")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Immunomodulatory effects of 17-O-acetylacuminolide in RAW264.7 cells and HUVECs: involvement of MAPK and NF-κB pathways
Authors Achoui M, Heyninck K, Looi CY, Mustafa AM, Haegeman G, Mustafa M
Received 31 May 2014
Accepted for publication 14 July 2014
Published 20 October 2014 Volume 2014:8 Pages 1993—2007
DOI https://doi.org/10.2147/DDDT.S68659
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mouna Achoui,1 Karen Heyninck,2 Chung Yeng Looi,1 Ali Mohd Mustafa,1 Guy Haegeman,2 Mohd Rais Mustafa1
1Department of Pharmacology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia; 2Department of Physiology, Laboratory of Eukaryotic Gene Expression and Signal Transduction, Ghent, Belgium
Abstract: The terpenoid 17-O-acetylacuminolide (AA) was shown to inhibit the production of several inflammatory mediators. However, the mechanisms by which this compound elicited its anti-inflammatory activity remain to be elucidated. In this study, we analyzed the effects of AA on inflammatory gene expression in two different cell types with primordial importance in the inflammatory processes – endothelial cells and macrophages. In human umbilical vein endothelial cells, AA inhibited the expression of inflammatory proteins including the adhesion molecules intercellular adhesion molecule 1; vascular cell adhesion molecule 1; and E-selectin, as well as the release of the chemokine interleukin-8. Additionally, AA hindered the formation of capillary-like tubes in an in vitro model of angiogenesis. AA’s effects in endothelial cells can be attributed at least in part to AA’s inhibition of tumor necrosis factor alpha-induced nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-κB)’s translocation. Also, in lipopolysaccharide-stimulated macrophage-like RAW264.7 cells, AA was able to downregulate the expression of the genes cyclooxygenase 2, inducible nitric oxide synthase, interleukin-6, and chemokine (C-C motif) ligand 2. Moreover, AA inhibited the phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor-alpha (IκBα), IκB kinase (IKK), and the mitogen-activated protein kinases JNK, ERK, and p38. In conclusion, the present results further support the anti-inflammatory potential of AA in different models of inflammation.
Keywords: 17-O-acetylacuminolide, angiogenesis, HUVEC, MAPK, NF-κB
Introduction
Of particular importance during an inflammatory response is the vascular endothelium. By increasing endothelial cells’ permeability, blood vessels are responsible for the transportation and dissemination of monocytes and monocyte-derived tissue macrophages, which play an essential role in both acute and chronic inflammation.1 Moreover, activation of the endothelium with stimuli, such as lipopolysaccharide (LPS) and cytokines, leads to the expression of chemokines, such as interleukin (IL)-8,2 as well as adhesion molecules,3 including intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.4 The expression of these molecules plays an important role in adhesion and extravasation of leukocytes in an inflammatory response,5,6 as well as in the attachment and metastasis of cancer cells.7
One of the main mediators in inflammatory responses is the transcription factor nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-κB). NF-κB not only influences the expression of cytokines and inducible inflammatory enzymes, but it also modulates the expression of survival genes, chemokines, adhesion molecules, and angiogenesis.8 Under physiological conditions, NF-κB is found in the cytoplasm, where it is bound to a protein called the inhibitor of NF-κB (IκBα). The IκBα inhibits the translocation of NF-κB into the nucleus. However, stimulation by a wide range of proinflammatory mediators including cytokines (tumor necrosis factor alpha [TNF-α] and IL-1) and infections with bacteria or viruses leads to NF-κB activation.9 This is achieved via the phosphorylation of IκBα by the IκB kinase complex (IKK). The IKK complex is composed of the catalytic IKKα and IKKβ subunits and a regulatory protein termed IKKγ. This complex is activated via cascades of signals depending on the initial stimulus.10 The LPS stimulation will activate toll-like receptor 4 signaling, leading to IKK complex activation. Once the IKK complex phosphorylates IκBα, the latter is eventually degraded by the proteasome, and NF-κB is free to translocate to the nucleus, where it carries out its transcriptional activities.11
In addition to the NF-κB activation, LPS can also activate mitogen-activated protein kinases (MAPKs).12 The MAPKs of interest in this study are three subgroups: extracellular signal-regulated kinases (ERK1/2); c-jun N-terminal or stress-activated protein kinase (JNK); and p38 protein kinase. These kinases play key roles in regulating numerous cellular functions, including cell growth, apoptosis, and cellular responses to inflammation.13
Several studies explored the various biological activities of terpenoids,14 a class of secondary metabolites to which the compound 17-O-acetylacuminolide (AA) belongs.15 We have recently described the isolation and some anti-inflammatory effects of this diterpene labdane.16 The current study looks into additional anti-inflammatory effects of this compound, as well as possible mechanisms of action involved. We explored its effects on the expression of adhesion molecules, cytokines, and other NF-κB target genes in TNF-α-stimulated human umbilical vein endothelial cells (HUVECs) and the LPS-induced macrophage cell line RAW264.7.
Additionally, due to the role NF-κB plays in angiogenesis,17,18 AA’s effect on tube formation was tested. The cytotoxicity of this compound was investigated in parallel with the different assays to avoid interference of cell death. AA was also tested in the human hepatic cell line (WRL-68) as a preliminary indicator of compound safety.19,20
Materials and methods
Cell lines and reagents
Cell lines RAW264.7 and WRL-68 were obtained from American Type Culture Collection (ATCC), Manassas, VA, USA. HUVEC and the endothelial cell growth medium (EGM™-2) Bulletkit™ containing basal medium, supplements, and growth factors were obtained from Lonza Group Ltd. (Basel, Switzerland). Suramin sodium salt (with a purity of ≥95%), curcumin (≥94%), BAY11-7085 (≥98%) and human TNF-α were obtained from Sigma-Aldrich Co., St Louis, MO, USA. The NF-κB translocation and cytoskeletal rearrangement kits were from Thermo Fisher Scientific (Waltham, MA, USA). For electrophoretic mobility shift assay (EMSA) reagents, Poly-deoxy-inosinic-deoxy-cytidylic acid (Poly[d(I-C)]) was from Hoffman-La Roche Ltd. (Basel, Switzerland), and [α-32P] deoxycytidine 5’-triphosphate (dCTP) was from PerkinElmer Inc. (Waltham, MA, USA).
The Nuclear Extraction Kit was from Cayman Chemical Company (Ann Arbor, MI, USA). Monoclonal antibodies for β-actin, phosphorylated NFκB inhibitor (P-IκBα), IκBα, mouse immunoglobulin G (IgG), phosphorylated IKK (P-IKK)α/β (Ser-176)-R, rabbit IgG, and the peroxidase-conjugated secondary antibodies goat antimouse IgG–HRP, and goat antirabbit IgG-horseradish peroxidase (HRP) were from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The Amersham enhanced chemiluminescence (ECL™) Western Blotting Detection Reagent was from Amersham Biosciences Corp. (GE Healthcare UK Ltd., Little Chalfont, UK). The Mitogen-activated protein kinase kinase (MEK), MAPK, and p38 inhibitors U0126 (with a purity of 95%), PD98059 (>95%), SB203580 (98%), ICAM-1, VCAM-1, and E-selectin human enzyme-linked immunosorbent assay (ELISA) kits were obtained from Thermo Fisher Scientific (Waltham, MA, USA). The Human CXCL8/IL-8 Quantikine ELISA Kit was obtained from R&D Systems, Inc., (Minneapolis, MN, USA).
The AA (with a purity of >98.5% as determined by high-performance liquid chromatography) was isolated and purified from Neouvaria foetida in our labs, as described previously,16 and was dissolved in dimethyl sulfoxide (DMSO) and diluted in Dulbecco’s Modified Eagle’s Medium or phosphate buffered saline (PBS) before it was used for testing its biological activity. BD Matrigel™ Matrix (phenol red-free) was from BD Biosciences (San Jose, CA, USA). The RNeasy Mini Kit for RNA extraction was from Qiagen GmbH (Hilden, Germany). For the quantitative polymerase chain reaction (PCR) analysis, oligo-dT and ribonuclease inhibitor were obtained from New England Biolabs (Ipswich, MA, USA). The Moloney murine leukemia virus (MMLV) reverse transcriptase, MMLV reverse transcriptase buffer (5×), and deoxyribonucleotides were from Promega Corporation (Madison, WI, USA), murine primers were from Sigma-Aldrich Co., and Thermo Fisher Scientific, and SYBR® Green was from Bio-Rad Laboratories Inc., Hercules, CA, USA.
Cell culture
Cell lines were maintained in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum (FBS). HUVECs were maintained as recommended in complete endothelial growth medium with 2% FBS. The cells were cultured every 2–3 days using standard aseptic techniques.
Cell viability and cytotoxicity
The viability of the cells was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay as described,21 with some modifications. WRL-68 cells were seeded at 10,000 cells/well overnight and were either left untreated (UT) or were treated with AA (0.08–50 μg/mL) for either 24 or 72 hours. After MTT treatment, the optical density of the samples was measured and compared to that of the negative control to obtain the percentage viability as follows:
Cell viability (%) = [(OD570 (sample)/OD570 (negative control)) ×100], | (1) |
where OD570 is the optical density or absorbance at 570 nm.
Real-time quantitative PCR analysis of selected murine inflammatory genes: cell treatment and RNA isolation
For quantitative PCR analysis, the protocol was as previously described,22 with some modifications. The RAW264.7 cells were seeded overnight at 1×106 cells/well in 6-well plates. The cells were then either left UT or were pretreated with the indicated concentrations of AA for 30 minutes. Subsequently, the cells were either left with no LPS treatment or were stimulated with 1 μg/mL of LPS for either 1 hour (IL-6 and chemokine [C-C motif] ligand 2 [CCL-2]) or 4 hours (cyclooxygenase 2 [COX-2] and inducible nitric oxide synthase [iNOS]). The cells were washed with cold PBS, and total cellular RNA was isolated using TRIzol® (Thermo Fisher Scientific). The RNA quantity and quality were assessed using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific), and the RNA was stored at −70°C until use.
Reverse transcription
In addition, 500 ng of RNA sample was mixed with 2.5 mM of oligo(deoxy-thymine [dT]) and brought to a final volume of 16.5 μL with nuclease-free water per reaction. The samples were heated for 5 minutes at 70°C and were subsequently cooled on ice. These mixtures were further supplemented with MMLV-reverse transcriptase and incubated at 37°C for 1 hour, followed by heating to 70°C for 10 minutes. The prepared complementary DNA was stored at −20°C until analysis.
Real-time quantitative PCR with SYBR® Green
SYBR® Green, complementary DNA samples, and forward and reverse primers of the gene of interest were used for quantitative PCR analyses to detect gene expression of CCL-2, IL-6, iNOS, COX-2, and cyclophilin (as a housekeeping gene), using the Roche LightCycler® 480 II instrument and LightCycler® 480 (Hoffman-La Roche Ltd.) Software Version 5. The protocol used was SYBR® fast-run protocol. The sequences of the primers used in this study are listed in Table 1.
 | Table 1 Sequences of the primers used for quantitative PCR analysis |
Nuclear extraction and EMSA
RAW264.7 macrophages cells were plated in 6-well plates (1×106 cells/well). The cells were either pretreated with 5 μg/mL AA for 30 minutes or with complete medium. The cells were then either stimulated with 1 μg/mL LPS for 30 minutes, or they were left UT. Nuclear extracts were prepared and frozen at −70°C until use.
The EMSA protocol conducted was as previously described23 with some minor modifications. Also, 10 μg of the nuclear extract proteins were mixed with 12 μL of a mixture containing binding buffer, poly-deoxy-inosinic-deoxy-cytidylic acid (Poly[d(I-C)]), bovine serum albumin (BSA), and an NFκB-specific 32P-labeled oligonucleotide comprising the sequence 5′-AGCTATGTGGGTTTTCCC ATGAGC-3′, in which the single IL-6 promoter-derived NFκB motif is bold and underlined. The mixture was incubated for 15 minutes at room temperature and then loaded onto native 6% polyacrylamide gel and run in tris(hydroxymethyl)aminomethane (Tris) borate-ethylene-diamine-tetraacetic acid buffer (pH 8). After electrophoresis, the gels were fixed in triacetic acid solution for 5 minutes and vacuum-dried for 2 hours at 70°C and exposed to a Phosphor imager screen (Amersham Biosciences, GE Healthcare UK Ltd.).
Sodium dodecyl sulfate – polyacrylamide gel electrophoresis and Western blotting
RAW264.7 cells were grown to confluence and for MAPK analysis were then starved in medium containing 2% FBS for 18 hours. Cells were pretreated with either AA (1.25–10 μg/mL) or the medium containing the corresponding DMSO concentrations or the MAPK inhibitors U0126, PD98059, and SB203580 at 50 μM for 1 hour. The cells were then stimulated with 1 μg/mL LPS for 15 minutes or were left unstimulated. Also, 20 μg of the cytoplasmic lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto nitrocellulose membranes.
The membrane was blocked overnight in a 5% milk solution in PBS with 0.05% Tween 20 (PBST) at 4°C. The membranes were then washed with PBST and incubated with the primary antibodies of interest for 2 hours at room temperature. After washing with the blocking buffer, the membranes were incubated with the appropriate secondary antibodies (1:2,000) for 1 hour. Depending on the secondary antibody used, the membranes were either developed using the ODYSSEY® CLx Imager (LI-COR, Inc., Lincoln, NE, USA) or the Amersham ECL™ Western Blotting Detection Reagent, according to the manufacturer’s protocol. The infrared-dye fluorescence was detected using the Odyssey V3 software (LI-COR, Inc.), while chemifluorescence was detected using the BioSpectrum® Imaging System from UVP, LLC (Upland, CA, USA). Where necessary, the membranes were stripped, blocked, and reprobed with the appropriate primary antibodies of the unphosphorylated forms of the proteins of interest.
ELISA assays
HUVECs were seeded overnight and were then either left UT or stimulated with the indicated concentrations of AA in combination with 1 μg/mL of LPS for 4 hours or 5 ng/mL human TNF-α overnight. As a positive control, the cells were also treated with 10 μM of the positive control BAY 11-7085.
For all assays, cell supernatant was aspirated and frozen at −80°C until analysis, while cell viability was evaluated with the MTT assay as described in “Cell viability and cytotoxicity”. The IL-8 concentrations were determined using the Human IL-8 ELISA kit, and the Human ELISA kits for the respective adhesion molecules were used to measure the concentrations of released adhesion molecules.
Tube formation assay
Matrigel was thawed overnight at 4°C, and 50 μL of the solution was added to a 96-well plate. The gel was allowed to polymerize at 37°C for 30 minutes. HUVECs were seeded at 20,000 cells/well on either a regular 96-well plate for testing viability, or on a clear-bottom black plate coated with matrigel. The cells were left UT in medium, or they were treated with the positive control suramin (50 μM) or indicated concentrations of AA, for 16–18 hours.24,25 Tube formation was evaluated using the cytoskeletal rearrangement kit from Cellomics (Thermo Fisher Scientific), according to the manufacturer’s protocols of fixing and staining. The Tube Formation BioApplication (Thermo Fisher Scientific) was used to quantify tubes formed. Viability was determined using MTT, as described earlier.
NF-κB translocation assay
HUVECs were seeded overnight in a 96-well plate at 6,000 cells/well. The cells were either pretreated for 1 hour with either the positive control curcumin (6.25 μg/mL), the different concentrations of AA, or were left UT. The cells were stimulated with 2 ng/mL TNF-α for 30 minutes. The medium was discarded and cells were fixed and stained using Cellomics® NF-κB activation kit from Thermo Fisher Scientific, according to the manufacturer’s instructions. The assay plate was evaluated on the ArrayScan high content screening (HCS) Reader. The Cytoplasm to Nucleus Translocation Bio-Application software (Thermo Fisher Scientific) was used to calculate the ratio of cytoplasmic and nuclear NF-κB intensity.26 The ratios were then compared among stimulated, treated, and UT cells.
Statistical analyses
The assays were conducted in at least three separate experiments, unless otherwise specified. Data are expressed as the mean ± standard deviation. Data were analyzed using GraphPad Prism® statistical software (version 4; GraphPad Software Inc., La Jolla, CA, USA) for one-way analysis of variance with Tukey’s post hoc test or Student’s t-test, as indicated. Differences were considered significant at P<0.05. The half maximal inhibitory concentration (IC50) was calculated using a sigmoidal dose-response (variable slope) equation under nonlinear regression (curve fit) utilizing the same software. Where inhibition is calculated, values from UT, unstimulated cells were considered as the maximum inhibition (100%). Inhibition is calculated as follows:
% inhibition =100× [([control] − [sample])/[control]]. | (2) |
Results
We have previously demonstrated that treating RAW264.7 cells with AA for 24 hours had no significant effect on their viability with concentrations ranging from 0.01–5 μg/mL.16 For short-duration assays (<2 hours) required for the evaluation of NF-κB and MAPKs activation, moderately high doses were used for which no significant toxicity was observed. Except for the highest concentration tested of 50 μg/mL, AA did not elicit a significant cytotoxic effect when tested at doses ranging from 0.08–5 μg/mL after 24 and 72 hours’ treatment in the human hepatic cell line WRL-68 (Figure 1).
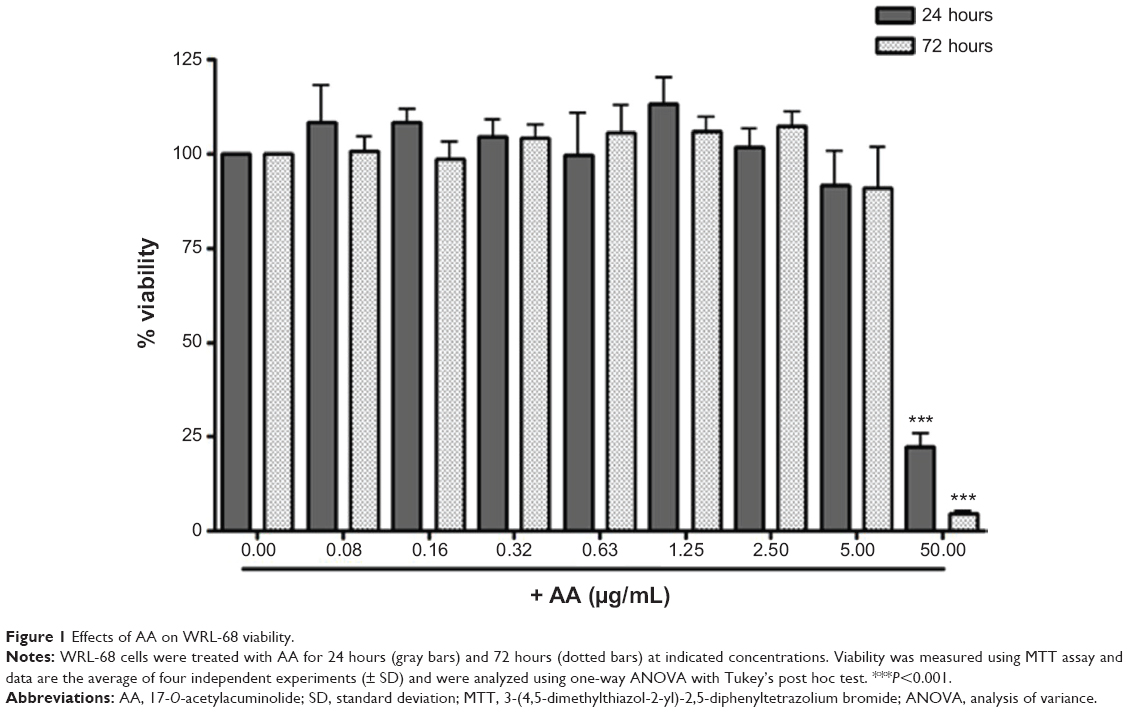 | Figure 1 Effects of AA on WRL-68 viability. |
Triggering of endothelial cells and macrophages with proinflammatory stimuli leads to profound induction of a wide range of NF-κB-controlled genes, including adhesion molecules, cytokines, chemokines, as well as proinflammatory enzymes. The anti-inflammatory effect of AA in RAW264.7 cells was analyzed by exploring its effects on the LPS-induced transcription of NF-κB regulated genes. Treatment of cells with LPS for 1 hour led to a significant increase in the expression of the IL-6 gene and the monocyte chemotactic protein 1 gene CCL-2; whereas, a significant increase in iNOS and COX-2 gene expression was observed following 4 hours of LPS stimulation. Additionally, AA at concentrations of 1.25 and 2.5 μg/mL significantly inhibited the LPS-induced expression of all four genes (Figure 2).
 | Figure 2 Inhibitory effect of AA on LPS-induced expression of indicated genes in RAW264.7 cells. |
Activation of the transcription factor NF-κB is tightly regulated and requires several steps of protein–protein interactions and phosphorylation events finally leading to the nuclear translocation of NF-κB from the cytoplasm to the nucleus. There, NF-κB will bind to its specific recognition sequences in the promoter of NF-κB-regulated genes. We analyzed the effect of AA on DNA binding and the nuclear translocation of NF-κB. The EMSA analysis indicated enhanced binding to the NF-κB DNA consensus sequence in the LPS-treated nuclear extracts of RAW264.7 cells compared to UT cells (Figure 3). Pretreatment of cells with vehicle (DMSO) did not affect NF-κB DNA binding. On the other hand, pretreatment with AA at 5 μg/mL significantly suppressed the NF-κB complex band intensity (Figure 3).
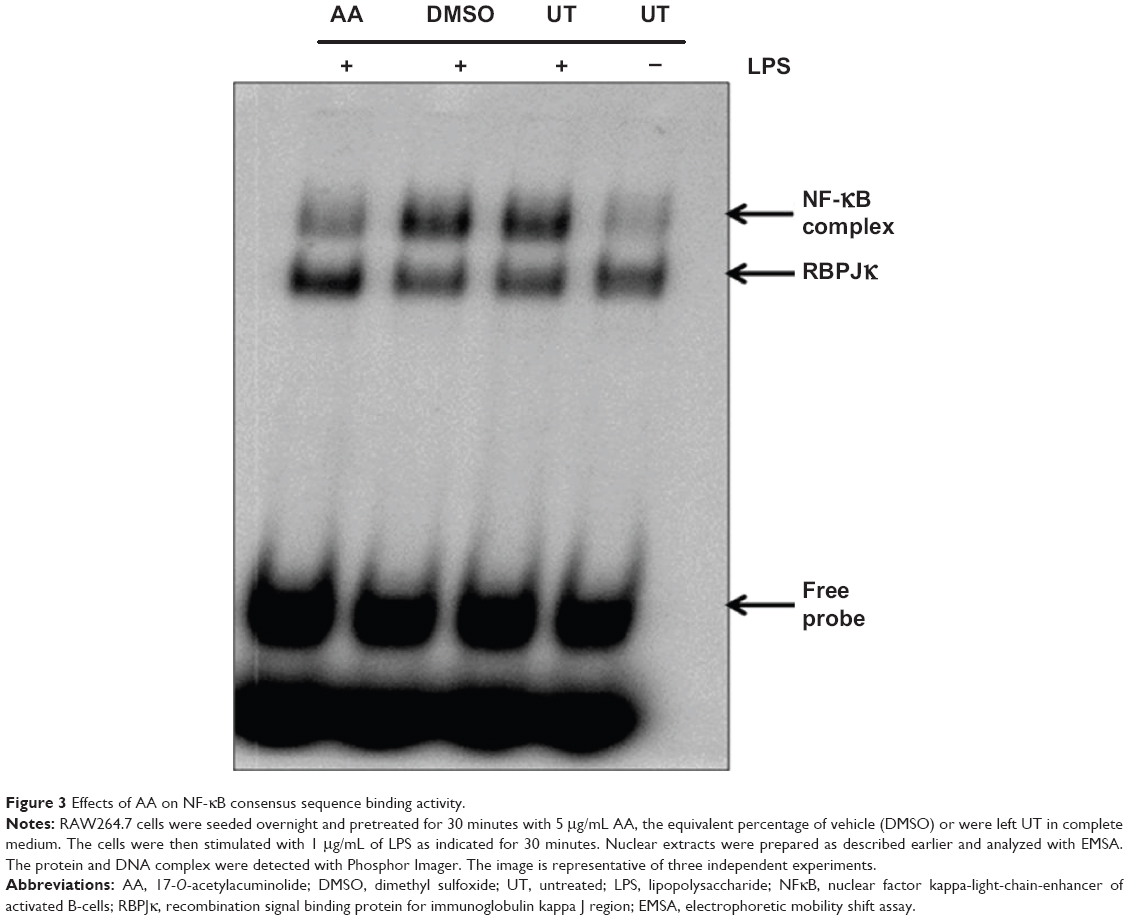 | Figure 3 Effects of AA on NF-κB consensus sequence binding activity. |
Phosphorylation and activation of the IKK complex followed by phosphorylation and degradation of IκBα precedes the nuclear translocation of NF-κB. We analyzed the effects of AA on these events to further determine the mechanism of action by which AA inhibited NF-κB translocation. As depicted in Figure 4, stimulation with LPS for 15 minutes caused the degradation and phosphorylation of the IκBα protein (Figure 4A and B, respectively). Moreover, increased phosphorylation of IKKα/β was observed in cells stimulated with LPS, and pretreatment with AA was able to significantly reduce this effect of LPS (Figure 4C).
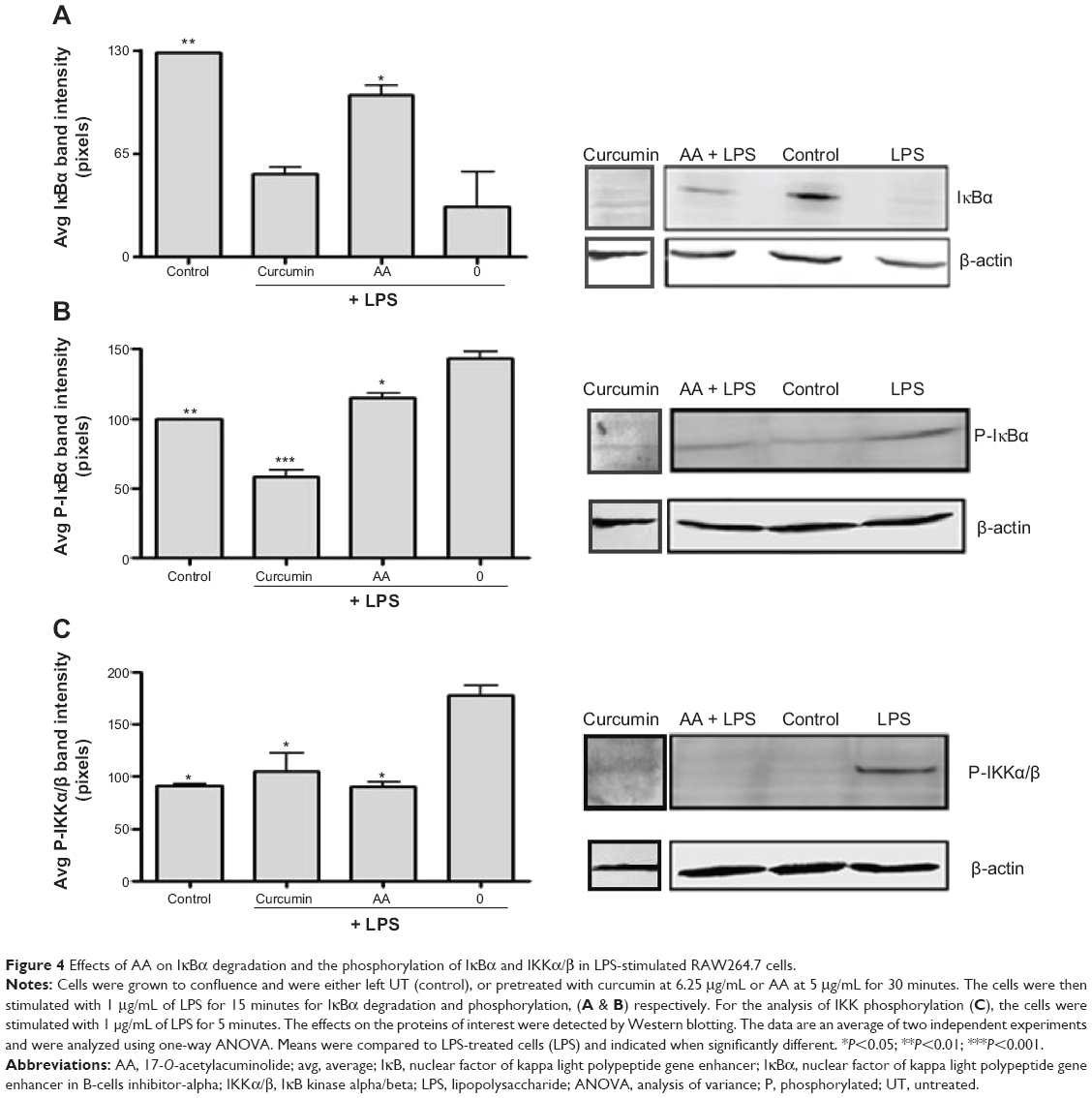 | Figure 4 Effects of AA on IκBα degradation and the phosphorylation of IκBα and IKKα/β in LPS-stimulated RAW264.7 cells. |
Because the MAPKs (p38, JNK, and ERK1/2) play essential roles in cell signaling pathways that regulate inflammation, the effects of AA on the activation of MAPKs were evaluated using Western blotting. The stimulation of RAW264.7 cells with LPS for 30 minutes caused prominent phosphorylation of all MAPKs as a mark for their activation, as manifested by the increased band intensity in vehicle pretreated and UT cells (0 μg/mL).
However, AA pretreatment (1.25–5 μg/mL) decreased LPS-induced MAPKs phosphorylation, as manifested by the lower band intensities of the phosphorylated form of the MAPKs with no effect on the total expression level of these MAPKs (Figures 5 and 6). On the other hand, treatment of cells with AA alone at high concentrations caused significant phosphorylation of JNK and p38, but it had no effect on ERK activation (Figure 7).
 | Figure 5 Effects of different doses of AA pretreatment on LPS-induced MAPKs phosphorylation. |
 | Figure 6 Quantified effects of different AA concentrations on LPS-induced MAPKs phosphorylation in RAW264.7 cells. |
 | Figure 7 Effects of AA alone on MAPKs phosphorylation. |
To gain further insight on the anti-inflammatory action of AA, we analyzed the TNF-α- or LPS-mediated release of adhesion molecules ICAM-1, VCAM-1, and E-selectin, as well as the IL-8 in endothelial HUVEC cells by specific ELISA. Pretreatment with AA significantly reduced the TNF-α-induced expression of these molecules without affecting the viability. Regulation of protein expression with no effect on cell viability was observed at different concentrations of AA: 0.01–0.32 μg/mL for ICAM-1 (Figure 8); 0.005–1.25 μg/mL for VCAM-1; and 0.01–0.08 μg/mL for E-selectin (Figure 9A and B). Moreover, LPS induced IL-8 production was significantly inhibited by AA under conditions where no effect on cell viability can be detected (Figure 10).
 | Figure 8 Effects of AA on HUVEC viability and soluble ICAM-1 inhibition. |
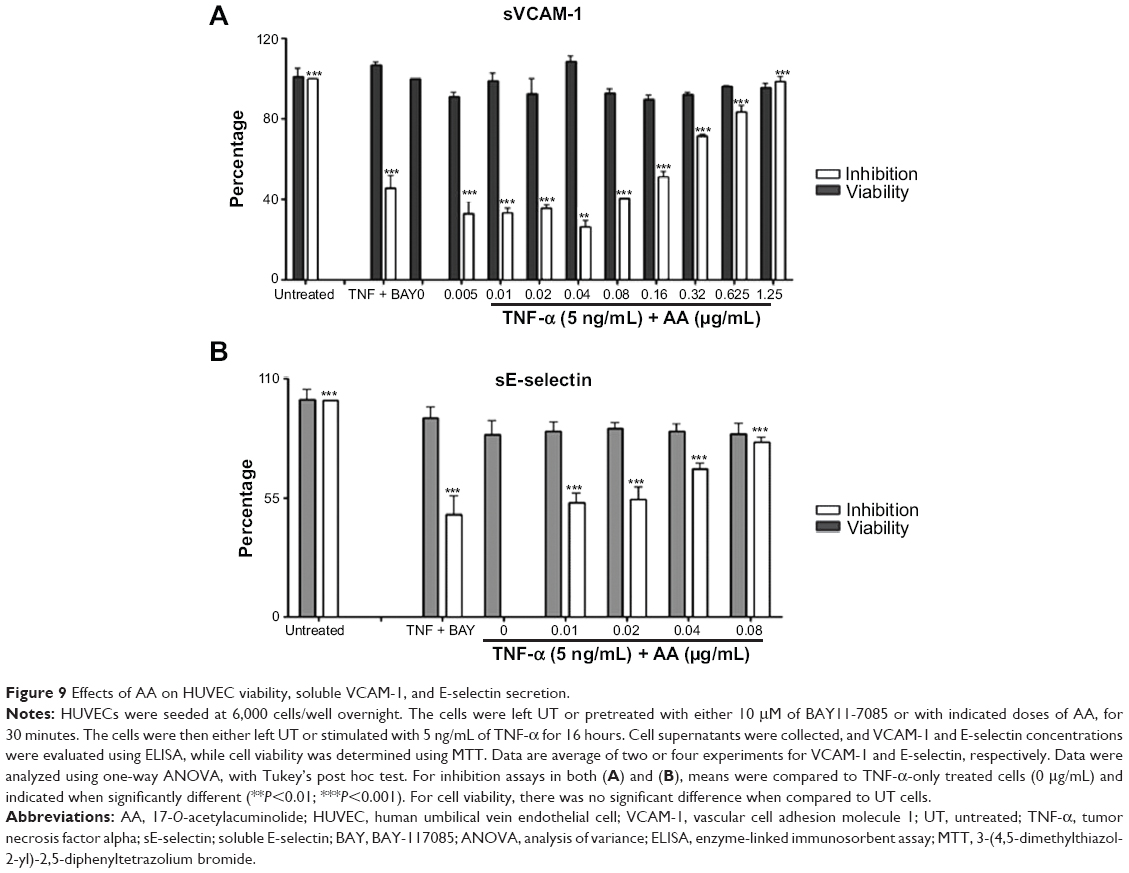 | Figure 9 Effects of AA on HUVEC viability, soluble VCAM-1, and E-selectin secretion. |
 | Figure 10 Effects of AA on IL-8 production and viability of HUVECs. |
Angiogenesis, the formation of new blood vessels from existing blood vessels, underlies the pathophysiology of several chronic inflammatory diseases, such as rheumatoid arthritis and atherosclerosis. To further elucidate the anti-inflammatory capacity of AA, we evaluated its effects on the endothelial tube formation, in an in vitro model of angiogenesis. HUVECs were grown overnight on matrigel to form a capillary-like network of tubes.
Treatment with AA at indicated concentrations significantly inhibited tube formation (Figure 11A). While high concentrations of 5 μg/mL and 10 μg/mL AA were cytotoxic to the cells when tested in a viability assay, the lowest concentration tested (2.5 μg/mL) had no effect on cell viability and effectively inhibited tube formation (Figure 11B and C). Since NF-κB activation is involved with both cytokines’ production and angiogenesis, we performed immunofluorescence staining for the NF-κB subunit p65. This analysis demonstrated a 1.8-fold increase in ratio nucleus:cytoplasm levels of NF-κB in TNF-α-stimulated HUVEC cells. AA pretreatment significantly reduced NF-κB translocation with an IC50 of 2.8 μg/mL (Figure 12).
 | Figure 11 Effects of AA on in vitro angiogenesis. |
 | Figure 12 Inhibitory effect of AA on the increase of nuclear NF-κB intensity in TNF-α-stimulated HUVECs. |
Discussion
This study looked into the various immunological effects of AA. Due to the fact that this compound was initially characterized as a cytotoxic compound,15 it was particularly important to look into its effects on cell viability to avoid attributing the anti-inflammatory effects to direct cellular toxicity. Moreover, by testing the compound in the human hepatic cell line,27 WRL-68, preliminary conclusions about the safety of the compound may be deduced.19,20 The compound seemed to be relatively safe when tested in WRL-68, with no significant toxic effects observed. Additionally, the compound’s cytotoxicity was assessed simultaneously with the in vitro inflammatory cell models used, and only noncytotoxic concentrations were subsequently evaluated.
In addition to the expression of cytokines and inducible inflammatory enzymes, the transcription factor NF-κB modulates the expression of survival genes, chemokines, and adhesion molecules.8 Therefore, NF-κB plays an important role in several processes including inflammation, angiogenesis, and carcinogenesis.28 We have previously demonstrated that AA inhibited the production of several inflammatory cytokines, both in vitro and in vivo.16
Looking into the additional anti-inflammatory effects of AA revealed its ability to inhibit IL-6 messenger ribonucleic acid, which was significantly induced upon LPS stimulation in macrophage-like RAW264.7 cells. The same was true for the messenger ribonucleic acid of the inflammatory chemokine monocyte chemotactic protein 1, CCL-2. In addition to downregulating several inflammatory cytokines, AA dose-dependently inhibited the transcription of the inducible inflammatory enzymes iNOS and COX-2, both of which play important roles in inflammation and are targets of several anti-inflammatory drugs.29,30
Due to the fact that the expression of COX-2, iNOS,31 and proinflammatory cytokines and mediators32 is regulated, at least in part, by the transcription factor NF-κB, the role AA plays in inhibiting the translocation of NF-κB and its DNA-binding were explored. The EMSA analysis confirmed the inhibition of NF-κB activation as the compound inhibited binding of NF-κB to its DNA recognition sequence. The inhibition of NF-κB/DNA binding indicates either no or little NF-κB translocation to the nucleus, or an impaired binding of NF-κB to its binding site on the DNA sequence, both of which would inhibit NF-κB-mediated transcriptional activity. To delineate the mechanism of action by which AA inhibited the NF-κB activation, the effects of the compound on upstream kinases were explored. The AA significantly inhibited the LPS-induced phosphorylation and degradation of IκBα, hence sequestering NF-κB in the cytoplasm.33 The mechanism of action was further elucidated by revealing the inhibitory effect of AA on LPS-induced IKKα/β phosphorylation, which is required for IκBα phosphorylation, and ultimately NF-κB activation.34 Cysteine 179 (Cys179) of the enzyme IKKβ plays a critical role in enzyme activation by promoting the phosphorylation of activation-loop serines (Ser-177 and Ser-181).35 Cys179 in the activation loop of IKKβ is known to be the target site for IKK inhibitors, including several terpenoids.36–38 It was shown previously that the Michael acceptor groups, such as that present in AA, can attack critical cysteines in target proteins.14 This presents a possible mechanism by which AA inhibits the IKK complex and exerts its anti-inflammatory effects.
In addition to NF-κB activation, LPS stimulation was also shown to activate MAPKs.39 MAPKs are involved in the inflammatory response mainly by mediating gene expression and are, in fact, involved in regulating the activity of NF-κB40 and activator protein 1 (AP-1).41
In this study, protein analysis indicated for all three MAPKs that their activation by LPS can be inhibited by AA at concentrations of 1.25–5 μg/mL. However, although low concentrations of AA inhibited LPS-induced MAPK (JNK, p38, and ERK1/2) activation, higher doses of AA either alone or in combination with LPS seemed to activate JNK and p38 kinases. This may be due to the fact that the treatment with the compound is perceived as a cellular stress signal that can activate MAPKs.
Previously, the activation of JNK and p38 was shown to have an inhibitory effect on ERK.42 Indeed, ERK1/2 was not activated by AA – even at high doses. The rapid and transient activation of MAPKs in response to most stimuli indicate that MAPKs function as a biological switch that must be downregulated, a task that is carried out by dual specificity phosphatases (DUSPs).43 In fact, preliminary microarray analysis revealed that the AA pretreatment significantly downregulated the expression of DUSP2 and DUSP5 when compared to the LPS-treated cells (data not shown). It is thus possible that the AA’s inhibition of the MAPKs in the presence of LPS is due to the combined effects of these stressors that bring about the negative regulation of MAPKs by DUSPs. Crosstalk between the different MAPKs further complicates the ability to specify the point at which they are inhibited or activated.42
This study further demonstrates the ability of AA to inhibit the expression of the adhesion molecules ICAM-1, VCAM-1, and E-selectin as well as the chemokine IL-8 in HUVECs at the protein level. In addition to the role of adhesion molecules in inflammation, these proteins play a part in tumor metastasis and invasion by facilitating the attachment and migration of cancer cells. Since tumor metastasis is a multistep process, these provide pharmacological targets for anticancer agents. One of these steps is angiogenesis.44 In this study, preliminary testing for the inhibition of angiogenesis revealed AA’s ability to inhibit tube formation, albeit with a narrow window of activity. This is because tube formation inhibition at higher concentrations (5–10 μg/mL) was likely due to cytotoxicity (Figure 11C).
Recently, we showed that AA inhibited the translocation of NF-κB in LPS-treated RAW264.7 cells and TNF-α-treated L929 cells.16 In this study, we further confirmed that AA inhibited the nuclear translocation of NF-κB in HUVEC cells, which would explain the compound’s ability to reduce the expression of the tested adhesion molecules and tube formation.
Taken together, the present study described the anti-inflammatory and possible anticancer activities of AA in several assays. The mechanism of action of AA includes the inhibition of several inflammatory kinases, such as the enzyme complex IKKα/β and the MAPKs. This confirms the compound’s capacity for the development of an anti-inflammatory and/or anticancer agent by testing it in relevant in vivo models of disease.
Acknowledgments
This work was supported in part by the following grants: Postgraduate Research Fund (PPP) grant number PS194/2010B; UMRG RG231/10HTM; and the High Impact Research Grant (HIR account number E00002-20001, University of Malaysia). We also like to acknowledge the financial support from the Targeted Delivery of Dietary Flavanols for Optimal Human Cell Function: Effect on Cardiovascular Health (Nutrition) (FLAVIOLA) grant (FP7-KBBE-2008-2B). The funding sources had no involvement in the study design, in the writing, or in the submission of the manuscript for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803–815. | ||
Castellani ML, De Lutiis MA, Toniato E, et al. Impact of RANTES, MCP-1 and IL-8 in mast cells. J Biol Regul Homeost Agents. 2010;24(1):1–6. | ||
Israël A, Le Bail O, Hatat D, et al. TNF stimulates expression of mouse MHC class I genes by inducing an NF kappa B-like enhancer binding activity which displaces constitutive factors. EMBO J. 1989;8(12): 3793–3800. | ||
Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med. 1993;177(5):1277–1286. | ||
Springer TA. Adhesion receptors of the immune system. Nature. 1990; 346(6283):425–434. | ||
Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. | ||
Price JT, Thompson EW. Mechanisms of tumour invasion and metastasis: emerging targets for therapy. Expert Opin Ther Targets. 2002;6(2): 217–233. | ||
Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002; 109 Suppl:S81–S96. | ||
Zabel U, Baeuerle PA. Purified human I kappa B can rapidly dissociate the complex of the NF-kappa B transcription factor with its cognate DNA. Cell. 1990;61(2):255–265. | ||
DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388(6642):548–554. | ||
Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2(3):a003350. | ||
Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183. | ||
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. | ||
de las Heras B, Rodríguez B, Boscá L, Villar AM. Terpenoids: sources, structure elucidation and therapeutic potential in inflammation. Curr Top Med Chem. 2003;3(2):171–185. | ||
Lee IS, Ma X, Chai HB, et al. Novel cytotoxic labdane diterpenoids from Neouvaria acuminatissima. Tetrahedron. 1995;51(1–2):21–28. | ||
Achoui M, Appleton D, Abdulla MA, Awang K, Mohd MA, Mustafa MR. In vitro and in vivo anti-inflammatory activity of 17-O-acetylacuminolide through the inhibition of cytokines, NF-κB translocation and IKKβ activity. PLoS One. 2010;5(12):e15105. | ||
Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10(4):369–373. | ||
Elgass S, Cooper A, Chopra M. Lycopene inhibits angiogenesis in human umbilical vein endothelial cells and rat aortic rings. Br J Nutr. 2012;108(3):431–439. | ||
Reyes MR, Reyes-Esparza J, Angeles OT, Rodríguez-Fragoso L. Mutagenicity and safety evaluation of water extract of Coriander sativum leaves. J Food Sci. 2010;75(1):T6–T12. | ||
Chan JY, Cheung JY, Luk SC, Wu YJ, Pang SF, Fung KP. Anti-cancer and pro-apoptotic effects of an herbal medicine and Saccharomyces cerevisiae product (CKBM) on human hepatocellular carcinoma HepG2 cells in vitro and in vivo. Immunopharmacol Immunotoxicol. 2004; 26(4):597–609. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Bougarne N, Paumelle R, Caron S, et al. PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kappaB. Proc Natl Acad Sci U S A. 2009;106(18):7397–7402. | ||
Kaileh M, Vanden Berghe W, Heyerick A, et al. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J Biol Chem. 2007;282(7): 4253–4264. | ||
Lamy S, Lachambre MP, Lord-Dufour S, Béliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53(5–6): 200–208. | ||
Venkateswaran A, Reddy YT, Sonar VN, et al. Antiangiogenic properties of substituted (Z)-(±)-2-(N-benzylindol-3-ylmethylene)quinuclidin-3-ol/one analogs and their derivatives. Bioorg Med Chem Lett. 2010;20(4):7323–7326. | ||
Ding GJ, Fischer PA, Boltz RC, et al. Characterization and quantitation of NF-kappaB nuclear translocation induced by interleukin-1 and tumor necrosis factor-a. J Biol Chem. 1998;273(44):28897–28905. | ||
Gutiérrez-Ruiz MC, Bucio L, Souza V, Gómez JJ, Campos C, Cárabez A. Expression of some hepatocyte-like functional properties of WRL-68 cells in culture. In Vitro Cell Dev Biol Anim. 1994;30A(6):366–371. | ||
Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10): 749–759. | ||
Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54(4): 469–487. | ||
Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. Curr Opin Struct Biol. 2001;11(6):752–760. | ||
Lowenstein CJ, Alley EW, Raval P, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci U S A. 1993;90(20): 9730–9734. | ||
Cheon MS, Yoon T, Lee do Y, et al. Chrysanthemum indicum Linné extract inhibits the inflammatory response by suppressing NF-kappaB and MAPKs activation in lipopolysaccharide-induced RAW264.7 macrophages. J Ethnopharmacol. 2009;122(3):473–477. | ||
Régnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997; 90(2):373–383. | ||
Israël A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 2000;10(4):129–133. | ||
Byun MS, Choi J, Jue DM. Cysteine-179 of IkappaB kinase beta plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines. Exp Mol Med. 2006;38(5):546–552. | ||
Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J Biol Chem. 2006;281(47):35764–35769. | ||
Haridas V, Kim SO, Nishimura G, Hausladen A, Stamler JS, Gutterman JU. Avicinylation (thioesterification): a protein modification that can regulate the response to oxidative and nitrosative stress. Proc Natl Acad Sci U S A. 2005;102(29):10088–10093. | ||
Lee JH, Koo TH, Yoon H, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72(10):1311–1321. | ||
Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13(2):85–94. | ||
Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009;48(3):161–169. | ||
Fujioka S, Niu J, Schmidt C, et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol. 2004;24(17):7806–7819. | ||
Shen YH, Godlewski J, Zhu J, et al. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem. 2003;278(29):26715–26721. | ||
Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26(22): 3203–3213. | ||
Jiang YL, Liu ZP. Natural products as anti-invasive and anti-metastatic agents. Curr Med Chem. 2011;18(6):808–829. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.