Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
IgA Vasculitis in a Lung Cancer Patient During Chemoradiotherapy
Authors Taoka M, Ochi N ![]() , Mimura A, Ichiyama N, Nagasaki Y, Nakagawa N
, Mimura A, Ichiyama N, Nagasaki Y, Nakagawa N ![]() , Nakanishi H
, Nakanishi H ![]() , Yamane H
, Yamane H ![]() , Oshiro Y, Monobe Y, Takigawa N
, Oshiro Y, Monobe Y, Takigawa N ![]()
Received 3 March 2021
Accepted for publication 20 April 2021
Published 3 June 2021 Volume 2021:17 Pages 571—575
DOI https://doi.org/10.2147/TCRM.S308771
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Masataka Taoka,1 Nobuaki Ochi,1 Ayaka Mimura,1 Naruhiko Ichiyama,1 Yasunari Nagasaki,1 Nozomu Nakagawa,1 Hidekazu Nakanishi,1 Hiromichi Yamane,1 Yoshiyuki Oshiro,2 Yasumasa Monobe,3 Nagio Takigawa1
1Department of General Internal Medicine 4, Kawasaki Medical School, Okayama, Japan; 2Department of General Internal Medicine 3, Kawasaki Medical School, Okayama, Japan; 3Department of Pathology, Kawasaki Medical School, Okayama, Japan
Correspondence: Nobuaki Ochi
Department of General Internal Medicine 4, Kawasaki Medical School, 2-6-1, Nakasange, Kita-ku, Okayama, Japan
Tel +81-86-225-2111
Email [email protected]
Abstract: A 72-year-old man with locally advanced lung squamous cell carcinoma experienced red purpura on the lower legs and hematuria when the disease progressed during definitive chemoradiotherapy. He had renal dysfunction and proteinuria. Biopsy specimens of the skin lesion and kidney revealed immunoglobulin A vasculitis. Potential causes such as paraneoplastic syndrome and cancer treatment have been proposed. The administration of steroids rapidly improved the symptoms. The presentation of immunoglobulin A vasculitis is accompanied by malignancies. Clinicians should keep this syndrome in mind, even during curative-intent treatment.
Keywords: IgA vasculitis, lung cancer, squamous cell carcinoma, nephritis, paraneoplastic syndrome
Introduction
Immunoglobulin A vasculitis (IgAV), formally known as Henoch-Schönlein purpura, is a systemic vasculitis that involves small vessels with the manifestation of purpura on the limbs, abdominal symptoms, arthropathy, and renal disorder.1 IgAV is the most common systemic vasculitis syndrome of childhood. In adults, IgAV is uncommon but can occur at any age.2 The exact cause of IgAV remains unknown, although various triggers, including infection, toxins, and certain drugs, have been hypothesized. Malignancy has also been reported as a rare causative factor of IgAV;3,4 however, only one case of IgA vasculitis occurring during chemotherapy has been reported.5 We encountered a case of IgAV associated with tumor progression during definitive chemoradiotherapy for locally advanced lung cancer. Locally advanced lung cancer usually responds to chemoradiotherapy; therefore, we believe that paraneoplastic syndrome seldom occurs, at least during therapy. Thus, clinicians should consider the possibility of IgAV when finding purpura and/or hematuria in lung cancer patients receiving curative treatment.
Case Report
A 72-year-old man presented with a productive cough lasting for 1 month. His medical history included hypertension and childhood asthma. His medications included olmesartan, levofloxacin, carbocysteine, and brotizolam. He had smoked cigarettes at two packs-per-day for more than 44 years until 8 years ago. He drank socially.
Chest computed tomography showed a nodule measuring 25 mm × 18 mm in the left upper lobe with a bulky subcarinal lymph node penetrating the right main bronchus. Bronchoscopy revealed stenosis of the right main bronchus with tumor invasion. He had been diagnosed with stage IIIA lung squamous cell carcinoma (T1bN2M0, stage IIIA). The cancer cells were moderately differentiated, and immunohistochemical staining revealed p40 protein. No epidermal growth factor receptor gene mutation or anaplastic lymphoma kinase rearrangement was detected.
The combination chemotherapy of cisplatin (80 mg/m2 on day 1) and docetaxel (60 mg/m2 on day 1) every 3 weeks was administered for four cycles, with the best response assessed as “stable disease.” Severe myelosuppression was not observed during chemotherapy. When sequential definitive radiotherapy was planned, he was hospitalized because of a high fever. Chest computed tomography showed obstructive pneumonia in the right lower lobe because of tumor progression. He was immediately treated with an antimicrobial agent and chest definitive radiotherapy was initiated. Ten days after radiotherapy initiation, palpable purpura emerged on the lower extremities (Figure 1). The laboratory findings 2 days after palpable purpura emerged were as follows: white blood cell count of 7190/μL; hemoglobin concentration of 9.9 g/dL; serum C-reactive protein, 2.92 mg/dL; creatinine, 1.81 mg/dL; blood urea nitrogen, 37 mg/dL; serum albumin, 28 g/L; CH50, 50 U/mL; C3, 116 mg/dL; C4, 34.0 mg/dL; and D-dimer of 1.40 μg/dL.
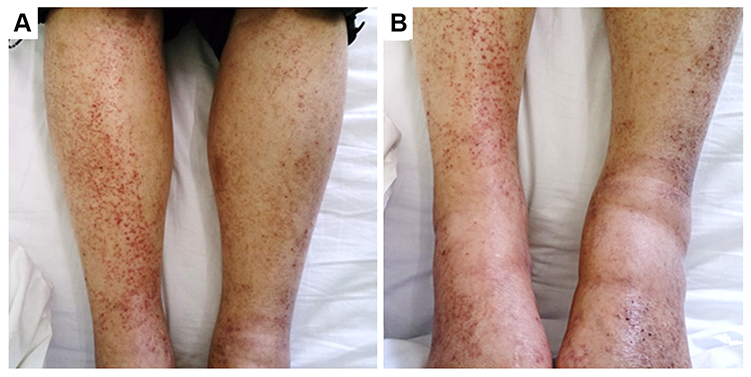 |
Figure 1 The photograph shows purpura on both legs. (A) Anterior view of the tibia. (B) Anterior view of the ankle. |
Urine analysis showed proteinuria (476 mg/mmol spot urine protein-to-creatinine ratio) and hematuria (>100 erythrocytes per high-power field). Platelet count, prothrombin time, activated partial thromboplastin time, and levels of transaminases were within the normal range. Findings were negative for antistreptolysin O, rheumatoid factor, antinuclear antibody, immunoglobulin A (IgA), antineutrophil cytoplasmic antibody, hepatitis B surface antigen, and hepatitis C virus antibody.
On the histopathologic examination of palpable purpura, vasculitis was observed with the infiltration of polymorphonuclear cells into the vessel walls of the dermis (Figure 2A).
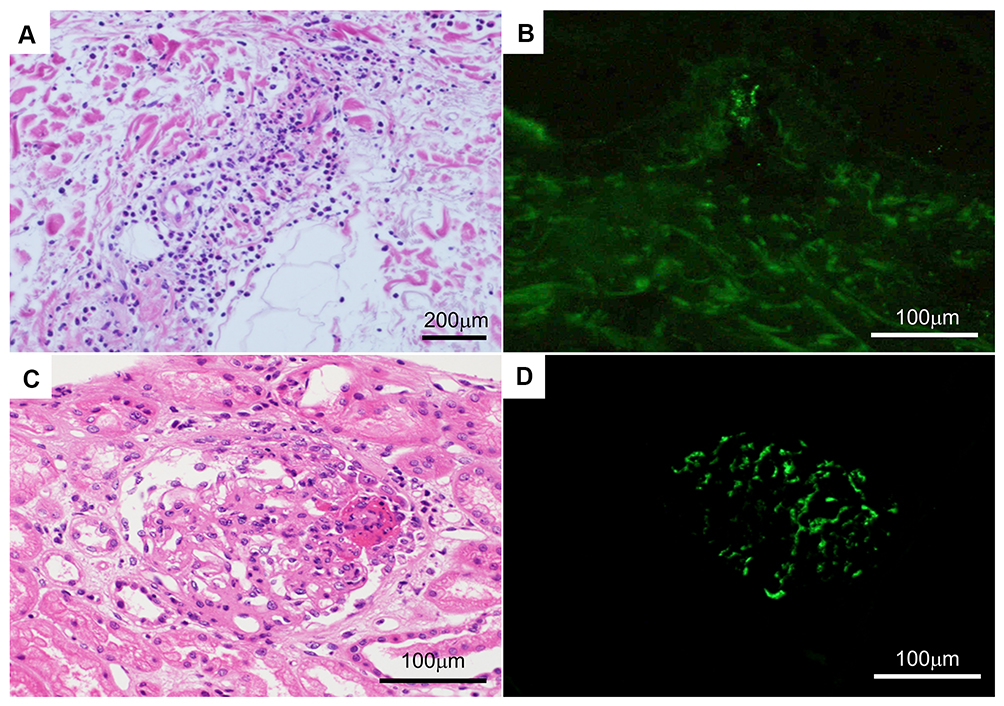 |
Figure 2 Histopathology of the palpable purpura and kidney. (A) The superficial dermis has perivascular infiltration of neutrophils, lymphocytes, and eosinophils. (B) Direct immunofluorescence using anti-IgA antibody reveals IgA deposition in the blood vessel walls of the dermal papillary layer. (C) The kidney has mesangial hypercellularity, increased mesangial matrix, and a cellular crescent. (D) Direct immunofluorescence using anti-IgA antibody shows mesangial IgA deposition. Abbreviation: IgA, immunoglobulin A. |
Vasculitis in the kidney specimen was similarly noted (Figure 2B). Immunofluorescent studies revealed mesangial deposition of IgA with C3 in the renal cells (Figure 2C) and in the superficial blood vessels of dermis (Figure 2D). In the present patient, treatment with methylprednisolone (500 mg/day) on days 1–3, followed by prednisolone (0.5 mg/kg/day), was initiated after a deterioration in renal function and a decline in urinary volume, as evidenced by a drop in the estimated glomerular filtration rate (eGFR) from 67.9 mL/min/1.73 m2 at the time of admission to 29.7 mL/min/1.73 m2 before starting prednisolone. We administered furosemide (20 mg/day) and trichlormethiazide (1 mg/day) to treat right pleural effusion, which emerged because of nephrotic syndrome (estimated urinary protein excretion, 800 mg/mmol), followed by hypoalbuminemia (serum albumin, 24 g/L). The clinical features of IgAV, including the rash, resolved within 2 weeks. The eGFR improved to 40.9 mL/min/1.73 m2. Right pleural effusion disappeared 4 weeks after the administration of steroid treatment. Prednisolone was tapered and maintained at 0.3 mg/kg/day.
Discussion
IgAV is a systemic vasculitis with deposition of IgA-containing immune complexes. It involves small vessels with the manifestation of purpura on the limbs, abdominal symptoms, arthritis, and renal disorder.1 IgAV occurs commonly in children (14 cases per 100,000), but can also affect adults (1.3 per 100,000) with a mean age of 50 years at the initial presentation. The clinical presentation of IgAV is more severe in adults than in children.2 According to the American College of Rheumatology diagnostic criteria for Henoch-Schönlein purpura (HSP), four criteria were identified, including palpable purpura unrelated to thrombocytopenia, age ≤20 years at disease onset, acute abdominal pain, and granulocytes in the walls of small arterioles and venules on biopsy.6 According to the revised criteria of the diagnosis of HSP were proposed by European League Against Rheumatism/the Paediatric Rheumatology International Trials Organization/Paediatric Rheumatology European Society, the presence of purpura or petechiae with lower limb predominance is mandatory, and at least one of the following four criteria should be met: (i) diffuse abdominal pain with acute onset, (ii) histopathology showing leukocytoclastic vasculitis or proliferative glomerulonephritis, with predominant IgA deposits, (iii) arthritis or arthralgia of acute onset, and (iv) renal involvement in the form of proteinuria or haematuria.7 Hočevar et al reported a diagnostic sensitivity of 99.2% and specificity of 86% in adult patients with HSP.8 Thus, the present case could be diagnosed as HSP. The clinical presentation of IgAV is more severe in adults than in children including renal disorder.9 The Japan Renal Biopsy Registry reports that 10.8% of adult patients with IgAV nephritis develop nephrotic syndrome and 18.8% in that of elderly patients (ie, ≥60 years old).10 For severe IgAV nephropathy patients, steroid pulse therapy (intravenous methylprednisolone 1 g per day on days 1–3) should be considered.11 Several alternative therapies such as mycophenolate, cyclophosphamide, cyclosporine, or rituximab have been used in patients with IgAV nephritis for whom initial therapy with glucocorticoids has not been effective. In one clinical practice, 19 patients who were diagnosed with IgAV with cancer received systemic treatment for vasculitis, including oral glucocorticoids (n=18), cyclophosphamide (n=1), and colchicine (n=2).12
The exact cause of IgAV in adults remains unknown, although malignancy has been reported as a causative factor. Mitsui et al13 report that 23 (43.4%) patients among 53 IgAV patients aged 41 years or older had a malignant tumor as an underlying disease. Zurada et al4 report that solid tumors accounted for most (61%) cases of coexisting malignancies and IgAV, and the most frequent tumor is non-small-cell lung cancer (25%). Mifune et al14 similarly report that the histology of tumors of 12 lung cancer patients with IgAV were squamous cell (n = 7), adenocarcinoma (n = 3), and small cell carcinoma (n = 2).
Some reports4,15 reveal that malignant tumors exist concurrently with or after a diagnosis of IgAV. Others mention drug-related IgAV in patients treated with vascular endothelial growth factor or immune checkpoint inhibitor.16,17 Three cases of solid-organ malignancy diagnosed as HSP were instructive. Two had complete remission with a combination of immunosuppressive therapies for HSP and treatment of the associated malignancy. One patient who never received treatment for his prostate cancer did not achieve complete remission of HSP. These suggest that HSP associated with malignancies might be resistant to immunosuppressive treatment if the comorbid malignancy is not treated simultaneously.18 Only one study5 has reported a case of the emergence of IgAV after cancer progression. Thus, IgAV occurs when a malignant tumor is not under control. A possible reason for this finding is that IgAV with malignant tumors may be correlated with the generation and expression of tumor associated antigens. Tumor-related antigens stimulate a patient’s immune system to produce abnormal antibodies. Immune complexes are formed and deposited on the blood vessel wall. In addition, tumor-related antigens decrease the clearance of circulating immune complexes, and lymphocyte dysfunction and cytokine release occur, thereby resulting in vascular inflammation.19 Meanwhile, tumor treatment may be associated with the expression and exposure of tumor-related antigens.
IgAV in our patient seemed to rapidly develop after the administration of cancer treatment, which included chemotherapy and radiation therapy. In the present patient, we diagnosed IgAV because the characteristic rash and renal dysfunction were observed within narrow intervals. The differential diagnosis of rash must exclude drug-induced factors, infections, and collagen diseases. The differential diagnosis of renal dysfunction with cancer patients must similarly exclude the toxicity of antineoplastic drugs (cisplatin in our patient), malignant obstructive uropathy, and oncologic emergency such as tumor lysis syndrome.20 Since this patient has already passed away due to the disease progression, informed consent was obtained from the patient’s next of kin for publication of this case report including the images. Institutional approval is not required to report the case report in our institution and to publish the case details.
When IgAV appears during cancer treatment, tumor exacerbation should be suspected and, conversely, if a rash appears during tumor exacerbation, IgAV should be suspected in addition to drug eruption.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11. doi:10.1002/art.37715.
2. Fervenza FC. Henoch-Schonlein purpura nephritis. Int J Dermatol. 2003;42:170–177. doi:10.1046/j.1365-4362.2003.01769.x.
3. Fain O, Hamidou M, Cacoub P, et al. Vasculitides associated with malignancies: analysis of sixty patients. Arthritis Rheum. 2007;57:1473–1480. doi:10.1002/art.23085.
4. Zurada JM, Ward KM, Grossman ME. Henoch-Schönlein purpura associated with malignancy in adults. J Am Acad Dermatol. 2006;55:S65–70. doi:10.1016/j.jaad.2005.10.011.
5. Ota S, Haruyama T, Ishihara M, et al. Paraneoplastic IgA vasculitis in an adult with lung adenocarcinoma. Intern Med. 2018;57:1273–1276. doi:10.2169/internalmedicine.9651-17.
6. Mills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheum. 1990;33:1114–1121. doi:10.1002/art.1780330809.
7. Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69:798–806. doi:10.1136/ard.2009.116657.
8. Hočevar A, Rotar Z, Jurčić V, et al. IgA vasculitis in adults: the performance of the EULAR/PRINTO/PRES classification criteria in adults. Arthritis Res Ther. 2016;18:58. doi:10.1186/s13075-016-0959-4.
9. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859–864. doi:10.1002/art.1780400513.
10. Komatsu H, Fujimoto S, Yoshikawa N, et al. Clinical manifestations of Henoch-Schonlein purpuranephritis and IgA nephropathy: comparative analysis of data from the Japan Renal Biopsy Registry (J-RBR). Clin Exp Nephrol. 2016;20:552–560. doi:10.1007/s10157-015-1177-0.
11. Pozzi C, Bolasco PG, Fogazzi GB, et al. Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet. 1999;353:883–887. doi:10.1016/s0140-6736(98)03563-6.
12. Hankard A, Michot JM, Terrier B, et al. New insights on IgA vasculitis with underlying solid tumor: a nationwide French study of 30 patients. Clin Rheumatol. 2020. doi:10.1007/s10067-020-05455-z.
13. Mitsui H, Shibagaki N, Kawamura T, et al. A clinical study of Henoch-Schönlein Purpura associated with malignancy. J Eur Acad Dermatol Venereol. 2009;23:394–401. doi:10.1111/j.1468-3083.2008.03065.x.
14. Mifune D, Watanabe S, Kondo R, et al. Henoch Schönlein purpura associated with pulmonary adenocarcinoma. J Med Case Rep. 2011;5:226. doi:10.1186/1752-1947-5-226.
15. Pertuiset E, Lioté F, Launay-Russ E, et al. Adult Henoch-Schönlein Purpura associated with malignancy. Semin Arhritis Rheum. 2000;29:360–367. doi:10.1053/sarh.2000.6988.
16. Endo Y, Negishi K, Hirayama K, et al. Bevacizumab-induced immunoglobulin A vasculitis with nephritis: a case report. Medicine (Baltimore). 2019;98:e17870. doi:10.1097/MD.0000000000017870.
17. Belkaid S, Berger M, Nouvier M, et al. A case of Schönlein-Henoch purpura induced by immune checkpoint inhibitor in a patient with metastatic melanoma. Eur J Cancer. 2020;139:169–172. doi:10.1016/j.ejca.2020.08.005.
18. Podjasek JO, Wetter DA, Pittelkow MR, et al. Henoch-Schönlein purpura associated with solid-organ malignancies: three case reports and a literature review. Acta Derm Venereol. 2012;92:388–392. doi:10.2340/00015555-1288.
19. Zhang X, Yang S, Li W, et al. Adult Henoch-Schonlein purpura associated with small cell lung cancer: a case report and review of the literature. Oncol Lett. 2013;5:1927–1930. doi:10.3892/ol.2013.1274.
20. Gallieni M, Cosmai L, Porta C. Acute kidney injury in cancer patients. Contrib Nephrol. 2018;193:137–148. doi:10.1159/000484970.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.