Back to Journals » ImmunoTargets and Therapy » Volume 10
Immune-Checkpoint Inhibitors Combinations in Metastatic NSCLC: New Options on the Horizon?
Authors Passiglia F, Reale ML ![]() , Cetoretta V, Novello S
, Cetoretta V, Novello S
Received 20 November 2020
Accepted for publication 13 January 2021
Published 5 February 2021 Volume 2021:10 Pages 9—26
DOI https://doi.org/10.2147/ITT.S253581
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Francesco Passiglia, Maria Lucia Reale, Valeria Cetoretta, Silvia Novello
Department of Oncology, University of Turin, S. Luigi Gonzaga Hospital, Orbassano (TO), Italy
Correspondence: Silvia Novello
Department of Oncology, University of Turin, San Luigi Gonzaga Hospital, Orbassano, Italy
Email [email protected]
Abstract: The therapeutic targeting of the programmed death-1 (PD-1)/programmed death ligand-1 (PD-L1) axis marked a milestone in the treatment of non-small cell lung cancer (NSCLC), leading to unprecedented response duration and long-term survival for a relevant subgroup of patients affected by non-oncogene-addicted, metastatic disease. However, the biological heterogeneity as well as the occurrence of innate/acquired resistance are well-known phenomena which significantly affect the therapeutic response to immunotherapy. To date, we are moving towards the second phase of the “immune-revolution”, characterized by the advent of new immune-checkpoint inhibitors combinations, aiming to target the main resistance pathways and ultimately increase the number of NSCLC patients who may derive long-term clinical benefit from immunotherapy. In this review, we provide an updated and comprehensive overview of the main PD-1/PD-L1 inhibitors’ combination approaches under clinical investigation in non-oncogene addicted, metastatic NSCLC patients, including checkpoints (other than CTLA-4) as well as “immune-metabolism” modulators, DNA repair pathway inhibitors, antiangiogenic agents, cytokines, and a new generation of vaccines, with the final aim of identifying the most promising options on the horizon.
Keywords: immune-checkpoint, PD-1/PD-L1, resistance, combinations, non-small cell lung cancer
Introduction
The application of immune-checkpoint inhibition paradigm to medical oncology is anticipated to improve all areas of cancer treatment, including the clinical management of lung cancer patients.
Since the introduction of programmed death-1 (PD-1)/programmed death ligand-1 (PD-L1) inhibitors in the treatment algorithm of metastatic non-small cell lung cancer (NSCLC), we have witnessed a progressive increase of long-term survival and quality of life for a relevant subgroup of patients, with about 30% of them now alive at 5 years, when receiving upfront pembrolizumab for high PD-L1 expressing tumors, as compared to 5.5% in the chemotherapy era.1
Alongside single-agent immune-checkpoint inhibitors (ICIs), different randomized Phase III studies consistently demonstrated as the addition of immunotherapy to upfront chemotherapy synergistically enhance the anti-tumor immune-response and prolong patients’ survival, regardless of tumor histotype and PD-L1 expression levels,2,3 thus becoming a new standard of care worldwide and ultimately extending the percentage of patients who may derive clinical benefit from ICI-therapies. Another investigated strategy included the development of “chemo-free” regimens, based on the combination of different ICIs with potential complementary effects, such as the PD1 and CTLA-4 inhibitors. Two main randomized phase III clinical trials compared a dual checkpoint blockade versus first-line platinum chemotherapy in patients with epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) wild-type advanced NSCLC, showing controversial results in terms of efficacy. Indeed, the checkmate-227 study4 demonstrated a significant increase in patients’ overall survival (OS) in favour of the nivolumab plus ipilimumab combination, regardless of tumor PD-L1 expression as well as mutation burden (TMB) levels, leading to the Food and Drug Administration (FDA) approval of such combination for the treatment of naïve advanced NSCLC patients with tumor PD-L1 >1%. Conversely, no survival advantages have been reported for the association of durvalumab and tremelimumab within the different PD-L1 expression subgroups analyzed in the MYSTIC trial,5 while the final results of the randomized NEPTUNE study (NCT02542293) are still pending. More recently a dual PD-1/CTLA-4 blockade combined with a short course (two cycles) of platinum-chemotherapy revealed to be more effective and equally tolerated than chemotherapy alone, increasing tumor response and OS of non-oncogene addicted, metastatic NSCLC patients included within the checkmate-9LA study,6 emerging as an additional potential upfront option for clinical use.
In this exciting scenario, characterized by the continuous advent of innovative drugs and treatment combinations, the occurrence of innate/acquired resistance to immunotherapy is a well-known phenomenon which significantly affect ICIs’ response at individual patients’ level.7 Therefore, developing new ICIs-based combination strategies targeting the main resistance pathways and identifying baseline predictive biomarkers for ICI response, beyond PD-L1, represent the areas of intense current investigation.
In this review, we provide an updated and comprehensive overview of the main ICIs-based combination approaches under clinical investigation in non-oncogene addicted, metastatic NSCLC patients, including checkpoints (other than CTLA-4) as well as “immune-metabolism” modulators, DNA repair pathway inhibitors, antiangiogenic agents, cytokines, and a new generation of vaccines, with the final aim of identifying the most promising treatment options on the horizon.
Combinations with Antiangiogenic Agents
It is well known that neoangiogenesis and immune-escape are interconnected processes. The irregular blood vessels enable immune evasion and decrease anti-cancer therapies’ efficacy by limiting the transportation of molecules, oxygen, and cytotoxic T cells from the bloodstream to the tumor environment. As consequence, the resulting hypoxia induces the upregulation of immune checkpoints as well as the infiltration of immunosuppressive components, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSC), within the tumor microenvironment (TME).8 Besides stimulating angiogenesis, vascular endothelial growth factor (VEGF) signaling acts itself as an immunosuppressive cytokine and its expression has been related to immunotherapy resistance. Indeed, VEGF signaling inhibits the binding of lymphocytes to endothelial cells and prevents T cell mobilization and tumor infiltration by upregulating Fas ligand. Furthermore, it promotes Tregs induction and proliferation and suppresses the development of dendritic cells.9
In this context, the use of antiangiogenic agents represents a valuable strategy to adjust tumor vascularization, expand immune cells’ infiltration, reduce hypoxia as well as the recruitment of immune-suppressive cells, thus ultimately turning into an immune-responsive TME.10
In preclinical NSCLC models, anti-PD-L1 in association with vascular endothelial growth factor receptor (VEGFR)2 inhibitors significantly reduces PD-L1 expression, increases tumor-infiltrating lymphocyte (TILs) and decreases Tregs and MDSC, thus showing synergistic and complementary immune-vascular interactions11 and ultimately supporting the combination of antiangiogenic agents and immunotherapy in the clinical setting. Different studies investigating the combinations of ICIs and antiangiogenics (both monoclonal antibodies targeting VEGF, such as bevacizumab, VEGFR, such as ramucirumab, and small molecule tyrosine kinase inhibitors, TKIs) have been conducted, others are currently ongoing (Table 1), and available preliminary data look promising.
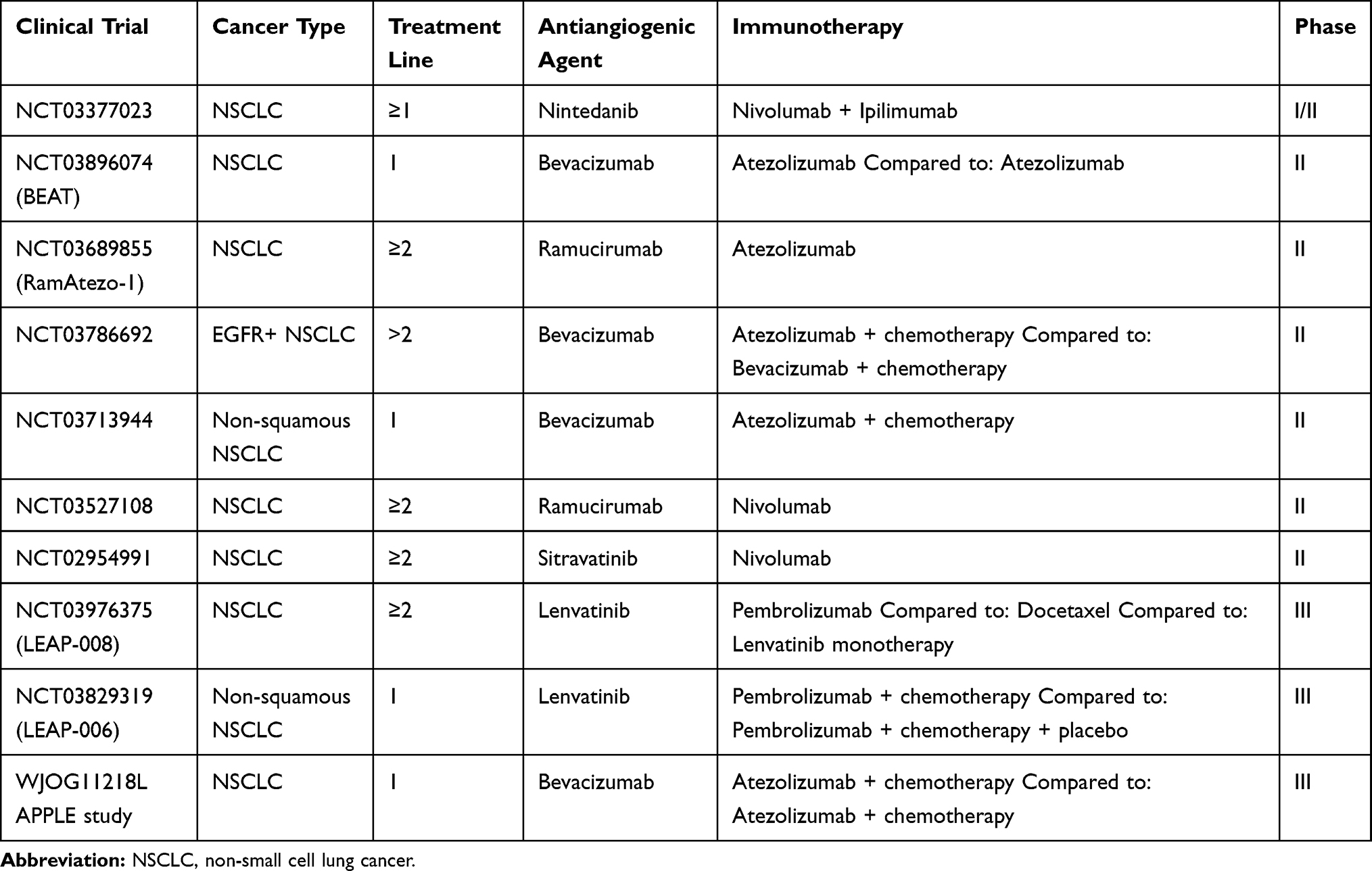 |
Table 1 Main Ongoing Clinical Trials of Antiangiogenics Agents Combined with ICIs |
The phase III randomized IMpower-150 trial first demonstrated as the upfront combination of Atezolizumab, chemotherapy and bevacizumab significantly improved OS compared to bevacizumab plus chemotherapy alone in patients with non-squamous advanced NSCLC (hazard ratio (HR): 0.78; 95% CI, 0.64 to 0.96).12 Importantly, this benefit was demonstrated despite PD-L1 expression, with tolerability profile consistent to the previously reported safety risks of the individual drugs, getting the regulatory approval for clinical use.
The efficacy of the combination of atezolizumab plus bevacizumab as first-line chemo-free regimen has been also demonstrated by the WJOG@Be study, including patients with non-squamous NSCLC and PD-L1 expression ≥50%. The positive results of this trial have been presented recently at the European Society of Medical Oncology (ESMO) 2020 meeting, showing a considerable ORR of 64.1% with a median duration of response of 10.4 months.13 A Phase II randomized trial comparing atezolizumab versus atezolizumab plus bevacizumab as first-line treatment in PD-L1-positive advanced NSCLC (BEAT trial) is currently ongoing.
The JVDJ trial is a Phase 1a/b single-arm, non-randomized, multi-cohort study of ramucirumab plus durvalumab in previously treated patients with NSCLC, gastric/gastro-esophageal junction adenocarcinoma (gastric/GEJ), or hepatocellular carcinoma (HCC). ORR was 11%, median PFS 2.7 months and OS 11.0 months in the NSCLC cohort. Patients with PD-L1–high tumours (TPS ≥ 25%) had longer median PFS (4.1 versus 2.6 months) and median OS (16.4 versus 7.5) than the patients in the PD-L1–low subgroup (TPS < 25%).14
Similarly, the JVDF trial, is a multicohort, non-randomized, phase 1a/b study, enrolling previously treated patients with gastric/GEJ adenocarcinoma, NSCLC or urothelial carcinoma. The combination of ramucirumab and the anti–PD-1 pembrolizumab in the NSCLC cohort exhibited durable activity, with higher responses in PD-L1 positive versus PD-L1–negative patients (unselected: PFS 9.7, OS 26.2 months; PD-L1–TPS ≥ 1%: PFS 6.9, OS 26.7 months; PD-L1 TPS < 1%: PFS 9.7, OS 17 months)15. Overall the preliminary data emerging from both studies are promising and revealed an impressing activity along with a manageable safety profile for the association of ramucirumab and pembrolizumab in NSCLC patients, which need to be confirmed within randomized clinical studies.
Recently, the number of studies exploring antiangiogenics plus ICIs combination has progressively increased if we consider also the wide spectrum of multi-target TKIs with antiangiogenic properties tested in the clinical setting (Table 1). During the 2019 World Conference on Lung Cancer (WCLC) Han et al presented the results of a multi-cohort phase Ib trial, testing the association of sintilimab (anti-PD-1 monoclonal antibody) with anlotinib (multi-target TKI) in a treatment-naïve cohort of patients with advanced NSCLC. The combination demonstrated encouraging anti-tumor activity regardless of PD-L1 expression, with an ORR of 72.7%, a disease control rate (DCR) of 100%, and a 6-month PFS rate of 93.8%. Interestingly, in the patient subgroups with high and low TMB, the ORR was 85.7% and 63.6%, respectively,16 while CCL2 (chemokine ligand 2) serological levels seem to correlate with anlotinib efficacy.17
In a phase Ib/II trial, the multitarget agent lenvatinib in combination with pembrolizumab showed considerable antitumor activity in the cohort of patients with advanced NSCLC, leading to a 24 weeks ORR of 33%, a median duration of response of 10.9 months and a median PFS of 5.9 months.18
The LEAP-006 is a two-part, phase III study evaluating upfront lenvatinib plus pembrolizumab plus platinum-based chemotherapy in patients with metastatic non-squamous NSCLC. The results of part 1 have been presented at the ESMO 2020 meeting, showing acceptable tolerability and preliminary evidence of antitumor activity: 13 patients were enrolled receiving a median of 10 cycles of combination therapy obtaining an ORR of 69.2%. Enrollment in part 2 is ongoing.19 Lenvatinib is being also tested in combination with pembrolizumab versus docetaxel within a phase III randomized trial (LEAP-008), including patients with metastatic NSCLC who failed prior immunotherapy and platinum-doublet chemotherapy, in order to evaluate the ability of this TKI to restore ICI sensitivity in this cohort of patients. Unfortunately, predictive biomarkers to select the best candidate to antiangiogenic and immunotherapy combination in the clinical setting represent still an unmet need.
Combinations with Other Checkpoint Inhibitors
The combination of anti-PD-1 agents with other ICIs is based on the assumption that non-responsive tumors use immunosuppressive pathways different than PD-1-PD-L1 axis, including both co-inhibitory and co-stimulatory receptors, according to their selective target.
LAG-3
Lymphocyte-activation Gene-3 (LAG-3 or CD223) is a 498 amino acid type I transmembrane protein binding MHC class II molecules.20,21 Alternative ligands, such as Galectin-3, LESCtin, alfa-synuclein fibers, were proposed to explain some LAG-3 suppressive effects in absence of MHC class II. Moreover, a recent work identified FGL-1 as a novel, high-affinity and cell-free, LAG-3 ligand, involved in the tumor immune evasion processes.22 Although both tissue distribution and functional roles of LAG-3 in human malignancies have been investigated within several studies,21 they have not been clearly defined yet. High PD-1 e LAG-3 expression by cytotoxic T CD8+ and NKT cells23 is consistent with the regulatory action expected from such molecules within effector cells. Both targets are also highly expressed by Tregs,23 suggesting that may have additional, cell-specific suppressive functions, and play a potential role in the development of immune tolerance. Of note, both PD-1 and LAG-3 are preferentially expressed on activated TILs, even though such molecules are considered exhaustion markers,24,25 confirming that coinhibitory receptors become upregulated upon T-cell stimulation in order to limit exaggerated responses and potential tissue damage.
The clinical interest towards these molecules resides in the negative association between the over-expression of LAG-3 and the survival of anti-PD-1-treated NSCLC patients. Furthermore, LAG-3 was found to be upregulated in NSCLC patients developing acquired resistance to PD-1 inhibitors. These data apparently suggest that tumors characterized by LAG-3-mediated immune-escape, are less sensitive to PD-1 inhibition, thus paving the way to the association of PD-1 and LAG3 ICIs as therapeutic strategy for non-oncogene addicted advanced NSCLC.
Recent results from a dose escalation phase I/II study of LAG525 ± Spartalizumab in metastatic malignancies (NCT02460224) showed that this combination was well tolerated with preliminary anti-tumor activity and immune profile modulation observed in the included population.26 Phase II studies are currently exploring the efficacy and safety profiles of either Eftilagimod alpha plus Pembrolizumab (NCT03625323) or BMS-986,016 plus Nivolumab (NCT01968109) combinations in both treatment naïve and ICI-resistant metastatic NSCLC patients and the results will provide us relevant information about the perspectives of LAG-3 targeting in NSCLC.27
Several trials are currently being conducted with the aim of evaluating the role of anti-LAG-3 agents in patients with advanced solid tumors, including NSCLC (Table 2).
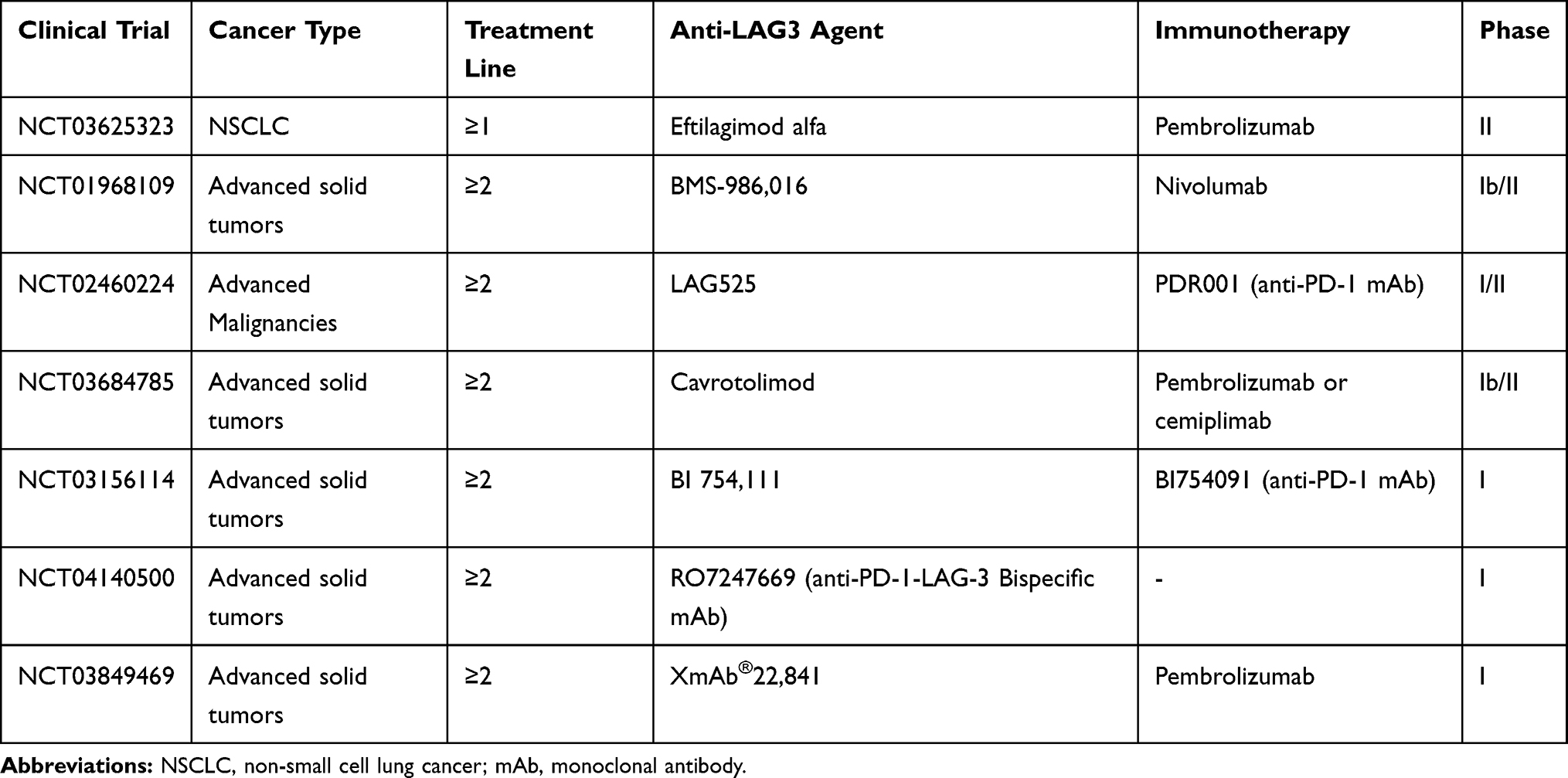 |
Table 2 Main Ongoing Clinical Trials of Anti-LAG-3 Agents Combined with ICIs |
TIM-3
T-cell immunoglobulin and mucin domain 3 (Tim-3) is an immunoglobulin (Ig) containing cell surface molecule which is expressed among distinct immune-cells populations, including CD4+, CD8+28 T regulatory,29 Th17 cells,30 dendritic cells, B cells, macrophages, NK and mast cells. Differently from other checkpoints receptors, as PD-1 and TIGIT, Tim-3 lacks classical immunoreceptor tyrosine-based switch signaling motifs in its cytoplasmatic tail. So far, four distinct ligands for Tim-3 have been identified: galectin-9, phosphatidylserine (PtdSer), high-mobility group protein B1 (HMGB1) and CECAM-1. Tim-3 is certainly co-regulated and co-expressed alongside other immune checkpoint receptors (PD-1, LAG-3 and TIGIT) on both CD4+ and CD8+ T cells,31,32 specifically marking the most dysfunctional and terminally exhausted subset of CD8+ T cells.33,34
Moreover, Tim-3 has been detected on both TILs and NSCLC cells, and its high levels have been related to poor patients’ prognosis,35 particularly when it was expressed by CD4+ cells.29 Pre-clinical evidence showed that Tim-3 overexpression is associated with PD-1 inhibitor resistance, suggesting that selective Tim-3 blockers may likely represent a valid therapeutic option at the time of anti-PD-1 treatment failure.36 Specifically, concomitant inhibition of PD-1 and Tim-3 looks promising,34 with several ongoing clinical trials investigating anti-Tim-3 in combination with anti-PD-1 inhibitors in advanced solid tumors (Table 3). A Phase II dose-expansion trial (NCT02608268), exploring the combination of MBG453 and spartalizumab, showed good tolerance but limited efficacy in patients with melanoma and NSCLC who failed prior anti-PD-1/PD-L1 therapy. In this trial 33 patients (melanoma: n=16; NSCLC: n=17) received MBG453 (800 mg, Q4W) plus spartalizumab (400 mg, Q4W) until unacceptable toxicity, progression disease (PD), or investigator’s/patients’ decision. On prior anti–PD-1/PD-L1 therapy, 6 (37.5%) melanoma and 7 (41.2%) NSCLC patients experienced durable clinical benefit, defined as complete, partial response, or stable disease (SD) for more than 6 months since treatment initiation. Common treatment-related adverse events (TRAEs) were reported in about 9% of the cases, while only 11.8% of NSCLC patients experienced grade 3–4 TRAEs.37 Further evaluation of MBG453 in other indications/combinations is warranted to assess the clinical relevance of Tim-3 inhibition in the clinical setting.
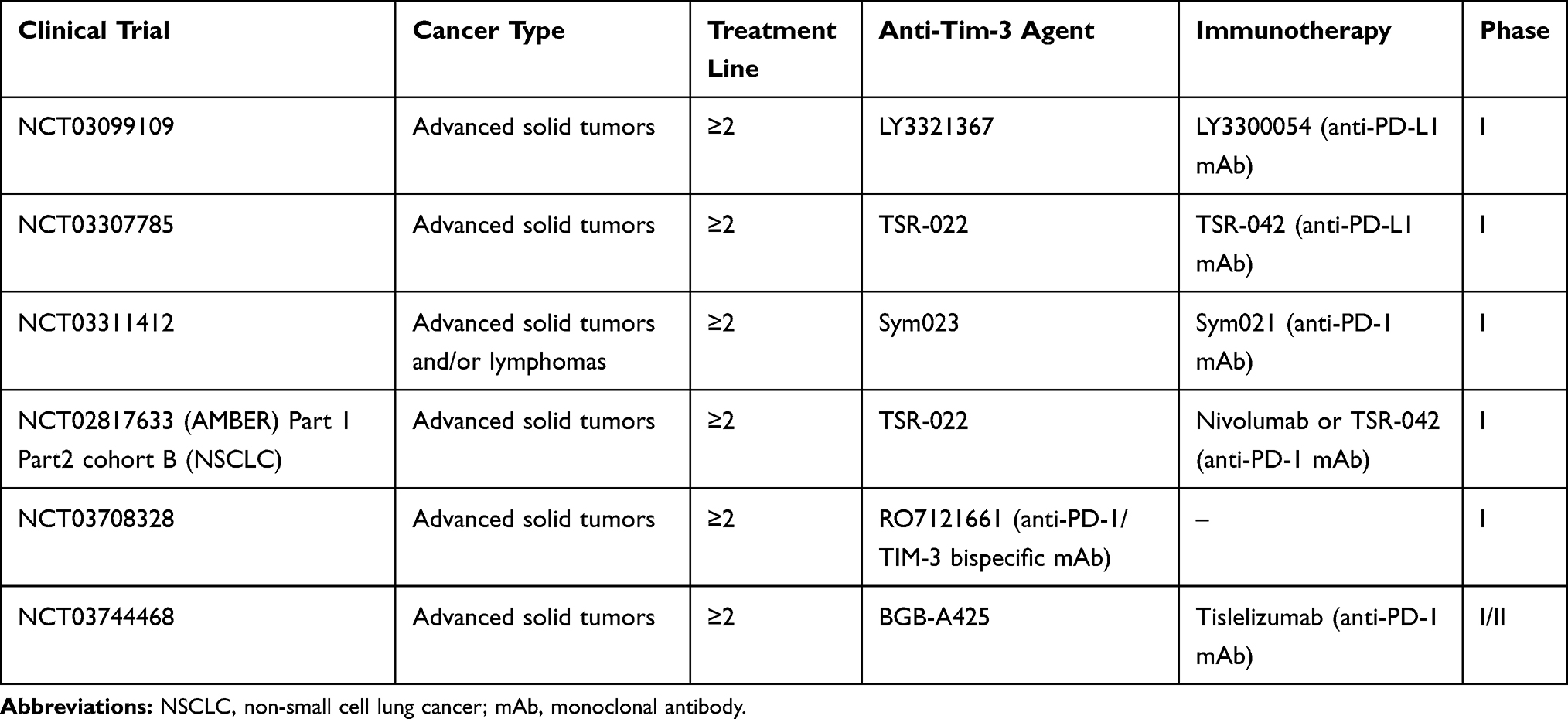 |
Table 3 Main Ongoing Clinical Trials of Anti-Tim-3 Agents Combined with ICIs |
Other clinical trials are currently being conducted with the aim of evaluating the role of anti-Tim-3 drugs in patients with advanced solid tumors, including NSCLC (Table 3).
TIGIT
T cell immunoglobulin and ITIM domain (TIGIT) is a T cell and natural killer (NK) inhibitor receptor38 critical to the immune tolerance development. It seems to preserve suppressor functions of Treg cells,39 while TIGIT-positive NK cells have been associated with tumor progression.40 In pre-clinical models of both cancer and chronic viral infection, antibody co-blockade of TIGIT and PD-L1 synergistically enhanced CD8+ T cell effector function, resulting in significant tumor and viral clearance responses,41 which makes TIGIT a new interesting therapeutic target for the clinical setting.
According to phase II data coming from the CITYSCAPE study, presented at the American Society for Clinical Oncology (ASCO) 2020 Meeting, the addition of the anti-TIGIT antibody Tiragolumab to the anti-PD-L1 atezolizumab resulted in a clinically meaningful improvement of both ORR and PFS, in patients with treatment naïve, PD-L1 high, non-oncogene addicted advanced NSCLC. In this prospective, randomized, placebo-controlled trial, 135 patients with tumor PD-L1 ≥ 1%, were randomized 1:1 to receive Tiragolumab plus atezolizumab (n=67) or placebo plus atezolizumab (n=68). At a median follow-up of 10.9 months, a significant improvement in ORR (37.3% versus 20.6%) and PFS (5.6 versus 3.9 months) was observed in favour of the experimental arm. Interestingly, when patients were analyzed according to their tumor PD-L1 expression levels, the clinical benefit was limited to those with TPS  50%, with ORR of 66% for Tiragolumab plus atezolizumab compared to 24% for the placebo arm. All grade TRAEs occurred in 80.6% versus 72% of cases, with grade
50%, with ORR of 66% for Tiragolumab plus atezolizumab compared to 24% for the placebo arm. All grade TRAEs occurred in 80.6% versus 72% of cases, with grade  14.9% and 19.1%, respectively.42 These data overall suggested that Atezolizumab plus Tiragolumab combination may represent an attractive option for the treatment of PD-L1 high NSCLC patients, with clinical efficacy to be confirmed within ongoing randomized phase III clinical studies (SKYSCRAPER-01, SKYSCRAPER-03) (Table 4).
14.9% and 19.1%, respectively.42 These data overall suggested that Atezolizumab plus Tiragolumab combination may represent an attractive option for the treatment of PD-L1 high NSCLC patients, with clinical efficacy to be confirmed within ongoing randomized phase III clinical studies (SKYSCRAPER-01, SKYSCRAPER-03) (Table 4).
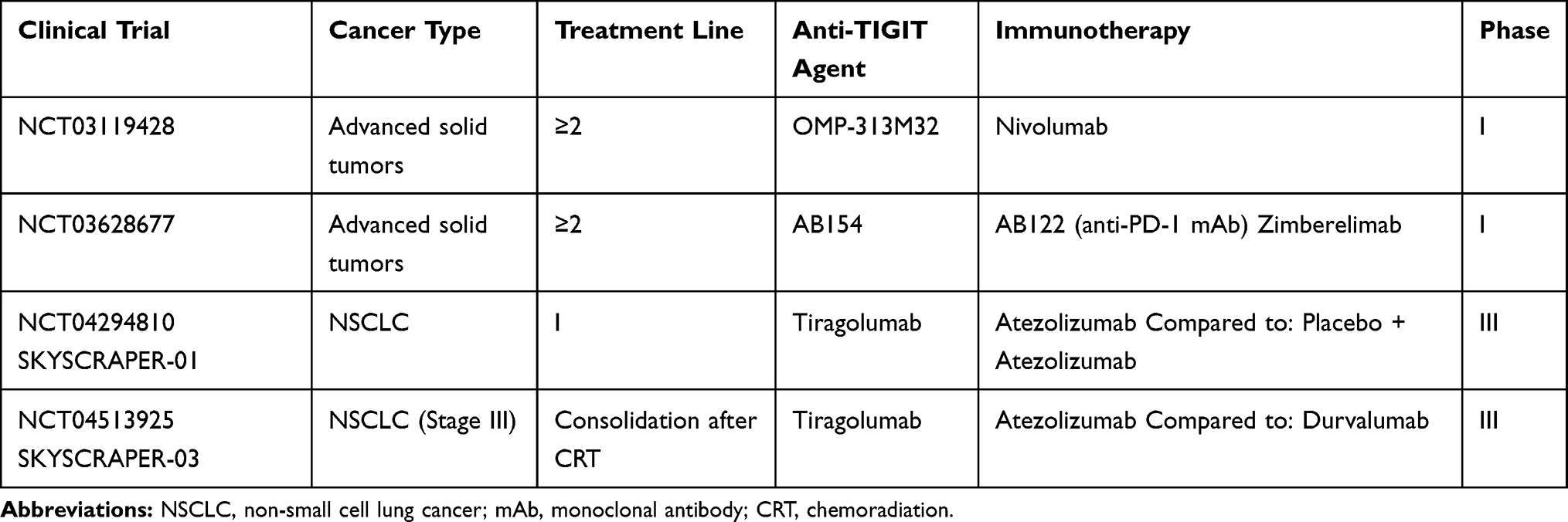 |
Table 4 Main Ongoing Clinical Trials of Anti-TIGIT Agents Combined with ICIs |
OX-40
OX-40 is a type1 transmembrane receptor, member of the tumor necrosis factor receptor (TNFR) family, expressed by activated CD4+ and CD8+ T cells during the antigen-specific priming.43–45 The activation of OX-40/OX-40L axis works as a co-stimulatory signal for CD4+ and CD8+ T cells activation, clonal division, cytokine production and establishment of T cells memory.46 In particular, OX-40 pathway promotes the transformation of CD4-positive T cells into T helper cells, by enhancing interleukin and interferon secretion,47–50 with preclinical data indicating its crucial role in the long-term survival of CD4+ and CD8+ T cells.51
He et al recently found that low levels of OX40 expression on TILs correlated with longer OS and recurrence-free survival (RFS) of NSCLC patients who underwent early-stage tumor resection. In their case series OX40 expression correlated with PD-L1 expression on tumor cells.52 Kashima et al observed that high OX40 and OX40L levels were associated with poor prognosis and may reflect the immune-exhausted status against lung adenocarcinoma.53 Moreover, Massarelli et al found that high OX40 expression within tumor immune infiltrate was associated to favorable prognosis in surgically resected stage I–III NSCLC patients, suggesting a potential role for the selection of candidate to OX40 agonist antibodies.54
Due to its biological functions and according to the existing evidences, the OX40-OX40L axis is expected to be a potential target of immunotherapy-based strategies for solid tumors.52,55–57
At the ASCO 2020 meeting Goldman et al showed encouraging Phase I trial results of MEDI0562, a humanized IgG1k OX40 monoclonal antibody (mAb) in combination with durvalumab (anti-PD-L1 mAb) or tremelimumab (anti-CTLA-4 mAb) in patients with previously treated advanced solid tumors (NCT02705482). Median OS was 17.4 and 11.9 months for MEDI0562 plus durvalumab and tremelimumab, respectively, with SD seen in 9 patients from each group (34.6% versus 29.0%, respectively). The safety profile of MEDI0562 in combination with durvalumab or tremelimumab was similar between groups.58
In a phase I trial of advanced solid tumors, Infante et al showed a tolerable profile for vonlerolizumab plus atezolizumab combination, with evidence of PD-L1 induction and immune activation in tumor-paired biopsies (NCT02410512).59 In the same target population, another ongoing phase 1 trial of GSK3174998 administered as monotherapy or combined with pembrolizumab (anti-PD-1 Ab) showed no dose-limiting toxicities (NCT02528357).60
Table 5 shows currently ongoing phase I/II trials testing OX40 agonists in combination with ICIs, other than those discussed above.
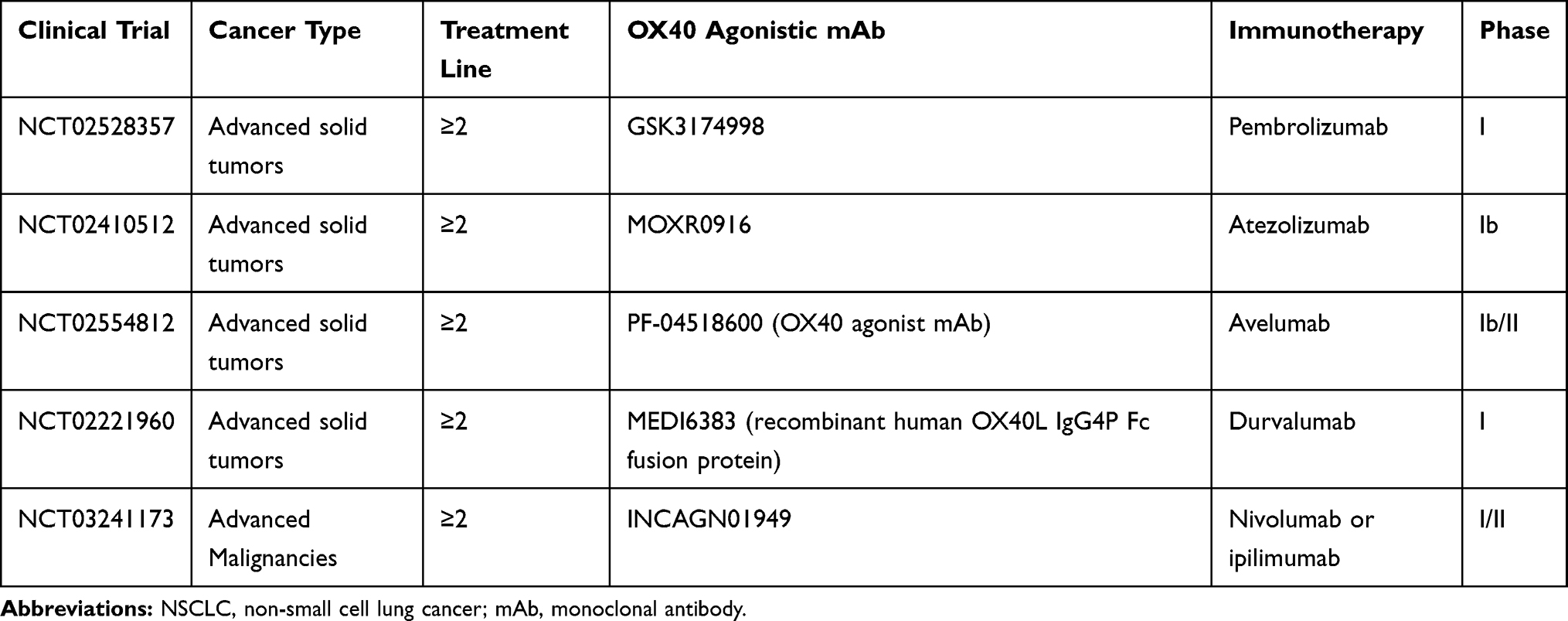 |
Table 5 Main Ongoing Clinical Trials of OX40 Agonistic Agents Combined with ICIs |
Combinations with Immune-Modulators
Immune-Metabolism Pathways: IDO-1
Nowadays, it is emerging that immune response may be influenced by different metabolic pathways, involving both tumor and effector CD8+ cells, which partially compete for the same resources within the tumor TME.61 Hence, new combination strategies which are able to manipulate the immunosuppressive TME and to enhance antitumor T cell responses are under investigation.
Preclinical studies identified multiple immunosuppressive mechanisms in the T-cell-inflamed tumors, including metabolic mechanisms of immunosuppression, such as the tryptophan-kynurenine-aryl hydrocarbon receptor (Trp-Kyn-AhR) pathway, which is the primary route for tryptophan catabolism.62,63 Kynurenine acts as a powerful AhR agonist, a ligand-gated transcription factor expressed by immune cells, which regulates a wide range of immunomodulatory effects.64,65 In preclinical models, heightened activities of the Trp-Kyn-AhR pathway have been related to the impairment of antitumor immunity and tumor growth.66,67 Three enzymes catalyze tryptophan conversion into kynurenine: IDO1, IDO2 and TDO.68 In murine cancer models, high IDO levels were associated with a reduced T lymphocytes infiltration. Elevated IDO1 and TDO activity as well as Kyn levels were associated to an increased tumor grade as well as a poor prognosis in different tumor types.69 Preclinical series showed that 40% of NSCLC express IDO1 with no significant differences between squamous and non-squamous subtype. Finally, IDO-1 overexpression has been observed after PD-1/PD-L1 ICIs treatment in NSCLC patients, suggesting its potential role in the context of acquired resistance, and has been correlated to worse patients’ prognosis.70
Based on these considerations, this pathway represents an attractive therapeutic target for cancer treatment, with different molecules currently under investigation, but phase I/II/III clinical trial results available only for IDO-1 inhibitors. Particularly, the anti-IDO-1 epacadostat has reached the most advanced stage of clinical development, with the recent publication of the ECHO-301/Keynote-252 phase III trial results showing no survival advantage from the addition of epacadostat to pembrolizumab in metastatic melanoma.71 These negative results were totally unexpected, since early phase trials of this combination in as many as 14 different solid tumors had previously shown promising activity.72 Significant differences among treatment populations, as well as inappropriately low dosing of epacadostat, with subsequent incomplete suppression of intratumoral Kyn pathway, may only partially explain the observed discrepancy. It is supposed that IDO-1 inhibition alone is likely inadequate to lowering intratumoral Kyn levels, thus paving the way to new combinations.73–76
Even though the combination of epacadostat and pembrolizumab have largely disattended previous expectations in melanoma, a phase II clinical trial is currently investigating its potential activity in treatment-naïve NSCLC patients characterized by high tumor PD-L1 expression (NCT03322540). Furthermore, there is still a great interest towards the investigation of Trp-Kyn-AhR pathway inhibition, with selective IDO-1 inhibitors (BMS-986,205, NLG-919 (navoximod/GDC-0919), dual IDO/TDO inhibitors (RG70099 and IOM-D) as well as indoximod plus anti-PD-1 combinations, currently in early stages of clinical development (NCT03343613, NCT03322540, NCT02298153, NCT03562871).
Cytokines: TGF-ß
Transforming growth factor- ß (TGF-ß) is a pleiotropic cytokine with dual role in cancer progression, acting both as tumor cells plasticity inducer and antitumor immune response suppressor.
TGF-ß trouble immune balance by holding the cytotoxic activity of NK cells77 and by favouring Tregs differentiation.78,79 On the other hand, TGF-ß acts as a key regulator of epithelial-mesenchymal transition (EMT) process, thus playing a role to the development of drug resistance among different cancer types. Particularly recent studies demonstrated the importance of TGF-β as a mechanism of resistance to ICI-therapies, leading to the clinical development of numerous new drugs targeting the TGF-β pathway, including small molecule inhibitors, antibodies and receptor-based TGF-β traps.80
Galunisertib is an oral small molecule inhibitor of TGF-β kinase receptor type I (TGF-β RI/ALK5)81 which selectively inhibits the serine/threonine activity, thereby preventing the phosphorylation of downstream proteins, SMAD2 and SMAD381. The antitumor activity of galunisertib has been demonstrated in three different in vivo tumor models, including NSCLC.82,83 Results from the phase II trial exploring the combination between galunisertib and nivolumab in recurrent or refractory NSCLC (NCT 02423343) are not available yet.
Several TGF-β-directed mAbs are currently under clinical evaluation. Among these, fresolimumab (GC1008), a fully human anti-TGF-β mAb targeting all TGF-β isoforms, has recently completed phase I clinical trial (NCT00356460), showing acceptable safety signals in renal cell carcinoma, melanoma and glioma84 and is currently being investigated in phase I/II trials including advanced solid tumors and mesothelioma.
Bintrafusp alfa is an antibody–drug conjugate (ADC) designed to simultaneously target both TGF-β and PD-L1 suppressive pathways that are commonly used by cancer cells to evade the immune system.
The dose-escalation portion of a Phase I, open-label clinical trial of bintrafusp alfa (MSB0011359C) has been completed, showing promising antitumor efficacy in heavily pretreated patients with metastatic solid tumors.85
Most recently, Paz-Ares et al showed encouraging efficacy and manageable tolerability associated with this agent within an expansion cohort of a phase I study, including 80 pretreated patients with metastatic NSCLC receiving bintrafusp alfa 500 or 1200 mg (n=40 each). At a median follow-up of 51.9 weeks, the ORR was 17.5% and 25% for the 500 mg and the 1200 mg doses, respectively, while reaching about 86% in the subgroup of patients with very high tumor PD-L1 expression (TPS  80%), while TRAEs occurred in about 69% of the patients.86 Based on these data, Bintrafusp alfa is currently being compared with pembrolizumab as an initial treatment for patients with advanced NSCLC and high tumor PD-L1 expression (NCT03631706).
80%), while TRAEs occurred in about 69% of the patients.86 Based on these data, Bintrafusp alfa is currently being compared with pembrolizumab as an initial treatment for patients with advanced NSCLC and high tumor PD-L1 expression (NCT03631706).
Stimulator of Interferon Genes: STING
STING (Stimulator of Interferon Genes) is a component of the endoplasmic reticulum (ER) which is essential for the production of type I interferon (IFN) in fibroblast, macrophages and dendritic cells (DCs), in response to cytoplasmic double-strand DNA ds(DNA) breaks as well as selected DNA viruses and intracellular bacteria.87,88 Type I IFN functions in a paracrine or autocrine manner, enhancing DCs cross-presentation activity and T cell activation. Particularly STING works as an adaptor protein that enhances the immune signaling following pathogen DNA detection by cytoplasmic DNA sensors (DAI, DHX9, DHX36, IFI204 etc)89. The recognition of microbial nucleic acids is one of the major mechanisms by which the immune system detects pathogens and STING plays a crucial role in the induction of immune response following DNA detection. Several studies identified type I IFN as a critical mediator in the spontaneous priming of antitumor CD8+ T cell responses.90 Accordingly, Woo and colleagues reported that the spontaneous CD8+ T cell priming against tumor cells was defective in mice lacking STING expression. Moreover, STING-deficient mice were unable to generate efficient antitumor T cell responses and prevent melanoma growth.
Della Corte et al demonstrated that STING pathway activation in NSCLC predicted features of immunotherapy response and was enhanced under cisplatin chemotherapy.91
On this basis, the combination of STING agonists with ICIs is emerging as a promising option, considering that STING pathway activation could “heat up” an immunologically cold tumor and trigger infiltration by immune cells, which in turn can be unleashed by the checkpoint blockade. Therefore, this kind of combination would allow to successfully target specific tumors that are usually refractory to single-agent immune checkpoint blockade. The development of STING agonists has rapidly grown as a novel class of immunotherapy, either alone or in combination with ICIs. STING agonists may be grouped into two main subtypes: nucleotidic or non-nucleotidic agonists.
Encouraging preclinical results have brought ADU-S100 forward to Phase I clinical trials, where it is administered by intratumoral injection to patients with advanced metastatic solid tumors or lymphomas, alone or in combination with ipilimumab (NCT02675439) or with anti-PD1 agents (NCT03172936).
Preliminary results of the phase 1 MK-1454, multicenter, dose escalation study exploring the MK-1454 as monotherapy (Arm 1) or in combination with pembrolizumab (Arm 2) in patients with solid tumors showed encouraging efficacy and an acceptable safety profile supporting the subsequent development of this combination regimen. TRAEs occurred in 83% and 82% of the patients in Arms 1 and 2, respectively, with only 9% and 14% of grade ≥3, 7% discontinuation in Arm 2 (0% in Arm 1), and no TRAEs-related deaths. PR were observed in 6 of 25 (24%) patients in Arm 2 (0% in Arm 1), while DCR was 20% in Arm 1 and 48% in Arm 2. Dose escalation trial is currently ongoing (NCT03010176),92 with final results expected within the end of 2021. Results from the Phase Ib dose escalation study exploring MIW815 (ADU-S100) plus spartalizumab in patients with advanced solid tumors supported synergistic antitumor effects when MIW815 (ADU-S100) was combined with ICIs (NCT03172936)93 but no specific data on NSCLC are available yet. The 5,6-dimethylxanthenone-4-acetic acid (DMXAA), also known as vadimezan or ASA404, is now recognized as a non-nucleotidic STING agonist and has rapidly entered clinical trials. However, DMXAA is a poor agonist of human STING and ultimately failed within phase III NSCLC clinical trials in combination with chemotherapy.94 Currently, non-nucleotidic STING agonists have not yet reached the clinical trials stage.
Despite all this encouraging evidence showing the rationale for implementing STING targeting therapy into the clinic, further characterization of the STING pathway is necessary for the development of tailored and effective combination treatments.
Combinations with PARP Inhibitors
The capacity of cancer to restore DNA damage is a crucial determinant of clinical response to ICIs. Indeed, genetic unstable and highly mutated cancers are usually characterized by increased tumor neoantigens load, thus likely to better respond to immunotherapy. Hsiehchen et al demonstrated that nucleotide excision repair as well as the homologous recombination (HR) gene defects are correlated with longer survival in cancer patients receiving ICIs.95 In the context of DNA repair pathways, poly (ADP-ribose) polymerase inhibitors (PARPi) represent a novel treatment strategy to selectively kill a subset of HR-deficient cancer cells by inducing synthetic lethality.96 Recent data suggested that besides its direct cytotoxic activity, PARP inhibition can have also immunomodulating effects, by increasing TMB, IFN-1 release, PD-L1 expression, and T-cell infiltration, thus potentially improving clinical response to immunotherapy.97,98 These data provided the biological rationale for the design of clinical trials investigating the combinations of PARPi and ICIs therapies in different tumor types, including lung cancer. Considering the prevalence of germline and/or somatic defects in the HR genes is reported to be around 5% in NSCLC,99 several clinical trials are currently assessing the efficacy and tolerability of this kind of combinations in different settings (Table 6).
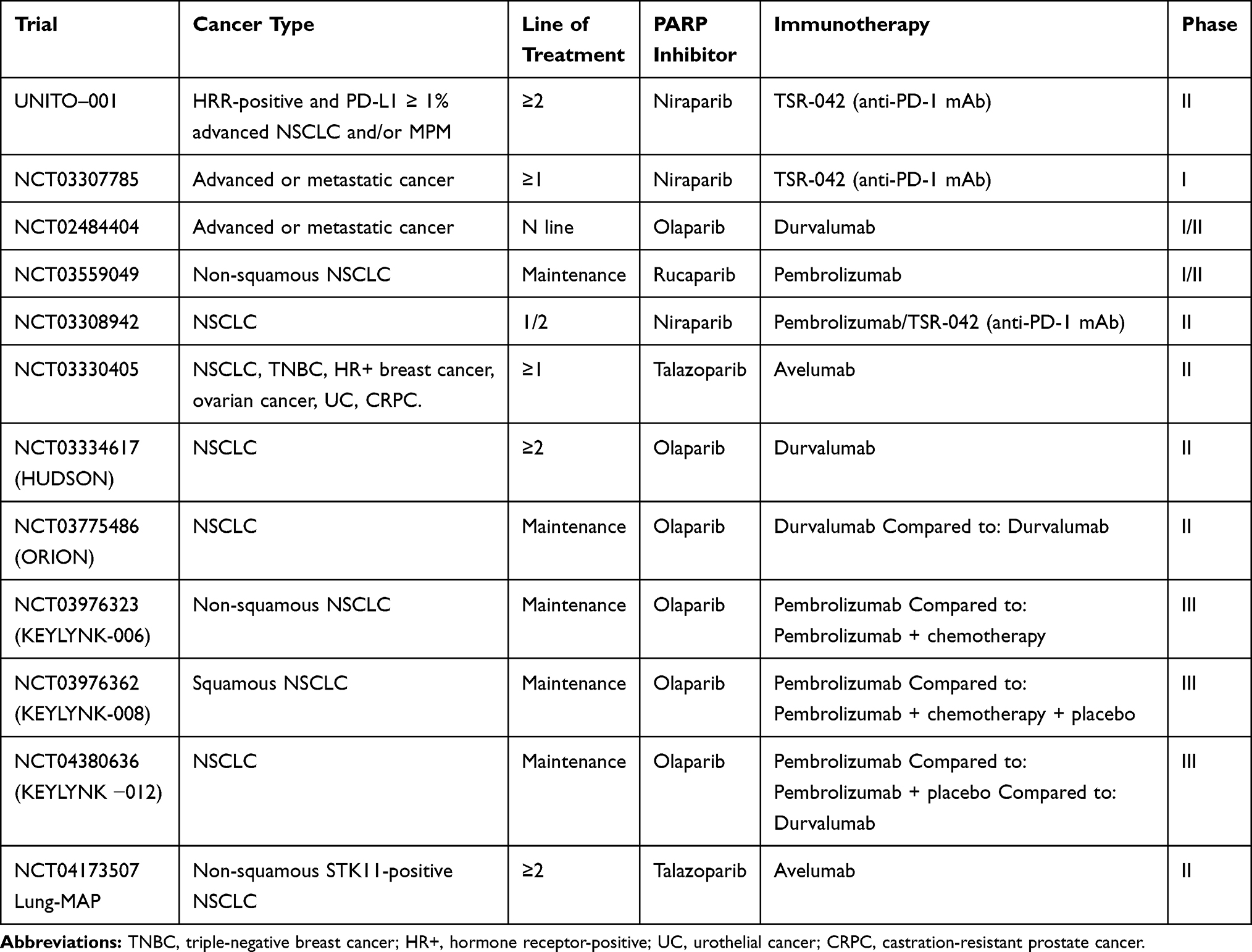 |
Table 6 Main Ongoing Trials of PARPi Combined with ICIs |
The ongoing phase II ORION trial (NCT03775486) was designed to evaluate the efficacy and safety of durvalumab plus olaparib versus durvalumab alone as maintenance therapy in patients affected by stage IV NSCLC not progressing after a first-line platinum-based chemotherapy plus durvalumab.100 Moreover, two phase III trials are evaluating the superiority of pembrolizumab plus maintenance olaparib versus maintenance pemetrexed in non-squamous NSCLC [KEYLYNK-006, NCT03976323] and of pembrolizumab with or without maintenance olaparib for squamous NSCLC [KEYLYNK-008, NCT03976362].
The HUDSON international, multi-arm, umbrella trial is specifically investigating the role of biomarker-directed combination strategies (durvalumab + PARP/STAT3/ATR/mTORC inhibitors) for NSCLC patients progressed on ICIs. Particularly patients with HR repair defects and LKB1 aberration will be treated with durvalumab and olaparib. Similarly, the NCT04173507 is a phase II trial investigating the efficacy of talazoparib plus avelumab combination in metastatic non-squamous NSCLC harboring STK11 gene mutation, while UNITO-001, is a Phase II, single-arm study, investigating the safety and antitumor activity of the PARPi niraparib plus dostarlimab (anti-PD-1 agent) in HRR-positive and PD-L1 ≥1% advanced NSCLC and/or malignant pleural mesothelioma patients. The results of these studies will likely provide interesting evidence about the ability of PARPi to restore ICI sensitivity in this molecularly selected cohort of patients.101
The interest in PARPi and ICIs combination involves also earlier clinical stages: the KEYLYNK-012 trial is a Phase 3, placebo-controlled study, in which pembrolizumab with concurrent chemoradiation therapy followed by pembrolizumab with or without olaparib will be compared to concurrent chemoradiation therapy followed by durvalumab in stage III NSCLC patients.102
The results of the aforementioned randomized studies are eagerly awaited and will be fundamental to evaluate the real benefit of the association of PARPi and immunotherapy in NSCLC patients.103
Combinations with Cancer Vaccines
Cancer vaccines represent an attractive treatment option for different tumor types, since their ability to sensitize host immune system and ultimately induce a specific T-cell mediated immune response against tumor antigens.104 Vaccination may be based on peptides derived from cancer antigens, recombinant cancer antigen proteins, recombinant viral vectors, tumor antigen-loaded dendritic cells, or DNA/RNA-encoding tumor antigens. Nowadays particular attention is reserved to personalized vaccines targeting individually selected shared antigens, in different cancer settings.104 Moreover, vaccines can upregulate PD-L1 expression within the TME, favoring the transition of immunologically cold into hot tumors, thus providing a strong rationale for the association to ICIs.105 Interestingly, ICIs themselves could increase the efficacy of vaccines in different ways: CTLA-4 inhibition can boost the amplitude of the priming phase by enhancing T-cell response, and can inhibit T-regs in the TME, while anti-PD-1/PD-L1 agents promote T-cells activity inside the TME increasing the amplitude of vaccine-mediated T cell responses.106 Based on several preclinical studies showing the additive and synergistic effects of vaccines and ICIs association, different clinical trials are ongoing.
Viagenpumatucel-L (HS-110) is an allogeneic cellular vaccine derived from a human lung adenocarcinoma cell line. The phase I/II DURGA trial (NCT02439450) is testing its combination with nivolumab in patients with previously treated advanced NSCLC. Preliminary results in the ICI-resistant cohort showed a good tolerability and responsiveness with a DCR of 55% and a PFS of 2.7 months (95% CI, 1.8–4.0 months).102
TG4010 is a cancer vaccine based on a viral vector, a Modified Vaccinia Virus Ankara (MVA) that codifies for MUC-1 (an antigen found in NSCLC) and for interleukin-2.107 Two different phase II studies are ongoing, evaluating the association of TG4010 with ICIs. One of them is testing the combination of TG4010 and nivolumab in previously treated non-squamous NSCLC patients (NCT02823990), the other one is evaluating the efficacy of first-line TG4010, nivolumab and chemotherapy in patients with PD-L1<50% (NCT03353675). DIRECT-01 is an ongoing open-label phase I/IIa trial, aiming to explore the safety, immunogenicity and efficacy of VB10.NEO, a DNA plasmid vaccine with intrinsic adjuvant effect designed for efficient delivery of personalized neoepitopes, in association with immunotherapy in patients with locally advanced or metastatic solid tumors including NSCLC.108 DSP-7888 is a therapeutic cancer vaccine composed of two synthetic peptides derived from Wilms’ tumor 1 (WT1) that can promote both cytotoxic and helper T-lymphocyte-mediated immune responses against WT1-expressing tumors. WT1 is overexpressed in various solid tumors. A phase Ib/II study is being conducted to evaluate DSP-7888 in combination with nivolumab or pembrolizumab in advanced solid tumors, including NSCLC patients (NCT03311334). Other several combination trials are ongoing and the results are awaited (Table 7). As for ICIs, not all patients respond to vaccines and the detection of predictive biomarkers represents a crucial point to select the best candidate for this treatment strategy. As known, lung cancer is often characterized by lack of HLA class I expression that is important for peptide recognition by lymphocytes. Thus, HLA expression is currently under investigation as predictive biomarker in clinical studies with cancer vaccines.109 Besides the high scientific appealing for these combinations, to date, clinical data are mostly lacking or still immature, at least in lung cancer, therefore the use of vaccines, remains experimental. Hopefully, progress in immune system knowledge and biological technologies will help to make vaccines plus ICIs combination, an effective, safety and accessible treatment option, tailored on every NSCLC patient.
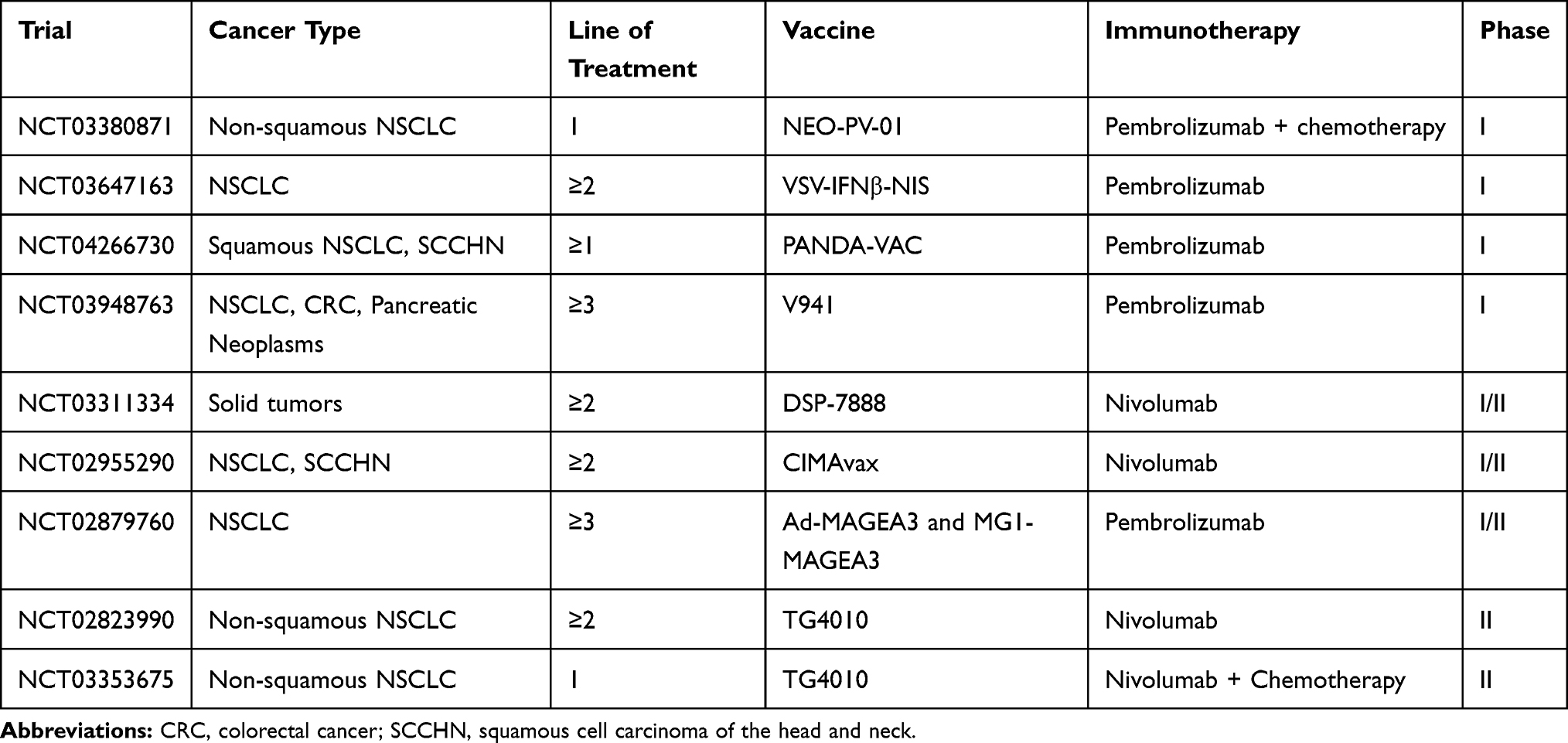 |
Table 7 Main Ongoing Trials of Cancer Vaccines Combined with ICIs |
Discussion
The introduction of ICIs targeting the PD-1/PD-L1 axis in clinical practice marked a milestone in the treatment of NSCLC, leading to unprecedented response duration and long-term survival for a relevant subgroup of patients affected by non-oncogene-addicted, metastatic disease. To date, we are facing the second phase of the “immune-revolution”, which is largely dominated by the advent of combination strategies. Different chemotherapy-ICIs regimens, have already entered in our current practice, allowing to extend the clinical benefits of immunotherapy to a larger fraction of NSCLC patients whose tumor harbor low or negative PD-L1 expression. As regards dual checkpoint blockade treatment, the combination of PD-1/PD-L1 plus CTLA-4 inhibitors was investigated within randomized trials, leading to regulatory approval of nivolumab plus ipilimumab either alone or in combination with chemotherapy as first-line treatment in metastatic NSCLC patients. However, the high rate of severe immune-related toxicities posed a major limitation to the clinical use of such combination, whose specific place in our real-word treatment algorithm remains still to be defined. Although the advent of the “first round” of ICIs combinations allowed to offer upfront immunotherapy to almost all patients with newly diagnosed, metastatic, non-oncogene addicted NSCLC, only a limited number of them still achieve durable response, with innate/acquired resistance occurring in most cases during the treatment course. On this basis, the “second round” of combinations (Figure 1) has been specifically designed to target the main resistance mechanisms occurring at the different time points of the cancer immunity cycles, in order to delay patients’ disease progression and ultimately increase the bar of long-term survivors under ICI therapies. Among the different combinations reaching the most advanced stages of clinical development, the upfront association of the anti-TIGIT mAb, Tiragolumab, with the anti-PD-L1 agent, atezolizumab, has recently emerged as a promising approach to further increase the survival benefit reached with single-agent PD-1 mAb in the subgroup of patients harboring high tumor PD-L1 expression, and is currently under evaluation within phase III randomized trials. Conversely, the combination of epacadostat and pembrolizumab has largely disattended previous expectations, considering the negative results observed in melanoma patients, and the potential role of IDO inhibitors as enhancer of PD-1 therapy activity in lung cancer remains undefined. The simultaneous inhibition of angiogenesis and PD-L1 pathways either alone or in combination with chemotherapy has already proven to be a valid strategy in non-squamous NSCLC, with the VEGF mAb, bevacizumab, approved for clinical use, and the VEGFR mAb, ramucirumab, emerging as an additional promising effective option in this setting. Pre-clinical data provided a strong rationale for the use of both PARPi and cancer vaccines in combination with ICIs, since their ability to enhance/restore PD-1 treatment activity against tumor-associated antigens. Although clinical evidence coming from early phase I studies seem to support this hypothesis, they are very preliminary and experimental data. The results of the ongoing phase II/III studies will be crucial to establish whether these treatments will be able to turn immunologically cold/excluded tumors into hot/inflamed ones and ultimately enhance ICIs-response in NSCLC patients. An actual and controversial issue related to the upcoming advent of these treatment approaches regards their tolerability, since the risk of adverse events related to the immunization significantly increase with the use of combination strategies, especially when considering immune-checkpoint agonist molecules or direct stimulators of immune-system activity.
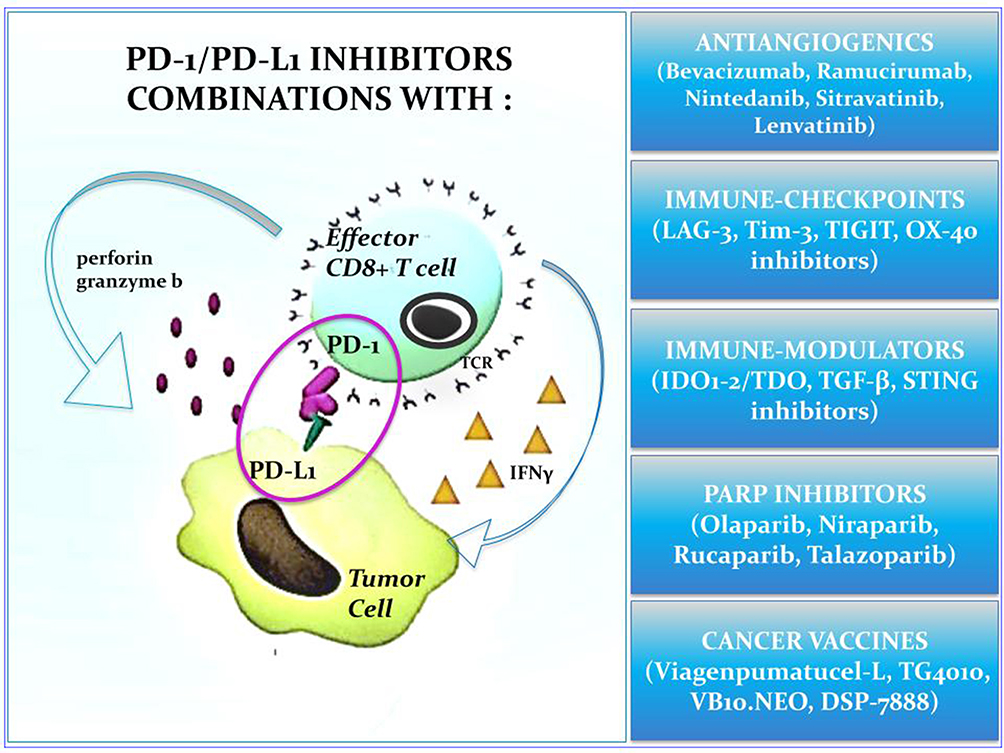 |
Figure 1 Immune-checkpoint inhibitors combinations in non-oncogene addicted advanced NSCLC. |
In this exciting scenario, characterized by the rapid development of innovative drugs and the advent of new combinations, personalizing ICIs-based approaches is emerging as the main objective of the third phase of the “immune-revolution”, in order to identify reliable biomarkers allowing to select the right treatment for the right patients at the right moment of his cancer history. Since tumor PD-L1 expression is considered as a reliable biomarker for single-agent first-line ICI activity, its predictive role with the new ICI-combinations is currently unknown. Several ongoing studies exploring different therapeutic approaches, like TIGIT, IDO1, and TGF-β inhibitors, are currently recruiting patients with high tumor PD-L1 expression, suggesting a potential role for such biomarker even in the upcoming years. The detection of germline and/or somatic mutation in the HRR genes is currently being prospectively investigated as potential predictor of ICI plus PARPi combinations benefit, while the HLA expression is the most promising biomarker in clinical studies with cancer vaccines. A deeper understanding of tumor biology has revealed that lung cancer is the result of complex dynamic networks involving tumor cells’ signaling pathways and tumor microenvironment, suggesting that only an integrated evaluation of both cancer genomics and immunomodulating processes will likely provide us the most reliable biomarkers to effectively personalize ICI-based combination approaches. Interestingly, liquid biopsy is recently emerging as an additional potential source of reliable information, for the real-time monitoring of both patients’ disease and immune system status, as well as to guide early treatment decisions under ICI therapy. In detail Nabet et al provided recent evidence about the DIREct-On circulating score, including high pre-treatment blood TMB, low peripheral CD8 T-cells count and ctDNA decrease after a single ICI infusion, as a potential non-invasive predictor of ICI combinations benefit to be validated in the context of upcoming clinical trials.110 We are now at the beginning of a new period of bottom-up research activity, including both pharma-designed clinical trials and spontaneous academic projects, from which a higher level of biomarker knowledge as well as personalized treatment practices is going to progressively emerge.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
Francesco Passiglia declared consultant’s fee from MSD, Astra Zeneca, Boehringer Ingelheim, BMS and Pfizer. Maria Lucia Reale declared consultant’s fee from Eli Lilly. Silvia Novello declared speaker bureau/advisor’s fee from Eli Lilly, MSD, Roche, BMS, Takeda, Pfizer, Astra Zeneca and Boehringer Ingelheim. The authors have no other conflict of interest to declare.
References
1. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-l1 tumor proportion score of 50% or greater. J Clin Oncol. 2019;37(7):537–546. doi:10.1200/JCO.18.00149
2. Dafni U, Tsourti Z, Vervita K, Peters S. Immune checkpoint inhibitors, alone or in combination with chemotherapy, as first-line treatment for advanced non-small cell lung cancer. A systematic review and network meta-analysis. Lung Cancer. 2019;134:127–140. doi:10.1016/j.lungcan.2019.05.029
3. Addeo A, Banna GL, Metro G, Di Maio M. Chemotherapy in combination with immune checkpoint inhibitors for the first-line treatment of patients with advanced non-small cell lung cancer: a systematic review and literature-based meta-analysis. Front Oncol. 2019;9:264. doi:10.3389/fonc.2019.00264
4. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi:10.1056/NEJMoa1910231
5. Rizvi NA, Cho BC, Reinmuth N, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6(5):661–674. doi:10.1001/jamaoncol.2020.0237
6. Reck M, Ciuleanu T-E, Dols MC, et al. Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): checkMate 9LA. J Clin Oncol. 2020;38(15_suppl):9501. doi:10.1200/JCO.2020.38.15_suppl.9501
7. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37(4):443–455. doi:10.1016/j.ccell.2020.03.017
8. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ. 2019;(39):147–164. doi:10.1200/edbk_240837
9. Khan AK, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol. 2018;15(5):310–324.
10. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–340. doi:10.1038/nrclinonc.2018.29
11. Manegold C, Dingemans AMC, Gray JE, et al. The potential of combined immunotherapy and antiangiogenesis for the synergistic treatment of advanced NSCLC. J Thorac Oncol. 2017;12(2):194–207. doi:10.1016/j.jtho.2016.10.003
12. Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–2301. doi:10.1056/NEJMoa1716948
13. Seto T, Nosaki K, Shimokawa M, et al. LBA55 WJOG @Be study: a phase II study of atezolizumab (atez) with bevacizumab (bev) for non-squamous (sq) non-small cell lung cancer (NSCLC) with high PD-L1 expression. Ann Oncol. 2020;31:S1185–S1186. doi:10.1016/j.annonc.2020.08.2288
14. Bang Y-J, Golan T, Dahan L, et al. Ramucirumab and durvalumab for previously treated, advanced non–small-cell lung cancer, gastric/gastro-oesophageal junction adenocarcinoma, or hepatocellular carcinoma: an open-label, phase Ia/b study (JVDJ). Eur J Cancer. 2020;137:272–284. doi:10.1016/j.ejca.2020.06.007
15. Herbst RS, Arkenau H-T, Santana-Davila R, et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced non-small-cell lung cancer, gastro-oesophageal cancer, or urothelial carcinomas (JVDF): a multicohort, non-randomised, open-label, phase 1a/b trial. Lancet Oncol. 2019;20(8):1109–1123. doi:10.1016/S1470-2045(19)30458-9
16. Han B, Chu T, Zhong R, et al. JCSE01.11 efficacy and safety of sintilimab with anlotinib as first-line therapy for advanced non-small cell lung cancer (NSCLC). J Thorac Oncol. 2019;14(10):S129. doi:10.1016/j.jtho.2019.08.269
17. Lu J, Zhong H, Chu T, et al. Role of anlotinib-induced CCL2 decrease in anti-angiogenesis and response prediction for nonsmall cell lung cancer therapy. Eur Respir J. 2019;53(3):1801562. doi:10.1183/13993003.01562-2018
18. Taylor MH, Lee C-H, Makker V, et al. Phase IB/II trial of lenvatinib plus pembrolizumab in patients with advanced renal cell carcinoma, endometrial cancer, and other selected advanced solid tumors. J Clin Oncol. 2020;38(11):1154–1163. doi:10.1200/JCO.19.01598
19. Hui R, Nishio M, Reck M, et al. Randomized, double-blind, phase 3 trial of first-line pembrolizumab + platinum doublet chemotherapy (chemo) ± lenvatinib in patients (pts) with metastatic nonsquamous non–small-cell lung cancer (NSCLC): LEAP-006. J Clin Oncol. 2019;37(15_suppl):TPS9118–TPS9118. doi:10.1200/JCO.2019.37.15_suppl.TPS9118
20. Woo S-R, Turnis ME, Goldbergz MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi:10.1158/0008-5472.CAN-11-1620
21. Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. 2017;276(1):80–96. doi:10.1111/imr.12519
22. Wang J, Sanmamed MF, Datar I, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019;176(1–2):334–347.e12. doi:10.1016/j.cell.2018.11.010
23. Datar I, Sanmamed MF, Wang J, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res. 2019;25(15):4663–4673. doi:10.1158/1078-0432.CCR-18-4142
24. Giordano M, Henin C, Maurizio J, et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 2015;34(15):2042–2058. doi:10.15252/embj.201490786
25. Wherry EJ, Ha S-J, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. doi:10.1016/j.immuni.2007.09.006
26. Hong DS, Schoffski P, Calvo A, et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J Clin Oncol. 2018;36(15_suppl):3012. doi:10.1200/JCO.2018.36.15_suppl.3012
27. Peguero JA, Bajaj P, Carcereny E, et al. A multicenter, phase II study of soluble LAG-3 (Eftilagimod alpha) in combination with pembrolizumab (TACTI-002) in patients with advanced non-small cell lung cancer (NSCLC) or head and neck squamous cell carcinoma (HNSCC). J Clin Oncol. 2019;37(15_suppl):TPS2667–TPS2667. doi:10.1200/JCO.2019.37.15_suppl.TPS2667
28. Monney L, Sabatos CA, Gaglia JL, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536–541. doi:10.1038/415536a
29. Gao X, Zhu Y, Li G, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. doi:10.1371/journal.pone.0030676
30. Hastings WD, Anderson DE, Kassam N, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39(9):2492–2501. doi:10.1002/eji.200939274
31. Chihara N, Madi A, Kondo T, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558(7710):454–459. doi:10.1038/s41586-018-0206-z
32. DeLong JH, O’Hara Hall A, Rausch M, et al. IL-27 and TCR stimulation promote T cell expression of multiple inhibitory receptors. ImmunoHorizons. 2019;3(1):13 LP- 25. doi:10.4049/immunohorizons.1800083
33. Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–2186. doi:10.1084/jem.20100637
34. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi:10.1084/jem.20100643
35. Jia K, He Y, Dziadziuszko R, et al. T cell immunoglobulin and mucin-domain containing-3 in non-small cell lung cancer. Transl Lung Cancer Res. 2019;8(6):895–906. doi:10.21037/tlcr.2019.11.17
36. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7(1):10501. doi:10.1038/ncomms10501
37. Mach N, Curigliano G, Santoro A, et al. Phase (Ph) II study of MBG453 + spartalizumab in patients (pts) with non-small cell lung cancer (NSCLC) and melanoma pretreated with anti–PD-1/L1 therapy. Ann Oncol. 2019;30:v491–v492. doi:10.1093/annonc/mdz253.028
38. Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of nectin-2 by the natural killer cell receptor T cell immunoglobulin and ITIM domain (TIGIT). J Biol Chem. 2017;292(27):11413–11422. doi:10.1074/jbc.M117.786483
39. Lucca LE, Axisa -P-P, Singer ER, Nolan NM, Dominguez-Villar M, Hafler DA. TIGIT signaling restores suppressor function of Th1 tregs. JCI Insight. 2019;4(3). doi:10.1172/jci.insight.124427
40. Zhang Q, Bi J, Zheng X, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–732. doi:10.1038/s41590-018-0132-0
41. Johnston RJ, Comps-Agrar L, Hackney J, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923–937. doi:10.1016/j.ccell.2014.10.018
42. Rodriguez-Abreu D, Johnson ML, Hussein MA, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J Clin Oncol. 2020;38(15_suppl):9503. doi:10.1200/JCO.2020.38.15_suppl.9503
43. Baumann R, Yousefi S, Simon D, Russmann S, Mueller C, Simon H-U. Functional expression of CD134 by neutrophils. Eur J Immunol. 2004;34(8):2268–2275. doi:10.1002/eji.200424863
44. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105(7):2845–2851. doi:10.1182/blood-2004-07-2959
45. Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J. 1990;9(4):1063–1068. doi:10.1002/j.1460-2075.1990.tb08211.x
46. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. doi:10.1016/j.immuni.2014.10.004
47. Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J Immunol. 2005;175(6):3534–3541. doi:10.4049/jimmunol.175.6.3534
48. Song J, So T, Croft M. Activation of NF-κB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol. 2008;180(11):7240LP- 7248. doi:10.4049/jimmunol.180.11.7240
49. So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci U S A. 2006;103(10):3740–3745. doi:10.1073/pnas.0600205103
50. Qui HZ, Hagymasi AT, Bandyopadhyay S, et al. CD134 plus CD137 dual costimulation induces eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J Immunol. 2011;187(7):3555–3564. doi:10.4049/jimmunol.1101244
51. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15(3):445–455. doi:10.1016/s1074-7613(01)00191-1
52. He Y, Zhang X, Jia K, et al. OX40 and OX40L protein expression of tumor infiltrating lymphocytes in non-small cell lung cancer and its role in clinical outcome and relationships with other immune biomarkers. Transl Lung Cancer Res. 2019;8(4):352–366. doi:10.21037/tlcr.2019.08.15
53. Kashima J, Okuma Y, Hosomi Y, Hishima T. High serum OX40 and OX40 ligand (OX40L) levels correlate with reduced survival in patients with advanced lung adenocarcinoma. Oncol. 2020;98(5):303–310. doi:10.1159/000505975
54. Massarelli E, Lam VK, Parra ER, et al. High OX-40 expression in the tumor immune infiltrate is a favorable prognostic factor of overall survival in non-small cell lung cancer. J Immunother Cancer. 2019;7(1):1–8. doi:10.1186/s40425-019-0827-2
55. Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res. 2014;2(2):142–153. doi:10.1158/2326-6066.CIR-13-0031-T
56. Jahan N, Talat H, Curry WT. Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF-expressing tumor cells. Neuro Oncol. 2018;20(1):44–54. doi:10.1093/neuonc/nox125
57. Linch SN, Kasiewicz MJ, McNamara MJ, Hilgart-Martiszus IF, Farhad M, Redmond WL. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc Natl Acad Sci U S A. 2016;113(3):E319–27. doi:10.1073/pnas.1510518113
58. Goldman JW, Piha-Paul SA, Curti BD, et al. Safety and tolerability of MEDI0562 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. J Clin Oncol. 2020;38(15_suppl):3003. doi:10.1200/JCO.2020.38.15_suppl.3003
59. Infante JR, Hansen AR, Pishvaian MJ, et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J Clin Oncol. 2016;34(15_suppl):101. doi:10.1200/JCO.2016.34.15_suppl.101
60. Infante JR, Ahlers CM, Hodi FS, et al. ENGAGE-1: a first in human study of the OX40 agonist GSK3174998 alone and in combination with pembrolizumab in patients with advanced solid tumors. J Clin Oncol. 2016;34(15_suppl):TPS3107–TPS3107. doi:10.1200/JCO.2016.34.15_suppl.TPS3107
61. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31(1):259–283. doi:10.1146/annurev-immunol-032712-095956
62. Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol. 2017;28(suppl_12):xii18–xii32. doi:10.1093/annonc/mdx238
63. Gajewski TF, Meng Y, Blank C, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213(1):131–145. doi:10.1111/j.1600-065X.2006.00442.x
64. Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36(2):189–204. doi:10.1016/s1357-2725(03)00211-5
65. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci. 2018;39(3):307–325. doi:10.1016/j.tips.2017.11.007
66. Munn DH, Sharma MD, Hou D, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114(2):280–290. doi:10.1172/JCI21583
67. Brandacher G, Perathoner A, Ladurner R, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006;12(4):1144–1151. doi:10.1158/1078-0432.CCR-05-1966
68. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–kynurenine–aryl hydrocarbon axis. Clin Cancer Res. 2019;25(5):1462–1471. doi:10.1158/1078-0432.CCR-18-2882
69. Reznik E, Luna A, Aksoy BA, et al. A landscape of metabolic variation across tumor types. Cell Syst. 2018;6(3):301–313.e3. doi:10.1016/j.cels.2017.12.014
70. Botticelli A, Cerbelli B, Lionetto L, et al. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J Transl Med. 2018;16(1):219. doi:10.1186/s12967-018-1595-3
71. Long GV, Dummer R, Hamid O, et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: results of the phase 3 ECHO-301/KEYNOTE-252 study. J Clin Oncol. 2018;36(15_suppl):108. doi:10.1200/JCO.2018.36.15_suppl.108
72. Mitchell TC, Hamid O, Smith DC, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J Clin Oncol. 2018;36(32):3223–3230. doi:10.1200/JCO.2018.78.9602
73. Kenison-white J, Wang Z, Sherr DH. The Aryl Hydrocarbon Receptor (AHR) as a Driver of Cancer Immunosuppression ABSTRACT.:163. 2018.
74. Joseph J, Gonzalez-Lopez M, Galang C, et al. Abstract 4719: small-molecule antagonists of the aryl hydrocarbon receptor (AhR) promote activation of human PBMCs in vitro and demonstrate significant impact on tumor growth and immune modulation in vivo. Cancer Res. 2018;78(13 Supplement):4719. doi:10.1158/1538-7445.AM2018-4719
75. Tchaicha J, McGovern K, Campesato LF, et al. Abstract 4723: targeting the IDO and TDO pathway through inhibition of the aryl hydrocarbon receptor. Cancer Res. 2018;78(13 Supplement):4723. doi:10.1158/1538-7445.AM2018-4723
76. Yamada T, Horimoto H, Kameyama T, et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat Immunol. 2016;17(6):687–694. doi:10.1038/ni.3422
77. Cortez VS, Cervantes-Barragan L, Robinette ML, et al. Transforming growth factor-β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity. 2016;44(5):1127–1139. doi:10.1016/j.immuni.2016.03.007
78. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50(4):924–940. doi:10.1016/j.immuni.2019.03.024
79. Tran DQ. TGF-β: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol. 2012;4(1):29–37. doi:10.1093/jmcb/mjr033
80. de Gramont A, Faivre S, Raymond E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology. 2017;6(1):1–5. doi:10.1080/2162402X.2016.1257453
81. Sawyer JS, Anderson BD, Beight DW, et al. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46(19):3953–3956. doi:10.1021/jm0205705
82. Maier A, Peille A-L, Vuaroqueaux V, Lahn M. Anti-tumor activity of the TGF-β receptor kinase inhibitor galunisertib (LY2157299 monohydrate) in patient-derived tumor xenografts. Cell Oncol. 2015;38(2):131–144. doi:10.1007/s13402-014-0210-8
83. Zhang M, Kleber S, Röhrich M, et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71(23):7155–7167. doi:10.1158/0008-5472.CAN-11-1212
84. Morris JC, Tan AR, Olencki TE, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFΒ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. doi:10.1371/journal.pone.0090353
85. Bang Y-J, Doi T, Kondo S, et al. Updated results from a phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with pretreated recurrent or refractory gastric cancer. Ann Oncol. 2018;29:viii222–viii223. doi:10.1093/annonc/mdy282.045
86. Paz-Ares L, Kim TM, Vicente D, et al. Bintrafusp Alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in second-line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol. 2020;15(7):1210–1222. doi:10.1016/j.jtho.2020.03.003
87. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. doi:10.1038/nature08476
88. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi:10.1038/nature07317
89. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38(5):870–880. doi:10.1016/j.immuni.2013.05.004
90. Dunn GP, Bruce AT, Sheehan KCF, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722–729. doi:10.1038/ni1213
91. Della Corte CM, Morgillo F. Early use of steroids affects immune cells and impairs immunotherapy efficacy. ESMO Open. 2019;4(1):2018–2019. doi:10.1136/esmoopen-2018-000477
92. Harrington KJ, Brody J, Ingham M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29:viii712. doi:10.1093/annonc/mdy424.015
93. Meric-Bernstam F, Sandhu SK, Hamid O, et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J Clin Oncol. 2019;37(15_suppl):2507. doi:10.1200/JCO.2019.37.15_suppl.2507
94. Lara PN, Douillard J-Y, Nakagawa K, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non–small-cell lung cancer. J Clin Oncol. 2011;29(22):2965–2971. doi:10.1200/JCO.2011.35.0660
95. Hsiehchen D, Hsieh A, Samstein RM, et al. HHS public access. JAMA Intern Med. 2020;1(3):1–18. doi:10.1016/j.xcrm.2020.100034.DNA
96. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84(2):137–146. doi:10.1016/j.bcp.2012.03.018
97. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–3720. doi:10.1158/1078-0432.CCR-16-3215
98. Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675–693. doi:10.1158/2159-8290.CD-17-0226
99. Kadouri L, Rottenberg Y, Zick A, et al. Homologous recombination in lung cancer, germline and somatic mutations, clinical and phenotype characterization. Lung Cancer. 2019;137(August):48–51. doi:10.1016/j.lungcan.2019.09.008
100. Ahn M-J, Liu Y, Improta T, Marcovitz M, DiPiazza K, Lanasa MC. ORION: a Phase 2, randomized, multicenter, double-blind study to assess efficacy and safety of durvalumab+olaparib vs durvalumab alone as maintenance therapy in stage IV non-small cell lung cancer (NSCLC). J Clin Oncol. 2019;37(15_suppl):TPS9126–TPS9126. doi:10.1200/JCO.2019.37.15_suppl.TPS9126
101. Heymach J, Thomas M, Besse B, et al. An open-label, multidrug, biomarker-directed, multicentre phase II umbrella study in patients with non-small cell lung cancer, who progressed on an anti-PD-1/PD-L1 containing therapy (HUDSON). J Clin Oncol. 2018;36(15_suppl):TPS3120–TPS3120. doi:10.1200/JCO.2018.36.15_suppl.TPS3120
102. Jabbour S, Cho BC, Bria E, et al. 1256TiP Phase III study of pembrolizumab (Pembro) with concurrent chemoradiation therapy (CCRT) followed by pembro with or without olaparib (Ola) vs CCRT followed by durvalumab (Durva) in unresectable, locally advanced, stage III non-small cell lung cancer. Ann Oncol. 2020;31:S810–S811. doi:10.1016/j.annonc.2020.08.151
103. Takahashi N, Surolia I, Thomas A. Targeting DNA repair to drive immune responses: it’s time to reconsider the strategy for clinical translation. Clin Cancer Res. 2020;26(11):2452–2456. doi:10.1158/1078-0432.CCR-19-3841
104. Solomon BJ, Beavis PA, Darcy PK. Promising immuno-oncology options for the future: cellular therapies and personalized cancer vaccines. Am Soc Clin Oncol Educ. 2020;(40):e253–e258. doi:10.1200/edbk_281101
105. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–1360. doi:10.1126/science.aar7112
106. Vermaelen K. Vaccine strategies to improve anticancer cellular immune responses. Front Immunol. 2019;10(JAN):1–17. doi:10.3389/fimmu.2019.00008
107. Limacher JM, Quoix E. TG4010: a therapeutic vaccine against MUC1 expressing tumors. Oncoimmunology. 2012;1(5):791–792. doi:10.4161/onci.19863
108. Krauss J, Krackhardt A, Jager E, et al. Abstract CT217: An Open-Label, Phase I/Iia Study of VB10.NEO (DIRECT-01) in Combination with Checkpoint Blockade in Patients with Locally Advanced or Metastatic Solid Tumors Including Melanoma, NSCLC, Renal Cell Carcinoma, Urothelial Cancer or SSCHN. 2019:CT217–CT217. doi:10.1158/1538-7445.am2019-ct217
109. Nakahara Y, Kouro T, Igarashi Y, Kawahara M, Sasada T. Prospects for a personalized peptide vaccine against lung cancer. Expert Rev Vaccines. 2019;18(7):703–709. doi:10.1080/14760584.2019.1635461
110. Nabet B-Y, Esfahani MS, Moding EJ. Noninvasive Early identification of therapeutic benefit from immune checkpoint inhibition. Cell. 2020;183(2):363–376.e13. doi:10.1016/j.cell.2020.09.001
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.