Back to Journals » Journal of Inflammation Research » Volume 13
Immune-Mediated Necrotizing Myopathy Initially Presenting as Erythema Nodosum
Authors Ying S, Li S, Tang S, Sun Q, Fang D, Li Y, Zhu D, Fang H, Qiao J
Received 17 July 2020
Accepted for publication 11 August 2020
Published 24 August 2020 Volume 2020:13 Pages 471—476
DOI https://doi.org/10.2147/JIR.S270114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Shuni Ying, Sheng Li, Shunli Tang, Qingmiao Sun, Deren Fang, Yali Li, Dingxian Zhu, Hong Fang, Jianjun Qiao
Department of Dermatology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, People’s Republic of China
Correspondence: Jianjun Qiao; Hong Fang
Department of Dermatology, The First Affiliated Hospital, Zhejiang University School of Medicine, No. 79, Qingchun Road, Hangzhou 310003, People’s Republic of China
Tel +86-571-87236385
Email [email protected]; [email protected]
Abstract: Immune-mediated necrotizing myopathy (IMNM) is a type of autoimmune myopathy characterized by severe diffuse proximal myofiber necrosis in the context of inflammatory myopathy. Autoantibodies of anti-signal recognition particle and anti-hydroxy-3-methylglutaryl-CoA reductase are two antibodies specific to IMNM. Erythema nodosum (EN) is often accompanied by various systemic diseases, such as autoimmune diseases. Herein, we report a female patient with signal recognition particle-associated IMNM, with EN as the first presentation. She showed significant clinical improvement after the initiation of glucocorticoids, intravenous immunoglobulin, rituximab, and mycophenolate mofetil. This case indicates that IMNM can initially present as EN. IMNM and EN might have overlapping pathogeneses.
Keywords: immune-mediated necrotizing myopathy, erythema nodosum, autoantibody
Introduction
Immune-mediated necrotizing myopathy (IMNM), also known as necrotizing autoimmune myopathy, is a type of immune-mediated myopathy (IMM) characterized by subacute and predominantly proximal muscle weakness.1–4 IMNM comprises up to 10% of inflammatory myopathies.2 It may also involve muscles of the respiratory and cardiovascular systems, which can cause severe morbidity and mortality.2,5-7
The recognized risk factors for IMNM include drug exposure (statins, pembrolizumab, and nivolumab),8 connective tissue diseases, cancer (commonly gastrointestinal adenocarcinomas, and lung cancer)9,10 and rarely, human immunodeficiency virus infection.4
The muscular histopathology of IMNM shows significant myofiber necrosis and minimal lymphocytic infiltrates without perifascicular atrophy.2,4,11 MRI can be used to investigate soft tissue abnormalities, such as muscle edema, muscle atrophy, fascial edema and fatty replacement, in patients with myositis.12 Combining with the findings of two myositis-specific antibodies, immune mechanisms are considered to account for the pathogenesis widely.13 Around two-thirds of patients with IMNM have autoantibodies recognizing either signal recognition particle (SRP) or 3-hydroxy-3-methylglutaryl-coenzyme-A reductase (HMGCR).11,14
Skin manifestations occur in less than 10% of patients with IMNM,2 including heliotrope rash, Gottron papules, V-sign, Shawl sign, Mechanic’s hands, and skin hyperpigmentation.15–17
Erythema nodosum (EN) is a common form of acute nodular septal panniculitis without signs of vasculitis.18–22 It is clinically characterized by bilaterally symmetrical red and painful subcutaneous nodules, mainly localized to the lower extremities, pretibial areas especially.18,19 EN is often associated with multisystemic diseases, including autoimmune diseases and hypersensitivity reactions in response to antigens or triggers. Including infection (eg, Streptococcus spp, Mycobacterium tuberculosis), drug, malignancy, inflammatory bowel disease, autoimmune atrophic gastritis, sarcoidosis, pregnancy and other causes.19–23
Here we present a case of IMNM that started with acute EN before the development of a subtype of IMNM identified by anti-SRP antibody, followed by significant clinical improvement after the initiation of glucocorticoids, intravenous immunoglobulin (IVIG), rituximab and mycophenolate mofetil. The report was approved by the ethics committee of the First Affiliated Hospital, Zhejiang University School of Medicine (Approved number: 2020-ITT-356) prior to study commencement. A signed copy of consent form for publication was obtained from the patient.
Case Presentation
In June 2019, a 34-year-old woman presented to her primary care provider with painful, swollen erythematous nodules located on the bilateral pretibial areas (Figure 1). The patient denied any other symptoms such as fever, cough, dyspnea, oral ulcer, abdominal pain, diarrhea, arthralgia, muscular weakness, fatigue, or weight loss. She denied past medical history and CT scanning of the lungs was normal. Laboratory tests were unremarkable, except for serum positive SSA and SSA-52. The histopathology of a biopsy taken from an erythematous nodule on the right leg showed interlobular septal panniculitis, and infiltration with predominately lymphocytes and histiocytes (Figure 2). Her skin rash gradually subsided with treatment of hydroxychloroquine, 0.2 g twice a day for a month.
 |
Figure 1 Erythema nodosum of the patient. Erythematous subcutaneous nodules in the pretibial area of bilateral lower extremities. |
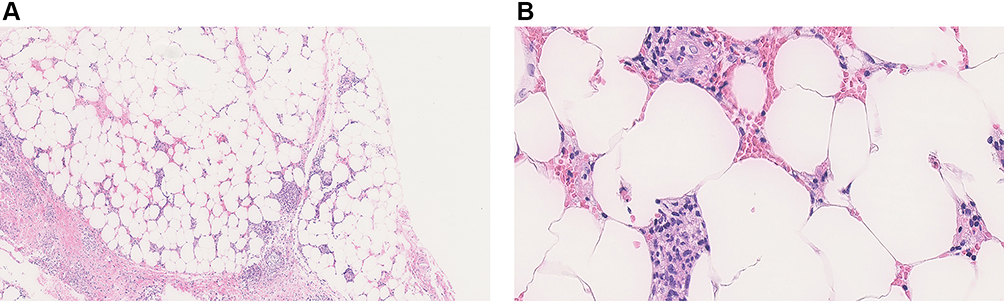 |
Figure 2 Histopathology of erythema nodosum. (A) Low-power (original magnification ×50) and (B) high-power microphotographs of the biopsy showing lymphocyte, neutrophil, and histiocyte infiltration of the interlobular septal in subcutaneous adipose tissue (original magnification ×400). |
One month later, she felt fatigued and developed progressive proximal muscle weakness, and myalgias involving her upper and lower limbs. Her weakness was so severe that she had difficulty climbing stairs, rising from a chair, and getting out of bed without assistance. In addition, she complained of an unintentional weight loss of 5 kg. She denied any arthralgia, ptosis, vision changes, dysphagia, dysarthria, dyspnea, Raynaud’s phenomenon, numbness or tingling, paresthesia, or other rashes except for erythematous nodules on the legs.
On physical examination, she had 3/5 strength in bilateral upper extremities and 2/5 strength in bilateral lower extremities using a Medical Research Council scale. No neck weakness or muscle atrophy were found. Deep tendon reflexes and sensation were normal. The erythematous nodules on the legs were poorly demarcated, 1–3 cm in diameter, and mildly painful on palpation.
The results of laboratory investigations are listed in Table 1. The electromyogram revealed myopathic changes including fibrillation potentials and positive sharp waves in biceps, triceps, deltoid, iliopsoas and quadriceps. Findings from PET-CT, chest radiography, and heart and abdominal ultrasound were unremarkable.
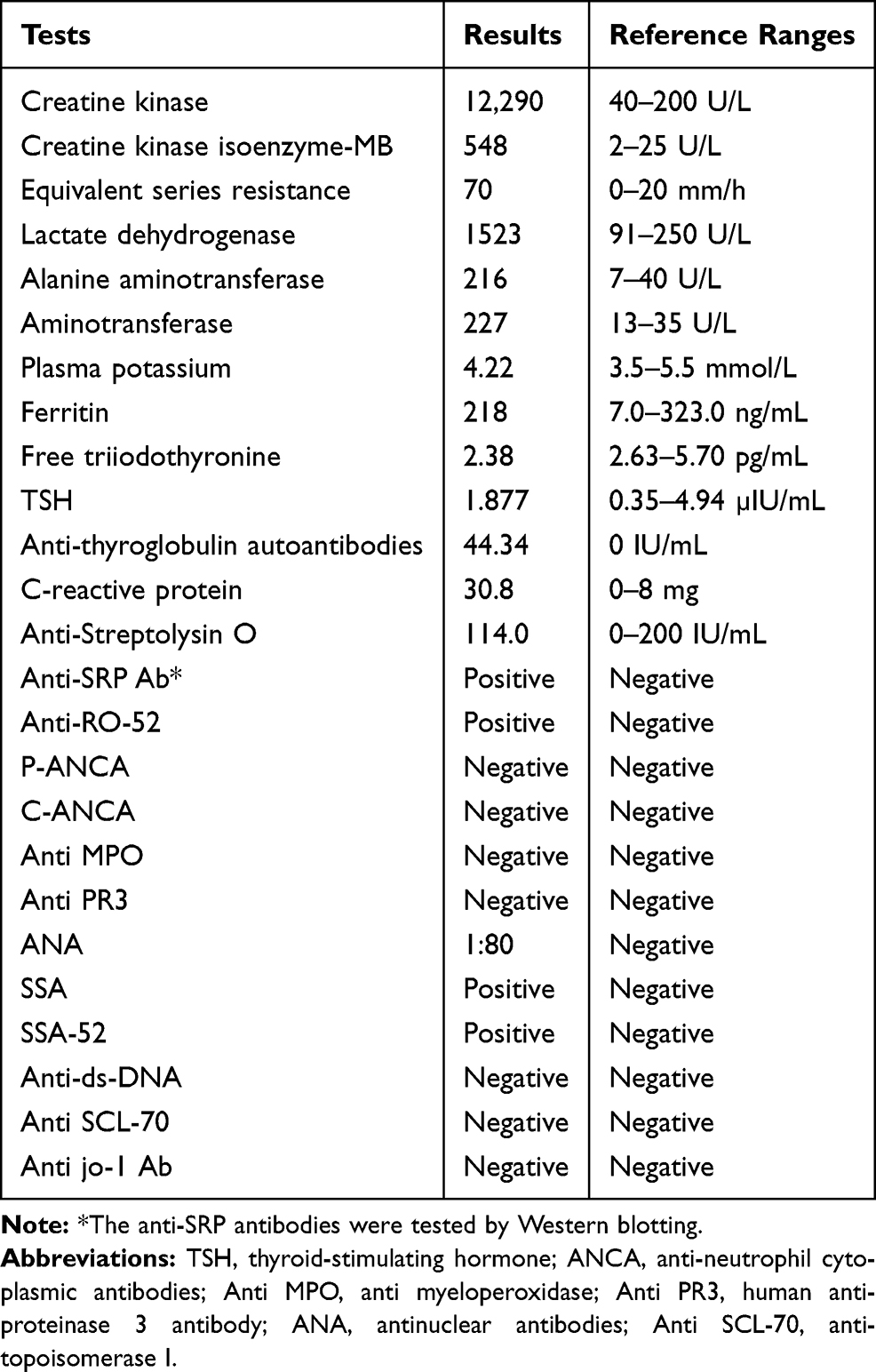 |
Table 1 Laboratory Test Results of the Patient with Anti-SRP-Associated Immune-Mediated Necrotizing Myopathy |
In view of the proximal muscle weakness, increased muscle enzyme levels, myopathic patterned electromyogram, and presence of positive anti-SRP autoantibody, a diagnosis of anti-SRP-associated IMNM was made.
Treatment protocol for this patient was showed in Figure 3. Treatment with methylprednisolone, 40 mg daily, was initiated for 9 days then the dose was increased to 80 mg daily, with less improvement in weakness. At day 26, due to progression of muscle weakness and development of dysphagia, 20 g of intravenous immunoglobulin, daily, and 200 mg of hydroxychloroquine, twice a day, were added to the regime. At day 32, the patient had 4/5 strength in neck muscles and choking, and was given 500 mg of methylprednisolone, daily for 6 days. At day 39, the strength in bilateral lower extremities was 3/5, she received 100 mg rituximab once and methylprednisolone 80mg per day. At day 40, she received rituximab 500 mg, resulting in a prompt decrease in her B-cell count, from 941/μL to 3/μL. At day 47, 250 mg of mycophenolate mofetil, twice a day, was added to support the treatment.
 |
Figure 3 Treatments of the patient with anti-SRP-associated immune-mediated necrotizing myopathy. The patient was not responsive to the usual dose of glucocorticoids. Hip flexion power and serum creatine kinase level gradually decreased to normal after rituximab and mycophenolate mofetil were added to the therapeutic schedule. |
After treatment, the patient’s strength in bilateral lower extremities increased to 4/5, creatine kinase (CK) reduced to 129 U/L, alanine aminotransferase reduced to 33 U/L, and aminotransferase reduced to 15 U/L. She was subsequently discharged from the hospital and continued long-term treatment with methylprednisolone and mycophenolate mofetil.
Discussion
Anti-SRP antibodies have been reported in up to 16% of IMNM cases,10 and patients who are positive for anti-SRP autoantibodies are correlated with more severe and aggressive diseases such as cardiac complications,24 respiratory insufficiency (eg, interstitial lung disease),1 muscle weakness (eg, dysphagia),6,11 and less skin lesions,11 in comparison with anti-HMGCR myopathy.
In our patient, EN preceded the occurrence of IMNM. Very recently, it was reported that EN can also present with necrotizing myopathy.25 Autoantibodies, including SRP antibody, can induce muscle atrophy with high levels of inflammatory cytokines, including Interleukin-6 (IL-6), tumor necrosis factor α (TNF-α) and reactive oxygen species. In addition, lower levels of IL-4 and IL-13 to impair myoblast fusion.26 Both IMNM and EN can present with high levels of IL-6 and TNF-α.20,26 The shared inflammatory cytokines might explain the concurrence of the two entities.
The optimal treatment strategy in IMNM has not been determined clinically, therefore it must be individualized. This patient’s CK began to reduce and her muscle power returned slowly after methylprednisolone infusion, but dysphagia and choking occurred. After administration of intravenous immunoglobulin and rituximab, the strength in bilateral lower extremities gradually improved to 4/5. Immunotherapy is the main treatment for patients with IMNM. First-line immunotherapy is glucocorticoids, such as oral glucocorticoids at 1 mg/kg/day.14 If the patient has severe muscle weakness, 0.5 to 1 g/day of intravenous glucocorticoids can be given for 3–5 days.13,14 But the addition of an appropriate high-dose second-line agent (eg, methotrexate, azathioprine, and mycophenolate mofetil) is often required because prednisone monotherapy is rarely sufficient, especially for patients with anti-SRP myopathy.5,6 Rituximab or IVIG could be used first line in anti-SRP-associated or anti-HMGCR-associated IMNM.14 If initial treatments are ineffective, all patients should use rituximab and IVIG at 6 months to get better prognosis.13,14 A retrospective review of a large cohort of patients at the Mayo Clinic suggested that a combination of high-dose glucocorticoids, an immunosuppressant and IVIG for more than 3 months, and then, followed by a steroid-sparing agent for a long time would have a significantly better clinical response.2,4 Correlation between antibody seropositivity and clinical outcome had not been found.4
Due to predominantly proximal muscle weakness and positive laboratory tests, especially the presence of myositis-specific autoantibody, our patient declined to do a muscle biopsy. Recently, the new European Neuromuscular Centre recommendations indicated that muscle biopsy is not strictly required to diagnose anti-HMGCR or anti-SRP myopathy,2,14 and autoantibodies should be evaluated early when serum CK >1000 IU/L.1 However, it is still needed to guide medication if necessary, as serological assay results may take several weeks. For those that are autoantibody-negative, to exclude alternative diagnoses, a muscle biopsy must be performed to establish a diagnosis of IMNM.11,14
In conclusion, we report a case of IMNM associated with EN. EN may correlate with various inflammatory diseases.19–23 In clinical practice, IMNM can be listed as an EN-associated entity. Our case also indicates that the IMNM and EN might have overlapping pathogeneses.
Ethics and Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. The report was approved by the ethics committee of the First Affiliated Hospital, Zhejiang University School of Medicine (Approved number: 2020-ITT-356) prior to study commencement.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest and report no conflicts of interest for this work.
References
1. Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry. 2016;87(10):1038–1044. PubMed PMID: 27147697. doi:10.1136/jnnp-2016-313166
2. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep. 2018;20(4):21. PubMed PMID: 29582188; PubMed Central PMCID: PMCPMC6019613. doi:10.1007/s11926-018-0732-6
3. Tiniakou E, Christopher-Stine L. Immune-mediated necrotizing myopathy associated with statins: history and recent developments. Curr Opin Rheumatol. 2017;29(6):604–611. PubMed PMID: 28857949. doi:10.1097/bor.0000000000000438
4. Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol. 2015;72(9):996–1003. PubMed PMID: 26192196. doi:10.1001/jamaneurol.2015.1207
5. Liang E, Rastegar M. Immune-mediated necrotising myopathy: a rare cause of hyperCKaemia. BMJ Case Rep. 2018;2018. PubMed PMID: 29691272. doi:10.1136/bcr-2017-223870
6. Milone M. Diagnosis and management of immune-mediated myopathies. Mayo Clin Proc. 2017;92(5):826–837. PubMed PMID: 28473041. doi:10.1016/j.mayocp.2016.12.025
7. Pitlick M, Ernste F. Anti-HMGCR myopathy presenting with acute systolic heart failure. BMJ Case Rep. 2019;12(5):e230213. PubMed PMID: 31068355. doi:10.1136/bcr-2019-230213
8. Mammen AL. Statin-associated autoimmune myopathy. N Engl J Med. 2016;374(7):664–669. PubMed PMID: 26886523. doi:10.1056/NEJMra1515161
9. Allenbach Y, Keraen J, Bouvier AM, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain. 2016;139(Pt 8):2131–2135. PubMed PMID: 27086869. doi:10.1093/brain/aww054
10. McGrath ER, Doughty CT, Amato AA. Autoimmune myopathies: updates on evaluation and treatment. Neurotherapeutics. 2018;15(4):976–994. PubMed PMID: 30341597; PubMed Central PMCID: PMCPMC6277300. doi:10.1007/s13311-018-00676-2
11. Selva-O’Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JM, Mammen AL. Classification and management of adult inflammatory myopathies. Lancet Neurol. 2018;17(9):816–828. PubMed PMID: 30129477. doi:10.1016/s1474-4422(18)30254-0
12. Pinal-Fernandez I, Casal-Dominguez M, Carrino JA, et al. Thigh muscle MRI in immune-mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann Rheum Dis. 2017;76(4):681–687. PubMed PMID: 27651398. doi:10.1136/annrheumdis-2016-210198
13. Day JA, Limaye V. Immune-mediated necrotising myopathy: a critical review of current concepts. Semin Arthritis Rheum. 2019;49(3):420–429. PubMed PMID: 31109639. doi:10.1016/j.semarthrit.2019.04.002
14. Allenbach Y, Mammen AL, Benveniste O, Stenzel W. 224th ENMC International Workshop:: clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul Disord. 2018;28(1):87–99. PubMed PMID: 29221629. doi:10.1016/j.nmd.2017.09.016
15. Suzuki S, Nishikawa A, Kuwana M, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: case series of 100 patients. Orphanet J Rare Dis. 2015;10:61. PubMed PMID: 25963141; PubMed Central PMCID: PMCPMC4440264. doi:10.1186/s13023-015-0277-y
16. Yongchairat K, Tanboon J, Waisayarat J, et al. Clinical spectrums and outcomes of necrotizing autoimmune myopathy versus other idiopathic inflammatory myopathies: a multicenter case-control study. Clin Rheumatol. 2019;38(12):3459–3469. PubMed PMID: 31446540. doi:10.1007/s10067-019-04756-2
17. Patil AK, Prabhakar AT, Sivadasan A, Alexander M, Chacko G. An unusual case of inflammatory necrotizing myopathy and neuropathy with pipestem capillaries. Neurol India. 2015;63(1):72–76. PubMed PMID: 25751473. doi:10.4103/0028-3886.152642
18. Chowaniec M, Starba A, Wiland P. Erythema nodosum - review of the literature. Reumatologia. 2016;54(2):79–82. PubMed PMID: 27407284; PubMed Central PMCID: PMCPMC4918048. doi:10.5114/reum.2016.60217
19. Inamadar AC, Adya KA. The rash with painful and erythematous nodules. Clin Dermatol. 2019;37(2):129–135. PubMed PMID: 30981293. doi:10.1016/j.clindermatol.2018.12.006
20. De Simone C, Caldarola G, Scaldaferri F, et al. Clinical, histopathological, and immunological evaluation of a series of patients with erythema nodosum. Int J Dermatol. 2016;55(5):e289–e294. PubMed PMID: 26917228. doi:10.1111/ijd.13212
21. Leung AKC, Leong KF, Lam JM. Erythema nodosum. World J Pediatr. 2018;14(6):548–554. PubMed PMID: 30269303. doi:10.1007/s12519-018-0191-1
22. Greuter T, Navarini A, Vavricka SR. Skin manifestations of inflammatory bowel disease. Clin Rev Allergy Immunol. 2017;53(3):413–427. doi:10.1007/s12016-017-8617-4
23. Eugénio G, Tavares J, Ferreira JF, Malcata A. Unusual association between erythema nodosum and autoimmune atrophic gastritis. BMJ Case Rep. 2018;2018:bcr2017223638. PubMed PMID: 29545439. doi:10.1136/bcr-2017-223638
24. Rider LG, Shah M, Mamyrova G, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013;92(4):223–243. PubMed PMID: 23877355; PubMed Central PMCID: PMCPMC3721421. doi:10.1097/MD.0b013e31829d08f9
25. Sato K, Takahashi Y, Yamashita T, et al. A unique case of hemi-tongue pseudohypertrophy, necrotizing myopathy, and erythema nodosum. Neurol Int. 2018;10(4):7852. PubMed PMID: 30687466; PubMed Central PMCID: PMCPMC6322047. doi:10.4081/ni.2018.7852
26. Arouche-Delaperche L, Allenbach Y, Amelin D, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol. 2017;81(4):538–548. PubMed PMID: 28224701. doi:10.1002/ana.24902
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.