–15 fragment. Preclinical studies in APP-V717IxPS-1 (APPxPS-1) double transgenic mice restored cognitive memory and reduced brain amyloid load and microbleeds. In humans, ACI-24 stimulated the immune system to produce β-sheet conformation-specific antibodies. A Phase I trial has been designed to identify the best dose of vaccine to be used in a Phase II trial.64 No results have been posted as yet.
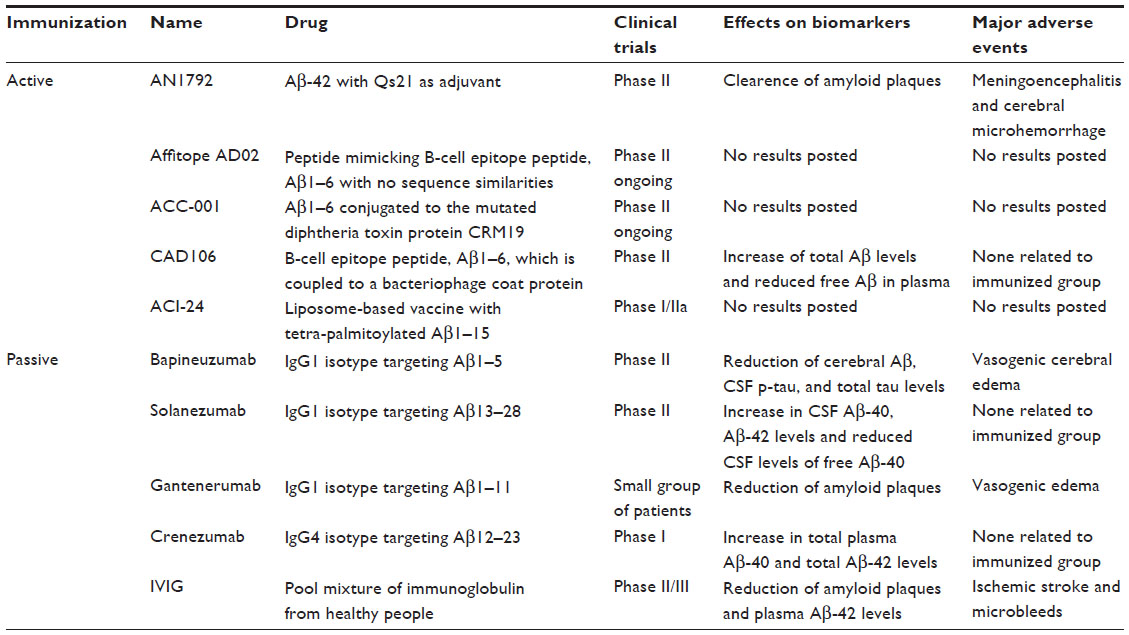 | Table 1 Immunotherapeutic strategies for the treatment of Alzheimer’s disease |
Recent advances in preclinical studies in animals have encouraged the development of humanized antibodies for clinical trials. These antibodies have been developed as a promising safe alternative strategy in clinical trials. One of them, bapineuzumab, is a humanized monoclonal immunoglobulin G1 (IgG1) antibody that recognizes the Aβ1–5 region. Preclinical studies showed a reduced plaque burden in transgenic mice.40 Phase II studies showed that immunization with bapineuzumab significantly reduced cerebral Aβ levels, hyperphosphorylated tau in cerebrospinal fluid, and total tau levels when compared with placebo-treated patients.65–67 However, this antibody did not show significant differences in AD patients compared with the placebo group. Post hoc analysis suggested a modest effect in apolipoprotein (Apo)ε4 noncarriers, although 9.7% of patients showed vasogenic cerebral edema.68 In Phase III clinical trials, neither AD patients carrying the Apoε4 allele nor those who were noncarriers showed improvement.69 A second antibody that has been used in clinical trials is solanezumab, a humanized monoclonal IgG1 antibody that recognizes the Aβ13–28 region (266 mouse monoclonal antibody), and has little affinity for fibrillar material. Although the antibody was found to be safe, with no features of meningoencephalitis, microhemorrhage, or vasogenic edema observed in Phase II clinical trials,70 treatment with solanezumab did not induce improvement in cognition.70,71 A Phase III study reported a 34% slowing of decline on the ADAS-cog and Mini-Mental State Examination and a slowing of functional decline on the Alzheimer’s Disease Cooperative Study-Activities of Daily Living (P=0.057) at 80 weeks in patients with mild AD. The treatment caused an increase in total Aβ1–40 and Aβ1–42 in cerebrospinal fluid, and lower cerebrospinal fluid levels of free Aβ1–40. However, positron emission tomography (PET) scan did not show a significant reduction of amyloid plaques. New results from a Phase III clinical trial did not show significant differences in biomarkers of neuronal damage in cerebrospinal fluid.69
Gantenerumab is a humanized monoclonal IgG1 antibody (Aβ1–11) that binds specifically to amyloid plaques. Treatment with this antibody reduced amyloid plaques in patients with mild to moderate AD, but no cognitive improvement was reported. Furthermore, treatment given to AD patients who were Apoε4 carriers resulted in reversible vasogenic edema.72 A Phase III clinical trial is now underway. Another monoclonal humanized antibody is crenezumab, an IgG4 that recognizes the Aβ12–23 region. Preclinical research using this antibody in AD mouse models showed reduction of amyloid plaque pathology and cognitive improvement as assessed by the novel object recognition test. More relevantly, the IgG4 isotype of crenezumab induced milder activation of microglia and less release of the proinflammatory cytokine, tumor necrosis factor-alpha, compared with the IgG1 isotype of the same antibody. Finally, a Phase I study did not show signs of vasogenic edema even in Apoε4 carriers.73 This antibody is now in a Phase II clinical trial.
Although some clinical trials are still ongoing, it is clear from the results observed thus far that this approach does not succeed in stopping disease progression in AD patients, and this may be the consequence of starting the treatment when pathologic events had already developed in the brain. Previous studies showed that changes in the brain start two decades before the onset of clinical symptoms,74 and this finding suggests that therapeutic intervention earlier in the course of the disease may provide more clinical benefits.
Upcoming human studies will evaluate prevention of the disease in individuals at risk for AD. These include the Dominantly Inherited Alzheimer Network study, the Alzheimer Prevention Initiative, and the study for Treatment of Asymptomatic Alzheimer’s Disease. The Dominantly Inherited Alzheimer Network study will analyze individuals from families with autosomal dominant AD who are carriers of mutations in APP, PSEN1, or PSEN2 genes. Patients will be treated with solanezumab, gantenerumab, or a beta secretase inhibitor (LY2886721).74,75 The Alzheimer Prevention Initiative study will investigate a group of families in Antioquia, Colombia, who are carriers of a rare autosomal dominant mutation in the PS1 gene (E280A) responsible for familial Alzheimer’s disease. In one clinical trial, cognitively normal individuals carrying the mutation in PSEN1 will be treated with the monoclonal antibody crenezumab. The second trial will investigate cognitively normal individuals homozygous for the Apoε4 allele associated with late-onset AD.76,77 For the Treatment of Asymptomatic Alzheimer’s Disease study, older patients who are not carriers of genetic mutation but whose brains have Aβ deposition as measured by PET scan, will be immunized with the solanezumab antibody.78
Aβ oligomers
Although amyloid plaques are a hallmark of AD, oligomers are considered to be mediators of the early state of the disease. The toxicity of oligomers has been widely demonstrated in cells both in culture and in vivo.7,8,10 Different oligomeric entities have been found in the AD brain, including dimer, trimer, dodecamer (Aβ*56), and high order oligomers. Synaptic dysfunction is one of the effects induced by dimers79 and Aβ*5680 when injected into the brain. These findings indicate that oligomers interfere with normal synaptic function, inducing cognitive deficit. Analyses of human brain and cerebrospinal fluid indicate that Aβ*56 levels correlate with neuronal dysfunction before onset.81 These findings suggest that removal of oligomeric assemblies is important to prevent interaction of Aβ with synapses.
Immunization targeting Aβ oligomers has been performed by vaccination of Tg2576 mice with amyloid oligomimics, prepared from a nonhuman random sequence assembled in to amyloid oligomer like structures, immunization with this antigen improved cognitive function, reduced total plaque load with a much lower incidence of microhemorrhage compared with Aβ antigens. These findings suggest that other alternatives may be used in order to avoid the autoimmune side effects produced by Aβ peptide.82 Furthermore, chronic immunization as well as acute treatment with m266 antibody in PDAPP mice prevented age-related memory deficits, but no effect on Aβ plaque load was observed either in the cortex or hippocampus, suggesting that removal of soluble Aβ, but not plaques, is sufficient to induce improvement in cognition.83 Exogenous and endogenous antibodies against Aβ oligomers prevented long-term potentiation in vivo.84 Additionally, vaccination of SAMP8 mice with A8 monoclonal antibody reduced low molecular weight Aβ oligomers and tau phosphorylation (pSer404) while improving cognitive function.85 Immunization with an antibody that recognizes dimers, small oligomers, and mature plaques also improved learning and memory deficits in Tg2576 mice.45 Treatment of APP mice with globulomer-specific antibody improved cognitive function and spine density.86
Treatment for Aβ oligomers has also been performed in humans. A small group of AD patients was treated with a pooled mixture of immunoglobulin from healthy people87 containing antibodies against Aβ oligomers and fibrils.88 In a pilot study, one of five AD patients treated with intravenous immunoglobulins showed stabilization and modest cognitive improvement. Aβ levels were reduced in cerebrospinal fluid and increased in plasma.89 Intravenous immunoglobulins were also used in eight patients with mild AD for 6 months, stopped for 3 months, and then resumed for another 9 months. Treated patients showed cognitive improvement as assessed by Mini-Mental State Examination at 6 months. After each treatment, Aβ decreased in cerebrospinal fluid and increased in plasma. Importantly, no serious adverse effects were reported in this study.90 However, in a Phase II clinical trial, evaluation of 58 AD patients did not reveal significant cognitive improvement. Moreover, no changes in concentrations of Aβ-40, Aβ-42, total tau, or p-tau were observed in cerebrospinal fluid. Further, one patient had an ischemic stroke (a known side effect of intravenous immunoglobulins), while 14% of patients had incident microbleeds, which were not seen in the placebo group.91 In a randomized, double blind, placebo-controlled Phase III clinical trial, treatment with intravenous immunoglobulins for 18 months in people with mild to moderate AD did not showed significant cognitive improvement in ADAS-Cog or the Alzheimer’s Disease Cooperative Study-Activities of Daily Living tests. However, an Apo-ε4 carrier subgroup receiving intravenous immunoglobulins at 400 mg/kg every 2 weeks (n=87) showed cognitive improvement on the Modified Mini-Mental State Examination and Trails B test. PET scan analysis with florbetapir showed a reduction in brain fibrillar amyloid in patients treated with an intravenous immunoglobulin preparation at 400 mg/kg every 2 weeks. Significant reductions in plasma Aβ-42 levels, but not Aβ-40 levels, were observed in patients treated with intravenous immunoglobulins. This group showed higher levels of anti-oligomer and anti-fibril antibodies in cerebrospinal fluid and plasma but no effect was observed for tau and phosphorylated tau levels in cerebrospinal fluid.90,91 The results obtained failed to meet the primary endpoints of slowing cognitive and functional decline.
Implication of tau pathology in AD
An unsolved question in AD is the relationship between Aβ and tau pathology. Tau has long been considered to be the secondary effect of Aβ pathology. However, new evidence suggests that tau pathology may appear early in life, and perhaps before Aβ.93 Tau pathology is another important hallmark of AD and perhaps the most promising target. It is widely expressed in the central nervous system, and is required for microtubule assembly, axonal transport, and neurite outgrowth.94,95 Further, it is known that tau pathology alone can cause neurodegenerative disease. Mutations in the tau gene, microtubule-associated protein tau (MAPT), can cause autosomal dominant frontotemporal dementia,96–98 directly implicating tau dysfunction in neurodegeneration. In AD, tau loses its affinity for microtubules and aggregates, forming oligomers, paired helical filaments (PHF) and NFTs. Growing evidence suggests that neuronal loss precedes formation of NFTs and indicates that tau oligomers are the most toxic species.99–102
Recent findings have revealed that tau pathology mediates Aβ toxicity, as demonstrated in animal models. Suppression or reduction of tau in mice prevents or reduces the toxic effects of Aβ.103 Indeed, hippocampal neurons from tau knockout mice are resistant to Aβ-induced cell death, implicating a role of tau in Aβ-related neurodegeneration in AD.104 Moreover, reduction of tau levels prevented behavioral deficits in an AD mouse model without altering Aβ levels.105 Thus, removal of tau by immunization should be beneficial. On the other hand, immunotherapy targeting Aβ reduced amyloid pathology but cognitive decline persisted. Perhaps this is a consequence of the fact that treatment has little or no effect on tau pathology. This was demonstrated in human subjects immunized with Aβ peptide (AN1792), in whom removal of plaques had some effect in reducing phosphorylated tau in neuronal processes,106 but the poor effect on tau pathology was not enough to stop cognitive decline.107 This suggests that once tau pathology is initiated, it can self-propagate and removal of Aβ is insufficient. All of these findings highlight the need to target tau as an alternative treatment for AD.
Tau immunotherapy
Immunomodulation to clear tau pathology is an exciting approach for the treatment of AD.108,109 Currently, few reports on targeting tau by immunotherapy have been published. Active and passive immunizations were performed in AD mouse models. Chronic treatment with tau peptide as well as an antibody against phosphosites Ser396/404 cleared NFTs in P301L110 and htau/PS1 (M146L) mice.111 Further, vaccination of tau mutant mice with other phosphoepitopes such as Tau195-213 (P-202/205), Tau207-220 (P-212/214), Tau224-238 (P-231),112 and tau peptide (KSPVVSGDTSPR) phosphorylated at serine S396/404,113 effectively reduced NFT pathology. Immunization targeting Phospho-Ser422 was also demonstrated in THY-Tau22 mice. Cognitive improvement was observed using the Y-maze, as was reduction of soluble tau from the brain.114
Conformational antibody-mediated clearance of aggregated tau has also been addressed. PHF1 and MC1 antibodies were used to treat JNPL3 and P301S mice. PHF1 recognizes phosphorylation at serine (396 and 404) and MC1 is a conformation-specific antibody that recognizes the amino acids 312–342 of tau protein. Treatment of 2-month-old mice showed a reduction of NFTs in the cortex/forebrain in the JNPL3 model, and lowered neurospheroids in P301S.115 Additionally, chronic treatment with MC1 and a sequence-specific antibody, DA31, was recently performed in the P301L mouse. MC1 significantly reduces total tau and insoluble tau in the brain, but not the DA31 antibody.116 These findings highlight the advantage of using conformational antibodies rather than sequence-specific antibodies. Further, given that tau is an endogenous protein with important functions, it is essential to select the correct target to avoid removal of functional protein.
Although immunization targeting tau aggregates effectively removes NFTs, the effect of toxic tau oligomers has not been evaluated. Recently, we engineered an anti-tau oligomer-specific antibody (TOMA) that does not recognize monomeric functional tau or mature NFTs and has high affinity for tau oligomers. Immunization with the TOMA antibody reversed behavioral deficits observed in the P301L mouse.117 Additionally, we used TOMA antibody to evaluate the effect of removal of tau oligomers by immunotherapy in Tg2576 mice. Preliminary results show that reduction of tau oligomers improves cognitive deficits in an AD mouse model. Interestingly, Aβ*56 was also reduced in mice treated with TOMA.118 Our findings suggest that oligomeric tau may be interacting with Aβ*56 in this mouse model.
So far, there is still much to investigate about the toxic relationship between Aβ and tau. Moreover, it is important to consider the stage of the disease at the time of treatment. Recent developments in the field suggest that therapeutic interventions may be more effective in the asymptomatic phase of AD,119,120 and understanding how Aβ is involved at a very early stage may lead to better preventive treatments.
Perspectives
Important findings in the field have been made since Alois Alzheimer described the pathology of AD for the first time 100 years ago. Unfortunately, there is still no treatment to stop or reverse progression of the disease. Therapeutic approaches to treat AD are currently under investigation. So far, the results obtained targeting Aβ in preclinical studies did not correspond with those observed in clinical trials, and many factors should be considered, including the stage of disease at the time of therapy. The lack of substantial cognitive improvement obtained by targeting Aβ raises the question of whether or not this is the correct target. Aβ may initiate a cascade of events that at a certain stage becomes irreversible, thereby making removal of Aβ insufficient for halting cognitive decline. This could be the case of tau pathology, once initiated it is able to self-propagate, affecting neighboring or synaptically connected cells and inducing neuronal death. Tau is also necessary to mediate Aβ-induced toxicity, highlighting the potential of tau oligomers as a therapeutic target for AD.
Acknowledgment
The authors are grateful to Julia Gerson and Urmi Sengupta for their useful suggestions in the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9(10):768–778. | |
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. | |
Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–223. | |
Bourgeat P, Chételat G, Villemagne VL, et al. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010;74(2):121–127. | |
Baglioni S, Casamenti F, Bucciantini M, et al. Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci. 2006;26(31):8160–8167. | |
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. | |
Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–6453. | |
Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. | |
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27(11):2866–2875. | |
Walsh DM, Selkoe DJ. A beta oligomer – a decade of discovery. J Neurochem. 2007;101(5):1172–1184. | |
Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1(3):213–216. | |
Braak H, Braak E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl. 1996;165:3–12. | |
Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155(3):853–862. | |
McLean CA, Cherny RA, Fraser FW, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–866. | |
Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol. 1999;158(2):328–337. | |
Dovey HF, John V, Anderson JP, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76(1):173–181. | |
Arbel M, Yacoby I, Solomon B, et al. Inhibition of amyloid precursor protein processing by beta-secretase through site-directed antibodies. Proc Natl Acad Sci U S A. 2005;102(21):7718–7723. | |
Arbel M, Solomon B. A novel immunotherapy for Alzheimer’s disease: antibodies against the beta-secretase cleavage site of APP. Curr Alzheimer Res. 2007;4(4):437–445. | |
Martone RL, Zhou H, Atchison K, et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther. 2009;331(2):598–608. | |
Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24(4):219–224. | |
Wisniewski T, Sadowski M. Preventing beta-amyloid fibrillization and deposition: beta-sheet breakers and pathological chaperone inhibitors. BMC Neurosci. 2008;9 Suppl 2:S5. | |
Eckman EA, Eckman CB. Abeta-degrading enzymes: modulators of Alzheimer’s disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans. 2005;33(Pt 5):1101–1105. | |
Shah P, Lal N, Leung E, Traul DE, Gonzalo-Ruiz A, Geula C. Neuronal and axonal loss are selectively linked to fibrillar amyloid-[beta] within plaques of the aged primate cerebral cortex. Am J Pathol. 2010;177(1):325–333. | |
Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451(7179):720–724. | |
Montarolo F, Parolisi R, Hoxha E, Boda E, Tempia F. Early enriched environment exposure protects spatial memory and accelerates amyloid plaque formation in APP(Swe)/PS1(L166P) mice. PLoS One. 2013;8(7):e69381. | |
Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. | |
Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408(6815):979–982. | |
Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982–985. | |
Matsuda J, Kaminaka K, Nozaki C. Amyloid beta peptides with an additional cysteine residue can enhance immunogenicity and reduce the amyloid beta burden in an Alzheimer’s disease mouse model. Biochem Biophys Res Commun. 2009;382(1):149–152. | |
Guan X, Yang J, Gu H, Zou J, Yao Z. Immunotherapeutic efficiency of a tetravalent Abeta1-15 vaccine in APP/PS1 transgenic mice as mouse model for Alzheimer’s disease. Hum Vaccin Immunother. 2013;9(8):1643–1653. | |
Davtyan H, Ghochikyan A, Petrushina I, et al. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J Neurosci. 2013;33(11):4923–4934. | |
Liu B, Frost JL, Sun J, et al. MER5101, a novel Abeta1-15:DT conjugate vaccine, generates a robust anti-Abeta antibody response and attenuates Abeta pathology and cognitive deficits in APPswe/PS1DeltaE9 transgenic mice. J Neurosci. 2013;33(16):7027–7037. | |
Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–193. | |
Gandy S, DeMattos RB, Lemere CA. Alzheimer A beta vaccination of rhesus monkeys (Macaca mulatta). Alzheimer Dis Assoc Disord. 2004;18(1):44–46. | |
Lemere CA, Beierschmitt A, Iglesias M, et al. Alzheimer’s disease abeta vaccine reduces central nervous system abeta levels in a non-human primate, the Caribbean vervet. Am J Pathol. 2004;165(1):283–297. | |
Kofler J, Lopresti B, Janssen C, et al. Preventive immunization of aged and juvenile non-human primates to beta-amyloid. J Neuroinflammation. 2012;9(3):84. | |
Austin L, Arendash GW, Gordon MN, et al. Short-term beta-amyloid vaccinations do not improve cognitive performance in cognitively impaired APP + PS1 mice. Behav Neurosci. 2003;117(3):478–484. | |
Joseph-Mathurin N, Dorieux O, Trouche SG, et al. Amyloid beta immunization worsens iron deposits in the choroid plexus and cerebral microbleeds. Neurobiol Aging. 2013;34(11):2613–2622. | |
Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides. 2002;23(12):2223–2226. | |
Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. | |
Bard F, Fox M, Friedrich S, et al. Sustained levels of antibodies against Abeta in amyloid-rich regions of the CNS following intravenous dosing in human APP transgenic mice. Exp Neurol. 2012;238(1):38–43. | |
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98(15):8850–8855. | |
Kotilinek LA, Bacskai B, Westerman M, et al. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22(15):6331–6335. | |
Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25(26):6213–6220. | |
Lee EB, Leng LZ, Zhang B, et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281(7):4292–4299. | |
Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281(51):39413–39423. | |
Chen G, Chen KS, Kobayashi D, et al. Active beta-amyloid immunization restores spatial learning in PDAPP mice displaying very low levels of beta-amyloid. J Neurosci. 2007;27(10):2654–2662. | |
Beckmann N, Gérard C, Abramowski D, Cannet C, Staufenbiel M. Noninvasive magnetic resonance imaging detection of cerebral amyloid angiopathy-related microvascular alterations using superparamagnetic iron oxide particles in APP transgenic mouse models of Alzheimer’s disease: application to passive Abeta immunotherapy. J Neurosci. 2011;31(3):1023–1031. | |
Zago W, Buttini M, Comery TA, et al. Neutralization of soluble, synaptotoxic amyloid beta species by antibodies is epitope specific. J Neurosci. 2012;32(8):2696–2702. | |
Zago W, Schroeter S, Guido T, et al. Vascular alterations in PDAPP mice after anti-Abeta immunotherapy: implications for amyloid-related imaging abnormalities. Alzheimers Dement. 2013;9(Suppl 5):S105–115. | |
Demattos RB, Lu J, Tang Y, et al. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron. 2012;76(5):908–920. | |
Pfeifer M, Boncristiano S, Bondolfi L, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298(5597):1379. | |
Wilcock DM, Rojiani A, Rosenthal A, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1(1):24. | |
Racke MM, Boone LI, Hepburn DL, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25(3):629–636. | |
Biron KE, Dickstein DL, Gopaul R, Fenninger F, Jefferies WA. Cessation of neoangiogenesis in Alzheimer’s disease follows amyloid-beta immunization. Sci Rep. 2013;3:1354. | |
Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562. | |
Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54. | |
Glaser R, Kiecolt-Glaser JK, Malarkey WB, Sheridan JF. The influence of psychological stress on the immune response to vaccines. Ann N Y Acad Sci. 1998;840:649–655. | |
Monsonego A, Zota V, Karni A, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112(3):415–422. | |
Schneeberger A, Mandler M, Otawa O, Zauner W, Mattner F, Schmidt W. Development of AFFITOPE vaccines for Alzheimer’s disease (AD) – from concept to clinical testing. J Nutr Health Aging. 2009;13(3):264–267. | |
Wilcock DM, Colton CA. Anti-amyloid-beta immunotherapy in Alzheimer’s disease: relevance of transgenic mouse studies to clinical trials. J Alzheimers Dis. 2008;15(4):555–569. | |
Ryan JM, Grundman M. Anti-amyloid-beta immunotherapy in Alzheimer’s disease: ACC-001 clinical trials are ongoing. J Alzheimers Dis. 2009;17(2):243. | |
Winblad B, Andreasen N, Minthon L, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11(7):597–604. | |
Pihlgren M, Madani R, Hickman DT, et al. The safety profile of ACI-24, an oligo-specific amyloid beta vaccine, demonstrated decrease of large microbleedings in brain of aged Alzheimer’s disease mouse model. Alzheimers Dement. 2009;5:P425–P426. | |
Rabinovici GD, Jagust WJ. Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol. 2009;21(1):117–128. | |
Laskowitz DT, Kolls BJ. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2010;74(24):2026. | |
Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–372. | |
Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;(24):2061–2070. | |
Vellas B, Carrillo MC, Sampaio C. Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement. 2013;9(4):438–444. | |
Farlow M, Arnold SE, van Dyck CH, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012;8(4):261–271. | |
Samadi H, Sultzer D. Solanezumab for Alzheimer’s disease. Expert Opin Biol Ther. 2011;11(6):787–798. | |
Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207. | |
Adolfsson O, Pihlgren M, Toni N, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;32(28):9677–9689. | |
Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. | |
Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: the Dominantly Inherited Alzheimer Network. Clin Investig (Lond). 2012;2(10):975–984. | |
Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. | |
Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26 Suppl 3:321–329. | |
Carrillo MC, Brashear HR, Logovinsky V, et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimers Dement. 2013;9(2):123–131. | |
Klyubin I, Betts V, Welzel AT, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28(16):4231–4237. | |
Lesné S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. | |
Lesné SE, Sherman MA, Grant M, et al. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain. 2013;136(Pt 5):1383–1398. | |
Rasool S, Albay R 3rd, Martinez-Coria H, et al. Vaccination with a non-human random sequence amyloid oligomer mimic results in improved cognitive function and reduced plaque deposition and micro hemorrhage in Tg2576 mice. Mol Neurodegener. 2012;7:37. | |
Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5(5):452–457. | |
Klyubin I, Walsh DM, Lemere CA, et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11(5):556–561. | |
Zhang Y, He JS, Wang X, et al. Administration of amyloid-beta42 oligomer-specific monoclonal antibody improved memory performance in SAMP8 mice. J Alzheimers Dis. 2011;23(3):551–561. | |
Hillen H, Barghorn S, Striebinger A, et al. Generation and therapeutic efficacy of highly oligomer-specific beta-amyloid antibodies. J Neurosci. 2010;30(31):10369–10379. | |
Mullane K, Williams M. Alzheimer’s therapeutics: continued clinical failures question the validity of the amyloid hypothesis- but what lies beyond? Biochem Pharmacol. 2013;85(3):289–305. | |
Szabo P, Relkin N, Weksler ME. Natural human antibodies to amyloid beta peptidae. Autoimmun Rev. 2008;7(6):415–420. | |
Dodel RC, Du Y, Depboylu C, et al. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75(10):1472–1474. | |
Relkin NR, Szabo P, Adamiak B, et al. 18-month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30(11):1728–1736. | |
Dodel R, Rominger A, Bartenstein P, et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol. 2013;12(3):233–243. | |
Relkin N, Gessert D, Stokes K, et al. The Gammaglobulin Alzheimer Partnership Study (GAP): design, screening, enrollment and futility analysis results. Alzheimers Dement. 2013;8(Suppl 4):P456. | |
Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171–181. | |
Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3(10):1141–1154. | |
Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33(32):9511–9522. | |
Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. | |
Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–825. | |
Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8(2):387–402. | |
Meraz-Ríos MA, Lira-De León KI, Campos-Peña V, De Anda-Hernández MA, Mena-López R. Tau oligomers and aggregation in Alzheimer’s disease. J Neurochem. 2010;112(6):1353–1367. | |
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. | |
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. | |
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012;26(5):1946–1959. | |
Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481. | |
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99(9):6364–6369. | |
Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. | |
Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133(Pt 5):1312–1327. | |
Boche D, Donald J, Love S, et al. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer’s disease. Acta Neuropathol. 2010;120(1):13–20. | |
Kayed R, Jackson GR. Prefilament tau species as potential targets for immunotherapy for Alzheimer disease and related disorders. Curr Opin Immunol. 2009;21(3):359–363. | |
Kayed R. Anti-tau oligomers passive vaccination for the treatment of Alzheimer disease. Hum Vaccin. 2010;6(11):931–935. | |
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27(34):9115–9129. | |
Boutajangout A, Quartermain D, Sigurdsson EM, et al. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30(49):16559–16566. | |
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010; 224(2):472–485. | |
Bi M, Ittner A, Ke YD, Götz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One. 2011;6(12):e26860. | |
Troquier L, Caillierez R, Burnouf S, et al. Targeting phospho-Ser422 by active tau immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res. 2012;9(4):397–405. | |
Chai X, Wu S, Murray TK, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286(39):34457–34467. | |
d’Abramo C, Acker CM, Jimenez HT, Davies P. Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One. 2013;8(4):e62402. | |
Castillo-Carranza DL, Lasagna-Reeves CA, Kayed R. Tau aggregates as immunotherapeutic targets. Front Biosci (Schol Ed). 2013;5: 426–438. | |
Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, et al. Reduction of tau oligomers by immunotherapy, improves cognition in an AD mouse model. AAIC. 2013;325–326. | |
Golde TE, Das P, Levites Y. Quantitative and mechanistic studies of Abeta immunotherapy. CNS Neurol Disord Drug Targets. 2009;8(1):31–49. | |
Zahs KR, Ashe KH. Beta-amyloid oligomers in aging and Alzheimer’s disease. Front Aging Neurosci. 2013;5:28. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.