")
Back to Journals » International Journal of General Medicine » Volume 15
Identifying Potential Gene Defect Patterns Related to COVID-19 Based on Pharmacological and Bioinformatics Analysis for Lung Adenocarcinoma
Authors Lou H, Li X, Gao S, Zhang Y, Chen H, Zhai X
Received 30 December 2021
Accepted for publication 18 March 2022
Published 21 April 2022 Volume 2022:15 Pages 4285—4301
DOI https://doi.org/10.2147/IJGM.S356444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Hongqiang Lou, Xusheng Li, Suhua Gao, Yanfang Zhang, Haohao Chen, Xia Zhai
Medical Molecular Biology Laboratory, School of Medicine, Jinhua Polytechnic, Jinhua City, Zhejiang Province, 330700, People’s Republic of China
Correspondence: Xia Zhai, Medical Molecular Biology Laboratory, School of Medicine, Jinhua Polytechnic, 1188 Wuzhou Street, Jinhua City, Zhejiang Province, 321000, People’s Republic of China, Email [email protected]
Abstract:
Background: Coronavirus disease 2019 (COVID-19) greatly affects cancer patients, especially those with lung cancer. This study aimed to identify potential drug targets for lung adenocarcinoma (LUAD) patients with COVID-19.
Methods: LUAD samples were obtained from public databases. Differentially expressed genes (DEGs) related to COVID-19 were screened. Protein–protein interactions among COVID-19-related genes, the traditional Chinese medicine (TCM) and TCM target genes were analyzed by CytoScape. The correlation between tumor microenvironment and COVID-19 target genes were assessed by Pearson correlation analysis. Unsupervised consensus clustering was conducted to categorize molecular subtypes.
Results: We filtered 26 COVID-19 target genes related to TCM for LUAD. Interleukin (IL)-17 signaling pathway and tumor necrosis factor (TNF) signaling pathway were significantly enriched in these 26 genes. A strong correlation was found between COVID-19 target genes and tumor microenvironment (TME), cell death. Importantly, interleukin-1beta (IL1B) was identified as a core gene in the protein–protein interactions (PPI) network. Based on the 26 target genes, two molecular subtypes showing distinct overall survival, TME and response to target therapy were developed.
Conclusions: This study explored 26 COVID-19 target genes, which could serve as potential therapeutic drug targets for LUAD. IL1B was verified as a critical target for developing new molecular drugs. Furthermore, two novel molecular subtypes showed the potential to guide personalized therapies in clinical practice.
Keywords: COVID-19, lung adenocarcinoma, traditional Chinese medicine, drug targets, tumor microenvironment, molecular subtypes
Introduction
Studies reported that cancer patients are more susceptible to coronavirus disease 2019 (COVID-19) and may develop severe illness due to suppressed immunity, a higher frequency to health-care centers, and older ages.1 In addition, cancer patients infected with COVID-19 are more likely to deteriorate.2 A normal health care and management of cancer patients could be disrupted in the pandemic as a result of the limitations and re-arrangement of medical resources. Clinical investigations also supported the conclusion that cancer patients with COVID-19 are associated with worse outcomes than with cancer only.3,4 The mortality of cancer patients with COVID-19 is approximately 25% to 30%.5,6 Moreover, patients who have recently received therapeutic management show a higher risk to develop worse outcomes, such as complications or even death.7
Lung cancer is a malignant tumor posing great threat to human health. In 2020, the number of lung cancer cases was about 2.21 million in the world, ranking the second among all new cancer cases of that year and the highest in all cancer-related mortality. It is estimated that in 2040, the number of lung cancer cases worldwide will reach about 3.6 million.8,9 Study showed that lung cancer is the most common cancer type among patients infected with COVID-19.10 Lung cancer patients face a higher death risk because of the possibilities such as smoking-induced pulmonary injury and insufficiency of respiratory healthcare during the pandemic.10,11 To alleviate the adverse effects resulted from COVID-19, professional organizations have proposed a series of recommendations to guide decision-making for managing patients with COVID-19. In China, the traditional Chinese medicine (TCM) plays a positive role in inhibiting COVID-19 progression in patients either in mild or severe condition.12–14 TCM protocol in diagnosis and treatment protocol for COVID-19 (trial version 8) developed by the National Health Commission in China proposes nine different TCM prescriptions for different stages of COVID-19 infection.15
As TCM is effective in treating COVID-19 and the unfavorable situations of cancer patients with COVID-19, we, therefore, identified TCM drug targets related to both COVID-19 and lung adenocarcinoma (LUAD).16–18 By using data from public databases, pharmacologic analysis and bioinformatics analysis, we want to identify the main COVID-19 target genes both associated with TCM and LUAD. These target genes may be new molecular targets for treating lung cancer patients with COVID-19. Furthermore, based on target genes, we also would like to investigate novel molecular subtypes capable of guiding targeted therapies such as immunotherapy for LUAD patients.
Materials and Methods
Data Source and Data Preprocessing
The Cancer Genome Atlas (TCGA)-LUAD dataset including RNA-seq data and clinical information of LUAD samples was downloaded from TCGA database on June 30, 2021. For RNA-seq data, samples without survival information or follow-up information were excluded. Ensembl ID was transferred into gene symbols. Median expression value was taken when one gene had multiple gene symbols. After preprocessing, 472 LUAD samples in the TCGA-LUAD dataset remained. Data of gene mutation and genomic variation including single nucleotide variation (SNV) and copy number variation (CNV) were independently downloaded from TCGA database. The GSE31210 dataset was downloaded from Gene Expression Omnibus (GEO) database on June 30, 2021. In the GSE31210 dataset, samples without survival information or follow-up information were excluded. Probes were transferred into gene symbols. One probe corresponded to multiple genes was excluded. Median value was taken when multiple probes corresponded to one gene. After preprocessing, 226 LUAD samples remained.
Identification of Differentially Expressed Genes Associated with COVID-19
COVID-19-related genes with a total of 1566 genes were downloaded from GeneCards database (https://www.genecards.org) in June 30, 2021. According to “gencode.v32.annotation.gff3” file, protein-coding genes (PCGs) were annotated. Limma R package was applied to identify differentially expressed genes (DEGs) between tumor samples and normal samples in the TCGA-LUAD dataset.19 |log2(fold change)| >1 and false discovery rate (FDR) <0.05 were determined as the threshold to screen DEGs. Venn analysis was used to show DEGs associated with COVID-19.
Identification of COVID-19 Target Genes
According to the TCM protocol in diagnosis and treatment protocol for COVID-19 (trial version 8), nine TCM prescriptions were obtained. For each prescription, main active ingredients were identified using the Traditional Chinese Medicine Database and Analysis Platform (TCMSP, https://tcmsp-e.com/). Effective molecules of active ingredients and drug targets were screened under the criteria of oral bioavailability (OB) >30% and drug-likeness (DL) >0.18. UniProt database (https://www.uniprot.org/) was used to annotate drug targets. CytoScape (V3.8.0)20 was introduced to develop a network among TCM, molecules and drug targets related to COVID-19. NetworkAnalyzer tool in CytoScape was employed to analyze topology for each interaction. The top 10 molecules and drug targets of FPQXZ were listed based on the degree and BetweennessCentrality. Finally, drug targets interacting with molecules and COVID-19 were confirmed as COVID-19 target genes.
Construction of Protein–Protein Interaction (PPI) Networks for COVID-19 Target Genes
Interactions among the 26 COVID-19 target genes were predicted by String online tool (https://cn.string-db.org/) and visualizing by CytoScape. NetworkAnalyzer tool in CytoScape was used to calculate the degree and determine the importance of one gene in the network. Under conditions of degree cutoff = 2, node score cutoff = 0.2, k-core = 2 and max depth = 100, MCODE and Haircut algorithms were applied to identify gene modules.
Functional Analysis
WebGestalt, which is a popular tool for interpreting functional categories based on the enrichment of gene sets, was employed to annotate gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.21 The top 10 enriched terms and pathways were visualized.
Assessment of Immune Features
ESTIMATE was employed to calculate immune score of tumor microenvironment (TME) in the TCGA-LUAD dataset based on immune-related gene sets.22 CIBERSORT was used to calculate the enrichment score for the 22 immune cells.23 Pearson correlation rank analysis was performed to assess the relation between the immune score and COVID-19 target genes, between the enrichment of immune cells and COVID-19 target genes.
Mutation Analysis of 26 COVID-19 Target Genes
Mutect2 algorithm was applied to detect SNVs, including missense, nonsense, splice site mutations, frame-shift insertions and deletions.24 CNVs were identified by Segment_Mean, where Segment_Mean >0.2 indicates amplification, −0.2 < Segment_Mean <0.2 indicates diploid, and Segment_Mean < −0.2 indicates deletion.
Correlation Analysis Between COVID-19 Target Genes and Cell Death
Gene sets of cell death including pyroptosis, ferroptosis and necroptosis were downloaded from Molecular Signatures Database v7.4 (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/). Single sample gene set enrichment analysis (ssGSEA) was conducted to calculate the enrichment score of the three types of cell death for each sample.25 Pearson correlation analysis was used to evaluate the association between COVID-19 target genes and cell death.
Unsupervised Consensus Clustering
Unsupervised consensus clustering in ConsensusClusterPlus R package is a popular method for an efficient and automatic clustering of samples based on gene expression patterns into different groups with different molecular features.26 Expression profiles were centralized and normalized using scale. PAM algorithm and Spearman was the basis to conduct 500 times of bootstraps, with 90% samples in the TCGA-LUAD dataset included in each bootstrap. Cluster number k was set from 2 to 10. Cumulative distribution function (CDF) and consensus matrix were used to confirm the optimal cluster number.
Prediction of Sensitivity to Immunotherapy and Chemotherapy
To predict the response of different molecular subtypes to immunotherapy, a treatment dataset IMvigor210 was introduced as a reference and SubMap analysis was used for comparing the similarity of expression profiles.27 P values between IMvigor210 and the TCGA-LUAD or the GSE31210 represent the similarity, where lower P values indicate a higher similarity. pRRophetic R package was applied to assess the biochemical half-maximal inhibitory concentration (IC50) of chemotherapeutic drugs including cisplatin, erlotinib, rapamycin, sunitinib and MG-132.28
Statistical Analysis
Student's t-test was conducted to detect statistical significance between the two groups. Log-rank test was used in Kaplan–Meier survival analysis. P < 0.05 was defined as significant. ns, no significance. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Statistical analysis was performed in R software (4.1.1). Parameters of tools without introduction were set as default.
Results
Identifying Dysregulated Genes Related to Both COVID-19 and LUAD
Firstly, we compared gene expression profiles between normal and tumor samples in the TCGA-LUAD dataset. Two thousand six hundred and seventeen DEGs (1012 upregulated and 1605 downregulated) were screened under the threshold of |log2 (fold change)| >1 and FDR < 0.05 (Figure 1A). Two hundred and ninety-one genes (167 downregulated and 124 upregulated) were found in the intersection of the 2617 DGEs with COVID-19-related genes (Figure 1B), indicating that these 291 genes may be potential drug targets for treating LUAD patients with COVID-19. To further understand the function of these genes, we analyzed enriched KEGG pathways with respect to upregulated and downregulated genes, respectively. Upregulated genes were significantly enriched to 5 KEGG pathways (FDR < 0.05), including drug metabolism, biosynthesis of amino acids, Fanconi anemia pathway, Interleukin (IL)-17 signaling pathway, and pyrimidine metabolism (Figure 1C). Downregulated genes were significantly enriched to 50 KEGG pathways, and the top 5 enriched pathways, namely, Malaria, Staphylococcus aureus infection, Rheumatoid arthritis, AGE-RAGE signaling pathway in diabetic complications, and hematopoietic cell lineage, were visualized (Figure 1D).
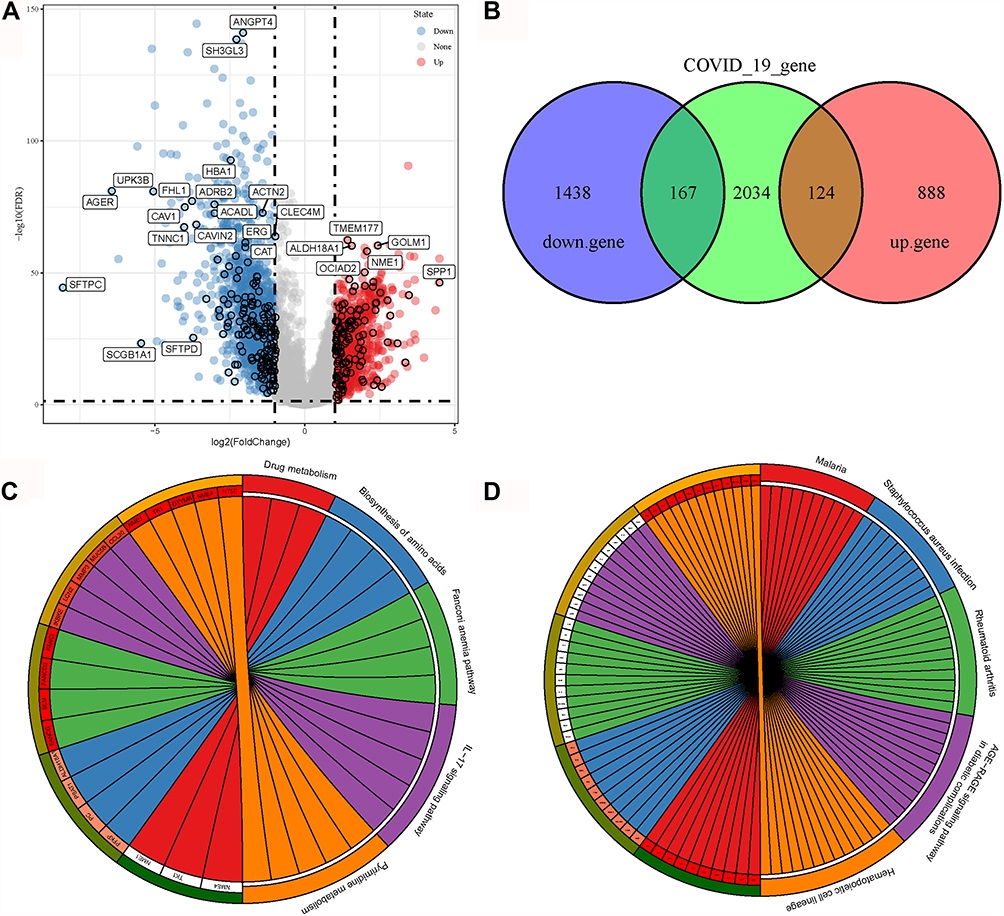 |
Figure 1 Identification of COVID-19-related target genes. (A) Volcano plot of upregulated (blue) and downregulated (red) genes in the TCGA-LUAD dataset. Vertical dashed lines indicate |log2(fold change)| = 1. Horizontal dashed line indicates FDR < 0.05. (B) Venn plot among downregulated genes (purple), upregulated genes (red) and COVID-19-related genes (green). (C and D) Enriched KEGG pathways on upregulated (C) and downregulated (D) COVID-19-related genes. Different colors indicate different pathways, and each radiation lines indicate one gene in one pathway. |
Constructing Networks of “TCM-Chemical Compounds-Target Genes” in Combination COVID-19 with LUAD
According to the TCM protocol in diagnosis and treatment protocol for COVID-19 (trial version 8), we obtained nine TCM prescriptions. For each TCM, the main active ingredients were analyzed using TCMSP. The effective molecules and drug targets were screened with criteria of OB ≥ 30% and DL ≥ 0.18. Drug targets were annotated by UniProt database. Next, CytoScape was applied to develop networks among drug targets, TCMs and ingredients grouped by each TCM prescription. For YDBFZ, 12 TCMs were included, and a number of effective ingredients of 12 TCMs were presented in the left of the network (Figure 2). A large number of targets of these molecules were visualized, and 18 targets were related to COVID-19, as shown in the right of the network. Networks of other eight TCM prescriptions were also presented, and COVID-19-related genes were identified (Supplementary Figures S1–S8). According to the degree of these molecules and COVID-19-related genes in the networks, the top 10 of them such as FPQXZ were screened (Tables 1 and 2). The one-to-one correspondence among molecules, COVID-19 targets, and FPQXZ were listed (Supplementary Table S1).
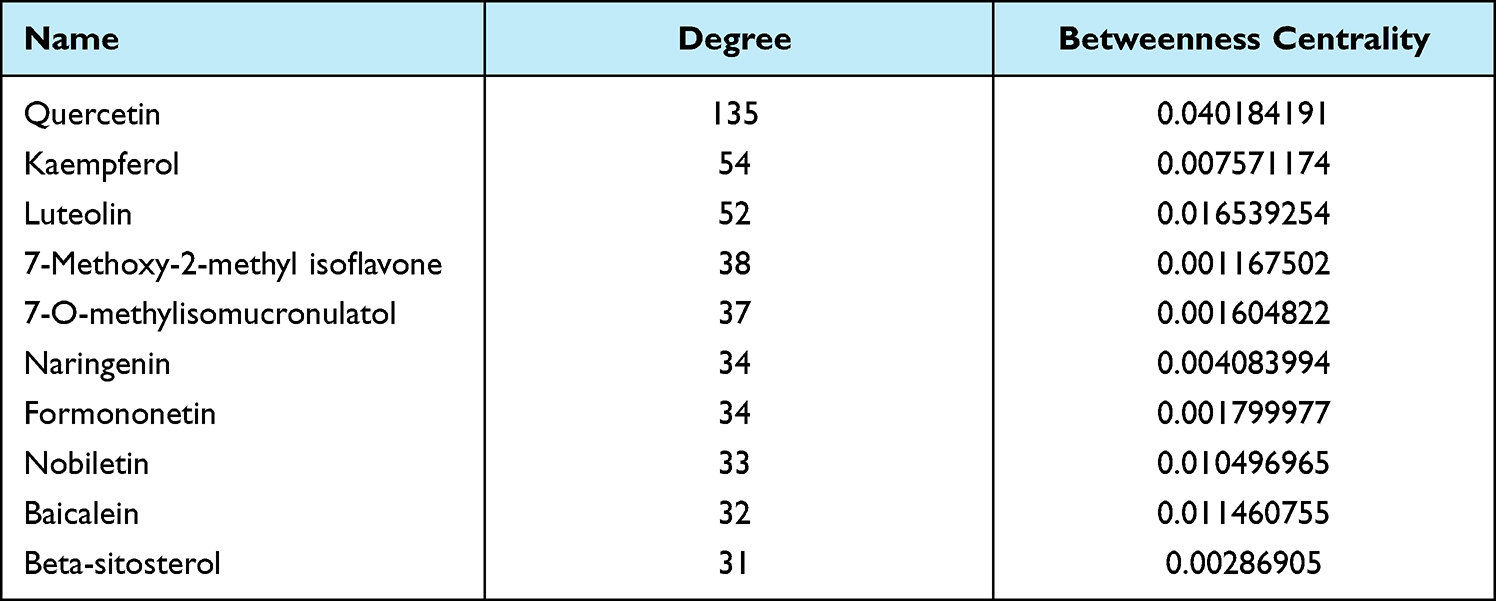 |
Table 1 The Top 10 Effective Molecules of FPQXZ Associated with COVID-19-Related Target Genes |
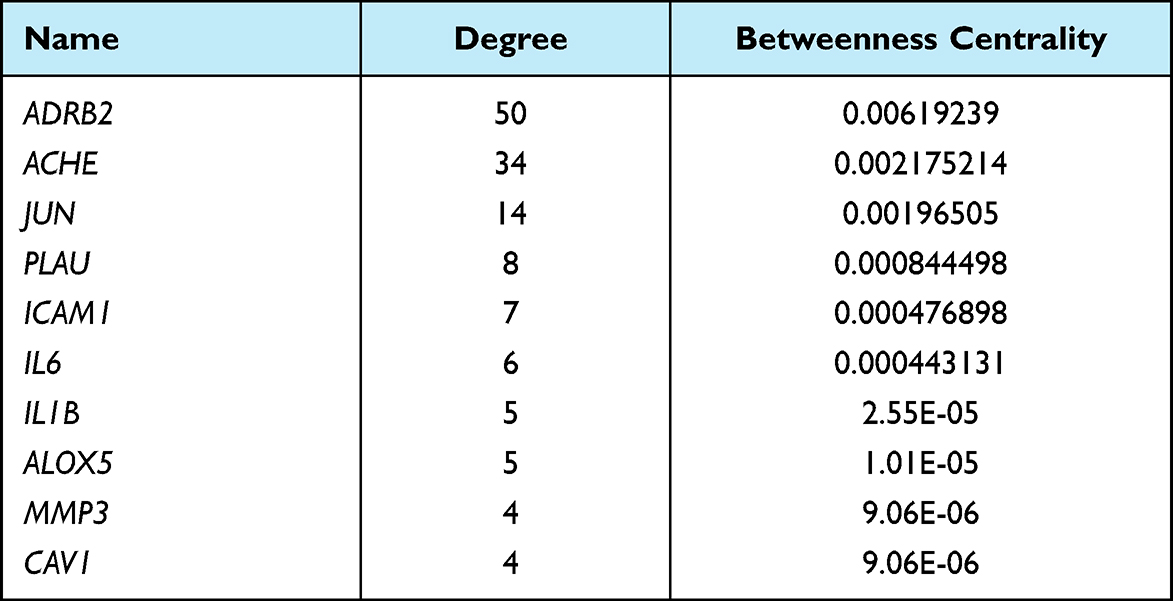 |
Table 2 The Top 10 COVID-19-Related Target Genes Associated with FPQXZ |
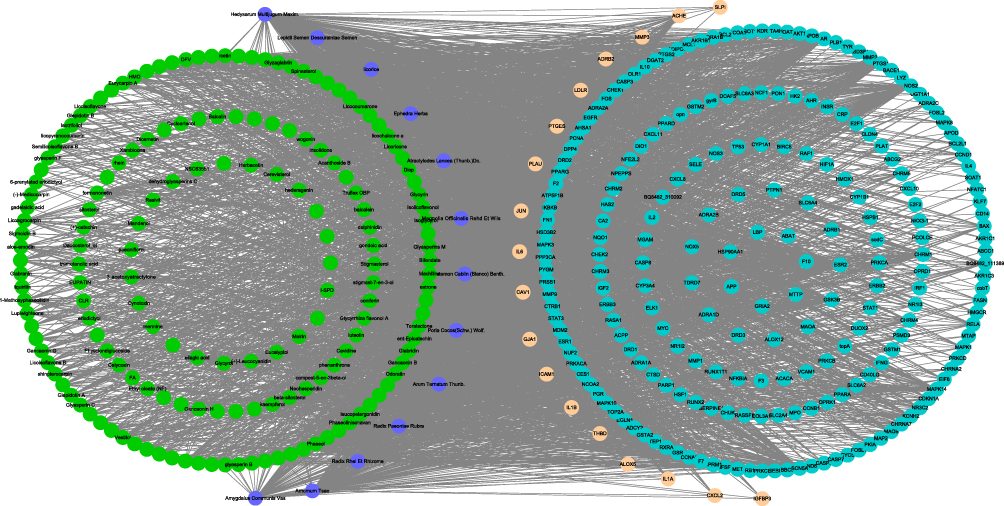 |
Figure 2 A network among YDBFZ, molecules of YDBFZ, and COVID-19-related target genes. Purple indicates TCMs of YDBFZ. Green indicates effective molecules in TCMs. Blue indicates target genes of molecules. Light Orange indicates COVID-19-related target genes. |
Genomic Features and Functional Analysis of COVID-19 Target Genes
In total, we identified 26 genes associated with both COVID-19 and TCMs. In tumor samples, these genes were noticeably mutated, particularly, the missense mutation type was the majority (Figure 3A). CD163 gene, which was reported as a marker of M2 macrophages, presented the highest mutations among others (Figure 3B). Apart from SNVs, a large number of CNVs, such as SELP (52.06% CNVs), IL6 (43.32 CNVs), and IGFBP3 (40.00% CNVs), were observed in tumor samples (Supplementary Table S2). The expression of these genes significantly changed with amplifications and/or deletions (Supplementary Figure S9).
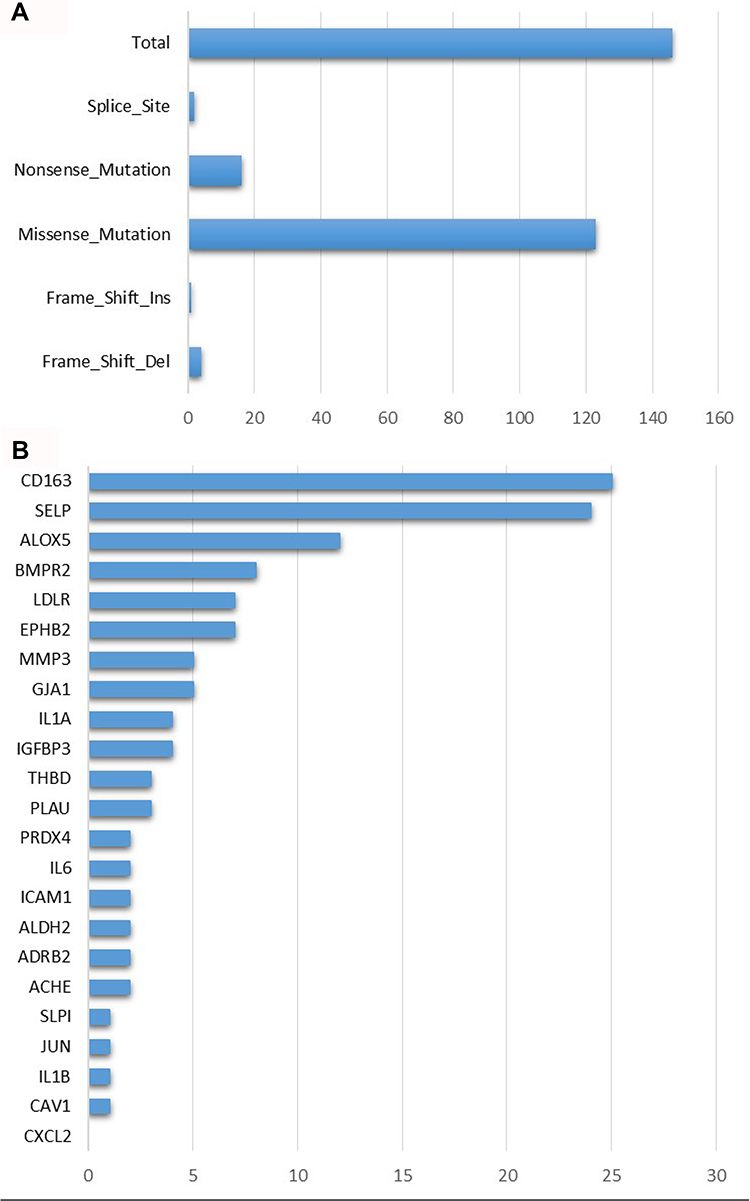 |
Figure 3 Mutation features of 26 COVID-19 target genes in the TCGA-LUAD dataset. (A) Mutation numbers of different types of mutations including splice site, nonsense, missense, frame-shift insertions and deletions. (B) Mutation counts of 26 COVID-19 target genes. |
To understand the function of these identified COVID-19 target genes, WebGestaltR was used to annotate GO terms and KEGG pathways for them. A total of 1610, 61 and 128 terms of biological process, cellular component, and molecular function were annotated, respectively, with FDR < 0.05, and the top 10 of each group were visualized (Figure 4A–C). In addition, we identified 156 KEGG pathways (FDR < 0.05). Tumor-related pathways such as prostate cancer, tumor necrosis factor (TNF) signaling pathway and pathways in cancer were annotated, and IL-17 signaling pathway related to the immune system was also identified (Figure 4D).
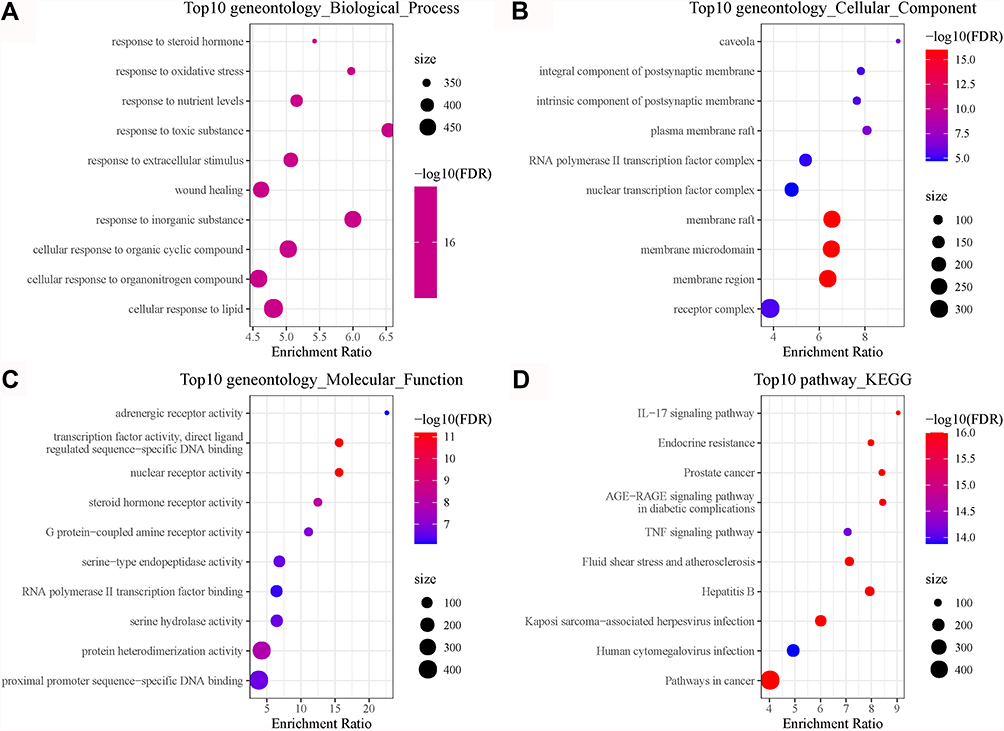 |
Figure 4 Functional analysis of 26 COVID-19 target genes in the TCGA-LUAD dataset. (A–C) The top 10 enriched terms of biological process, cellular component and molecular function. (D) The top 10 enriched KEGG pathways. Size indicates gene counts. FDR, false discovery rate. |
The Relation Between COVID-19 Target Genes and Immune Feature
As COVID-19 target genes were reported to be closely related to immunity, we further investigated their correlations in the TCGA-LUAD dataset through ESTIMATE and CIBERSORT tools. ESTIMATE score was calculated for each gene. Spearman correlation analysis revealed that ESTIMATE score was correlated with the expression of most COVID-19 target genes (Figure 5A), especially with ADRB2 (R = 0.40, P = 2.31e-21), ALOX5 (R = 0.65, P = 2.61e-63), Interleukin-1beta (IL1B) (R = 0.48, P = 1.82e-30), CD163 (R = 0.64, P = 2.05e-60), and SELP (R = 0.42, P = 6.56e-23). Eleven of 26 genes showed significant positive correlations with ESTIMATE score, indicating that COVID-19 target genes may be closely associated with the regulation of tumor immune microenvironment. Furthermore, we introduced CIBERSORT tool to calculate enrichment score of 22 immune cells for each sample. Apart from CD4 naive T cells, other 21 immune cells all obtained corresponding scores. Using Pearson correlation analysis between enrichment score and the expression of 16 genes, a heatmap was showing strong correlations between the two was plotted (Figure 5B). In general, plasma cells, follicular helper T cells, regulatory T cells and activated NK cells were negatively correlated with COVID-19 target genes, while neutrophils were positively correlated with COVID-19 target genes. Different immune cells manifested different correlation patterns with the 26 genes, suggesting that COVID-19 target genes played different roles on these immune cells.
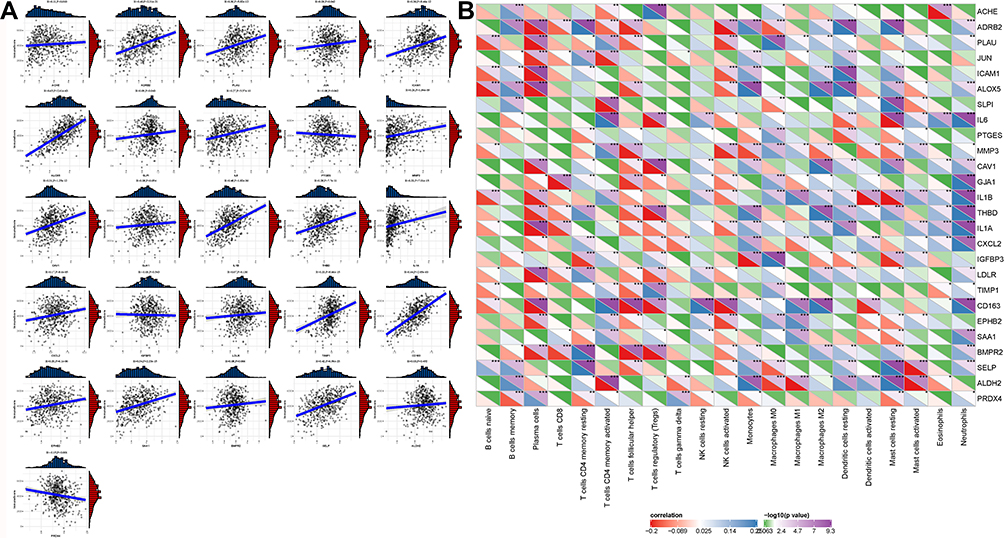 |
Figure 5 Correlation analysis between immune score and COVID-19 target genes in the TCGA-LUAD dataset. (A) Spearman correlation analysis between immune score and the expression of 26 COVID-19 target genes. Vertical axis represents immune score and horizontal axis represents gene expression. (B) Pearson correlation analysis between the enrichment of 21 immune cells and the expression of 26 COVID-19 target genes. Colors from red to blue indicate negative to positive correlations. Colors from green to purple indicate P values from high to low. *P < 0.05, **P < 0.01, ***P < 0.001. |
Identifying Key COVID-19 Target Genes and Pathways Through PPI Analysis
To better understand the interactions among these 26 COVID-19 target genes, String online tool was used to analyze their correlations and CytoScape was used to visualize the interactions (Figure 6A). Based on the degree of each gene and the analysis of MCODE module, two clusters were identified to be the key components by Haircut algorithm (Figure 6B). Specifically, cluster 1 included IL6, IL1B, ICAM1, PLAU, IGFBP3, LDLR and ALOX5, and cluster 2 included TIMP1, CAV1, MMP3, SAA1, JUN, SELP, CXCL2, and IL1A. The remained 8 genes were dissociative from the two clusters. For the 15 genes within the two clusters, we enriched 31 KEGG pathways including tumor-related pathways (such as NF-kappa B signaling, NOD-like receptor signaling, TNF signaling pathways, and transcriptional misregulation in cancer) and immune-related pathways (such as cytokine–cytokine receptor interaction, IL-17 signaling and Th17 cell differentiation) (Figure 6C).
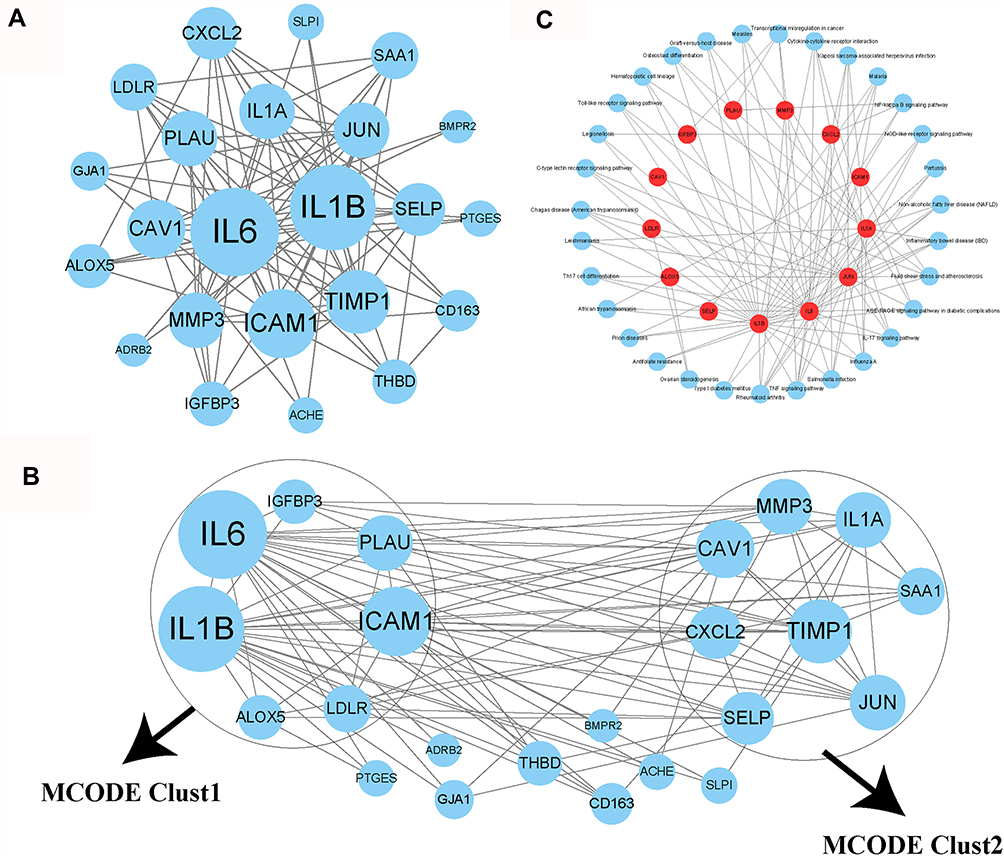 |
Figure 6 Analysis of interactions among 26 COVID-19 target genes. (A) A PPI network among the 26 genes. Higher dot size indicates more interactions with other genes. (B) Three groups (clust1, clust2 and genes outside clusters) identified by MOCODE. (C) Enrichment analysis of genes in clust1 and clust2. Red indicates genes in clust1 and clust2. Blue dots indicate enriched KEGG pathways. |
COVID-19 Target Genes are Associated with Cell Death
Subsequently, we tried to know if there was a relation between COVID-19 target genes and cell death. Three classes of genes related to cell death were obtained from MsigDB database, including pyroptosis, necroptosis and ferroptosis. The enrichment score of three types of cell death was calculated by ssGSEA. Spearman correlation analysis showed that 25 of 26 genes (except for ALDH2) were markedly correlated with at least one type of cell death (Figure 7). Especially, 13 of 26 genes, including ARDB2, PLAU, ICAM1, IL6, MMP3, CAV1, IL1B, THBD, IL1A, TIMP1, CD163, EPHP2 and SAA1, were significantly related to either one type of cell death (Figure 7 and Supplementary Figures S10–S12).
 |
Figure 7 Pearson correlation analysis between 26 COVID-19 target genes and cell death including ferroptosis, necroptosis and pyroptosis. Blue indicates negative correlation and red indicates positive correlation. *P < 0.05, **P < 0.01, ***P < 0.001. |
Constructing Two Molecular Subtypes Based on 26 COVID-19 Target Genes
As 26 COVID-19 target genes were highly involved in tumor-related pathways, immune response and cell death in LUAD, we then attempted to construct molecular subtypes based on their expression. ConsensusClusterPlus was used to conduct unsupervised consensus clustering. Cluster number k was set from 2 to 10, and CDF and consensus matrix were used to determine the optimal cluster number (Figure 8A and B). In the TCGA-LUAD dataset, samples were clearly clustered into the two groups when k = 2 (Figure 8C). Survival analysis showed that the two groups (Clust1 and Clust2) had differential overall survival with Clust1 had a better prognosis in the TCGA-LUAD dataset (P = 0.01, Figure 8D). In another independent dataset (the GSE31210), the same results were presented (P = 0.00026, Figure 8E).
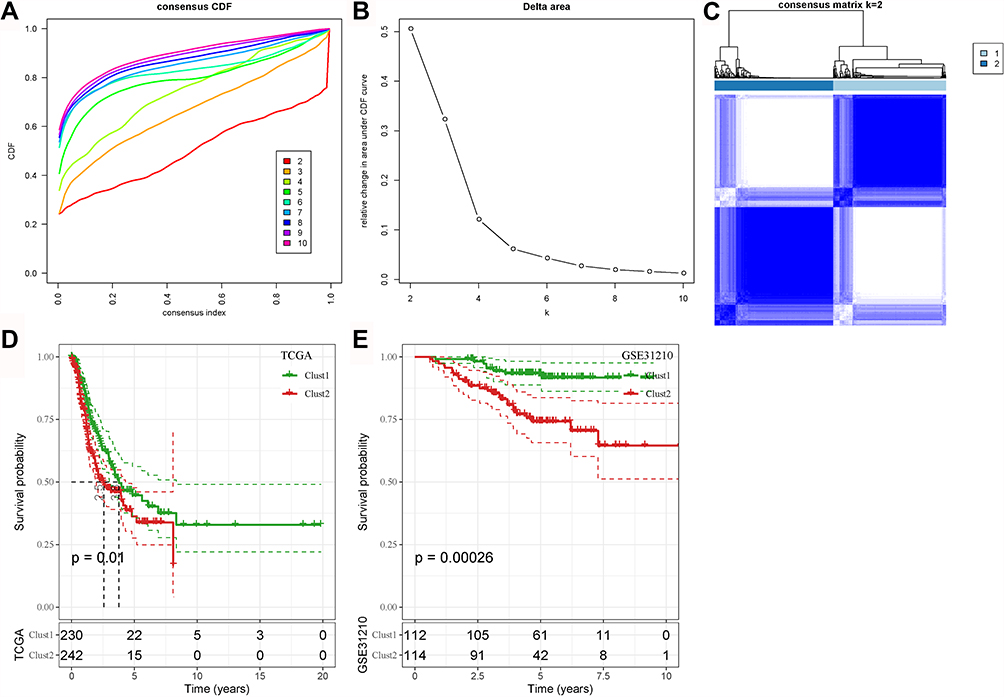 |
Figure 8 Unsupervised consensus clustering based on 26 COVID-19 target genes. (A and B) Consensus CDF curve and relative change in area under CDF curve when cluster number k = 2–10. (C) Consensus matrix when k = 2. (D and E) Kaplan–Meier survival analysis of Clust1 and Clust2 in the TCGA-LUAD (D) and the GSE31210 (E) datasets. Log-rank test was conducted. |
TME of Two Molecular Subtypes
To understand whether 26 COVID-19 target genes had an effect on TME, we analyzed the proportion of 22 immune cells in the two groups. As a result, 12 of 21 immune cells (naïve CD4 T cells with no expression were excluded) had differential enrichment scores in the TCGA-LUAD dataset (P < 0.05, Figure 9A and B). No significant difference in CD8 T cells was shown between the two groups. However, M0 and M1 macrophages were more enriched in Clust2, and M2 macrophages were more enriched in Clust1. Within 10 oncogenic pathways, 7 pathways were differentially enriched between Clust1 and Clust2 (P < 0.05, Figure 9C). Interestingly, except for the cell cycle and PI3K signaling pathway, other 5 pathways were all lower enriched in Clust2, which seemed contradictory with its unfavorable prognosis.
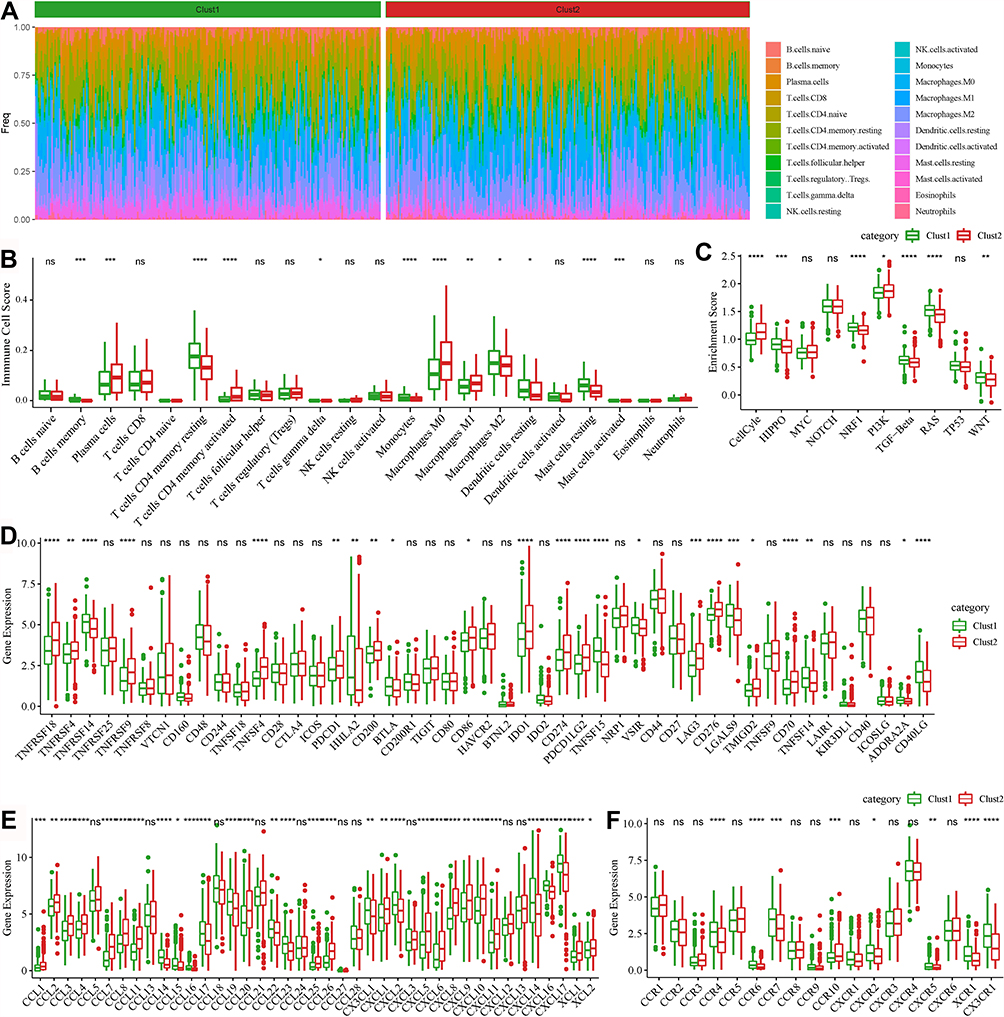 |
Figure 9 TME features of two molecular subtypes in the TCGA-LUAD dataset. (A) A heatmap of the proportion of 22 immune cells in two subtypes. (B) The enrichment score of the 22 immune cells in two subtypes. (C) The enrichment score of 10 oncogenic pathways. (D) Expression of 47 immune checkpoints in two subtypes. (E and F) Expression of chemokines (E) and chemokine receptors (F) in the two subtypes. Student t-test was conducted between the two groups. ns, no significance. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
To find out the reason for the above observations, we further assessed 47 immune checkpoints identified to play an important role in cell–cell communication in TME. The result showed that nearly half of the immune checkpoints were differentially expressed between two Clust1 and Clust2 (P < 0.05, Figure 9D). Notably, PDCD1 (PD-1), CD274 (PD-L1), IDO1 and LAG3, which were considered as immunosuppressive factors, were significantly upregulated in Clust2, and this may contribute to a worse prognosis of Clust2. In addition, we evaluated the expression of chemokines and chemokine receptors. It was observed that most of chemokines were more expressed in Clust2 compared with Clust1 (Figure 9E). However, chemokine receptors apart from CCR10 were more enriched in Clust1 in 8 differentially expressed receptors (Figure 9F). We also observed similar results on the GSE31210 dataset (Supplementary Figure S13), demonstrating that COVID-19 target genes could affect TME.
Sensitivity of Two Subtypes to Immunotherapy and Chemotherapy
As the TME was different in Clust1 and Clust2, we next explored if there was a difference in immunotherapy as well. SubMap analysis was applied to compare the similarity of expression patterns between the non-treated dataset and the treated dataset (IMvigor210). The result showed that the expression pattern of Clust1 was more similar to the anti-PD-1-treated samples with a status of complete response (CR) and partial response (PR) (P = 0.091), while Clust1 was more similar to stable disease (SD) and progressive disease (PD) (P = 0.001, Figure 10A). It could be speculated that Clust1 may benefit more from immunotherapy than Clust2. In addition, we assessed the sensitivity of the two groups to five chemotherapeutic drugs, including cisplatin, erlotinib, rapamycin, sunitinib and MG-132. Compared to Clust1, Clust2 showed a lower estimated IC50 of the five drugs, indicating that Clust2 was more sensitive to chemotherapy (P < 0.01, Figure 10B–F). The results were further verified in the GSE31210 dataset (Figure 10G–L), indicating that two subtypes had differential responses to targeted therapies.
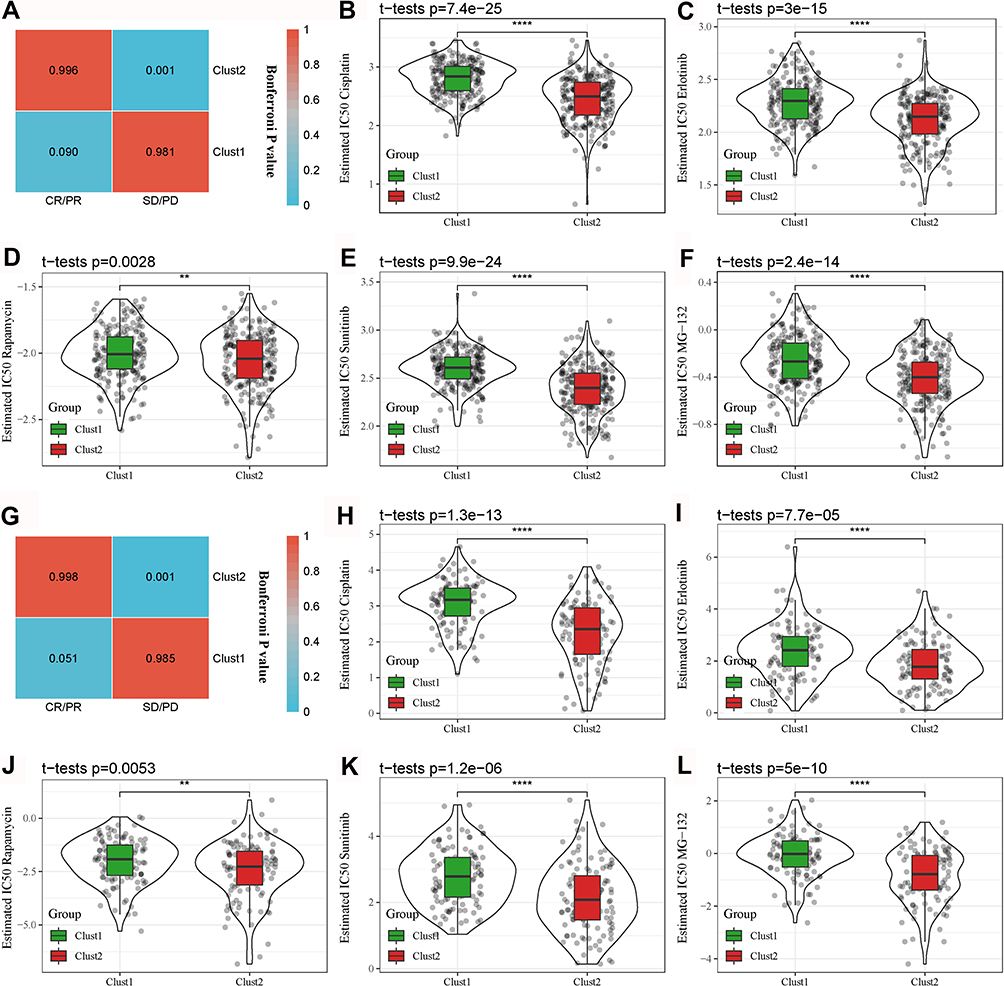 |
Figure 10 Differential sensitivity of the two subtypes to immunotherapy and chemotherapy. (A) SubMap analysis for comparing expression patterns between the TCGA-LUAD and IMvigor210 datasets grouped by CR/PR and SD/PD. (B–F) Comparison of estimated IC50 of five chemotherapeutic drugs between Clust1 and Clust2 in the TCGA-LUAD dataset. (G) SubMap analysis for comparing expression patterns between the GSE31210 and IMvigor210 datasets grouped by CR/PR and SD/PD. (H–L) Comparison of estimated IC50 of five chemotherapeutic drugs between Clust1 and Clust2 in the GSE31210 dataset. The gray dots in the figure represent tumor samples. Student t-test was conducted between the two groups. **P < 0.01, ****P < 0.0001. |
Discussion
The present identified 26 COVID-19 target genes related to TCM, COVID-19 and LUAD. Functional analysis showed that these genes were significantly enriched in tumor-related pathways including IL-17 signaling, TNF signaling, prostate cancer and pathways in cancer. Numerous studies showed that IL-17 plays an important role in tumor development, especially in TME regulation.29,30 IL-17 secreted from type 17 cells mediates the recruitment of immunosuppressive cells dependent on the collaboration with chemokines.30 Moreover, IL-17 signaling in TME can lead to epithelial-mesenchymal transition (EMT) via STAT3 and NF-κB signaling pathways in LUAD.31,32 Those data indicated that the 26 genes may be involved in the development of LUAD.
As for the relation between the 26 genes and TME, Pearson correlation analysis revealed that many genes were strongly correlated with immune infiltration and the proportion of certain immune cells, especially ADRB2, ALOX5, IL1B, CD163, and SELP. IL1B and CD163 have been widely reported to be associated with tumor development and TME. A high expression of IL1B could increase the risk of non-small cell lung cancer (NSCLC).33 CD163 is considered as one of the markers of M2 macrophages related to worse outcomes.34,35 Our result also showed a positive correlation between CD163 expression and M2 macrophages. PPI analysis presented that IL6 and IL1B showed the most interactions with other genes, indicating that they played central roles in regulation network among these genes.
Combining the above two results, we found that IL1B may be an important gene, the expression level of which could largely affect the interactions in the network and TME. Th17 cells are one of the major cells secreting IL-17. IL1B, a pro-inflammatory cytokine, drives Th17 differentiation together with transforming growth factor-beta (TGF-β), IL-6, and IL-21.36 Chen et al have found that lung cancer microparticles (L-MPs) induce macrophages to release IL1B, which has a tumor-promoting effect.37 Mature IL1B is released from immune cells through pyroptosis-mediated cell death.38 Active IL1B promotes tumor cell growth and metastasis through vascular endothelial growth factor (VEGF), prostaglandin E2 (PGE2) and TGF-β.39 Furthermore, through binding with IL-1, IL1B can activate MAPK and NF-κB pathways and induce downstream effects such as releasing granulocyte-macrophage colony-stimulating factor related to the activation of myeloid-derived suppressor cells (MDSCs) and M2 macrophages.40 Therefore, aberrant IL1B expression and signaling can alter the formation of TME and then promote tumor cell invasion and migration.
In addition, the close relation between cell death and 26 target genes were illustrated. Among the three types of cell death, necroptosis and pyroptosis were obviously shown to be positively correlated with the 26 target genes, especially PLAU, ALOX5, IL1B, IL1A and CD163. As indicated before, IL1B is one of the main products of pyroptosis,41 and IL-1β secretion can also be triggered by necroptosis,42 which is in accordant with our results. To some extent, cell death can affect the modulation of TME through inducing IL-1β. Kaplanov et al have found that IL1B inhibition can restore an effective immune response in breast cancer, and has a synergetic effect when combined with anti-PD-1 therapy in mice model.43 In IL1B-deficient mice, a high anti-tumor activity was shown, such as increased activated CD8 T cells and dendritic cells.43 We speculated that IL1B may be also a potential target for treating LUAD, but additional experiments are needed in future studies.
Based on the expression of 26 target genes, we constructed two novel molecular subtypes showing distinct prognosis. Notably, the two subtypes manifested differential TME, including the proportion of immune cells, expression of immune checkpoints, chemokines and chemokine receptors. The observations indirectly proved the role of 26 target genes in TME modulation and tumor development. Importantly, the two subtypes showed differential sensitivity to the immune checkpoint blockade and chemotherapy, which may guide clinical treatments for LUAD patients with COVID-19. Those data may prove worthwhile in developing new strategies for combating COVID-19 for LUAD.
Conclusions
Although potential COVID-19 targets predicted for LUAD therapy were identified from a large number of samples applying bioinformatics approaches, some limitations still exist in this study. Firstly, the sample lacked clinical follow-up information; thus, factors such as the presence of other health conditions were not considered during the identification of the biomarkers. Secondly, the results obtained by bioinformatics analysis alone were not convincing enough, which requires further experimental verifications. Genetic and experimental studies with larger sample sizes and experimental validation are also needed. In conclusion, this study proposed 26 potential COVID-19 targets, particularly IL1B, which may serve as a therapeutic drug target for developing effective molecular drugs for LUAD patients with COVID-19. The effective molecules in TCM interacting with IL1B may be considered as potential drugs, and can also guide the exploration of new therapeutic drugs for LUAD patients. In addition, the two new developed molecular subtypes can provide guidance for target therapies in clinical practice.
Data Sharing Statement
The dataset analyzed in this study is available in GSE31210 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31210.
Ethics Approval
This study has been exempted from ethical review by the Medical Ethics Committee of School of Medicine, Jinhua Polytechnic.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Science and Technology Planning Project of Jinhua City, P.R. China (grant no. 2020XG-10, and no. 2020-3-068).
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Addeo A, Friedlaender A. Cancer and COVID-19: unmasking their ties. Cancer Treat Rev. 2020;88:102041. doi:10.1016/j.ctrv.2020.102041
2. Bakouny Z, Hawley JE, Choueiri TK, et al. COVID-19 and cancer: current challenges and perspectives. Cancer Cell. 2020;38(5):629–646. doi:10.1016/j.ccell.2020.09.018
3. Giannakoulis VG, Papoutsi E, Siempos II. Effect of cancer on clinical outcomes of patients with COVID-19: a meta-analysis of patient data. JCO Glob Oncol. 2020;6:799–808. doi:10.1200/GO.20.00225
4. Kuderer NM, Choueiri TK, Shah DP, et al. Clinical impact of COVID-19 on patients with cancer (CCC19): a cohort study. Lancet. 2020;395(10241):1907–1918. doi:10.1016/S0140-6736(20)31187-9
5. Saini KS, Tagliamento M, Lambertini M, et al. Mortality in patients with cancer and coronavirus disease 2019: a systematic review and pooled analysis of 52 studies. Eur J Cancer. 2020;139:43–50. doi:10.1016/j.ejca.2020.08.011
6. Zhang H, Han H, He T, et al. Clinical characteristics and outcomes of COVID-19-infected cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst. 2021;113(4):371–380. doi:10.1093/jnci/djaa168
7. Liang W, Guan W, Chen R, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncol. 2020;21(3):335–337. doi:10.1016/S1470-2045(20)30096-6
8. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;41(1):1–24. doi:10.1016/j.ccm.2019.10.001
9. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.21654
10. Passaro A, Bestvina C, Velez Velez M, Garassino MC, Garon E, Peters S. Severity of COVID-19 in patients with lung cancer: evidence and challenges. J Immunother Cancer. 2021;9(3):e002266. doi:10.1136/jitc-2020-002266
11. Dai M, Liu D, Liu M, et al. Patients with cancer appear more vulnerable to SARS-CoV-2: a multicenter study during the COVID-19 outbreak. Cancer Discov. 2020;10(6):783–791. doi:10.1158/2159-8290.CD-20-0422
12. Li Q, Wang H, Li X, et al. The role played by traditional Chinese medicine in preventing and treating COVID-19 in China. Front Med. 2020;14(5):681–688. doi:10.1007/s11684-020-0801-x
13. Ren JL, Zhang AH, Wang XJ. Traditional Chinese medicine for COVID-19 treatment. Pharmacol Res. 2020;155:104743. doi:10.1016/j.phrs.2020.104743
14. Zhao Z, Li Y, Zhou L, et al. Prevention and treatment of COVID-19 using traditional Chinese medicine: a review. Phytomedicine. 2021;85:153308. doi:10.1016/j.phymed.2020.153308
15. National Health Commission in China. TCM protocol in diagnosis and treatment protocol for COVID-19 (trial version 8). 2020. Available from: http://www.gov.cn/zhengce/zhengceku/2020-08/19/5535757/files/da89edf7cc9244fbb34ecf6c61df40bf.pdf.
16. Wang Z, Yang L. Chinese herbal medicine: fighting SARS-CoV-2 infection on all fronts. J Ethnopharmacol. 2021;270:113869. doi:10.1016/j.jep.2021.113869
17. Yang L, Wang Z. Natural products, alone or in combination with FDA-approved drugs, to treat COVID-19 and lung cancer. Biomedicines. 2021;9(6):689. doi:10.3390/biomedicines9060689
18. Wang Z, Yang L. Turning the tide: natural products and natural-product-inspired chemicals as potential counters to SARS-CoV-2 infection. Front Pharmacol. 2020;11:1013. doi:10.3389/fphar.2020.01013
19. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
20. Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Curr Protocols Bioinform. 2014;47:
21. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199–w205. doi:10.1093/nar/gkz401
22. Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi:10.1038/ncomms3612
23. Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–259.
24. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi:10.1038/nbt.2514
25. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi:10.1038/nature08460
26. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi:10.1093/bioinformatics/btq170
27. Roh W, Chen PL, Reuben A, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9:379. doi:10.1126/scitranslmed.aah3560
28. Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 2014;9(9):e107468. doi:10.1371/journal.pone.0107468
29. Fabre J, Giustiniani J, Garbar C, et al. Targeting the tumor microenvironment: the protumor effects of IL-17 related to cancer type. Int J Mol Sci. 2016;17(9). doi:10.3390/ijms17091433
30. Kuen DS, Kim BS, Chung Y. IL-17-producing cells in tumor immunity: friends or foes? Immune Netw. 2020;20(1):e6. doi:10.4110/in.2020.20.e6
31. Huang Q, Han J, Fan J, et al. IL-17 induces EMT via Stat3 in lung adenocarcinoma. Am J Cancer Res. 2016;6(2):440–451.
32. Gu K, Li MM, Shen J, et al. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-κB/ZEB1 signal pathway. Am J Cancer Res. 2015;5(3):1169–1179.
33. Landvik NE, Hart K, Skaug V, Stangeland LB, Haugen A, Zienolddiny S. A specific interleukin-1B haplotype correlates with high levels of IL1B mRNA in the lung and increased risk of non-small cell lung cancer. Carcinogenesis. 2009;30(7):1186–1192. doi:10.1093/carcin/bgp122
34. Matsubara E, Komohara Y, Shinchi Y, et al. CD163-positive cancer cells are a predictor of a worse clinical course in lung adenocarcinoma. Pathol Int. 2021;71(10):666–673. doi:10.1111/pin.13144
35. Kwiecień I, Polubiec-Kownacka M, Dziedzic D, Wołosz D, Rzepecki P, Domagała-Kulawik J. CD163 and CCR7 as markers for macrophage polarization in lung cancer microenvironment. Cent Eur J Immunol. 2019;44(4):395–402. doi:10.5114/ceji.2019.92795
36. Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30(4):576–587. doi:10.1016/j.immuni.2009.02.007
37. Chen J, Sun W, Zhang H, et al. Macrophages reprogrammed by lung cancer microparticles promote tumor development via release of IL-1β. Cell Mol Immunol. 2020;17(12):1233–1244. doi:10.1038/s41423-019-0313-2
38. Kong H, Wang Y, Zeng X, Wang Z, Wang H, Xie W. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumour Biol. 2015;36(10):7501–7513. doi:10.1007/s13277-015-3473-4
39. Van Gorp H, Lamkanfi M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019;20(6). doi:10.15252/embr.201847575
40. Garon EB, Chih-Hsin Yang J, Dubinett SM. The role of interleukin 1β in the pathogenesis of lung cancer. JTO Clin Res Rep. 2020;1(1):100001. doi:10.1016/j.jtocrr.2020.100001
41. He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
42. Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep. 2015;11(10):1535–1548. doi:10.1016/j.celrep.2015.05.003
43. Kaplanov I, Carmi Y, Kornetsky R, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. 2019;116(4):1361–1369. doi:10.1073/pnas.1812266115
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.