Back to Journals » ImmunoTargets and Therapy » Volume 8
Immunochemotherapy for Richter syndrome: current insights
Authors Puła B ![]() , Salomon-Perzyński A, Prochorec-Sobieszek M, Jamroziak K
, Salomon-Perzyński A, Prochorec-Sobieszek M, Jamroziak K
Received 2 October 2018
Accepted for publication 28 December 2018
Published 5 February 2019 Volume 2019:8 Pages 1—14
DOI https://doi.org/10.2147/ITT.S167456
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Bartosz Puła,1 Aleksander Salomon-Perzyński,1 Monika Prochorec-Sobieszek,2,3 Krzysztof Jamroziak1
1Department of Hematology, Institute of Hematology and Transfusion Medicine, Warsaw, Poland; 2Department of Diagnostic Hematology, Institute of Hematology and Transfusion Medicine, Warsaw, Poland; 3Department of Pathology and Laboratory Medicine, Maria Sklodowska-Curie Institute – Oncology Center, Warsaw, Poland
Abstract: Richter syndrome (RS) is recognized as the development of a secondary and aggressive lymphoma during the clinical course of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Most of such histological transformations are from RS to diffuse large B-cell lymphoma (DLBCL-RS, 90%) and Hodgkin’s lymphoma (HL-RS, 10%). Histopathological examination is a prerequisite for diagnosis. It is crucial to assess the relationship between the RS clone and the underlying CLL/SLL because clonally related DLBCL-RS has a poor outcome, while clonally unrelated cases have a prognosis similar to de novo DLBCL. An anti-CD20 antibody-based immunochemotherapy is hitherto the frontline treatment of choice for DLBCL-RS; nonetheless, the results are unsatisfactory. Allogeneic stem cell transplantation should be offered to younger and fit patients as a consolidative treatment; however, the majority of the patients may not be qualified for this procedure. The HL-RS transformation has better outcomes than those of DLBCL-RS and can effectively be treated by the adriamycin, bleomycin, vinblastine, and dacarbazine regimen. Although novel agents are currently being investigated for RS, immunochemotherapy nevertheless remains a standard treatment for DLBCL-RS.
Keywords: Richter syndrome, Richter transformation, chronic lymphocytic leukemia, diffuse large B-cell lymphoma, Hodgkin’s lymphoma
Introduction
The development of a secondary and aggressive lymphoma during the clinical course of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is recognized as Richter syndrome (RS).1 This entity was originally described as a reticular cell sarcoma in 1928 by Maurice Richter in an autopsy report of a 46-year-old patient.2 The actual term “RS” was proposed by Lortholary et al,3 who in 1964 described a series of autopsy cases where CLL transformed into an aggressive lymphoma resembling the histology of “reticular cell sarcoma.” For decades, RS had been recognized as a poorly understood complication of CLL with a dismal prognosis; however, during recent years, the molecular pathogenesis of RS has been better characterized. Most cases of RS consist of a histologic transformation to diffuse large B-cell lymphoma (DLBCL) and less often to Hodgkin’s lymphoma (HL); however, the development of plasmablastic lymphoma (PBL) or B-lymphoblastic leukemia/lymphoma has also been observed.4 The evolution of CLL into prolymphocytic leukemia (PLL) should be considered as a separate entity and termed “PLL transformation.”5 As the DLBCL subtype of RS (ie, DLBCL-RS) occurs in the majority of the cases, this review will focus exclusively on this clinical entity, unless otherwise specified.
Biology and features of DLBCL-RS
Epidemiology
DLBCL-RS may occur at different stages of CLL during various times after diagnosis (1–4 years), regardless of the administered treatment constituting around 90% of such diagnosed transformations. It is believed that these transformations occur in up to 10% of all CLL patients with an annual rate estimated at ~0.5%–1%. However, with the introduction of novel agents, higher incidences of DLBCL-RS in heavily pretreated patients have also been reported.6,7 It is furthermore not uncommon for CLL and DLBCL-RS to be diagnosed simultaneously.8
It should be noted that the proportion of CLL/SLL patients undergoing transformation may be underestimated. Primarily, the rates of histologically confirmed cases of RS depend much on the hospital policy of a given medical center regarding lymph node biopsy in CLL patients with rapidly progressive lymphadenopathy and clinical deterioration. Second, the increased RS prevalence observed in the recent years may be due to the use of more intensively immunosuppressive therapies as well as the prolonged overall survival (OS) in patients with CLL.9
Histopathology and pathogenesis
DLBCL-RS should be differentiated from histologically aggressive lymphomas, the so-called accelerated CLL, by experienced pathologists because it is suspected that up to 20% of RS cases could be misdiagnosed.1,10 Figure 1 shows the histopathological examples of DLBCL-RS, whereas Figure 2 shows the histopathological examples of HL-RS. DLBCL-RS cases are characterized by the following: 1) the presence of large B-cells with nuclear sizes either equal to the nucleus of macrophages or more than twice that of a normal lymphocyte and 2) a diffuse growth pattern of large cells (Figure 1).11 Most of the DLBCL-RS cases represent a nongerminal center B-cell-like phenotype (90%).12 In the presented DLBCL-RS case, expression pattern of CD23 and BCL6 could distinguish between transformed and nontransformed CLL cells. Furthermore, high MIB1 antigen expression indicates the highly proliferative potential of DLBCL-RS cells (Figure 1). In 80% of the DLBCL-RS cases, clonal relationships based on IGHV analysis can be determined where such cases are regarded by some as being a true RS transformation. In the remaining 20% cases, the IGHV rearrangement differs from that of the CLL/SLL clone where such cases are defined as being clonally unrelated and resemble the occurrence of de novo DLBCL and have a significantly better prognosis, similar to that of de novo DLBCL.13–15
 | Figure 1 CLL transformation into DLBCL. Notes: (A) Large cells of DLBCL (lower left) next to infiltration by small CLL cells (upper right) HE, ×200 magnification. (B) DLBCL with centroblastic morphology (upper right); few small CLL cells (lower left); HE staining, ×400 magnification. (C) DLBCL cells reveal stronger membrane CD20 expression than that of CLL cells. (D) MIB1 staining in 80% of the DLBCL cells and in 3% of the CLL cells. (E) CD23 membrane expression in CLL cells; DLBCL cells are negative. (F) BCL6 nuclear expression in DLBCL cells; CLL is negative; EnVision staining, ×400 magnification. Abbreviations: CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; HE, hematoxylin and eosin. |
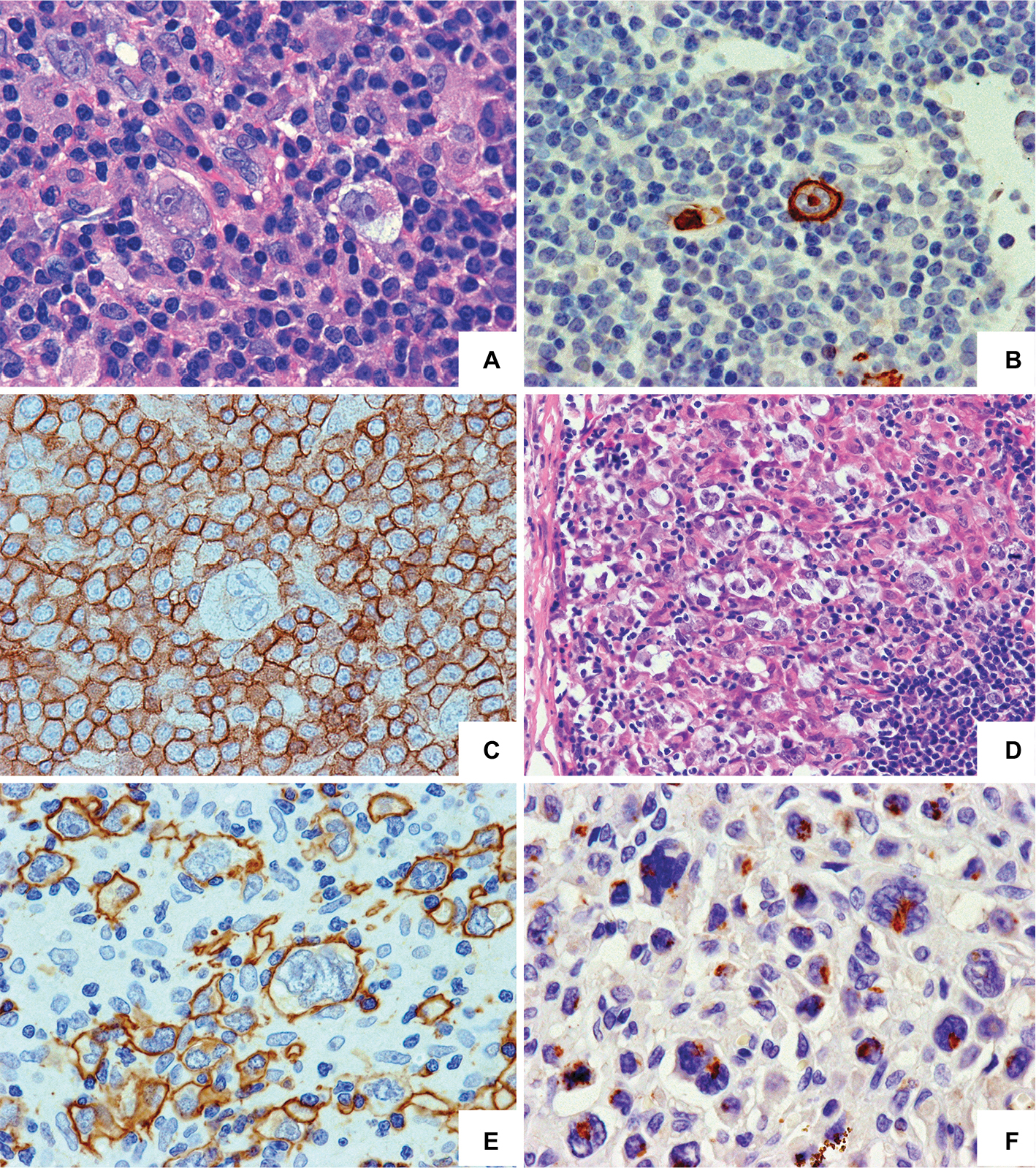 | Figure 2 Morphological and phenotypic spectrum of CLL transformation into HL may strongly differ upon histopathological examination. Notes: Type I – CLL with Hodgkin transformation (A–C). (A) Reed–Sternberg cells are sparsely dispersed in the background of small CLL cells; HE staining. (B) CD15 membrane and “dot-like” expression in HRS cell. (C) CD23 expression in CLL cells; the Reed–Sternberg cell is negative. EnVision staining, ×400 magnification. Type II – CLL transformation in HL (D–F). (D) The numerous HRS cells among histiocytes, eosinophils, and small lymphocytes in the background; a few CLL cells in the lower right; HE staining, ×200 magnification. The HRS cells reveal membrane CD30 expression (E) and “dot-like” expression of CD15 (F); EnVision staining, ×400 magnification. Abbreviations: CLL, chronic lymphocytic leukemia; HE, hematoxylin and eosin; HL, Hodgkin’s lymphoma; HRS cell, Hodgkin and Reed–Sternberg cell. |
Clonal relationships between the underlying CLL and the diagnosed DLBCL-RS are mostly diagnosed by sequencing immunoglobulin genes.16 The introduction of novel sequencing and molecular methods has allowed a better understanding of DLBCL-RS pathogenesis while also addressing the issue of its clonal evolution. It is recognized that most of the genetic alterations occur in a particular dominant CLL clone at the time of disease transformation, so giving a rise to a “linear” transformation model.4,13,15 A minority of DLBCL-RS cases develop from a common precursor cell that had acquired alterations early on, possibly leading to the rise of separate CLL and DLBCL-RS clones. Such a “branched” transformation model is a characteristic feature of leukemic RS cases and is associated with CDKN2A loss.4,17 Interestingly, although de novo DLBCL and DLBCL-RS present similar morphologies upon histopathological examination, significant genetic and epigenetic differences have been noted.15,17 Most molecular events associated with DLBCL-RS lead to the deregulation of cell cycle control, proliferation, and damage to DNA repair and target genes via somatic mutations of TP53 (60%–80%), CDKNA2 (30%), or MYC itself (30%) or by affecting their regulatory functions, eg, NOTCH1 (30%) and MGA (10%).17–22 Furthermore, DLBCL-RS lacks the typical recurrent mutations of de novo DLBCL affecting nuclear factor-κB (eg, CARD11, TNFAIP/A20, CD79A, CD79B, BCL6, BCL2, PRDM1, and EZH2) or genes associated with CLL chemorefractoriness and progression (eg, BIRC3, MYD88, DDX3X, SF3B1, and RPS15).14,15,17,23–27 Furthermore, aberrant somatic hypermutations occurring in PIM1, PAX5, RhoH/TTF, and MYC are rarely observed in DLBCL-RS, whereas in de novo DLBCL they are observed in over 50% of the analyzed cases.15,20 Besides the gene mutations, recurrent copy number alterations have also been reported comprising deletions of 7q31, 8p, 14q, and trisomy 12 and amplifications of 8q21, 13q, and 18q.19,28,29
The analysis of genetic alterations between CLL and DLBCL-RS has led to the proposal of existence of two main genetic pathways responsible for transformation. The first transformation route, in over 50% cases, was found to be associated with TP53 and CDKNA2 mutations. The second is regarded to be associated with trisomy 12 and NOTCH1 mutations in almost one third of instances.17 For the remaining DLBCL-RS cases, no clear genetic transformation profile could be distinguished. It is therefore suggested that B-cell receptor (BCR) signaling and cell–cell interactions may also be key factors leading to the development of DLBCL-RS. The BCR on CLL cells of the rare subset, #8, bearing the unmutated configuration IGVH4-39/IGHD6-13/IGHJ5, exhibits a broad polyreactivity to various antigens as compared to subsets #1 and #2.30 Furthermore, subset #8 cells were found to frequently bear NOTCH1 mutations or trisomy 12 and possess an increased risk of transforming into DLBCL.30–34 Both these features may be due to the 24-fold increase in the transformation risk of this subset. In addition, DLBCL-RS cases, especially those clonally related to the underlying CLL, were shown to express the programmed death-1 transmembrane protein (PD-1, CD279).35,36 Through the interaction with its ligands, PD-L1 and PD-L2 form a key immune checkpoint by downregulating T-cell activation and autoimmunity, thereby promoting self-tolerance, and by enabling malignant traits of CLL cells to be acquired, thus potentially leading to the development of RS.35–37
Risk factors for CLL transformation
Numerous clinical and molecular factors associated with an increased risk of RS development have hitherto been identified. The genotoxic and/or immunosuppressive activity of anti-CLL/SLL agents contributing to transformation into RS is still debatable. Purine analog-based therapy has been regarded as a risk factor for DLBCL-RS development; however, the results are still equivocal.23,38,39 Using alemtuzumab is considered a risk factor for developing clonally unrelated DLBCL-RS by reactivating Epstein–Barr virus (EBV) infection and inducting T-cell defects in immunocompromised CLL/SLL patients.40–42
Much attention has been focused on CLL/SLL transformation during treatment with novel agents (ibrutinib, idelalisib, and venetoclax). With such therapy, most RS transformations are diagnosed within the first year of treatment, while the risk seems to vary depending on the compound used.27 The incidence of RS transformation for ibrutinib ranges from 0.8% to 8%,43–47 while a similar incidence is observed for idelalisib (range 0%–7%).46,48,49 Moreover, an even higher RS incidence, of up to 16%, was reported during venetoclax monotherapy.7 It is however likely that such elevated RS rates, which are particularly apparent in observational studies, depend rather on the characteristics of heavily pretreated patients included in these studies where some individuals probably have RS when novel agents are initiated. Consistent with this, randomized Phase III studies have not reported any increased incidence of transformations in the experimental arms when using ibrutinib and idelalisib.50–53 An interesting question however arises as to whether ibrutinib may partially shift the direction of transformation to the HL variant of RS (HL-RS), as perhaps is suspected by some studies and case reports.47,54–56
Besides the use of toxic agents, some biological CLL/SLL factors were also demonstrated to increase the risk of developing DLBCL-RS. Among those associated with increased risks of transformation are a lymph node size ≥3 cm, the absence of del13q14, shortened telomere length ≤5,000 base pairs, the presence of stereotyped BCR (with subset #8 increasing the risk of transformation by 24-fold), unmutated IGHV status, and the expression of ZAP-70 as well as CD38.6,13,17,32,57,58 Furthermore, del9p, del11q, del17p, and trisomy 12, along with mutations of TP53, CDKNA2, NOTCH1, and MYC were also shown to increase the risk of RS.6,15,17,19 A latest research indicates that patients bearing/acquiring defects in the p53 signaling pathway (del17p/TP53 mutation), BCL6 abnormalities, a complex or near-tetraploid karyotype, and Bruton’s tyrosine kinase (BTK) mutations may be at a higher risk of transformation.44,59,60 Recently, based on the retrospective comparison of microRNA expression levels in groups of patients who underwent or did not undergo CLL transformation, it was suggested that a high expression of miR-125a-5p or a low expression of miR-34a-5p could predict the development of RS in around 50% of patients.61
Clinical and laboratory characteristics
Most commonly, DLBCL-RS clinically presents with rapidly enlarging lymph nodes (especially in one region), the presence of constitutional symptoms such as high-grade fevers and weight loss together with elevated lactate dehydrogenase (LDH) levels or hypercalcaemia.57 The involvement of extranodal sites is found in about 40% cases and should always raise the suspicion that RS is present.62,63 Such sites are usually the gastrointestinal tract, bone marrow, central nervous system, and skin. Laboratory findings show increased LDH activity levels in 50%–80% of patients. Cytopenias resulting from bone marrow involvement are also common including anemia with hemoglobin concentration <11 g/dL found in about 50% of cases and thrombocytopenia <100.000 G/L in 43% of patients.23,63
Diagnosis and differential diagnosis
The diagnosis of RS is exclusively histopathological through examining tissue biopsied from suspected lesions, most frequently an enlarged lymph node or a trephine bone marrow biopsy. Some authors propose that a flow cytometry of fine-needle aspiration biopsy material combined with cytological and cytogenetical analysis could also lead to the diagnosis of RS in urgent cases; however, this approach has not been so far verified in large-scale retrospective or prospective studies.64 The target of the biopsy should be appropriately selected, as histopathological features of the transformation may only be present in selected lymph nodes, while others may still show a typical CLL/SLL image.
Whenever RS is suspected, a positron emission tomography/computed tomography (PET-CT) scan is indicated, using 18-fluorodeoxyglucose uptake, where suitable biopsy sites for RS are demonstrated when maximum standardized uptake values (SUVmax) are ≥5.65–67 By adopting this cutoff value for making diagnostic decisions, a high negative predictive value of 97% is achieved; however, the positive predictive value is only 53%. Suspected sites should therefore be biopsied when making a differential diagnosis, and due consideration should be given to the possibilities of accelerated CLL and PLL being present as well as immunosuppressive therapy–related EBV-positive lymphoproliferative syndrome (post–allogeneic stem cell transplantation or alemtuzumab) or indeed any other solid tumors.23,67–70 With the introduction of kinase inhibitors and anti-Bcl-2 agents, PET/CT imaging may gain a significance and be crucial in patients’ monitoring and qualification for novel therapies due to the possibility of excluding RS during and upon treatment initiation. Mato et al reported the largest so far series of patients with PET-CTs prospectively performed at kinase inhibitor discontinuation (ibrutinib and idelalisib) concluding that PET-CT SUVmax ≥10 alone lacks both sensitivity and specificity to distinguish CLL progression from that of RS type.71 This lack of accuracy was ascribed to the fact that CLL progression following ibrutinib and idelalisib treatment seems to be more metabolically active than that following the failure of rituximab-based immunochemotherapy. Although RS was confirmed in eight of 57 (14%) cases suspected for progression, it is worth to mention that all of these cases were characterized by SUV max ≥5. The increase in SUV max values observed under BCR inhibitor treatment might be potentially used for disease monitoring under the circumstance of developing reliable diagnostic algorithms in the clinical setting of progression. Nevertheless, further studies are needed to address the clinical issue of distinguishing the type of progression under BCR inhibitor therapies using imaging methods alone.71
Principles of RS treatment
Figure 3 presents the algorithm of RS management. The clonal relationship between DLBCL and underlying CLL represents the most important prognostic factor for patients with RS, with significantly worse outcomes achieved by patients with clonally related DLBCL, median OS values being 14.2 vs 62.5 months, respectively.15 Given the favorable prognosis for patients with clonally unrelated DLBCL, the general consensus is to treat them in accordance with the guidelines developed for de novo DLBCL.18 In contrast, there are no established standards of care for patients with clonally related DLBCL-RS, and a clinical trial should thus always be considered. If a clinical trial is unavailable, the most appropriate therapeutic interventions currently appear to be induction treatment with immunochemotherapy followed by postremission consolidation with allogeneic or (if not feasible) autologous hematopoietic stem cell transplantation (HSCT).18,72,73 In a large retrospective analysis of the European Group for Blood and Marrow Transplantation, the median OS, relapse-free survival, and the cumulative incidences of relapse and nonrelapse mortality at 3 years for patients with RS undergoing allogeneic HSCT were 36%, 27%, 47%, and 26%, respectively, while they were 59%, 45%, 43%, and 12%, respectively, for RS patients undergoing autologous HSCT.73
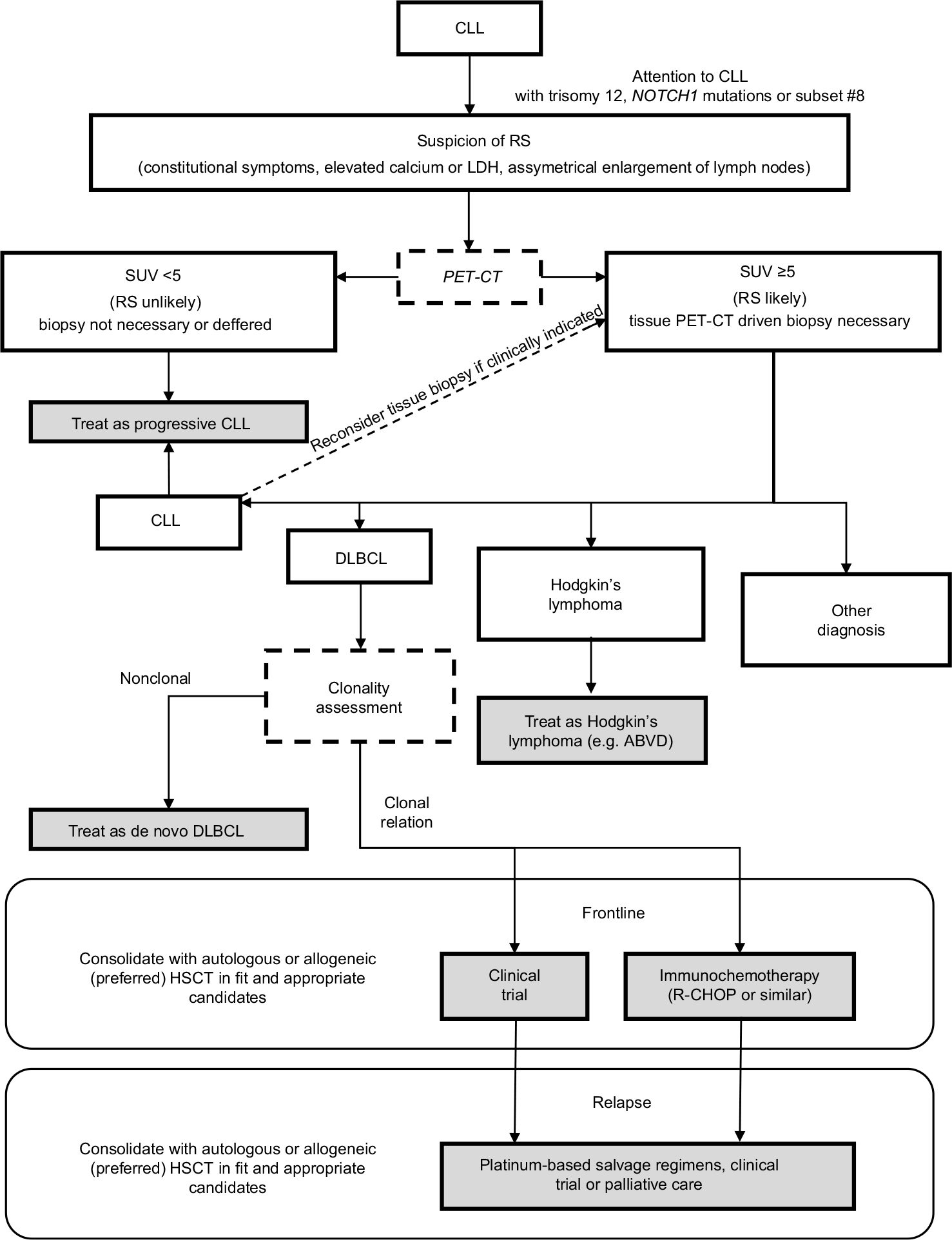 | Figure 3 Management algorithm for patients with a clinical suspicion of RS. Notes: In CLL patients characterized by chromosome 12 trisomy or NOTCH1 mutations, or classified as subset #8, and especially in those with the presence of constitutional symptoms or asymmetrical lymph node enlargement, caution is advised due to the increased risk of RS transformation. In case RS is suspected, a PET/CT should be performed and regions with SUV >5 were qualified for excisional biopsy. If the biopsy reveals no transformation, CLL treatment may be initiated. However, in cases with a strong suspicion of RS, a secondary biopsy should be considered. Once the histopathological diagnosis of RS-DLBCL is confirmed, a patient should be qualified for clonality assessment. In case of no clonal relationship between CLL and DLBCL, the patient should be treated as having de novo DLBCL. In contrast, for clonally related cases, the enrollment into clinical trial with novel compounds is advised due to poor prognosis with a standard immunochemotherapy. Fit and transplant-eligible patients with clonally related disease should be qualified for allogeneic stem cell transplantation or, if not possible, for autologous stem cell transplantation. In case of relapsed disease, platinum-based salvage regimens or participation in clinical trials is recommended. Transformation to Hodgkin’s lymphoma should be treated with ABVD regimen. Shading relates to administered therapies or treatment as final points of the algorithm. Other non-shaded fields represent the diagnostic activities and results. Abbreviations: ABVD, adriamycin, bleomycin, vinblastine, and dacarbazine; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; HSCT, hematopoietic stem cell transplantation; LDH, lactate dehydrogenase; PET/CT, positron emission tomography/computed tomography; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; RS, Richter syndrome; SUV, standardized uptake value. |
The emerging problem is finding an optimal therapy for patients who develop RS during CLL treatment with novel agents, in particular the BCR inhibitor – ibrutinib. This group has an extremely poor prognosis with a median OS of 2 months.43 If possible, patients who succumb to RS after ibrutinib should be treated in clinical trials with novel drugs, especially with checkpoint inhibitors such as pembrolizumab and PI3K inhibitor, idelalisib.74,75 Otherwise, it seems reasonable to apply the aforementioned strategy; nonetheless, consolidation with allogeneic HSCT should be considered in all patients who develop DLBCL-RS after treatment with ibrutinib.76
Role of immunochemotherapy for DLBCL-RS
A number of chemotherapy regimens used for aggressive non-HL have been evaluated in the treatment of DLBCL-RS patients including some retrospective analyses (Table 1) and prospective clinical trials (Table 2). It should be noted that the overall obtained responses are transient and the outcomes disappointing, particularly in patients without postremission consolidation with HSCT.
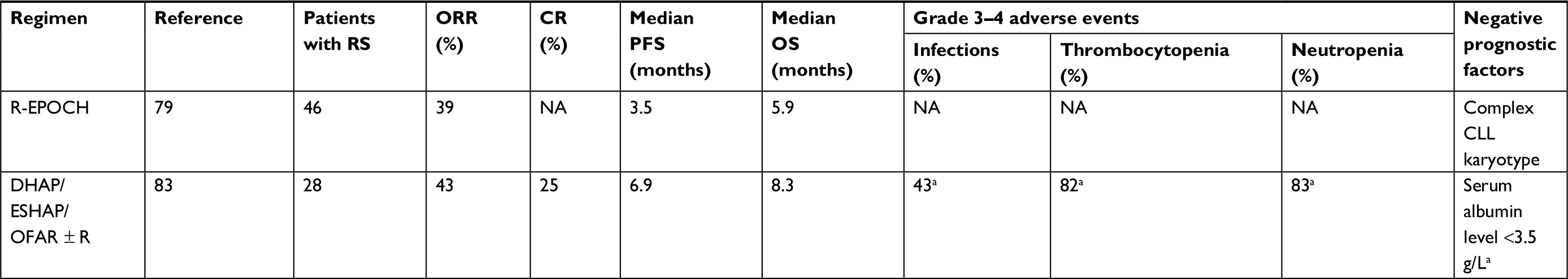 | Table 1 Summary of the published retrospective studies assessing the efficacy and safety of induction regimens for patients with DLBCL-RS Note: aData for the entire study population which included 47 patients with relapsed/refractory CLL and 28 patients with RS. Abbreviations: CLL, chronic lymphocytic leukemia; CR, complete response; DHAP, dexamethasone, cytarabine, and cisplatin; DLBCL, diffuse large B-cell lymphoma; ESHAP, etoposide, methylprednisolone, cytarabine, and cisplatin; NA, not available; OFAR, oxaliplatin, fludarabine, cytarabine, and rituximab; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; R-EPOCH, rituximab, etoposide, cyclophosphamide, doxorubicin, vincristine, and prednisone; RS, Richter syndrome. |
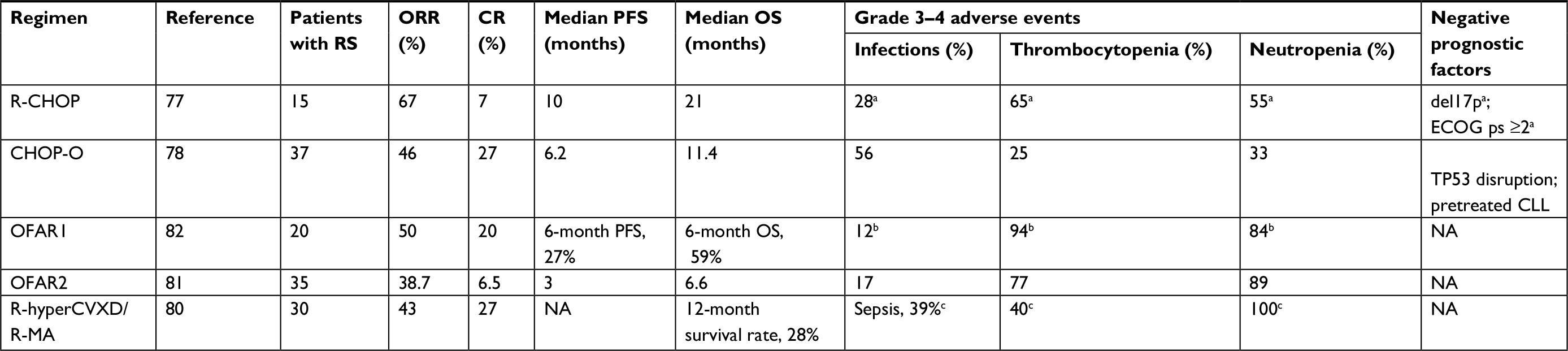 | Table 2 Summary of the published prospective studies assessing the efficacy and safety of induction regimens for patients with DLBCL-RS Note: aData for the entire study population which included 26 patients with high-risk relapsed/refractory CLL, 19 patients with CLL plus autoimmune cytopenia, and 15 patients with RS; bdata for the entire study population which included 30 patients with relapsed/refractory CLL and 20 patients with RS; cdata for the entire study population which included 19 patients with relapsed/refractory CLL and 30 patients with RS. Abbreviations: CHOP-O, cyclophosphamide, doxorubicin, vincristine, prednisone, and obinutuzumab; CLL, chronic lymphocytic leukemia; CR, complete response; del17p, deletion of chromosome 17p; DLBCL, diffuse large B-cell lymphoma; ECOG ps, the Eastern Cooperative Oncology Group performance status; FFS, failure-free survival; NA, not available; OFAR, oxaliplatin, fludarabine, cytarabine and rituximab; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-hyperCVXD/R-MA, rituximab, fractioned cyclophosphamide, vincristine, liposomal daunorubicin, and dexamethasone alternating with rituximab, methotrexate, and cytarabine; RS, Richter syndrome; TP53 disruption, TP53 deletion or mutation. |
An anthracycline-based regimen, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), is still the treatment of choice for the newly diagnosed de novo DLBCL, but has shown poor efficacy in a cohort study of 15 DLBCL-RS patients prospectively evaluated by a German CLL study group trial.77 The overall response rate (ORR) was 67% with only one (7%) complete response (CR). The median progression-free survival (PFS) and median OS were 10 and 21 months, respectively. Despite there being no separate data provided in this trial on the safety profile of R-CHOP for patients with DLBCL-RS, 15 of the 60 (27%) CLL patients enrolled in this study had their therapy discontinued earlier than planned because of the treatment-related toxicity.77
Substituting rituximab with the humanized monoclonal anti-CD20 antibody ofatumumab within cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) regimen has not demonstrated any substantial clinical benefits. In a Phase II National Cancer Research Institute, 37 RS patients were treated with six cycles of CHOP in combination with ofatumumab induction followed by 12 months of ofatumumab maintenance.78 For the six cycles, 27% and 19% patients achieved CR and partial response (PR), respectively, with an ORR of 46%. The median PFS was 6.2 months, and the median OS was 11.4 months. There were no treatment-related deaths; however, a significant number of grade 3 or 4 serious adverse events were noted (n=85 and n=50, respectively), which emphasizes that patients with RS treated with immunochemotherapy are at an increased risk of severe treatment-related toxicity. This study provided evidence that RS patients treated with immunochemotherapy are not a prognostically homogeneous group and that superior outcomes were achieved in both cases of treatment-naïve CLL patients at the time of transformation and those without TP53 deletion or mutation when compared to their counterparts.78
Recently, the anti-lymphoma efficacy of a more intensive regimen, rituximab, etoposide, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-EPOCH), has been retrospectively evaluated by Rogers et al.79 Patients with DLBCL-RS (n=46) were treated with first-line R-EPOCH in nonescalated doses. A PR was achieved in at least 17 patients (37%). The median PFS and median OS were 3.5 and 5.9 months, respectively. Importantly, poor safety profiles were observed with 13 deaths (30%) without confirmed or suspected lymphoma progression. The prognostic and predictive values of the complex CLL karyotype were found to be significant in patients with RS treated with immunochemotherapy. RS patients with the complex CLL karyotype treated with R-EPOCH had significantly inferior outcomes, where the estimated 1-year PFS and median OS were 11% and 18%, respectively, compared with 57% and 71% for those without a complex CLL karyotype.79
Attempts to improve outcomes by using intensive regimens (initially developed for highly aggressive B-cell non-HLs) have not provided any breakthrough in treating RS patients. In a Phase II MD Anderson Cancer Center study, 30 patients with DLBCL-RS received up to six cycles of fractionated cyclophosphamide, vincristine, liposomal daunorubicin, and dexamethasone in combination with rituximab alternating with methotrexate and cytarabine plus rituximab (R-hyper-CVXD/R-MA).80 CR was achieved in 27% patients, while the ORR was 43%. The 12-month survival rate was 28%. The substantial toxicity of this regimen should nevertheless be particularly stressed. Approximately 20% of the patients died during the first two cycles of hyper-CVXD/R-MA; the treatment safety data reported in this study concerned not only patients with RS but also those with fludarabine-refractory CLL.80
Platinum-containing regimens that are widely used for treating recurrent aggressive B-cell lymphomas have also been evaluated as a frontline therapy for RS patients. The clinical activity of the oxaliplatin, fludarabine, cytarabine, and rituximab (OFAR) regimen in two different dosing schedules was prospectively assessed by two Phase I-II MD Anderson Cancer Center trials, OFAR1 and OFAR2.81,82 Among the 55 DLBLC-RS patients treated in both the trials, the ORR ranged from 39%–50% with a CR achieved in 6%–20% of RS cases. The median OS was 6–8 months. Unfortunately, the high anti-lymphoma efficacy of the OFAR regimen was associated with a poor safety profile. Grade 3 or 4 neutropenia and thrombocytopenia occurred in ~90% and 80% of the patients. One in five patients suffered episodes of neutropenia complicated by a neutropenic fever.81,82
Similar efficacies in treating DLBCL-RS patients have been retrospectively shown by other platinum-based chemotherapy regimens, such as dexamethasone, cytarabine, and cisplatin (DHAP) and etoposide, methylprednisolone, cytarabine, and cisplatin, mostly combined with rituximab.83 The ORR was 43%, while the median PFS and median OS were 7 and 8 months, respectively. The main toxicity was myelosuppression with grade 4 neutropenia and thrombocytopenia found in ~80% of the patients; the treatment safety data reported in this study concerned not only patients with RS but also those with relapsed/refractory CLL.83
Novel agents
Bearing in mind that in addition to the underlying hematological cancers, the majority of CLL/SLL patients are also often at an advanced age and suffer from numerous other comorbidities, and therefore only ~10%–15% of patients can undergo the potentially curative allogeneic HSCT.18,23 Furthermore, intensive chemotherapy regimens are highly toxic to this population grouping and lead to excessive treatment-related morbidity.77,80–83 Enrolling DLBCL-RS patients in clinical trials is therefore justifiable, particularly those with clonally related disease. Due to the poor activity of immunochemotherapy, the possibility of using novel agents in the treatment of RS is of great interest.
BCR inhibitors: ibrutinib, acalabrutinib, and idelalisib
In a few case reports, ibrutinib, a first-in-class BCR inhibitor widely used for the treatment of R/R CLL, has been shown to have a promising activity in RS. In case series reported by Tsang et al, three patients had RS refractory to two lines of previous immunochemotherapy (R-CHOP as the frontline treatment and rituximab, ifosfamide, carboplatin and etoposide, R-DHAP, or R-EPOCH as a salvage regimen, respectively), while one patient had treatment-naive RS.84 Ibrutinib was able to induce at least PR in three patients, while in one clinical benefit during ibrutinib therapy was observed. The median duration of treatment with ibrutinib in these patients was 6.1 months (range, 2.8–10.8 months). Among two patients who achieved PR, one experienced CLL progression at 11 months, and the other relapsed with DLBCL 8 months after beginning the therapy. Ibrutinib was safe, and no patient required discontinuation as a result of adverse events.84 Recently, Visentin et al have reported treatment outcomes of patients with RS treated with single agent, ibrutinib, or other BCR inhibitor, idelalisib.75 Of the four patients treated with ibrutinib, one had PR and two stable disease. The ORR in patients treated with idelalisib (n=4) was 75% with one CR and two PR. After a median follow-up of 6 months, the median time to relapse for the patients treated with ibrutinib or idelalisib was 13.7 months.75 The observation that has been made in this case series that patients with RS refractory to ibrutinib can achieve significant clinical benefits from a salvage treatment with idelalisib requires a special attention and warrants further investigations.
According to the preliminary data from ACE-CL-001 Phase 1/2 trial (NCT02029443), acalabrutinib, a second-generation BTK inhibitor, has recently shown a clinically relevant efficacy used as monotherapy in RS patients.85 The ORR was 38%, including CR in 14% and PR in 24% of patients. The median duration of response was 5.7 months, while the median PFS for the entire study cohort was 3.2 months. Although the presented data may not seem impressive, it should be emphasized that patients treated with acalabrutinib in this study were heavily pretreated with the median number of prior systemic therapies for RS reaching four.85 Acalabrutinib in combination with other novel agents such as vistusertib and pembrolizumab in the treatment of RS is currently being investigated in Phase 1/2 studies (NCT03205046 and NCT02362035).
Bcl-2 inhibitor: venetoclax
Venetoclax, a Bcl-2 inhibitor with a significant efficacy in the treatment of R/R CLL, has been evaluated recently in Phase 1 study of R/R-non-HLs.86 In fact, venetoclax used as a single agent had a limited activity in DLBCL de novo (n=34) with an ORR of 38% and an estimated median PFS of 1 month; however, in the RS subgroup (n=7), the PR was achieved in three cases (ORR =43%) and the duration of venetoclax treatment in three patients with RS exceeded 12 months, suggesting that anti-Bcl-2 agents may be a useful option in some cases of RS. Venetoclax in combination with dose-adjusted R-EPOCH in the treatment of RS is currently being evaluated in the Phase 2 study (NCT03054896).
Checkpoint inhibitors: pembrolizumab and nivolumab
Pembrolizumab, a monoclonal antibody directed against PD-1 receptor (anti-PD-1 MoAb), was first shown to be clinically active in RS in the Phase 2 study.74 Although none of the patients with R/R CLL responded to immunotherapy with pembrolizumab, four of nine patients with RS achieved objective response (ORR=44%), including one CR. Importantly, all responses were observed in RS patients who experienced disease progression after prior therapy with ibrutinib. After a median follow-up time of 11 months, the median PFS in the RS cohort was 5.4 months. The median OS was 10.7 months in the whole study, but the median PFS was not reached in RS patients who progressed after prior ibrutinib.74
Recently, these results have been questioned by Rogers et al.87 In case series including ten patients with RS, most of which (n=6) were treated with anti-PD-1 MoAb, pembrolizumab (n=3) and nivolumab (n=7), the median time to treatment failure and the median OS from the first dose of anti-PD-1 MoAb was 1.2 and 2 months, respectively. Importantly, all of the patients in this case series were previously exposed to ibrutinib.87 Currently, pembrolizumab alone (NCT02576990) or in combination with ublituximab and TGR1202 (NCT02535286) or acalabrutinib (NCT02362035) or ibrutinib and idelalisib (NCT02332980) is being evaluated in RS patients in prospective studies, and the results in the context of the existing controversies described above are highly expected.
Selective exportin-1 (XPO1) inhibitor: selinexor
Selinexor, a selective inhibitor of nuclear export protein XPO1 that leads to retention of p53 within the nucleus enhancing its anti-neoplastic activity in tumor cells, is another agent with a potential activity in the treatment of RS patients. In Phase 1 study of patients with a heavily pretreated R/R-non-HL, selinexor used in monotherapy was able to induce PR in two of five patients with RS while in other two patients stable disease was observed.88 The Phase 2 study to assess the efficacy of selinexor monotherapy exclusively in the group of RS patients was terminated due to enrollment challenges in this rare disease (NCT02138786).
Other novel therapies of interest
For relapsed DLBCL-RS cases, blinatumomab (a bispecific antibody directing cytotoxic T cells toward CD19+ B cells) and chimeric antigen receptor-modified T-cells could be interesting therapeutic options; however, further studies are required to better assess their efficacy and toxicity including a greater number of patients.89–92
HL-RS
HL-RS is a relatively rare but distinct subtype of RS accounting for ~5%–15% of cases.93–95 Upon histopathological examination (Figure 2), particular HL-RS cases may vary strongly ranging from ones where the Reed–Stenberg cells are rarely observed within rich CLL infiltrate (type I transformation) to ones characterized by typical HL morphology with the Reed–Stenberg cells dispersed among histiocytes, eosinophils, and small lymphocytes in the background (type II transformation). In our retrospective analysis of the CLL disease course in 786 patients from a single center, we found that 40 (5.1%) patients had developed RS including 33 (4.2%) DLBCL-RS, six (0.8%) HL-RS, and one PBL.96
Although the total numbers of HL-RS cases reported in the World Medical Literature are low, some distinct features of this type of transformation are apparent.97 When compared to the more common DLBCL-RS form, HL-RS occurs predominantly within the context of CLL with mutated IgVH genes, whereas clonal relationships of transformed cells with CLL cells are less frequently observed, 40%–50% vs 80%–90%.12 Both these biological features may partially explain the relatively better prognosis for HL-RS. Additionally, the involvement of EBV is also more frequent in this RS variant.23,97 In contrast, our incidental observation that HL-RS may be a more common type of RS in the context of ibrutinib and may not be supported by the larger series of patients with CLL transformation occurring during ibrutinib therapy.44,54
The prognosis for HL-RS is better than for DLBCL-RS, although still significantly inferior to the outcomes for de novo HL.98 Patients with HL-RS typically present with disseminated disease and general symptoms and cannot be clinically distinguished from those with DLBCL-RS.93–96 Some factors were identified that affect the survival of HL-RS patients as follows. Similarly to DLBCL-RS cases, patients in whom the transformed cells are clonally related with the preceding CLL have worse outcomes.57,97 Furthermore, the International Prognostic Score (IPS), originally developed for de novo HL, was found to stratify prognoses among HL-RS patients from the Mayo Clinic.98 An Italian multicenter retrospective analysis on 33 HL-RS cases further demonstrated that an IPS4 adversely influenced the OS.99 The outcomes of HL-RS may also depend on previous treatments used for the underlying CLL, with significantly worse results achieved in patients with a history of exposure to purine analogs fludarabine or cladribine and probably also to ibrutinib.44,47,54,96,97
Since there have been no prospective randomized trials performed on this rare disease entity, treatment decisions need to be based on the retrospective evidence from the reported case series. The most active therapy for HL-RS appears to be combination regimens employed in patients with de novo classical HL, most often adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD), resulting in almost half of the patients responding in the largest series.57,96 In those patients not achieving CR with such approaches, second-line intensive protocols are applied and, if feasible, optimally followed by reduced-intensity conditioning allogeneic HSCT.23
Interestingly, CD20-based immunochemotherapies such as those used for DLBCL-RT are also possible treatments for HL-RS as tumor cells in a half of the cases express CD20.97 Based on retrospective data, immunochemotherapy regimens nevertheless seem to be less active than HL-type therapies such as ABVD.57 Novel immunotherapy strategies that are already in use for relapsed or refractory de novo HL, such as brentuximab and anti-PD-1 and anti-PD-L1 antibodies, may potentially improve the outcomes of HL-RS in the future.
Future directions
CLL/SLL affects mostly elderly patients often suffering from numerous underlying comorbidities, thus limiting the administration of intensive immunochemotherapy regimens followed by allogeneic HSCT in the majority of patients with RS. It is therefore highly reasonable to qualify such patients for treatment with novel agents within clinical trials. At present, immune checkpoint inhibitors or BCR inhibitors in monotherapy or in combination with immunochemotherapy appear to be a potentially viable option for more effectively treating DLBCL-RS and HL-RS patients. Nevertheless, further studies on larger cohorts of patients are warranted to assess the efficacy and safety of these novel compounds.
Acknowledgment
This study was supported by a scientific grant from National Science Center in Poland (grant number: UMO-2016/21/D/NZ5/02569).
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Campo E, Ghia P, Montserrat E, Harris N. Chronic lymphocytic leukemia/Small lymphocytic leukemia. In: Swerdlow S, Campo E, Harris N, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2017:216–220. | ||
Richter MN. Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. Am J Pathol. 1928;4(4):285–292. | ||
Lortholary P, Boiron M, Ripault P, Levy JP, Manus A, Bernard J. Leucémie lymphoide chronique secondairement associée à une réticulopathie maligne, syndrome de Richter [Chronic lymphoid leukemia secondarily associated with a malignant reticulopathy: Richter’s syndrome]. Nouv Rev Fr Hematol. 1964;4:621–644. French. | ||
Agbay RL, Jain N, Loghavi S, Medeiros LJ, Khoury JD. Histologic transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma. Am J Hematol. 2016;91(10):1036–1043. | ||
Reiniger L, Bödör C, Bognár A, et al. Richter’s and prolymphocytic transformation of chronic lymphocytic leukemia are associated with high mRNA expression of activation-induced cytidine deaminase and aberrant somatic hypermutation. Leukemia. 2006;20(6):1089–1095. | ||
Parikh SA, Rabe KG, Call TG, et al. Diffuse large B-cell Lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol. 2013;162(6):774–782. | ||
Roberts AW, Davids MS, Pagel JM, et al. Targeting Bcl2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322. | ||
Linden MA. Case study interpretation-New Orleans: case 4. Richter syndrome characterized by diffuse large B cell lymphoma in the context of prior chronic lymphocytic leukemia/small lymphocytic lymphoma. Cytometry B Clin Cytom. 2013;84(5):350–353. | ||
Cwynarski K, van Biezen A, de Wreede L, et al. Autologous and allogeneic stem-cell transplantation for transformed chronic lymphocytic leukemia (Richter’s syndrome): a retrospective analysis from the chronic lymphocytic leukemia Subcommittee of the chronic leukemia Working Party and lymphoma Working Party of the European Group for blood and marrow transplantation. J Clin Oncol. 2012;30(18):2211–2217. | ||
Giné E, Martinez A, Villamor N, et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica. 2010;95(9):1526–1533. | ||
Soilleux EJ, Wotherspoon A, Eyre TA, Clifford R, Cabes M, Schuh AH. Diagnostic dilemmas of high-grade transformation (Richter’s syndrome) of chronic lymphocytic leukaemia: results of the phase II National Cancer Research Institute CHOP-OR clinical trial specialist haemato-pathology central review. Histopathology. 2016;69(6):1066–1076. | ||
Mao Z, Quintanilla-Martinez L, Raffeld M, et al. IgVH mutational status and clonality analysis of Richter’s transformation: diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol. 2007;31(10):1605–1614. | ||
Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008;142(2):202–215. | ||
Rossi D, Spina V, Forconi F, et al. Molecular history of Richter syndrome: origin from a cell already present at the time of chronic lymphocytic leukemia diagnosis. Int J Cancer. 2012;130(12):3006–3010. | ||
Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391–3401. | ||
Cherepakhin V, Baird SM, Meisenholder GW, Kipps TJ. Common clonal origin of chronic lymphocytic leukemia and high-grade lymphoma of Richter’s syndrome. Blood. 1993;82(10):3141–3147. | ||
Chigrinova E, Rinaldi A, Kwee I, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122(15):2673–2682. | ||
Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–2772. | ||
Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273–2288. | ||
Rossi D, Berra E, Cerri M, et al. Aberrant somatic hypermutation in transformation of follicular lymphoma and chronic lymphocytic leukemia to diffuse large B-cell lymphoma. Haematologica. 2006;91(10):1405–1409. | ||
Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389–1401. | ||
Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. | ||
Jamroziak K, Tadmor T, Robak T, Polliack A. Richter syndrome in chronic lymphocytic leukemia: updates on biology, clinical features and therapy. Leuk Lymphoma. 2015;56(7):1949–1958. | ||
Rinaldi A, Mensah AA, Kwee I, et al. Promoter methylation patterns in Richter syndrome affect stem-cell maintenance and cell cycle regulation and differ from de novo diffuse large B-cell lymphoma. Br J Haematol. 2013;163(2):194–204. | ||
Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–6908. | ||
Scandurra M, Rossi D, Deambrogi C, et al. Genomic profiling of Richter’s syndrome: recurrent lesions and differences with de novo diffuse large B-cell lymphomas. Hematol Oncol. 2010;28(2):62–67. | ||
Jamroziak K, Puła B, Walewski J. Current treatment of chronic lymphocytic leukemia. Curr Treat Options Oncol. 2017;18(1):5. | ||
Ahn IE, Underbayev C, Albitar A, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129(11):1469–1479. | ||
Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7(1):11589. | ||
Gounari M, Ntoufa S, Apollonio B, et al. Excessive antigen reactivity may underlie the clinical aggressiveness of chronic lymphocytic leukemia stereotyped subset #8. Blood. 2015;125(23):3580–3587. | ||
Ghiotto F, Fais F, Valetto A, et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113(7):1008–1016. | ||
Rossi D, Spina V, Cerri M, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15(13):4415–4422. | ||
Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121(8):1403–1412. | ||
Athanasiadou A, Stamatopoulos K, Gaitatzi M, Stavroyianni N, Fassas A, Anagnostopoulos A. Recurrent cytogenetic findings in subsets of patients with chronic lymphocytic leukemia expressing IgG-switched stereotyped immunoglobulins. Haematologica. 2008;93(3):473–474. | ||
Behdad A, Griffin B, Chen YH, et al. PD-1 is highly expressed by neoplastic B-cells in Richter transformation. Br J Haematol. Epub 2018 Jul 20. | ||
He R, Ding W, Viswanatha DS, et al. PD-1 expression in chronic lymphocytic Leukemia/Small lymphocytic lymphoma (CLL/SLL) and large B-cell Richter transformation (DLBCL-RT): a characteristic feature of DLBCL-RT and potential surrogate marker for clonal relatedness. Am J Surg Pathol. 2018;42(7):843–854. | ||
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236(1):219–242. | ||
Solh M, Rai KR, Peterson BL, et al. The impact of initial fludarabine therapy on transformation to Richter syndrome or prolymphocytic leukemia in patients with chronic lymphocytic leukemia: analysis of An intergroup trial (CALGB 9011). Leuk Lymphoma. 2013;54(2):252–254. | ||
Rossi D. Richter syndrome and fludarabine: a controversial relationship. Leuk Lymphoma. 2013;54(2):213–214. | ||
O’Brien SM, Kantarjian HM, Thomas DA, et al. Alemtuzumab as treatment for residual disease after chemotherapy in patients with chronic lymphocytic leukemia. Cancer. 2003;98(12):2657–2663. | ||
Karlsson C, Norin S, Kimby E, et al. Alemtuzumab as first-line therapy for B-cell chronic lymphocytic leukemia: long-term follow-up of clinical effects, infectious complications and risk of Richter transformation. Leukemia. 2006;20(12):2204–2207. | ||
Lepretre S, Aurran T, Mahé B, et al. Excess mortality after treatment with fludarabine and cyclophosphamide in combination with alemtuzumab in previously untreated patients with chronic lymphocytic leukemia in a randomized phase 3 trial. Blood. 2012;119(22):5104–5110. | ||
Jain P, Thompson PA, Keating M, et al. Long-term outcomes for patients with chronic lymphocytic leukemia who discontinue ibrutinib. Cancer. 2017;123(12):2268–2273. | ||
Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80–87. | ||
Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16(2):169–176. | ||
Mato AR, Nabhan C, Barr PM, et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood. 2016;128(18):2199–2205. | ||
Iskierka-Jaz·dz·ewska E, Hus M, Giannopoulos K, et al. Efficacy and toxicity of compassionate ibrutinib use in relapsed/refractory chronic lymphocytic leukemia in Poland: analysis of the Polish adult leukemia group (PALG). Leuk Lymphoma. 2017;58(10):2485–2488. | ||
Barrientos JC, Coutre S, De Vos S. Long-term follow-up of a phase Ib trial of idelalisib (IDELA) in combination with chemoimmunotherapy (CIT) in patients (PTS) with relapsed/refractory (R/R) CLL including PTS with del17p/TP53 mutation. J Clin Oncol. 2015;33(15 Suppl):7011–7011. | ||
Puła B, Budziszewska BK, Rybka J, et al. Comparable efficacy of idelalisib plus rituximab and ibrutinib in relapsed/refractory chronic lymphocytic leukemia: a retrospective case matched study of the Polish adult leukemia group (PALG). Anticancer Res. 2018;38(5):3025–3030. | ||
Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223. | ||
Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–2437. | ||
Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (Helios): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17(2):200–211. | ||
Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007. | ||
Jamroziak K, Szymczyk A, Hus M, et al. Hodgkin’s variant of Richter’s transformation during ibrutinib therapy in a series of CLL patients; the Polish adult leukemia Group report (PALG). Eur J Haematol. 2018;100(4):389–391. | ||
Glavey S, Quinn J, McCloy M, et al. Emergence of Bruton’s tyrosine kinase-negative Hodgkin lymphoma during ibrutinib treatment of chronic lymphocytic leukaemia. Eur J Haematol. 2017;99(4):378–380. | ||
Sachanas S, Pangalis GA, Moschogiannis M, et al. Hodgkin lymphoma transformation of chronic lymphocytic leukemia under ibrutinib therapy: chance association or therapy-related? Anticancer Res. 2017;37(6):3277–3280. | ||
Tsimberidou AM, O’Brien S, Khouri I, et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol. 2006;24(15):2343–2351. | ||
Rossi D, Lobetti Bodoni C, Genuardi E, et al. Telomere length is an independent predictor of survival, treatment requirement and Richter’s syndrome transformation in chronic lymphocytic leukemia. Leukemia. 2009;23(6):1062–1072. | ||
Kadri S, Lee J, Fitzpatrick C, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1(12):715–727. | ||
Miller CR, Ruppert AS, Heerema NA, et al. Near-tetraploidy is associated with Richter transformation in chronic lymphocytic leukemia patients receiving ibrutinib. Blood Adv. 2017;1(19):1584–1588. | ||
Balatti V, Tomasello L, Rassenti LZ, et al. miR-125a and miR-34a expression predicts Richter syndrome in chronic lymphocytic leukemia patients. Blood. 2018;132(20):2179–2182. | ||
Omoti CE, Omoti AE. Richter syndrome: a review of clinical, ocular, neurological and other manifestations. Br J Haematol. 2008;142(5):709–716. | ||
Robertson LE, Pugh W, O’Brien S, et al. Richter’s syndrome: a report on 39 patients. J Clin Oncol. 1993;11(10):1985–1989. | ||
Woroniecka R, Rymkiewicz G, Grygalewicz B, et al. Cytogenetic and flow cytometry evaluation of Richter syndrome reveals MYC, CDKN2A, IgH alterations with loss of CD52, CD62L and increase of CD71 antigen expression as the most frequent recurrent abnormalities. Am J Clin Pathol. 2015;143(1):25–35. | ||
Papajík T, Myslivecˇek M, Urbanová R, et al. 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography/computed tomography examination in patients with chronic lymphocytic leukemia may reveal Richter transformation. Leuk Lymphoma. 2014;55(2):314–319. | ||
Niemann CU, Polliack A, Hutchings M. Suspected Richter transformation: positron emission tomography/computed tomography tells us who should have a biopsy and where. Leuk Lymphoma. 2014;55(2):233–234. | ||
Bruzzi JF, Macapinlac H, Tsimberidou AM, et al. Detection of Richter’s transformation of chronic lymphocytic leukemia by PET/CT. J Nucl Med. 2006;47(8):1267–1273. | ||
Robak T, Malignancies S. Second malignancies and Richter’s syndrome in patients with chronic lymphocytic leukemia. Hematology. 2004;9(5–6):387–400. | ||
Robak T, Blonski JZ, Gora-Tybor J, et al. Second malignancies and Richter’s syndrome in patients with chronic lymphocytic leukaemia treated with cladribine. Eur J Cancer. 2004;40(3):383–389. | ||
Travis LB, Curtis RE, Hankey BF, Fraumeni JF. Second cancers in patients with chronic lymphocytic leukemia. J Natl Cancer Inst. 1992;84(18):1422–1427. | ||
Mato AR, Wierda WG, Davids MS, et al. Analysis of PET-CT to identify Richter’s transformation in 167 patients with disease progression following kinase inhibitor therapy. Blood. 2017;130(Suppl 1):834–834. | ||
Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123(11):1647–1657. | ||
Yun S, Vincelette ND, Abraham I, Puvvada S, Anwer F. Outcome comparison of allogeneic versus autologous stem cell transplantation in transformed low-grade lymphoid malignancies: a systematic review and pooled analysis of comparative studies. Acta Haematol. 2016;136(4):244–255. | ||
Ding W, Laplant BR, Call TG, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129(26):3419–3427. | ||
Visentin A, Imbergamo S, Scomazzon E, et al. BCR kinase inhibitors, idelalisib and ibrutinib, are active and effective in Richter syndrome. Br J Haematol. Epub 2018 Jul 5. | ||
Woyach JA. How I manage ibrutinib-refractory chronic lymphocytic leukemia. Blood. 2017;129(10):1270–1274. | ||
Langerbeins P, Busch R, Anheier N, et al. Poor efficacy and tolerability of R-CHOP in relapsed/refractory chronic lymphocytic leukemia and Richter transformation. Am J Hematol. 2014;89(12):E239–E243. | ||
Eyre TA, Clifford R, Bloor A, et al. NCRI phase II study of CHOP in combination with ofatumumab in induction and maintenance in newly diagnosed Richter syndrome. Br J Haematol. 2016;175(1):43–54. | ||
Rogers KA, Huang Y, Ruppert AS, et al. A single-institution retrospective cohort study of first-line R-EPOCH chemoimmunotherapy for Richter syndrome demonstrating complex chronic lymphocytic leukaemia karyotype as an adverse prognostic factor. Br J Haematol. 2018;180(2):259–266. | ||
Tsimberidou AM, Kantarjian HM, Cortes J, et al. Fractionated cyclophosphamide, vincristine, liposomal daunorubicin, and dexamethasone plus rituximab and granulocyte-macrophage-colony stimulating factor (GM-CSF) alternating with methotrexate and cytarabine plus rituximab and GM-CSF in patients with Richter syndrome or fludarabine-refractory chronic lymphocytic leukemia. Cancer. 2003;97(7):1711–1720. | ||
Tsimberidou AM, Wierda WG, Wen S, et al. Phase I-II clinical trial of oxaliplatin, fludarabine, cytarabine, and rituximab therapy in aggressive relapsed/refractory chronic lymphocytic leukemia or Richter syndrome. Clin Lymphoma Myeloma Leuk. 2013;13(5):568–574. | ||
Tsimberidou AM, Wierda WG, Plunkett W, et al. Phase I-II study of oxaliplatin, fludarabine, cytarabine, and rituximab combination therapy in patients with Richter’s syndrome or fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol. 2008;26(2):196–203. | ||
Durot E, Michallet AS, Leprêtre S, Le QH, Leblond V, Delmer A. Platinum and high-dose cytarabine-based regimens are efficient in ultra high/high-risk chronic lymphocytic leukemia and Richter’s syndrome: results of a French retrospective multicenter study. Eur J Haematol. 2015;95(2):160–167. | ||
Tsang M, Shanafelt TD, Call TG, et al. The efficacy of ibrutinib in the treatment of Richter syndrome. Blood. 2015;125(10):1676–1678. | ||
Hillmen P, Schuh A, Eyre TA. Acalabrutinib monotherapy in patients with Richter transformation from the phase 1/2 ACE-CL-001 clinical study. Blood. 2016;128(22):60–60. | ||
Davids MS, Roberts AW, Seymour JF, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol. 2017;35(8):826–833. | ||
Rogers KA, Huang Y, Dotson E, et al. Use of PD-1 (PDCD1) inhibitors for the treatment of Richter syndrome: experience at a single academic centre. Br J Haematol. Epub 2018 Jul 20. | ||
Kuruvilla J, Byrd JC, Flynn JM. The Oral Selective Inhibitor of Nuclear Export (SINE) selinexor (KPT-330) demonstrates broad and durable clinical activity in relapsed / refractory Non Hodgkin’s Lymphoma (NHL). Blood. 2014;124(21):396–396. | ||
Alderuccio JP, Mackrides N, Chapman JR, Vega F, Lossos IS. Rapid complete response to blinatumomab as a successful bridge to allogeneic stem cell transplantation in a case of refractory Richter syndrome. Leuk Lymphoma. Epub 2018 May 10. | ||
Evans A, Burack R, Rothberg PG, Porter D. Evolution to Plasmablastic Lymphoma (PBL) after CAR-T Cell Therapy in a Case of SLL/CLL with Richter’s Transformation. Blood. 2014;124(21):5660–5660. | ||
Chavez JC, Locke FL. CAR T cell therapy for B-cell lymphomas. Best Pract Res Clin Haematol. 2018;31(2):135–146. | ||
Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285–295. | ||
Tsimberidou AM, O’Brien S, Kantarjian HM, et al. Hodgkin transformation of chronic lymphocytic leukemia: the M. D. Anderson Cancer Center experience. Cancer. 2006;107(6):1294–1302. | ||
Brecher M, Banks PM. Hodgkin’s disease variant of Richter’s syndrome. Report of eight cases. Am J Clin Pathol. 1990;93(3):333–339. | ||
Tadmor T, Shvidel L, Goldschmidt N, et al. Hodgkin’s variant of Richter transformation in chronic lymphocytic leukemia; a retrospective study from the Israeli CLL Study Group. Anticancer Res. 2014;34(2):785–790. | ||
Jamroziak K, Grzybowska-Izydorczyk O, Jesionek-Kupnicka D, Gora-Tybor J, Robak T. Poor prognosis of Hodgkin variant of Richter transformation in chronic lymphocytic leukemia treated with cladribine. Br J Haematol. 2012;158(2):286–288. | ||
Bockorny B, Codreanu I, Dasanu CA. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: a retrospective analysis of world literature. Br J Haematol. 2012;156(1):50–66. | ||
Parikh SA, Habermann TM, Chaffee KG, et al. Hodgkin transformation of chronic lymphocytic leukemia: Incidence, outcomes, and comparison to de novo Hodgkin lymphoma. Am J Hematol. 2015;90(4):334–338. | ||
Mauro FR, Galieni P, Tedeschi A, et al. Factors predicting survival in chronic lymphocytic leukemia patients developing Richter syndrome transformation into Hodgkin lymphoma. Am J Hematol. 2017;92(6):529–535. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.