Back to Journals » Journal of Inflammation Research » Volume 19
Immunosenescence in Idiopathic Pulmonary Fibrosis
Authors Xiao S ![]() , Li Z, Zhang H, Cai W, Yu S, Song X
, Li Z, Zhang H, Cai W, Yu S, Song X
Received 8 December 2025
Accepted for publication 19 May 2026
Published 5 June 2026 Volume 2026:19 586167
DOI https://doi.org/10.2147/JIR.S586167
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Cynthia Koziol-White
Shiyu Xiao,1– 3,* Zhouzhou Li,1– 3,* Haoyu Zhang,1– 3 Wenjie Cai,1– 3 Shiwen Yu,1– 3 Xinyu Song1– 3
1Department of Respiratory and Critical Care Medicine, The First College of Clinical Medicine Science, China Three Gorges University, Yichang Central People’s Hospital, Yichang, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Yichang Central People’s Hospital, Yichang, People’s Republic of China; 3Clinical Medical Research Center for Precision Diagnosis and Treatment of Lung Cancer and Management of Advanced Cancer Pain of Hubei Province, Yichang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xinyu Song, Yichang Central People’s Hospital, No. 183, Yiling Avenue, Wujiaogang District, Yichang, Hubei, People’s Republic of China, Email [email protected]
抽象的: Idiopathic pulmonary fibrosis (IPF) is a common lung disease among the elderly, which has attracted increasing attention in recent years due to its poor prognosis and limited treatment options. Over the past decade, significant progress has been made in understanding the mechanisms of IPF, establishing it as an age-related disease in which cellular senescence plays a critical role in its pathogenesis and progression. Aging also induces structural and functional changes in the immune system, a process termed immunosenescence, which triggers systemic low-grade chronic inflammation and is likely involved in promoting pulmonary fibrosis. This highlights immunosenescence as a potential core driver of IPF. Consequently, targeting immunosenescence in IPF represents a novel avenue for disease exploration. Given the narrative nature of this review, we conducted a comprehensive but non-systematic literature search. We searched electronic databases (including PubMed and Web of Science) for publications published up to October 2025. The search used combinations of terms related to “IPF” and “Aging”. Due to the broad scope of this review, we did not adopt strict algorithmic inclusion or exclusion criteria; instead, we prioritized papers with pioneering significance, high impact studies, and publications that most clearly demonstrated the evolution of concepts and current debates in the field. This paper systematically reviews recent advances in research on immunosenescence in IPF and the roles of relevant immune cell subsets, discussing promising biomarkers for clinical diagnosis and related therapeutic strategies. In the future, further studies are required to elucidate the mechanisms by which immunosenescence contributes to IPF. Employing advanced technologies to identify sensitive biomarkers and develop promising therapeutic approaches is essential to address the current clinical challenges faced by IPF patients.
Keywords: aging, pulmonary fibrosis, immune system, inflammaging, SASP
Introduction
Interstitial lung disease is a chronic, progressive, and irreversible lung disorder, with IPF being the most common type. Current anti-fibrotic drugs like nintedanib and pirfenidone have limited effectiveness and cause side effects,1 underscoring the urgent necessity for novel therapeutic strategies.
Recent studies, have identified IPF as an age-associated chronic condition,2 with cellular senescence serving as a pivotal driving mechanism. Cellular senescence is defined as the permanent cessation of the cell cycle, influenced by various factors including telomere shortening, DNA damage, endoplasmic reticulum stress, mitochondrial dysfunction, and the senescence-associated secretory phenotype (SASP).3 Immune senescence, or age-related immune decline, results from this process, impairing the removal of senescent cells and promoting their buildup in lung tissue, leading to fibrosis. It also causes chronic inflammation, disrupting tissue repair and cytokine balance, thus contributing to IPF progression. Immune senescence is both an aging-related immune issue and a potential trigger for IPF.
Despite significant progress in understanding immune senescence and IPF in the last decade, the exact impact of immune senescence on IPF development remains unclear. This article reviews recent research on immune senescence in IPF, explores new markers, and consolidates existing therapies targeting immune senescence to suggest new approaches for IPF diagnosis and management.
Immunosenescence and Its Manifestations in IPF Lung Tissue
Idiopathic Pulmonary Fibrosis
IPF is a chronic interstitial lung disease with complex etiologies. Epidemiological studies have demonstrated a strong association with aging—the median age at diagnosis exceeds 65 years, and incidence increases with age.4 The pathological and physiological characteristics of IPF mainly manifest in the remodeling of structural units. At the tissue pathological level, IPF is characterized by the typical pattern of idiopathic interstitial pneumonia (UIP).5 The typical manifestation of UIP is the patchy distribution of interstitial fibrosis, with the lung tissue presenting areas of scarring and thickening, and the intensity of fibrosis varies. This patchy distribution and the associated temporal heterogeneity are the key pathological features for diagnosing IPF.6 In the later stage, honeycomb-like cysts form in the subpleural area.
Current research has shown that the pathogenesis of IPF involves repeated micro-injuries to alveolar epithelial cells, abnormal tissue repair, and the formation of a self-perpetuating fibrotic microenvironment.7
During the development of IPF, various aging characteristics are observed in the lung tissue, including telomere shortening, DNA damage accumulation, and mitochondrial dysfunction in alveolar epithelial cells, accompanied by the excessive release of SASP factors.8 Cellular aging can exert its core driving effect through three interwoven molecular levels: the abnormal reactivation of developmental-related signaling pathways, the alteration of epigenetic modifications, and the regulatory imbalance of non-coding RNAs. Aging manifests as age-related epigenetic drift, namely the random loss (hypomethylation) of DNA methylation patterns and the hypermethylation of promoter regions, which directly leads to the abnormal expression of dormant developmental-related genes. At the same time, non-coding miRNAs, as key components of the epigenetic network, also show significant dysregulation. It is noteworthy that the expression of these miRNAs is itself regulated by DNA methylation, thereby forming a multi-layer feedback loop between epigenetics and miRNAs. Moreover, cellular aging further amplifies the abnormal signals of the above developmental pathways by releasing SASP. In summary, epigenetic drift provides an upstream basis for the reactivation of developmental pathways and the dysregulation of miRNAs, and the latter mutually reinforce and synergize with the paracrine effect of cellular aging, jointly converting the molecular changes related to aging into a persistent fibrotic program in lung tissue.9
Immunosenescence
Aging affects all cell types in the human body, but the sensitivity to aging varies considerably among different types. Immune cells are among the most susceptible.10 Immunosenescence refers to the functional remodeling of the immune system that occurs with advancing age. This complex, multidimensional process manifests not only as a progressive decline in immune defense functions—including both innate and adaptive immunity—leading to increased susceptibility to infection, reduced vaccine efficacy, and weakened immune surveillance, but also as a chronic state of sterile low-grade inflammation.11 It is characterized by a persistent elevation of systemic pro-inflammatory factors (e.g., IL-6, IL-1β) and an imbalance of anti-inflammatory factors (e.g., IL-10, TGF-β), resulting in a concurrent decline in immune competence and a rise in inflammatory markers. Breakthrough research in recent years has revealed that the aging of immune cells may pose the greatest threat to the organism. Senescent immune cells accelerate the aging of other organs and thereby promote systemic aging by secreting a spectrum of pro-inflammatory and pro-fibrotic SASP molecules, including cytokines, chemokines, matrix metalloproteinases, and growth factors.9 Experimental evidence indicates that specifically inducing senescence in immune cells accelerates the aging phenotype in distal organs, such as the lungs, while eliminating senescent immune cells delays the overall aging process.12 This confirms that the senescence of the immune system can exert systemic propagating effects. This state of immune imbalance, marked by a loss of T-cell repertoire diversity, impaired B-cell function, and declined activity of innate immune cells, is closely associated with the pathological progression of IPF. Notably, significant features of immunosenescence have been detected in both the peripheral blood and the local pulmonary microenvironment of IPF patients, including: (1) a marked reduction in naïve T cells; (2) accumulation of senescence-associated B cells; and (3) polarization of alveolar macrophages, among others.13 In summary, these findings reveal that the lungs of IPF patients ultimately develop a vicious cycle that promotes fibrosis initiation and progression, strongly associated with immune senescence.
Immune Senescence in IPF: Focus on the SASP Inflammatory Network
In recent years, research on the pathogenesis of IPF has become increasingly extensive and in-depth; however, studies focusing on the immunosenescence pathway remain insufficient. Although the mechanisms of immunosenescence are not yet fully unified, three key features have been identified: the aging of immune organs such as thymic involution,the senescence of immune cells (discussed later), and the production of circulating SASP factors.14 Immunosenescence drives fibrotic progression through dual pathways: on one hand, the diminished clearance capacity of senescent immune cells leads to the abnormal accumulation of senescent lung epithelial cells within the pulmonary interstitium,15 which persistently secrete pro-fibrotic mediators; on the other hand, the inflammatory microenvironment associated with immunosenescence directly activates pulmonary fibroblasts, inducing excessive deposition of extracellular matrix. The chronic inflammatory network mediated by SASP cytokines, which accompanies immunosenescence, appears to be a potent driver of fibrosis.
The SASP factors serve as a central mechanism driving the inflammation-fibrosis vicious cycle in IPF by establishing a complex inflammatory network. Earlier research exploring the involvement of cellular senescence in IPF pathogenesis revealed that type II alveolar epithelial (AT2) cells, fibroblasts, and immune cells in the lungs of IPF patients enter a state of irreversible senescence under the combined effects of multiple stressors, including telomere shortening, oxidative stress, and epigenetic remodeling. These senescent cells subsequently secrete a spectrum of SASP cytokines, which promote fibrosis through three interrelated mechanisms. Firstly, the growth factors centered on TGF-β in SASP drive the activation of myofibroblasts and the synthesis of extracellular matrix (ECM). TGF-β induces fibrosis by influencing fibroblast gene expression and activating SMAD and Wnt/β-catenin pathways. It works with growth factors like connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), and endothelin-1 to boost myofibroblast proliferation and activation.16 Secondly, senescent cells secrete interleukin family, stimulating local inflammatory responses through complex signaling axes. The IL-1β-IL-17A-TGF-β axis is a key pro-fibrotic pathway.17 IL-4 and IL-13 promote fibrosis by activating JAK-STAT and PI3K-AKT signaling pathways in M2 macrophages, leading to the secretion of TGF-β,18,19 and thus the activation of fibroblasts and epithelial-mesenchymal transition (EMT). IL-11 directly stimulates the activation and EMT of fibroblasts.20 Beyond pro-inflammatory factors, SASP also contains anti-inflammatory molecules. For instance, IL-10 limits pulmonary fibrosis by suppressing IL-17A production.21 Elevated IL-10 levels detected in the bronchoalveolar lavage fluid (BALF) of IPF patients treated with pirfenidone further corroborate its anti-inflammatory effect.22 Most chemokine family members are secreted by senescent immune cells, mediating inflammatory responses alongside the interleukin family such as CCL-3, 4, 523 or directly inducing CTGF expression through Rac/ERK, JNK, and AP-1 pathways to promote fibrosis such as CXCL-1.24 Thirdly, matrix metalloproteinases (MMPs) and their inhibitor PAI-1 mediate abnormal extracellular matrix deposition. Pro-fibrotic MMP3 and MMP7 are activated by TGF-β,25 while MMP10 may exert anti-fibrotic effects by promoting collagen degradation.26 PAI-1 plays a key role in connecting senescence, inflammation, and fibrosis: it is induced by TGF-β, promotes the paracrine senescence of ATII cells, and directly inhibits the degradation of extracellular matrix.27 These three types of factors do not act independently but form a self-enhancing network. SASP factors induce the senescence of adjacent healthy cells through paracrine signal transduction, thereby promoting more SASP production. At the same time, pro-inflammatory factors triggered by SASP accelerate the senescence of immune cells, hindering the clearance of senescent cells and inflammatory mediators. This continuous accumulation of senescent cells maintains the release of SASP and forms a vicious cycle, ultimately promoting progressive remodeling of the lungs through fibroblast activation, epithelial-mesenchymal transition, and extracellular matrix deposition (Figure 1).
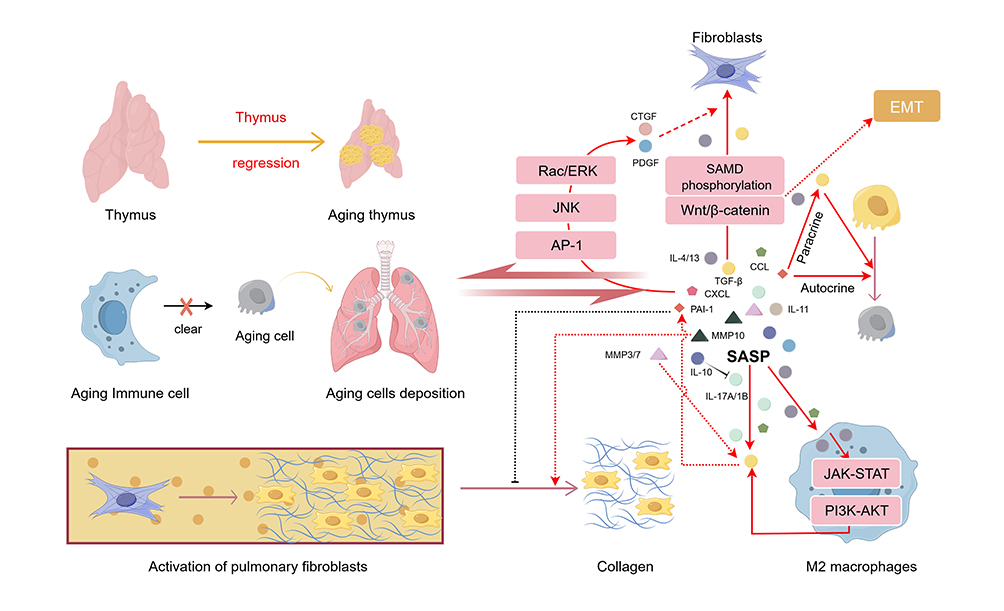 |
Figure 1 Immune aging plays a central role in the pathogenesis of IPF. This figure illustrates that thymus degeneration causes senescent cells to accumulate and secrete SASP, which drives fibroblast activation, ECM deposition, and a self-amplifying inflammation-fibrosis cycle. |
The Role of Immune Aging-Related Cell Subpopulations in IPF
Previous studies have demonstrated that immune cell aging primarily manifests as a reduction in the production of naive T and B lymphocytes, dysfunction of memory cells, accumulation of senescent cells, and dysregulated cytokine production. Beyond the critical homeostatic imbalance in adaptive immunity, the innate immune system also undergoes a series of alterations with aging. Innate immune cells exhibit various functional impairments that compromise their ability to promote tissue repair and mount appropriate protective responses.28 Current research on the immune mechanisms in IPF remains insufficiently comprehensive, and the role of the immune system in IPF is not yet fully elucidated. It is established that nearly every type of immune cell, including lymphocytes and macrophages, is involved in the disease. Fortunately, the rapid advancement of single-cell RNA sequencing (scRNA-seq) technology in recent years has opened new avenues for investigation. By analyzing the gene expression profiles of individual cells, researchers can uncover changes in immune cells within the lungs of IPF patients, providing novel insights into the underlying immune mechanisms.
Senescent T Cells in IPF
The primary hallmarks of T cell senescence include a marked reduction in naive T cells due to thymic involution, along with the heightened differentiation and functional impairment of expanded memory T cell populations.29 Among these, the decline in CD8+ T cells is most pronounced.30 The decrease in naive T cell populations leads to reduced T cell receptor repertoire diversity, increasing susceptibility in the elderly and impairing the effective initiation of immune responses. ScRNA-seq studies in recent years have corroborated these findings in progressive IPF, demonstrating a decreased proportion of naive T cells alongside an increased frequency of regulatory T cells (Tregs). T cell dysfunction is also manifested by an imbalance in co-stimulatory signaling molecules: T cells from IPF patients exhibit significantly reduced surface expression of CD28 and ICOS, while PD-1 expression is elevated. The loss of CD28/ICOS expression is directly correlated with declining lung function (reduced FVC and DLCO) and shorter patient survival.31 Conversely, high PD-1 expression exacerbates pulmonary inflammation and fibrosis by inducing TGF-β and IL-17A secretion.32 Tregs are a distinct subset of CD4+ T lymphocytes characterized by the expression of the transcription factor forkhead box P3 (FoxP3) and high levels of the interleukin-2 (IL-2) receptor α chain (CD25).33 Research indicates that this cell population is closely associated with IPF, exhibiting complex and often contradictory roles within the aging and IPF microenvironments. Their function is highly dependent on the disease stage and the heterogeneity of Treg subpopulations. Multiple single-cell RNA sequencing studies have revealed an enrichment of Tregs in IPF lung tissue, suggesting a potentially critical role. Studies have demonstrated that depleting Tregs during the early stage of bleomycin (BLM)-induced pulmonary fibrosis reduces fibrotic lesions, indicating a pro-fibrotic role for Tregs.34 This aligns with Treg secretion of cytokines such as TGF-β and TGF-β1. TGF-β can induce increased synthesis of matrix proteins and decreased activity of matrix proteases.35 Specifically, TGF-β1 promotes the aberrant differentiation of fibroblasts into myofibroblasts and ECM synthesis via the TGF-β1/Smad signaling pathway, establishing it as a crucial pro-fibrotic cytokine in IPF pathogenesis.36 Under homeostatic conditions, a Th2-dominant immune response drives pulmonary fibrosis,37 whereas the cytokine IFN-γ secreted by Th1 cells can ameliorate fibrosis by inducing the production of MMPs.38 An imbalance in the Th1/Th2 ratio plays a modulatory role during the inflammatory phase of IPF.39 However, during acute infections or acute exacerbations of IPF (AE-IPF), both Th1 and Th2 responses are suppressed, with Tregs and Th17 cells becoming dominant.40 Tregs can also convert into Th17 cells and, upon IL-6 stimulation, secrete IL-17A,41,42 which acts on alveolar epithelial cells (AECs) and myofibroblasts to ultimately exacerbate pulmonary inflammation and fibrosis.43 This is confirmed by AE-IPF mice with IL-17 knockout exhibiting milder pulmonary inflammation and fibrosis.44 Conversely, IL-10 is another factor secreted by Tregs. Experimental mouse models have shown that IL-10 can prevent pulmonary fibrosis by downregulating type I collagen synthesis in human scar tissue-derived fibroblasts, thereby directly inhibiting fibrosis.45 Furthermore, other studies have indicated that bone marrow-derived mesenchymal stem cells (BMSCs) alleviate fibrosis through Tregs,46 and that the activation of the aryl hydrocarbon receptor leading to Treg expansion can mitigate fibrosis.47 This body of evidence underscores the anti-fibrotic attributes of Tregs. In addition to their dual role, a review of the relevant literature reveals controversy regarding the abundance of Tregs in peripheral blood, with some studies reporting a decrease and others an increase in their numbers. Given the capacity of Tregs to interact with various immune and non-immune cell types, we speculate that Tregs might be recruited from the circulation to the lungs of patients, where they engage in interactions with diverse pulmonary cells. This hypothesis could explain the discrepant findings in peripheral blood Treg counts versus the consistently observed elevation of Tregs within lung tissue. Furthermore, the heterogeneity of Treg subsets, coupled with variations in measurement methodologies employed across studies, likely contributes to these discrepancies. Different Treg subsets are known to exert distinct functions. The relative proportions of these subsets are dynamically regulated during disease progression, which may underlie the context-dependent dual role of Tregs in IPF. Future research employing precise measurements of specific Treg subsets will be crucial for elucidating their contributions to the immunopathogenesis of IPF.(Figure 2)
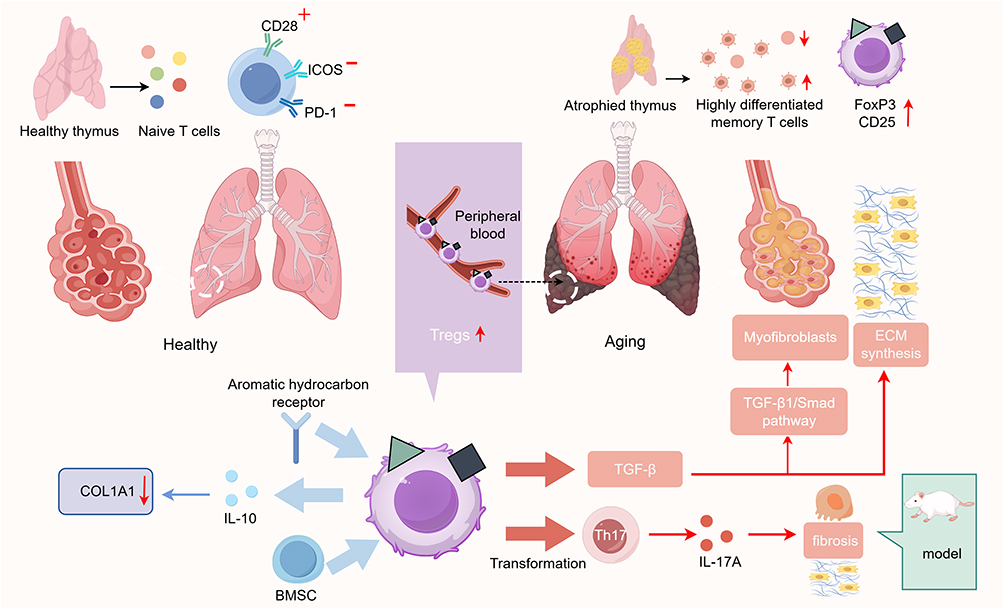 |
Figure 2 T cell aging plays a critical role in the pathogenesis of IPF. This figure illustrates that thymus degeneration drives T-cell senescence and alters Treg function in the lungs, promoting fibroblast activation and ECM deposition via the TGF-β1/Smad pathway to induce fibrosis. |
Senescent B Cells in IPF
The earliest research on the effects of aging on B cells was published by Cancro, who examined the impact of aging on B cell subsets, development, and function, proposing B cells as the initiators of immunosenescence.48 However, current studies on the role of B cells in the pathogenesis of IPF remain insufficient, with only limited evidence confirming that B cells, plasma cells derived from B cells, and the antibodies they produce are involved in the disease process. With advancing age, B lymphopoiesis in the bone marrow declines, and the capacity for class-switch recombination (CSR) becomes impaired, leading to a restricted B-cell receptor (BCR) repertoire. This diminishes the immune system’s ability to recognize and respond to neo-antigens.49 Additionally, the proportional distribution and functional capacity of B cell subsets are altered. B-cell enrichment has been observed in the lung tissue of IPF patients, alongside elevated levels of circulating B-cell activators and the B-cell chemokine CXCL13.50 These findings collectively demonstrate the role of B cells in IPF progression. Single-cell RNA sequencing analysis of peripheral blood from patients with progressive IPF has revealed a decreased proportion of B lymphocytes,51 while a distinct cell type—age-associated B cells (ABCs)—was enriched in aged mice. These cells have been demonstrated to exhibit a senescent phenotype, including telomere shortening and cell cycle arrest, and most importantly, they secrete SASP-associated inflammatory factors such as IL-6 and TNF-α8. IL-6 can participate in regulating fibroblast senescence and apoptosis via the JAK/STAT signaling pathway.52 It can also induce sustained proliferation and confer apoptosis resistance in IPF lung fibroblasts, thereby accelerating disease progression. TNF-α, a common pro-inflammatory cytokine and a key mediator of pulmonary fibrosis, can facilitate the initiation and progression of fibrosis through multiple pathways. In summary, B cells contribute to pulmonary fibrosis by mediating fibroblast activation and migration. Plasma cells originate from activated proliferating B cells. Studies have shown that plasma cell ablation significantly suppresses bleomycin-induced pulmonary fibrosis, though the precise mechanisms remain unclear.53 As early as many years ago, autoantibodies were reported in IPF, detectable in both lung tissue and bronchoalveolar lavage fluid, yet their pathogenic role remained unknown. A 2023 study experimentally elucidated the involvement of the autoantibody against periplakin (PPL) in IPF progression:54 PPL antigen-antibody complexes form and activate immune cells carrying Fcγ receptors (FcγR). The activated cells then release pro-inflammatory, chemotactic, and pro-fibrotic factors, such as CXCL13, which recruits B cells, T cells, and fibrocytes to sites of lung injury. This process fosters the development of inducible bronchus-associated lymphoid tissue (iBALT), enriched with B cells, which perpetuates the production of autoantibodies and pro-inflammatory/fibrotic factors. Ultimately, this leads to increasedECM deposition and the generation of new fibroblasts and myofibroblasts. In conclusion, B cells, plasma cells, and autoantibodies are all implicated in IPF disease progression, yet substantial gaps remain in understanding the underlying mechanisms.
Senescent Macrophages in IPF
Macrophages are the most critical immune cells in the healthy lung, playing a pivotal role in initiating innate immune responses through cytokine release. Pulmonary macrophages can be categorized into three subpopulations: monocyte-derived macrophages, interstitial macrophages (IMs), and alveolar macrophages (AMs).55 These cells contribute to fibrotic progression through three primary mechanisms: secretion of pro-inflammatory and pro-fibrotic cytokines, mediation of oxidative stress, and disruption of the M1/M2 polarization balance. During the development of IPF, senescent macrophages exhibit functional impairments and phenotypic transformations. With aging, macrophage metabolism shifts, leading to increased production of inflammatory cytokines and a concurrent decline in immune function. Studies have shown that TLR3 mutations in IPF patients reduce macrophage responses to microorganisms, triggering acute IPF exacerbations.56 Additionally, deficiency in the stimulator of interferon genes (STING) compromises macrophage antiviral defense.57 Elevated expression of SIRPα attenuates the phagocytic capacity of macrophages,58 while M2 macrophage polarization, characterized by low MHCII expression, impairs antigen presentation.59 Collectively, these macrophage alterations in IPF patients undermine pathogen clearance and exacerbate tissue damage. According to the classical definition, macrophages can be polarized into two major phenotypes: M1 and M2. M1 macrophages, polarized by Th1 pro-inflammatory cytokines (e.g., TNF, IL-2, IFN-γ), exhibit pro-inflammatory and immunostimulatory functions. In contrast, M2 macrophages, induced by Th2 cytokines (e.g., IL-4, IL-5, IL-13), mediate anti-inflammatory and pro-repair responses.60 The senescent microenvironment drives macrophages toward a pro-fibrotic phenotype, characterized by excessive M2 polarization. These macrophages activate fibroblasts and mediate the EMT of alveolar epithelial cells by secreting factors such as TGF-β and CCL18.61 Recent advances in single-cell omics have identified a novel M2-like macrophage subset: scar-associated macrophages (SAMs), characterized by high expression of osteopontin (SPP1)—a recognized biomarker for fibrotic progression in IPF patients—as well as CD36 and CD84.62 Research suggests that their precursor cells are likely myeloid-derived suppressor cells (MDSCs). Upon stimulation by GM-CSF/sB7H3, MDSCs expand63 and subsequently contribute, via SPP1 highly expressed in SAMs, to the SPP1-CD44-PI3K/Akt axis, which directly induces EMT and collagen deposition.64 Furthermore, senescent macrophages secrete SASP factors, contributing to the establishment of a chronic inflammatory microenvironment, among other effects.65 However, some researchers have recently proposed that viewing fibrosis merely as an “M2-driven” disease is overly simplistic. Consequently, macrophages are now categorized into CCR2+ (“inflammatory”) or CX3CR1+ (“repair”) phenotypes. This approach replaces the M1/M2 classification and provides a more comprehensive understanding of macrophage roles in IPF60 (Figure 3).
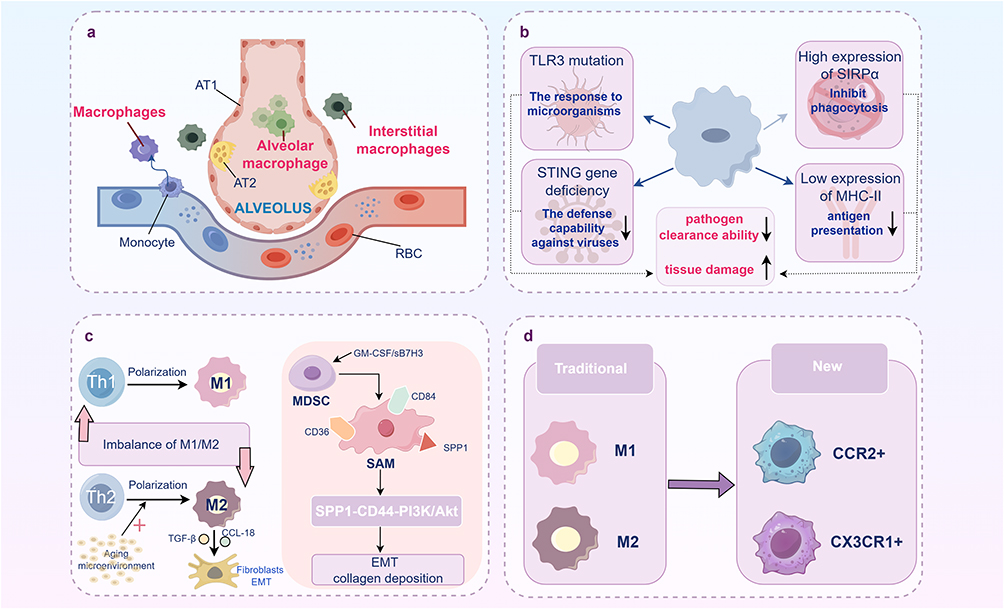 |
Figure 3 Macrophage aging plays a crucial role in the progression of IPF. (a) Three subsets of macrophages in lung tissue; (b). The core mechanism by which senescent macrophages drive IPF progression; (c). M2 polarization imbalance and the pro-fibrotic pathway of novel scar-associated macrophages; (d). A new classification of macrophage phenotypes. |
Senescent Neutrophils in IPF
With advancing age, neutrophil counts vary, but their functional activity—including antimicrobial capacity, phagocytosis, and chemotaxis—undergoes changes marked by a general decline.29 As one of the first responders to tissue injury or pathogen invasion in the lungs, neutrophils may influence IPF through multiple mechanisms. Interestingly, in IPF, the role of aged neutrophils exhibits a paradoxical state: their biological behavior displays dual characteristics that are both pro-fibrotic and potentially protective. Studies indicate that senescent neutrophils exhibit enhanced ROS production and drive pathological NETosis via PAD-4 activation. The released neutrophil extracellular traps (NETs) contain mediators such as elastase (NE) and myeloperoxidase (MPO).66 NE contributes to ECM deposition and promotes fibroblast proliferation and myofibroblast differentiation by activating TGF-β, thereby participating in fibrotic formation.67 Furthermore, neutrophils themselves can release pro-fibrotic cytokines and chemokines, such as TGF-β1, IL-8, and IL-17,68 directly contributing to IPF progression. Concurrently, focal hypoxia in IPF upregulates β2 integrin via HIF-1α, enhancing neutrophil recruitment, activation, and NET release within the pulmonary interstitium.69 Clinical studies have confirmed elevated levels of IL-8 and G-CSF in the BALF and sputum from IPF patients, indicating neutrophil recruitment and activation that predicts future disease worsening.70 In peripheral blood, neutrophil counts and the neutrophil-to-lymphocyte ratio (NLR) serve as markers for IPF progression.71 The aforementioned evidence underscores the pro-fibrotic role of senescent neutrophils. However, evidence also suggests anti-fibrotic and potentially protective functions of neutrophils in IPF. For instance, under specific conditions, NE can degrade type IV collagen and elastin to alleviate fibrosis; neutrophil depletion experiments have failed to attenuate fibrosis in animal models;72 and interestingly, increased BAL neutrophil counts are not consistently associated with elevated mortality.73 These findings imply that neutrophil infiltration might represent a host compensatory attempt to counteract fibrosis, rather than solely a pro-fibrotic driver. The essence of this contradiction likely lies in altered functional states. Aging transforms neutrophils from transient protectors during acute injury (exercising phagocytosis and pathogen clearance) into persistent destroyers during chronic phases (dominated by uncontrolled NETosis). This phenotypic shift may be influenced by factors within the microenvironment, such as TGF-β1 levels, hypoxia severity, and miR-155-5p epigenetic regulation. This insight also explains why anti-inflammatory therapies prove ineffective against IPF. It suggests that precisely targeting pathological pro-fibrotic functions while preserving their antimicrobial and ECM-degrading capacities holds greater therapeutic value than the wholesale elimination of neutrophils.
Senescent Mesenchymal Stem Cells in IPF
Mesenchymal stem cells (MSCs) are pivotal regulators of tissue repair and wound healing processes, possessing remarkable immunomodulatory functions. Preclinical pulmonary fibrosis (PF) models have demonstrated that MSCs can suppress inflammation, reduce fibrosis, and extend survival.74 Notably, MSCs preferentially localize to injury sites, primarily the lungs, followed by the liver. Furthermore, they mitigate pulmonary inflammation and fibrosis through various immunomodulatory mechanisms, including reducing the expression of co-stimulatory proteins in dendritic cells (DCs) and macrophages, attenuating T cell immune responses, promoting the generation of Tregs and IL-10,75 and concurrently diminishing TGF-β signaling and extracellular matrix (ECM) production.76 However, aged MSCs derived from IPF patients likely lose these functions. Studies have shown that bone marrow-derived MSCs (B-MSCs) from IPF patients exhibit typical hallmarks of senescence compared to healthy controls, including increased cell volume, morphological alterations, accumulated DNA damage, telomere shortening, and replicative senescence. This senescent state is characterized by significant mitochondrial dysfunction and impaired recovery capacity. More importantly, senescent B-MSCs display severe functional defects with markedly diminished paracrine capabilities. This not only compromises their ability to suppress inflammation (by reducing pro-inflammatory factors IL-1β and TNF-α while restoring anti-inflammatory factor IL-10) and promote tissue repair, but may also weaken their regulatory effects on myofibroblasts.77 This suggests that senescent B-MSCs, through the loss of their critical inhibitory paracrine signals, may contribute to the formation and maintenance of the aberrant fibrotic microenvironment in IPF. Consequently, MSC senescence in IPF patients represents not merely a functional alteration of cellular state under disease conditions but likely serves as a driving factor in disease progression—particularly in late-onset IPF. Further investigation into MSCs will provide a theoretical foundation for developing novel therapeutic strategies specifically targeting stem cell senescence.
Biological Markers of IPF
The identification of biomarkers for IPF is crucial not only for the early detection of patients but also for advancing mechanistic studies and clinical treatment. Multiple rapidly evolving technologies, such as genomics, transcriptomics, proteomics, and metabolomics, are now available to assist researchers in discovering and identifying biomarkers capable of predicting IPF progression and prognosis. Over the past few years, utilizing these technologies, researchers have uncovered numerous potential biomarkers. This section will summarize both representative established biomarkers and emerging markers identified in recent studies.
Traditional Biological Markers
Direct Markers of Immune Senescence: Epigenetics and Cell Cycle Regulation
Cellular senescence in IPF patients manifests as a pathological state, rendering the assessment of biological age far more significant than chronological age. For instance, studies have demonstrated an association between epigenetic aging and IPF. Analysis of DNA methylation levels in lung tissue from IPF patients revealed significant acceleration across multiple epigenetic clocks—including Horvath, Hannum, PhenoAge, and DunedinPACE—which correlated positively with disease severity.78 This validates the potential of DNA methylation as a biomarker for IPF. Matrix metalloproteinase-7 (MMP7) has long been established as a classical prognostic biomarker in IPF, with its plasma concentration independently correlating with declining lung function and poor outcomes. Research has provided additional insight from a different perspective: the co-upregulation of p16—a key regulator intimately associated with the cell cycle senescence pathway—with matrix remodeling proteins such as MMP7.79
Changes in Senescent Immune Cells
Given that the most central feature of immunosenescence is the aging of immune cells, the most intuitive biomarkers are the altered immune cell populations themselves. Among these, Tregs are considered to hold significant prognostic value, with multiple studies demonstrating that elevated Treg ratios correlate negatively with disease severity and mortality rates.80,81 Similarly, increased monocyte counts have been shown to predict mortality risk,82 and elevated neutrophil counts in BALF may serve as predictors of IPF deterioration.83 Current research predominantly focuses on the association between individual immune cell counts and IPF, albeit with limited accuracy. If future studies combine different immune cells with predictive potential, such as the ratio of Treg cells to monocytes, more accurate predictive methods may be developed.
SASP Factors
As previously discussed, SASP factors serve as pivotal mediators linking immune senescence and fibrosis. Moreover, compared to the measurement of immune cells, which requires fresh blood samples, these cytokines can be obtained from archived plasma samples and offer greater stability, making them promising candidates for soluble biomarkers of immunosenescence. One study demonstrated that GDF15 activates macrophages and fibroblasts via the ALK5-Smad2/3 pathway, driving a pro-fibrotic phenotype. It is significantly upregulated in both peripheral blood and bronchoalveolar lavage fluid, correlating positively with disease severity.84 The S100 calcium-binding protein family is associated with neutrophil activation. Among them, S100A12 has been recognized as a predictive serum biomarker for IPF, while blood concentrations of S100A8 have also been validated as important predictive biomarkers in IPF patients during acute exacerbation.85 Additionally, IL-6, which has been extensively studied, although not showing a significant difference between IPF patients and controls, is significantly correlated with a decline in DLCO, suggesting its potential as a sensitive indicator of deteriorating lung function.86
New Composite Markers
Composite biomarkers can overcome the limitations associated with single biomarkers. Studies have confirmed that individual biomarkers alone are insufficient to provide clinically reliable predictions. Currently, numerous studies are exploring the value of combined biomarkers. For instance, Justin M. Oldham’s team employed a machine learning model incorporating 96 plasma protein combinations, finding this model significantly outperformed single factors in predicting transplant-free survival (TFS).87 Another indicator, the neutrophil-to-lymphocyte ratio (NLR), which reflects systemic inflammatory status, has been shown to serve as an independent prognostic factor for shortened overall survival in both IPF andAE-IPF patients.88 Furthermore, the composite GAP index (cGAP2) model was developed by integrating immune parameters—such as monocyte count and red cell distribution width—into the established GAP index (comprising Gender, Age, and Physiology). This model demonstrated marked improvement in predicting disease progression and mortality.89 These advances underscore the translational potential of immunosenescence biomarkers in optimizing risk stratification, for instance by combining them with the GAP index, and in guiding personalized treatment strategies.
Exploring the Challenges and Clinical Significance of Biomarkers
The quest for an ideal IPF biomarker currently faces several major challenges. Firstly, as a progressive disease, the complex pathophysiology of IPF is challenging to encapsulate with a single biomarker. Moreover, the same biomarker may convey different implications at various disease stages or in distinct anatomical locations. Secondly, many currently identified biomarkers lack adequate specificity and sensitivity. For example, elevated MMP-7 levels are observed not only in IPF but also in non-ILD pulmonary diseases. Furthermore, most discoveries stem from retrospective studies or small-sample cohorts, which lack validation in large-scale, multicenter, prospective studies. Additionally, some assays are prohibitively expensive and methodologically intricate, hindering their widespread clinical application. Finally, disease progression and therapeutic interventions can induce dynamic fluctuations in biomarker levels, particularly in the context of immune cell exploration, where levels are highly susceptible to external influences, complicating the precise correlation of cell levels with disease progression. Addressing these challenges will likely require future advancements in developing composite models that integrate multidimensional data. This may involve combining clinical indicators with diverse biomarker panels, utilizing emerging technologies such as single-cell and spatial omics for discovery, and ultimately validating and standardizing these models through rigorously designed, large-scale prospective cohort studies.
IPF Treatment Strategies Targeting Immune Senescence
The treatment of IPF has long been a challenging and highly debated topic. From the limited therapeutic options of pirfenidone and nintedanib to agents targeting senescent cell clearance, such as dasatinib and quercetin, therapeutic strategies for IPF have evolved toward multi-targeted interventions. This paper focuses on elucidating the relationship between IPF and immune senescence, emphasizing immune reprogramming, metabolic microenvironment regulation, and precision delivery systems to summarize novel therapeutic approaches for reversing the fibrotic process.
Eliminate Senescent Immune Cells
Senescent immune cells, such as senescent T cells and macrophages, contribute to inflammation and fibrosis in IPF pathogenesis. Consequently, targeting these cells for elimination represents a promising therapeutic strategy. Studies have demonstrated that in liver fibrosis, administering CAR-T cells targeting uPAR on fibrogenic hepatic stellate cells significantly reduces ECM deposition and improves liver function. Interestingly, the collateral clearance of uPAR-expressing macrophages was found to amplify the efficacy of this CAR-T strategy.90 Although CAR-T cell technology has shown significant progress in liver fibrosis models, its direct application in pulmonary fibrosis remains in the early exploratory phase. Nevertheless, the underlying principles and target selection provide a clear direction for IPF therapy. For instance, in liver fibrosis, CAR-T cell technology can specifically recognize and eliminate senescent cells expressing uPAR or FAP. Similarly, NKG2D-CAR-T cells can target and clear senescent cells expressing NKG2D ligands,91 a strategy theoretically applicable to senescent cells within the lung. Furthermore, senolytic drugs, such as the combination of quercetin and dasatinib, can alleviate fibrosis by inducing apoptosis in senescent cells and have entered clinical trials to evaluate their safety and efficacy in pulmonary fibrosis.
Restore the Function of Senescent Immune Cells
A prominent characteristic of senescent immune cells is their functional dysregulation. Consequently, restoring the normal function of these cells represents a crucial therapeutic approach. As discussed earlier, T cell senescence is a major component of adaptive immune imbalance in IPF, particularly involving Tregs. Modulating the mitochondrial unfolded protein response (mitoUPR) can enhance mitochondrial integrity, thereby restoring Treg stability and function. This helps mitigate excessive inflammation and maintain immune homeostasis within the senescent microenvironment28. Beyond the development of novel drugs, repurposing existing medications may offer another viable avenue. Metformin, long established as a first-line drug for diabetes, has recently been shown to induce autophagy in CD4+ T cells and inhibit Th17 cell differentiation.92 From an anti-inflammatory and anti-senescence perspective, this suggests potential benefits for IPF treatment. Additionally, researchers have explored epigenetic interventions to reverse cellular senescence. The telomerase activator TAC promotes the transcription of telomerase reverse transcriptase (TERT), effectively repairing telomere damage and reducing DNA damage signaling, thereby delaying cellular senescence and supporting cellular proliferation and function.93 This provides a robust therapeutic strategy for delaying the senescence of immune cells such as T cells and restoring their function. In addition, TPCA-1 is a dual inhibitor of both the NF-κB and JAK-STAT pathways,94 both of which are implicated in IPF pathogenesis and cellular senescence. Salvianolic acid B (SAB), considered a promising agent for lung diseases, has been demonstrated to directly interfere with the transcription of senescence-associated genes. It inhibits the senescence program in macrophages and alveolar epithelial cells by impeding the binding of transcription factor SP1 to the promoters of key senescence genes like p16 and p21, thereby reducing the release of SASP factors such as IL-6 and IL-1β and alleviating pulmonary fibrosis progression upstream.95 Recent research suggests that specific nutrients can slow biological aging and enhance immune function by targeting shared pathways of senescence and immunoregulation. For example, plant extracts such as ginsenoside Rg1/Rb1 and Dendrobium officinale polysaccharides can enhance antioxidant capacity and modulate macrophage polarization by activating pathways like NRF2 and SIRT1, collectively improving the immunometabolic state.96,97
New Delivery System
To enhance therapeutic targeting and minimize systemic exposure and toxicity, various novel delivery systems have been developed for IPF treatment. Exosome therapy has emerged as a recent research focus, encompassing diverse sources that deliver distinct miRNAs to exert targeted effects and ultimately inhibit fibrosis progression. Mesenchymal stem cells (MSCs) are the most commonly used source due to their ease of isolation and immunomodulatory properties. Bone marrow MSC-derived extracellular vesicles (BMSC-EVs) carry miRNAs such as miR-186 and miR-29b-3p, which inhibit target genes like SOX4 and FZD6, block the Wnt/β-catenin pathway, and consequently suppress fibroblast activation and EMT. Adipose-derived MSC extracellular vesicles (ADSC-EVs) deliver miR-29c and miR-129 to target myofibroblasts and affect ECM deposition. They can also transfer let-7d-5p to suppress TGFBR1, reducing ROS levels and inflammation, thereby exerting anti-fibrotic effects. Human umbilical cord MSC extracellular vesicles (HUMSC-EVs) deliver miR-218 to inhibit endothelial-mesenchymal transition (EndMT) by modulating the MeCP2/BMP2 signaling axis or employ miR-26a-5p to block the Adam17/Notch pathway, significantly alleviating pulmonary fibrosis and promoting tissue repair.98 Synthetic nanoparticles enable efficient targeted delivery of small-molecule nucleic acid therapeutics. Recent studies have developed inhalable siRNA delivery systems, such as PEI-GBZA/siIL-11 and siIL11@PPGC nanoparticles. These particles effectively penetrate the mucus barrier, accumulate in fibrotic regions, and inhibit the ERK/SMAD pathway by targeting IL-11. This blocks fibroblast-to-myofibroblast differentiation and ECM deposition while reducing neutrophil and macrophage recruitment, demonstrating improved lung function without systemic toxicity in animal models.99 Beyond these two major systems, lipid nanoparticle (LNP) technology offers another novel approach for targeted in vivo immunomodulation. Studies indicate that systemic delivery of LNP-encapsulated mRNA encoding a chimeric antigen receptor (CAR) can directly reprogram T cells in vivo into CAR-T cells, enabling targeted clearance of aberrantly activated cells in the lungs. This strategy offers transient and dose-controllable effects, avoiding the potential long-term clearance risks associated with conventional CAR-T therapy, thereby maintaining high efficacy and safety.90
Other Collaborative Strategies
Beyond the targeted strategies discussed above, IPF management also encompasses a range of integrated approaches including immunomodulation, nutritional interventions, lifestyle modifications, and optimization of existing therapies, all aimed at reshaping the immune microenvironment and slowing the progression of fibrosis. While previous sections have addressed methods to either eliminate or restore the function of senescent immune cells in IPF, distinct immunomodulatory strategies are required for aberrantly activated immune cells. Studies indicate that B cells form tertiary lymphoid structures (TLS) in the lungs and secrete pro-fibrotic factors. Anti-CD20 monoclonal antibodies such as rituximab can effectively eliminate B cells; however, efficacy has so far been observed only in certain non-IPF-ILD patients, and their benefit in IPF patients remains undetermined.100 Furthermore, mast cells promote TGF-β secretion and fibrosis via the PAR-2/PKC-α pathway,101 positioning them as potential therapeutic targets. The JAK inhibitor tofacitinib can inhibit M2 macrophage polarization and reduce the CD206+ macrophage population, demonstrating clinical efficacy in slowing pulmonary fibrosis progression in RA-ILD patients.102 Specific nutrients and microbial metabolites exert adjunctive therapeutic effects by modulating immune metabolism. Short-chain fatty acids (SCFAs) can enhance alveolar macrophage function and suppress inflammatory cytokine release under homeostatic conditions.103,104 Additionally, vitamin D alleviates oxidative stress and immunosenescence by inhibiting Th17 differentiation.105 The combination of the polyphenol quercetin with dasatinib is currently undergoing clinical trials. Furthermore, ongoing research explores combining standard antifibrotic drugs like nintedanib or pirfenidone with polyphenols such as caffeic acid and epicatechin, which have been shown to regulate SASP factor production, thereby delaying both senescence and fibrotic progression.106 While immunomodulation and drug therapies offer greater precision, they inevitably carry side effects. The safest approach remains lifestyle intervention: reducing caloric intake reverses pro-inflammatory polarization of M1 macrophages, thereby decreasing age-related inflammation;107 regular physical activity alleviates chronic low-grade inflammation, helping reduce the incidence of age-related diseases;108 specific probiotic strains can modulate immune cell activity;109 zinc supplementation may alleviate fibrosis by downregulating the SLC39A8/ZIP8 transporter.110 These approaches are safe and easily implementable but generally act slowly, exhibit individual variability in effectiveness, and currently lack large-scale clinical evidence to support their independent efficacy. Therefore, they should be considered supplementary to foundational IPF management (Figure 4).
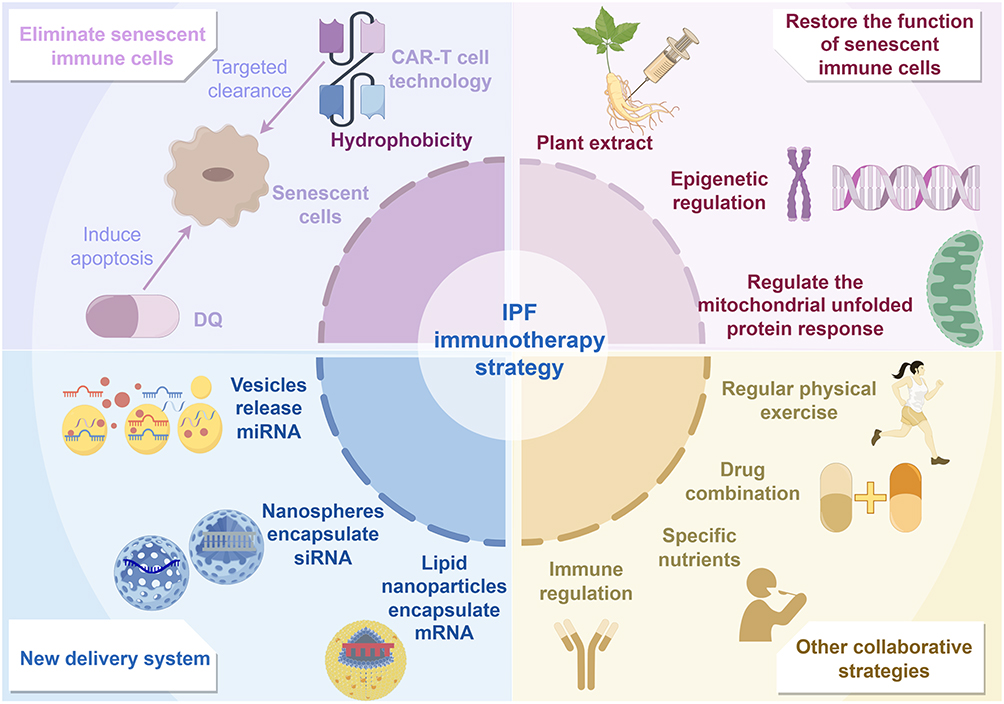 |
Figure 4 Therapeutic strategies for IPF from the perspective of immune aging. This figure presents four immunosenescence-based therapeutic strategies for IPF: eliminating senescent immune cells, restoring their function, utilizing novel delivery systems, and employing other synergistic approaches. |
Conclusions and Future Prospects
Immune senescence significantly contributes to IPF by fostering a fibrotic lung environment through senescent immune cells and SASP secretion, disrupting immune balance, activating fibroblasts, and increasing extracellular matrix deposition. The key question is whether immune senescence causes IPF, results from it, or forms a self-sustaining cycle. Evidence increasingly supports the latter, indicating a mutually reinforcing relationship. Early IPF diagnosis is challenging, and prognosis is poor, highlighting the need for biomarkers and therapies. This article explores potential biomarkers and treatments related to immune senescence, though patient variability and disease complexity limit their broad application.The bleomycin-induced fibrosis mouse model struggles to accurately mimic the human aging microenvironment, hindering the translation of findings to clinical settings. Future research should use advanced technologies for a detailed analysis of this complexity. Conditional knockout models can clarify the roles of SASP factors, while single-cell RNA sequencing and spatial transcriptomics can provide insights into the aging of immune cells and their spatial relationship with fibrotic areas in IPF lungs. Identifying IPF subtypes based on immune aging traits could enable targeted treatments. Longitudinal studies are needed to link immune aging markers with disease progression and prognosis. While removing senescent cells is promising, the risk of increased infection susceptibility must be carefully evaluated.
In summary, viewing immune aging as a cause of IPF offers new treatment possibilities. However, effective clinical application requires collaboration, combining research with advanced technology, and strong clinical validation. This comprehensive approach is crucial to overcoming challenges and realizing the potential of therapies targeting immune aging for IPF patients.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Acknowledgments
We sincerely thank Fei Xiang (Department of Pulmonary and Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China) for her insightful comments on this article. We also appreciate Figdraw (www.figdraw.com) for assistance in the creation of figures.
Author Contributions
Shiyu Xiao: Writing – original draft, Writing – review & editing, validation, formal analysis
Zhouzhou Li: Writing – original draft, Resources, Supervision, Data curation
Haoyu Zhang: Writing – original draft, Resources, Supervision, Data curation
Wenjie Cai: Writing – original draft, Resources, Supervision, Data curation
Shiwen Yu: Writing – review & editing, Resources, Supervision, Data curation
Xinyu Song: Methodology, Writing – review & editing, Funding acquisition, Formal analysis
All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the demonstration project of Technological Innovation of Hubei Province China (project number: 2022BCE031).
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Jiang M, Bu W, Wang X, et al. Pulmonary fibrosis: from mechanisms to therapies. Journal of Translational Medicine. 2025;23(1):515. doi:10.1186/s12967-025-06514-2
2. Cai W, Zhang H, Li Z, et al. Tissue fibrosis decoded via cellular senescence: mechanisms, treatments, and emerging technologies. Aging and Disease. 2025. doi:10.14336/ad.2025.0503
3. Liu Z, Zhang Y, Li D, Fu J. Cellular senescence in chronic lung diseases from newborns to the elderly: an update literature review. Biomedicine & Pharmacotherapy. 2024;173:116463. doi:10.1016/j.biopha.2024.116463
4. Jo HE, Randhawa S, Corte TJ, Moodley Y. Idiopathic pulmonary fibrosis and the elderly: diagnosis and management considerations. Drugs Aging. 2016;33(5):321–17. doi:10.1007/s40266-016-0366-1
5. Mancini M, Bargiacchi L, De Vitis C, et al. Histologic analysis of idiopathic pulmonary fibrosis by morphometric and fractal analysis. Biomedicines. 2023;11(5):1483. doi:10.3390/biomedicines11051483
6. Tian L, Wang Y, Qi W, et al. Pathophysiological insights and clinical management strategies for interstitial lung diseases. Biomolecules & Therapeutics. 2025;33(5):785–803. doi:10.4062/biomolther.2025.003
7. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annual Review of Pathology: Mechanisms of Disease. 2022;17(1):515–546. doi:10.1146/annurev-pathol-042320-030240
8. Liu S, Xi Q, Li X, Liu H. Mitochondrial dysfunction and alveolar type II epithelial cell senescence: the destroyer and rescuer of idiopathic pulmonary fibrosis. Front Cell Dev Biol. 2025;13:1535601. doi:10.3389/fcell.2025.1535601
9. Yousefzadeh MJ, Flores RR, Zhu Y, et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594(7861):100–105. doi:10.1038/s41586-021-03547-7
10. Sato Y. Immune aging and its implication for age-related disease progression. Physiology (Bethesda). 2025;40(4). doi:10.1152/physiol.00051.2024
11. Pangrazzi L, Meryk A. Molecular and cellular mechanisms of immunosenescence: modulation through interventions and lifestyle changes. Biology (Basel). 2024;14(1). doi:10.3390/biology14010017
12. Robinson AR, Yousefzadeh MJ, Rozgaja TA, et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 2018;17:259–273. doi:10.1016/j.redox.2018.04.007
13. Kim N-H, Sim S-J, Han H-G, Yoon J-H, Han Y-H. Immunosenescence and age-related immune cells: causes of age-related diseases. Arch Pharm Res. 2025;48(2):132–149. doi:10.1007/s12272-024-01529-7
14. Nikolich-žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19(1):10–19. doi:10.1038/s41590-017-0006-x
15. Wang S, Huo T, Lu M, et al. Recent advances in aging and immunosenescence: mechanisms and therapeutic strategies. Cells. 2025;14(7). doi:10.3390/cells14070499
16. Bascands J-L, Schanstra JP. Obstructive nephropathy: insights from genetically engineered animals. Kidney Int. 2005;68(3):925–937. doi:10.1111/j.1523-1755.2005.00486.x
17. Wilson MS, Madala SK, Ramalingam TR, et al. Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010;207(3):535–552. doi:10.1084/jem.20092121
18. Passalacqua G, Mincarini M, Colombo D, et al. IL-13 and idiopathic pulmonary fibrosis: possible links and new therapeutic strategies. Pulm Pharmacol Ther. 2017;45:95–100. doi:10.1016/j.pupt.2017.05.007
19. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31(1):317–343. doi:10.1146/annurev-immunol-032712-095906
20. Ng B, Huang KY, Pua CJ, et al. Interleukin-11 causes alveolar type 2 cell dysfunction and prevents alveolar regeneration. Nat Commun. 2024;15(1):8530. doi:10.1038/s41467-024-52810-8
21. Arai T, Abe K, Matsuoka H, et al. Introduction of the interleukin-10 gene into mice inhibited bleomycin-induced lung injury in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):L914–22. doi:10.1152/ajplung.2000.278.5.L914
22. Ronan N, Bennett DM, Khan KA, et al. Tissue and bronchoalveolar lavage biomarkers in idiopathic pulmonary fibrosis patients on pirfenidone. Lung. 2018;196(5):543–552. doi:10.1007/s00408-018-0140-8
23. Loetscher P, Uguccioni M, Bordoli L, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391(6665):344–345. doi:10.1038/34814
24. Lin C-H, Shih C-H, Tseng -C-C, et al. CXCL12 induces connective tissue growth factor expression in human lung fibroblasts through the Rac1/ERK, JNK, and AP-1 pathways. PLoS One. 2014;9(8):e104746. doi:10.1371/journal.pone.0104746
25. Maeda S, Dean DD, Gomez R, Schwartz Z, Boyan BD. The first stage of transforming growth factor β1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (MMP-3). Calcif Tissue Int. 2002;70(1):54–65. doi:10.1007/s002230010032
26. Murray MY, Birkland TP, Howe JD, et al. Macrophage migration and invasion is regulated by MMP10 expression. PLoS One. 2013;8(5):e63555. doi:10.1371/journal.pone.0063555
27. Rana T, Jiang C, Liu G, et al. PAI-1 regulation of TGF-β1–induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am J Respir Cell Mol Biol. 2020;62(3):319–330. doi:10.1165/rcmb.2019-0071OC
28. Lewis L, Valvi D, Gedaly R, Marti F. Mitochondrial unfolded protein response in regulatory T cell function: a protective mechanism in immune aging. Front Immunol. 2025;16:1621759. doi:10.3389/fimmu.2025.1621759
29. Faner R, Rojas M, Macnee W, Agustí A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(4):306–313. doi:10.1164/rccm.201202-0282PP
30. Ucar D, Márquez EJ, Chung C-H, et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J Exp Med. 2017;214(10):3123–3144. doi:10.1084/jem.20170416
31. Bonham CA, Hrusch CL, Blaine KM, et al. T cell Co-stimulatory molecules ICOS and CD28 stratify idiopathic pulmonary fibrosis survival. Respir Med X. 2019;1. doi:10.1016/j.yrmex.2019.100002
32. Celada LJ, Kropski JA, Herazo-Maya JD, et al. PD-1 up-regulation on CD4 + T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci Transl Med. 2018;10(460). doi:10.1126/scitranslmed.aar8356
33. Šileikienė V, Jurgauskienė L. Role of regulatory T cells in pulmonary ageing and COPD development. Int J Mol Sci. 2025;26(8):3721. (). doi:10.3390/ijms26083721
34. Boveda-Ruiz D, D’Alessandro-Gabazza CN, Toda M, et al. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology. 2013;218(2):245–254. doi:10.1016/j.imbio.2012.05.020
35. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi:10.1038/nm.2807
36. Wang M, Feng Y, Zhang P, et al. Jiawei Maxing Shigan Tang alleviates radiation-induced lung injury via TGF-β1/Smad signaling pathway mediated by regulatory T cells. Journal of Ethnopharmacology. 2024;320(320):117389. doi:10.1016/j.jep.2023.117389
37. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345(7):517–525. doi:10.1056/NEJMra003200
38. Roderfeld M, Rath T, Pasupuleti S, et al. Bone marrow transplantation improves hepatic fibrosis in Abcb4 −/− mice via Th1 response and matrix metalloproteinase activity. Gut. 2012;61(6):907–916. doi:10.1136/gutjnl-2011-300608
39. Ishida Y, Kuninaka Y, Mukaida N, Kondo T. Immune mechanisms of pulmonary fibrosis with bleomycin. Int J Mol Sci. 2023;24(4). doi:10.3390/ijms24043149
40. Kono M, Miyashita K, Hirama R, et al. Prognostic significance of bronchoalveolar lavage cellular analysis in patients with acute exacerbation of interstitial lung disease. Respir Med. 2021;186:106534. doi:10.1016/j.rmed.2021.106534
41. Komatsu N, Okamoto K, Sawa S, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–68. doi:10.1038/nm.3432
42. Shen X, Zhang H, Xie H, et al. Reduced CCR6+IL-17A+treg cells in blood and CCR6-dependent accumulation of IL-17A+treg cells in lungs of patients with allergic asthma. Front Immunol. 2021;12:710750. doi:10.3389/fimmu.2021.710750
43. Xiao H, Peng L, Jiang D, et al. IL-17A promotes lung fibrosis through impairing mitochondrial homeostasis in type II alveolar epithelial cells. J Cell Mol Med. 2022;26(22):5728–5741. doi:10.1111/jcmm.17600
44. Chen S, Zhang X, Yang C, Wang S, Shen H. Essential role of IL-17 in acute exacerbation of pulmonary fibrosis induced by non-typeable Haemophilus influenzae. Theranostics. 2022;12(11):5125–5137. doi:10.7150/thno.74809
45. Wangoo A, Laban C, Cook HT, Glenville B, Shaw RJ. Interleukin-10- and corticosteroid-induced reduction in type I procollagen in a human ex vivo scar culture. Int J Exp Pathol. 1997;78(1):33–41. doi:10.1046/j.1365-2613.1997.d01-241.x
46. Takao S, Nakashima T, Masuda T, et al. Human bone marrow-derived mesenchymal stromal cells cultured in serum-free media demonstrate enhanced antifibrotic abilities via prolonged survival and robust regulatory T cell induction in murine bleomycin-induced pulmonary fibrosis. Stem Cell Res Ther. 2021;12(1):506. doi:10.1186/s13287-021-02574-5
47. Takei H, Yasuoka H, Yoshimoto K, Takeuchi T. Aryl hydrocarbon receptor signals attenuate lung fibrosis in the bleomycin-induced mouse model for pulmonary fibrosis through increase of regulatory T cells. Arthritis Res Ther. 2020;22(1):20. doi:10.1186/s13075-020-2112-7
48. Gearhart PJ. Aging’s impact on the immune system. J Immunol. 2025;214(5):851–852. doi:10.1093/jimmun/vkaf029
49. Zhou H, Zheng Z, Fan C, Zhou Z. Mechanisms and strategies of immunosenescence effects on non-small cell lung cancer (NSCLC) treatment: a comprehensive analysis and future directions. Semin Cancer Biol. 2025;109:44–66. doi:10.1016/j.semcancer.2025.01.001
50. Yang H, Wu X, Xiao X, et al. Elucidating the causal associations and mechanisms between circulating immune cells and idiopathic pulmonary fibrosis: new insights from Mendelian randomization and transcriptomics. Front Immunol. 2025;15:1437984. doi:10.3389/fimmu.2024.1437984
51. Zhang T, Hou Z, Ding Z, Wang P, Pan X, Li X. Single cell RNA -Seq identifies cell subpopulations contributing to idiopathic pulmonary fibrosis in humans. J Cell Mol Med. 2025;29(3):e70402. doi:10.1111/jcmm.70402
52. Papaioannou I, Xu S, Denton CP, Abraham DJ, Ponticos M, Heldin C-H. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol Biol Cell. 2018;29(2):84–95. doi:10.1091/mbc.E17-06-0342
53. Prêle CM, Miles T, Pearce DR, et al. Plasma cell but not CD20-mediated B-cell depletion protects from bleomycin-induced lung fibrosis. Eur Respir J. 2022;60(5):2101469. doi:10.1183/13993003.01469-2021
54. Koether K, Besnard V, Sandig H, et al. Autoantibodies are associated with disease progression in idiopathic pulmonary fibrosis. Eur Respir J. 2023;61(5):2102381. doi:10.1183/13993003.02381-2021
55. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology. 2020;160(2):126–138. doi:10.1111/imm.13154
56. McElroy AN, Invernizzi R, Laskowska JW, et al. Candidate Role for Toll-like Receptor 3 L412F Polymorphism and Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2022;205(5):550–562. doi:10.1164/rccm.202010-3880OC
57. Qiu H, Weng D, Chen T, et al. Stimulator of interferon genes deficiency in acute exacerbation of idiopathic pulmonary fibrosis. Front Immunol. 2017;8:1756. doi:10.3389/fimmu.2017.01756
58. Yamaguchi R, Sakamoto A, Yamamoto T, et al. Surfactant protein D inhibits interleukin-12p40 production by macrophages through the SIRPα/ROCK/ERK signaling pathway. Am J Med Sci. 2017;353(6):559–567. doi:10.1016/j.amjms.2017.03.013
59. Murray PJ. Macrophage Polarization. Annual Review of Physiology. 2017;79(1):541–566. doi:10.1146/annurev-physiol-022516-034339
60. Kamiya M, Carter H, Espindola MS, et al. Immune mechanisms in fibrotic interstitial lung disease. Cell. 2024;187(14):3506–3530. doi:10.1016/j.cell.2024.05.015
61. Gibbons MA, MacKinnon AC, Ramachandran P, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184(5):569–581. doi:10.1164/rccm.201010-1719OC
62. Fabre T, Barron AM, Christensen SM, et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci Immunol. 2023;8(82):eadd8945. doi:10.1126/sciimmunol.add8945
63. Fang C, Rinke AE, Wang J, Flaherty KR, Phan SH, Liu T. B7H3 expression and significance in idiopathic pulmonary fibrosis. J Pathol. 2022;256(3):310–320. doi:10.1002/path.5838
64. Weng X, Maxwell-Warburton S, Hasib A, Ma L, Kang L. The membrane receptor CD44: novel insights into metabolism. Trends Endocrinol Metab. 2022;33(5):318–332. doi:10.1016/j.tem.2022.02.002
65. Tanaka T, Basisty N, Fantoni G, et al. Plasma proteomic biomarker signature of age predicts health and life span. Elife. 2020;9. doi:10.7554/eLife.61073
66. Adel RM, Helal H, Ahmed Fouad M, Sobhy Abd-Elhalem S. Regulation of miRNA-155-5p ameliorates NETosis in pulmonary fibrosis rat model via inhibiting its target cytokines IL-1β, TNF-α and TGF-β1. Int Immunopharmacol. 2024;127:111456. doi:10.1016/j.intimp.2023.111456
67. Gregory AD, Kliment CR, Metz HE, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. 2015;98(2):143–152. doi:10.1189/jlb.3HI1014-493R
68. Kruger P, Saffarzadeh M, Weber ANR, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015;11(3):e1004651. doi:10.1371/journal.ppat.1004651
69. Khawaja AA, Chong DLW, Sahota J, et al. Identification of a novel HIF-1α-αMβ2 integrin-NET axis in fibrotic interstitial lung disease. Front Immunol. 2020;11:2190. doi:10.3389/fimmu.2020.02190
70. Mendoza N, Casas-Recasens S, Olvera N, et al. Blood Immunophenotypes of Idiopathic Pulmonary Fibrosis: relationship with Disease Severity and Progression. Int J Mol Sci. 2023;24(18):13832. doi:10.3390/ijms241813832
71. Achaiah A, Rathnapala A, Pereira A, et al. Neutrophil lymphocyte ratio as an indicator for disease progression in idiopathic pulmonary fibrosis. BMJ Open Respir Res. 2022;9(1):e001202. doi:10.1136/bmjresp-2022-001202
72. Thrall RS, Phan SH, McCormick JR, Ward PA. The development of bleomycin-induced pulmonary fibrosis in neutrophil-depleted and complement-depleted rats. Am J Pathol. 1981;105(1):76–81.
73. Tabuena RP, Nagai S, Tsutsumi T, et al. Cell profiles of bronchoalveolar lavage fluid as prognosticators of idiopathic pulmonary fibrosis/usual interstitial pneumonia among Japanese patients. Respiration. 2005;72(5):490–498. doi:10.1159/000087673
74. Li D-Y, Li R-F, Sun D-X, Pu -D-D, Zhang Y-H. Mesenchymal stem cell therapy in pulmonary fibrosis: a meta-analysis of preclinical studies. Stem Cell Res Ther. 2021;12(1):461. doi:10.1186/s13287-021-02496-2
75. Cargnoni A, Romele P, Bonassi Signoroni P, et al. Amniotic MSCs reduce pulmonary fibrosis by hampering lung B-cell recruitment, retention, and maturation. Stem Cells Transl Med. 2020;9(9):1023–1035. doi:10.1002/sctm.20-0068
76. Li F, Han F, Li H, et al. Human placental mesenchymal stem cells of fetal origins-alleviated inflammation and fibrosis by attenuating MyD88 signaling in bleomycin-induced pulmonary fibrosis mice. Mol Immunol. 2017;90:11–21. doi:10.1016/j.molimm.2017.06.032
77. Cárdenes N, Álvarez D, Sellarés J, et al. Senescence of bone marrow-derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis. Stem Cell Res Ther. 2018;9(1):257. doi:10.1186/s13287-018-0970-6
78. Kurbanov DB, Ahangari F, Adams T, et al. Epigenetic age acceleration in idiopathic pulmonary fibrosis revealed by DNA methylation clocks. Am J Physiol Lung Cell Mol Physiol. 2025;328(3):L456–l462. doi:10.1152/ajplung.00171.2024
79. Aversa Z, Atkinson EJ, Carmona EM, et al. Biomarkers of cellular senescence in idiopathic pulmonary fibrosis. Respir Res. 2023;24(1):101. doi:10.1186/s12931-023-02403-8
80. Atanasova E, Milosevic D, Bornschlegl S, et al. Normal ex vivo mesenchymal stem cell function combined with abnormal immune profiles sets the stage for informative cell therapy trials in idiopathic pulmonary fibrosis patients. Stem Cell Res Ther. 2022;13(1):45. doi:10.1186/s13287-021-02692-0
81. Unterman A, Zhao AY, Neumark N, et al. Single-cell profiling reveals immune aberrations in progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2024;210(4):484–496. doi:10.1164/rccm.202306-0979OC
82. Kreuter M, Lee JS, Tzouvelekis A, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;204(1):74–81. doi:10.1164/rccm.202003-0669OC
83. Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE Jr. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest. 2008;133(1):226–232. doi:10.1378/chest.07-1948
84. Sawant H, Borthakur A. Disease-specific novel role of growth differentiation factor 15 in organ fibrosis. Int J Mol Sci. 2025;26(12):5713. doi:10.3390/ijms26125713
85. Tan Y, Qian B, Ma Q, Xiang K, Wang S. Identification and analysis of key immune- and inflammation-related genes in idiopathic pulmonary fibrosis. J Inflamm Res. 2025;18:1993–2009. doi:10.2147/jir.S489210
86. De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. The Journal of Rheumatology. 2013;40(4):435–446. doi:10.3899/jrheum.120725
87. Oldham JM, Huang Y, Bose S, et al. Proteomic biomarkers of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2024;209(9):1111–1120. doi:10.1164/rccm.202301-0117OC
88. Monteleone G, Passantino L, Simonetti J, et al. A simple ratio in a complex disease: exploring the neutrophil-to-lymphocyte ratio in idiopathic pulmonary fibrosis. J Clin Med. 2025;14(14):5100. doi:10.3390/jcm14145100
89. Kreuter M, Lee JS, Tzouvelekis A, et al. Modified blood cell GAP model as a prognostic biomarker in idiopathic pulmonary fibrosis. ERJ Open Res. 2024;10(4):00666–2023. doi:10.1183/23120541.00666-2023
90. Jaeckel E, Friedman SL, Hudecek M, Protzer U. Chimeric antigen receptor (CAR) T-cell therapy: engineering immune cells to treat liver diseases. J Hepatol. 2025;83:1156–1171. doi:10.1016/j.jhep.2025.06.007
91. Yang D, Sun B, Li S, et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci Transl Med. 2023;15(709):eadd1951. doi:10.1126/scitranslmed.add1951
92. Bharath LP, Agrawal M, McCambridge G, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020;32(1):44–55.e6. doi:10.1016/j.cmet.2020.04.015
93. Shim HS, Iaconelli J, Shang X, et al. TERT activation targets DNA methylation and multiple aging hallmarks. Cell. 2024;187(15):4030–4042.e13. doi:10.1016/j.cell.2024.05.048
94. Nan J, Du Y, Chen X, et al. TPCA-1 is a direct dual inhibitor of STAT3 and NF-κB and regresses mutant EGFR-associated human non–small cell lung cancers. Mol Cancer Ther. 2014;13(3):617–629. doi:10.1158/1535-7163.Mct-13-0464
95. Li Y, Chen R, Wu J, et al. Salvianolic acid B protects against pulmonary fibrosis by attenuating stimulating protein 1-mediated macrophage and alveolar type 2 cell senescence. Phytother Res. 2024;38(2):620–635. doi:10.1002/ptr.8070
96. Wang H, Zhang S, Zhai L, et al. Ginsenoside extract from ginseng extends lifespan and health span in Caenorhabditis elegans. Food Funct. 2021;12(15):6793–6808. doi:10.1039/d1fo00576f
97. Zhao Y, Sun Y, Wang G, Ge S, Liu H. Dendrobium officinale polysaccharides protect against MNNG-induced PLGC in rats via activating the NRF2 and antioxidant enzymes HO-1 and NQO-1. Oxid Med Cell Longev. 2019;2019:9310245. doi:10.1155/2019/9310245
98. Yang Y, Lv M, Xu Q, Wang X, Fang Z. Extracellular vesicles in idiopathic pulmonary fibrosis: pathogenesis, biomarkers and innovative therapeutic strategies. Int J Nanomedicine. 2024;19:12593–12614. doi:10.2147/ijn.S491335
99. Dong S, Fang H, Zhu J, et al. Inhalable siRNA targeting IL-11 nanoparticles significantly inhibit bleomycin-induced pulmonary fibrosis. ACS Nano. 2025;19(2):2742–2758. doi:10.1021/acsnano.4c15130
100. Keir GJ, Maher TM, Ming D, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. Apr. 2014;19(3):353–359. doi:10.1111/resp.12214
101. Wygrecka M, Dahal BK, Kosanovic D, et al. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: role of transmembrane SCF and the PAR-2/PKC-α/Raf-1/p44/42 signaling pathway. Am J Pathol. 2013;182(6):2094–2108. doi:10.1016/j.ajpath.2013.02.013
102. Pommerolle L, Beltramo G, Biziorek L, et al. CD206 + macrophages are relevant non-invasive imaging biomarkers and therapeutic targets in experimental lung fibrosis. Thorax. 2024;79(12):1124–1135. doi:10.1136/thorax-2023-221168
103. Machado MG, Patente TA, Rouillé Y, et al. Acetate improves the killing of streptococcus pneumoniae by alveolar macrophages via NLRP3 inflammasome and glycolysis-HIF-1α axis. Front Immunol. 2022;13:773261. doi:10.3389/fimmu.2022.773261
104. Park HJ, Yu D, Hong S-T, et al. Bifidobacterium longum RAPO attenuates dermal and pulmonary fibrosis in a mouse model of systemic sclerosis through macrophage modulation and growth of short-chain fatty acid producers. Immune Netw. 2024;24(6):e41. doi:10.4110/in.2024.24.e41
105. Zhao R, Zhang W, Ma C, et al. Immunomodulatory function of vitamin D and its role in autoimmune thyroid disease. Front Immunol. 2021;12:574967. doi:10.3389/fimmu.2021.574967
106. Godoy MCX, Monteiro GA, Moraes BH, Macedo JA, Gonçalves GMS, Gambero A. Addition of polyphenols to drugs: the potential of controlling “inflammaging” and fibrosis in human senescent lung fibroblasts in vitro. Int J Mol Sci. 2024;25(13):7163. doi:10.3390/ijms25137163
107. Ma S, Sun S, Geng L, et al. Caloric restriction reprograms the single-cell transcriptional landscape of rattus norvegicus aging. Cell. 2020;180(5):984–1001.e22. doi:10.1016/j.cell.2020.02.008
108. Chastin SFM, Abaraogu U, Bourgois JG, et al. Effects of regular physical activity on the immune system, vaccination and risk of community-acquired infectious disease in the general population: systematic review and meta-analysis. Sports Med. 2021;51(8):1673–1686. doi:10.1007/s40279-021-01466-1
109. Cristofori F, Dargenio VN, Dargenio C, Miniello VL, Barone M, Francavilla R. Anti-inflammatory and immunomodulatory effects of probiotics in gut inflammation: a door to the body. Front Immunol. 2021;12:578386. doi:10.3389/fimmu.2021.578386
110. Foster PS, Tay HL, Oliver BG. Deficiency in the zinc transporter ZIP8 impairs epithelia renewal and enhances lung fibrosis. J Clin Invest. 2022;132(11). doi:10.1172/jci160595
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Improvement in Cutaneous Conditions Can Benefit Some Health Conditions in the Elderly
Yang B, Man MQ
Clinical Interventions in Aging 2023, 18:2031-2040
Published Date: 1 December 2023
Targeting Cellular Senescence for Healthy Aging: Advances in Senolytics and Senomorphics

Alum EU, Izah SC, Uti DE, Ugwu OPC, Betiang PA, Basajja M, Ejemot-Nwadiaro RI
Drug Design, Development and Therapy 2025, 19:8489-8522
Published Date: 19 September 2025
OPG and TNFR1 as Potential Biomarkers of Inflammation in Older Adults with Acute COVID-19 and Indicators of Frailty in Post-COVID-19: A Pilot Study
Ramon-Luing LA, Flores-Gonzalez J, Cataneo-Piña DJ, Falfán-Valencia R, Pérez-Rubio G, Buendia-Roldan I, Selman M, Chavez-Galan L
Journal of Inflammation Research 2025, 18:14841-14855
Published Date: 25 October 2025