Back to Journals » Infection and Drug Resistance » Volume 19
Identification of ROS-Related Gene TNFSF13B as a Diagnostic Biomarker in Tuberculosis: Insights from WGCNA
Authors Shang X, Ji X, Dong J, Yao S, Zhang X, Zhao Y, Jia H, Huang M, Wu Y, Zhang L, Zhu C, Zhang Z, Pan L
Received 14 March 2026
Accepted for publication 15 May 2026
Published 8 June 2026 Volume 2026:19 589810
DOI https://doi.org/10.2147/IDR.S589810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sandip Patil
Xuetian Shang,1 Xiuxiu Ji,1 Jing Dong,1 Siyu Yao,2 Xinyue Zhang,1 Yuehan Zhao,1 Hongyan Jia,1 Mailing Huang,3 Yadong Wu,1 Lanyue Zhang,1 Chuanzhi Zhu,1 Zongde Zhang,1 Liping Pan1
1Department of Molecular Biology, Beijing Key Laboratory for Drug Resistant Tuberculosis Research, Beijing Chest Hospital, Capital Medical University, Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing, People’s Republic of China; 2Department of Critical Care Medicine, Beijing Chest Hospital, Capital Medical University; Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing, People’s Republic of China; 3Department of Tuberculosis, Beijing Chest Hospital, Capital Medical University; Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing, People’s Republic of China
Correspondence: Liping Pan, Email [email protected]
Background: Despite medical advances, active tuberculosis (TB) remains difficult to diagnose, particularly in asymptomatic or paucibacillary cases lacking detectable pathogen evidence. Elevated oxidative stress is a hallmark of Mycobacterium tuberculosis (Mtb) infection, and the genes driving this process represent promising diagnostic biomarkers. Transcriptomic profiling captures dynamic host immune responses preceding clinical manifestations, while weighted gene co-expression network analysis (WGCNA) identifies functionally coherent gene modules. Integrating WGCNA with immune infiltration analysis, this study aimed to discover oxidative stress-related hub genes as robust biomarkers to address critical gaps in active TB detection and differential diagnosis.
Methods: Microarray data were derived from 10 healthy donors (HCs) and 10 active TB patients recruited from Beijing Chest Hospital. Other external datasets for validation were obtained from the Gene Expression Omnibus database. Correlation analysis of gene expression and immune cell infiltration using the CIBERSORT approach. WGCNA was employed to identify the critical modules associated with immune cells.
Results: WGCNA and immune infiltration analysis identified a monocyte-associated module harboring five ROS-related genes. Among these, TNFSF13B exhibited strong diagnostic performance (AUC > 0.8), served as a key oxidative stress gene in TB macrophages, was tied to energy metabolism. Notably, TNFSF13B expression effectively distinguished TB patients from those with other pulmonary diseases (AUC = 0.892), addressing a critical diagnostic gap, and significantly decreased following anti-tuberculosis therapy (p = 0.0001), suggesting utility in treatment monitoring. These findings were further supported by in vitro data showing markedly elevated TNFSF13B in Mtb-infected THP-1 cells compared to uninfected controls (FC = 5.98, p < 0.001).
Conclusion: Our findings identify TNFSF13B as a macrophage-specific, oxidative stress-related biomarker with strong potential for active TB diagnosis, not only for differential diagnosis from other pulmonary diseases but also for monitoring TB treatment response.
Keywords: tuberculosis, oxidative stress, monocyte, weighted gene co-expression network analysis, biomarker
Introduction
Mycobacterium tuberculosis (Mtb), the causative pathogen of tuberculosis (TB), accounts for the highest mortality among all infectious diseases globally. According to the Global TB Report, 10.7 million new cases and 1.23 million fatalities were documented in 2024.1 The lack of sufficiently accurate diagnostics and effective treatment regimens poses a critical impediment to controlling TB. For diagnosis of active TB, although the development of rapid molecular diagnostics has significantly improved the etiological confirmation rate for TB recently, approximately half of suspected patients in clinical settings still cannot obtain a definitive result, particularly those extrapulmonary TB patients with paucibacillary samples and the pulmonary TB patients with no sputum samples.1 These critical gaps highlight the urgent need for rapid, unambiguous, non-sputum-based biomarkers of active TB.2
As an intracellular pathogen, Mtb triggers oxidative stress responses that can lead to cellular degeneration, gene mutations, tissue damage, and compromised immune function. Host redox-dependent physiological reactions play a pivotal role in shaping the course of mycobacterial infection.3
On one hand, oxidative stress serves as an effective antimicrobial mechanism. The host can enhance anti-TB immunity by modulating genes involved in redox regulation. For example, SIRT3 facilitates anti-mycobacterial immunity by modulating mitochondrial homeostasis and inducing autophagy via the PPARα signaling pathway.4 High expression of SLIT2 post-infection has also been shown to exacerbate oxidative stress and glutathione depletion, worsening hepatic TB caused by Mtb.5
On the other hand, to counteract host defenses, Mtb virulence proteins such as the acetyltransferase Rv3034c, which induces peroxisome formation to resist macrophage oxidative stress and promote intracellular survival.6 Another acetyltransferase known as CysE controls Mtb virulence and drug resistance by mediating oxidative stress-related responses.7 However, excessive oxidative stress can aggravate tissue damage. Recent studies have reported that ferroptosis—an iron-mediated form of regulated necrotic cell death characterized by lipid peroxidation—can facilitate Mtb survival within host cells.8
With the global rise of multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB), host-directed therapies (HDTs) targeting oxidative stress regulation have emerged as promising strategies. For instance, metformin induces iron deficiency and enhances mitochondrial reactive oxygen species (ROS) in macrophages, creating an unfavorable environment for intracellular Mtb. N-acetylcysteine (NAC), a precursor of the antioxidant glutathione, provides conditionally essential cysteine for glutathione synthesis. A recent clinical study showed that NAC supplementation as an adjunctive therapy improved lung function recovery in TB patients.9 These results highlight the pivotal involvement of oxidative stress in the pathogenesis of TB.
As the primary host cells for Mtb, macrophage-mediated ROS play a crucial role in anti-tuberculosis immunity. Thus, the ROS-related genes in macrophages may not only reflect immune responses to infection but also serve as indicators of disease status. Identifying ROS-related genes in macrophages may offer novel targets for host-directed therapies against TB. The weighted gene co-expression network analysis (WGCNA) is a topological network analysis method that can establish associations between gene modules and clinical traits, which aligns perfectly with our goal: to identify the key genes related to macrophage ROS for use in the diagnosis or treatment monitoring of TB.
Herein, we performed a microarray analysis using peripheral blood mononuclear cells (PBMCs) from 10 active TB patients and 10 healthy controls (HCs), and identified the ROS-related genes with significant differences between patients with active TB and HCs. Furthermore, WGCNA and immune infiltration analysis were also performed to identify the module significantly associated with monocytes. Within this module, five genes—SH2D2A, KIR2DL3, KIR2DL4, GNAL, and TNFSF13B—were selected as potential key genes associated with ROS in macrophages of TB patients. Among these, TNFSF13B exhibited promising diagnostic value for active TB, which was supported by our microarray data and further validated by other external datasets. Additionally, the TNFSF13B was significantly upregulated in Mtb-infected THP-1 cells. In summary, our investigation suggests a new ROS-related candidate biomarker with potential for diagnosing active TB.
Materials and Methods
Participants Enrollment
All procedures adhered to the ethical standards of the Declaration of Helsinki and were sanctioned by the Institutional Review Board of Beijing Chest Hospital, Capital Medical University (approval no. BJXK-2017-40-01). All participants have signed the informed consent forms. Participants with active TB were enrolled at Beijing Chest Hospital during the period from January to May 2019. From October to December 2019, the HCs were recruited from a TB system screening project in Changping District, Beijing. The active TB was defined on the basis of positive Mtb culture or positive Xpert/MTB test. Healthy controls (HCs) were defined as individuals who showed no clinical symptoms or imaging findings suggestive of TB, had no history of TB, and had a negative interferon-γ release assay (IGRA) result. All the individuals were confirmed to be free of HIV, HBV, HCV, diabetes, autoimmune diseases, immunotherapy, anti-tuberculosis treatment, or pregnant.10
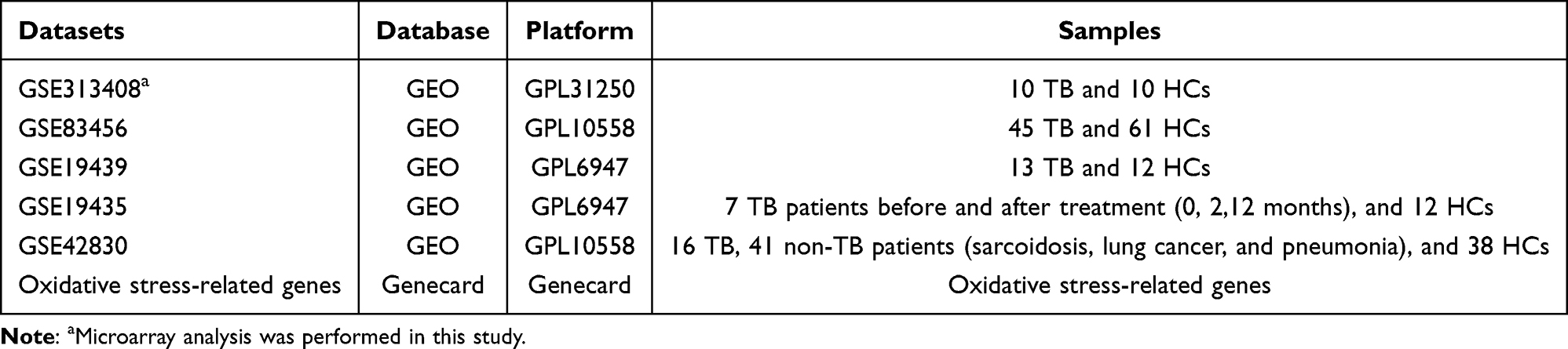 |
Table 1 Summary of the Datasets Utilized in This Study |
Blood Collection and PBMCs Separation
A total of 3 mL heparinized blood was collected, and PBMCs were separated using Lymphocyte Cell Separation Media within 4 hours. The PBMCs were immediately lysed by TRIzol lysis (Invitrogen, Carlsbad, CA, United States) and then frozen at −80 °C to avoid repeated freezing and thawing.
RNA Extraction and Microarray
RNA was purified from PBMC specimens utilizing the miRNeasy Mini Kit (QIAGEN, Germany). To remove genomic DNA contamination, the samples were treated with RNase-free DNase I (QIAGEN, Germany). RNA integrity and quality were evaluated on an Agilent 2100 Bioanalyzer, and only samples with an RNA integrity number (RIN) ≥ 7.0 and a 28S/18S ratio > 0.7 were subjected to microarray analysis. The microarray was performed in Shanghai Biotechnology Corporation, according to the manufacturer’s instructions. Briefly, 1.65 µg Cy3-cRNA was hybridized for 17 h on Agilent slides (Human ceRNA array V1.0 4×180K, Agilent Technologies), and then washed with a Gene Expression Wash Buffer kit (5188–5327, Agilent Technologies) and scanned using an Agilent Microarray Scanner (G2565CA, Agilent Technologies). Raw data were extracted (Feature Extraction 10.7) and normalized with the R-limma Quantile algorithm.11
External Data
External validation datasets with GEO accession numbers GSE83456, GSE19439, GSE19435, and GSE42830 were downloaded for this investigation. Additionally, we downloaded 13632 genes associated with oxidative stress from the Genecard database (https://www.genecards.org). The pertinent details were displayed in Table 1.
Identification of DEGs and DEOSGs
In our microarray results, the raw data were normalized using the limma package in R software, and differentially expressed genes were identified by fold-change (FC) analysis and T-test. The differentially expressed genes (DEGs) between the TB and HC groups were defined as those with |log2(FC)| > 1 and p < 0.05. The DEGs from GSE83456 and GSE19439 were identified utilizing “Limma” R package. Genes with log2(FC) > 1 and p < 0.05 were defined as upregulated, while genes with log2(FC) < −1 and p < 0.05 were classified as downregulated. The DEG set was subsequently overlapped with the oxidative stress gene list obtained from GeneCards database (https://www.genecards.org), generating differentially expressed genes related to oxidative stress (DEOSGs).
Integrative Analysis of WGCNA and Immune Cell Infiltration
Based on transcriptome data, CIBERSORT applies deconvolution methods to infer both the proportions and relative abundance of immune cell types in mixed cellular samples.12 We analyzed the immune infiltration using the CIBERSORT algorithm (https://www.bioinformatics.com.cn), an online platform for data analysis and visualization,13 and evaluated the proportion of 22 kinds of immune cells in PBMCs of HCs and TB patients. WGCNA was performed to identify the highly correlated gene modules, analyze the inter-module interactions and their associations with immune cells, and identify candidate biomarkers or therapeutic targets. Herein, WGCNA was performed with the WGCNA plug-in integrated in TBtools-II v2.0.14 Briefly, the fragments per kilobase of transcript per million mapped reads (FPKM) matrices were log2-transformed for further analysis. The top 8000 genes, selected based on the highest median absolute deviation (MAD), were retained for subsequent analysis. A signed co-expression network was constructed using the automatic one-step block-wise approach with the following key parameters: β = 18 (R2 = 0.85), minModuleSize = 30, mergeCutHeight = 0.25. Module–trait relationships were quantified by Pearson correlation between module eigengenes (MEs) and clinical metadata. The modules with the strongest correlation with the immune cells were selected as key module for subsequent analysis. Finally, the overlap between DEOSGs and key module genes was identified, with the resulting genes designated as differentially expressed immune-related oxidative stress genes (DEIOSGs) for further study.
Functional Enrichment and Pathway Activation Analysis
Gene ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes Genomes (KEGG) signal pathway analysis of DEGs were carried out by David (https://david.ncifcrf.gov/) online analysis tool. We generated the bubble chart using https://www.bioinformatics.com.cn.13 Single-gene GSEA (sgGSEA) was performed using the “fgsea” package to investigate the key signal pathways associated with TNFSF13B. In brief, all genes were ranked according to their Spearman coefficients with TNFSF13B, and then the ranked genes were compared with the locally downloaded KEGG files from the MSigDB database. To ensure consistency in gene symbols, they were harmonized using the clusterProfiler package and the human gene annotation database org.Hs.egdb.
Regulatory Network Construction
Transcription factor (TF)-target interactions were extracted from the Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining (TRRUST) (https://www.grnpedia.org/trrust/). Furthermore, we queried Mirwalk—a public database for miRNA-target interactions—to extract relevant miRNA-target pairs. Network construction was performed using Cytoscape.
Receiver Operating Characteristic (ROC) Analysis
ROC analysis was conducted using the “pROC” package in R, and the area under the curve (AUC) was computed to assess the sensitivity and specificity of the prediction model.
Macrophage Model for Mtb Infection
The human monocytic leukemia cell line THP-1 is frequently employed as an experimental system to explore macrophage response and mechanisms. THP-1 cells were differentiated using 100 ng/mL phorbol 12-myristate 13-acetate (PMA) for 36 hours. After washing with 1× PBS twice, they were infected with Mtb (H37Rv strain) at an MOI of 10 for 4 hours, 24 hours and 48 hours. Total RNA was extracted for subsequent analysis.
RT-qPCR Analysis
SYBR Green-based quantitative PCR was performed on the ABI 7500 Real-Time PCR System (Applied Biosystems, USA) with Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China). Relative quantification of target gene expression was performed using the comparative CT method (2−ΔCT), with GAPDH serving as the internal control for normalization. The cDNA samples were used as templates for the qPCR reactions. The primers of the TNFSF13B were below: F: 5′-aaagaagaagcgataagtggagtca-3′; R: 5′-gttaatccacattagcagcaacac-3′.
Statistical Analysis
All statistical evaluations were conducted in GraphPad Prism (version 8.0.2), with continuous variables presented as mean ± standard deviation (SD). Between-group differences were analyzed using two-tailed unpaired Student’s t-test for pairwise comparisons and one-way ANOVA for multi-group analyses. Non-parametric data were analyzed using the Mann–Whitney U-test. Statistical significance was defined as p < 0.05.
Results
Identification of ROS-Related DEGs
The experimental design and analytical pipeline are schematically presented in Figure 1. The high-throughput microarray test was conducted using 10 TB patients and 10 HCs matched by age and gender. However, due to the inadequate quality of the raw data, 2 TB patients and 1 HC were excluded from the final analysis. The demographic characteristics of the study participants are summarized in Table 2. Transcriptomic data from these individuals identified 1300 DEGs between the TB and HC groups. Among these, 874 genes were up-regulated, and 426 were down-regulated in TB patients (Figure 2A and B). The GO and KEGG enrichment analysis revealed that these DEGs were mainly enriched in the pathogen recognition and innate immune activation pathways of the infection-inflammation axis (Figure 2C and D). Among the DEGs, a total of 709 genes were identified as differentially expressed oxidative stress-related genes (DEOSGs), which were determined by intersecting the 1300 DEGs with the 13,632 known oxidative stress-related genes from the Genecard database (Figure 2E).
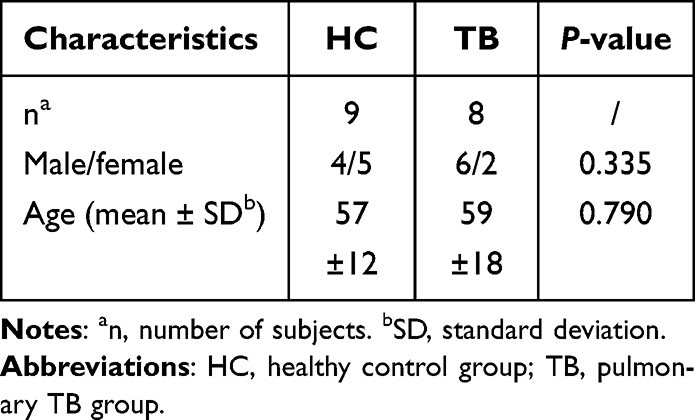 |
Table 2 Characteristics of the Microarray Cohort |
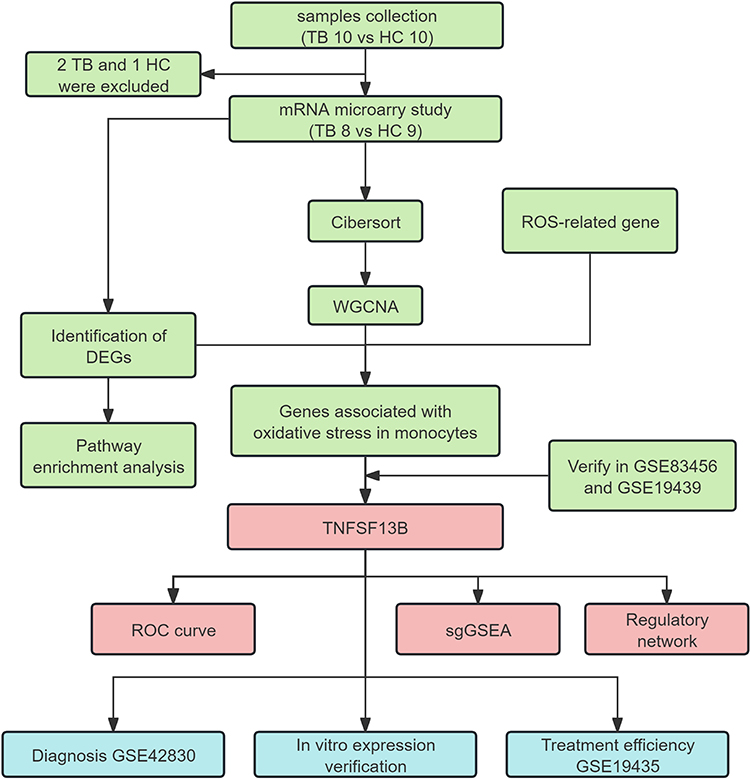 |
Figure 1 Flow diagram of this study. The color coding in the flowchart indicates three distinct research phases: green represents data collection and preliminary analysis; red denotes core findings and validation targets; blue signifies the independent validation stage. |
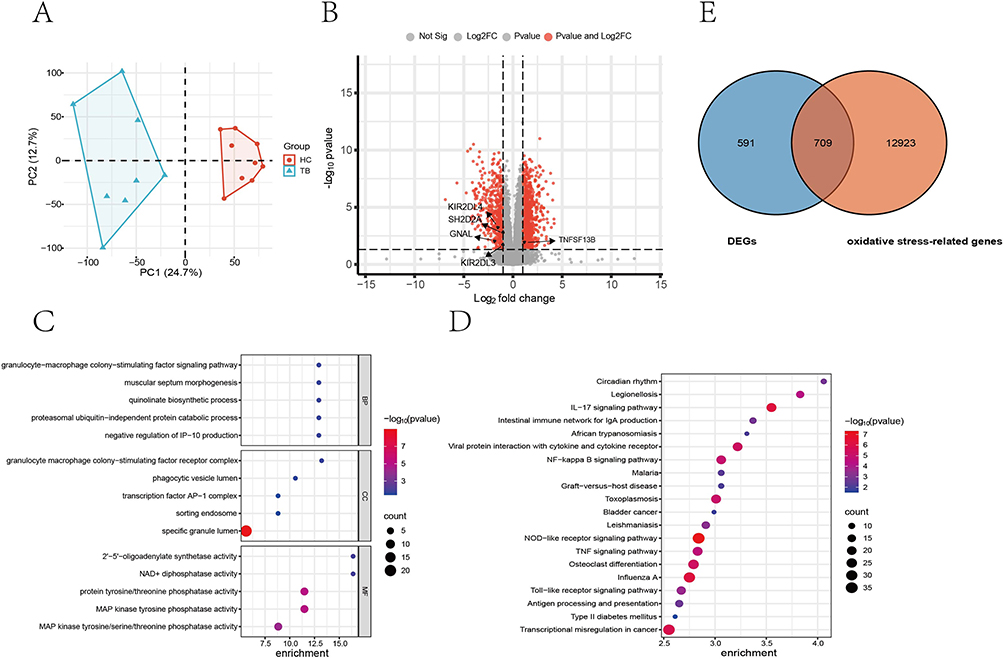 |
Figure 2 Screening of differentially expressed genes (DEGs) and oxidative stress-related DEGs in active TB patients. (A) Principal component analysis (PCA). (B) Volcano plot of the changes of gene expression in PBMCs derived from active TB patients compared with healthy controls (HCs). (C) GO enrichment analysis. (D) KEGG enrichment analysis. (E) Venn diagram showing the overlap between the DEGs and oxidative stress-related genes. Abbreviations: BP, biological process; MF, molecular function; CC, cellular component. |
Integrative Analysis of WGCNA and Immune Cell Infiltration
CIBERSORT algorithm analysis revealed significant differences in eight immune cell populations—memory B cells, eosinophils, monocytes, resting mast cells, activated NK cells, naive CD4 T cells, follicular helper T cells, and gamma delta T cells—between the TB and HC groups (Figure 3A).
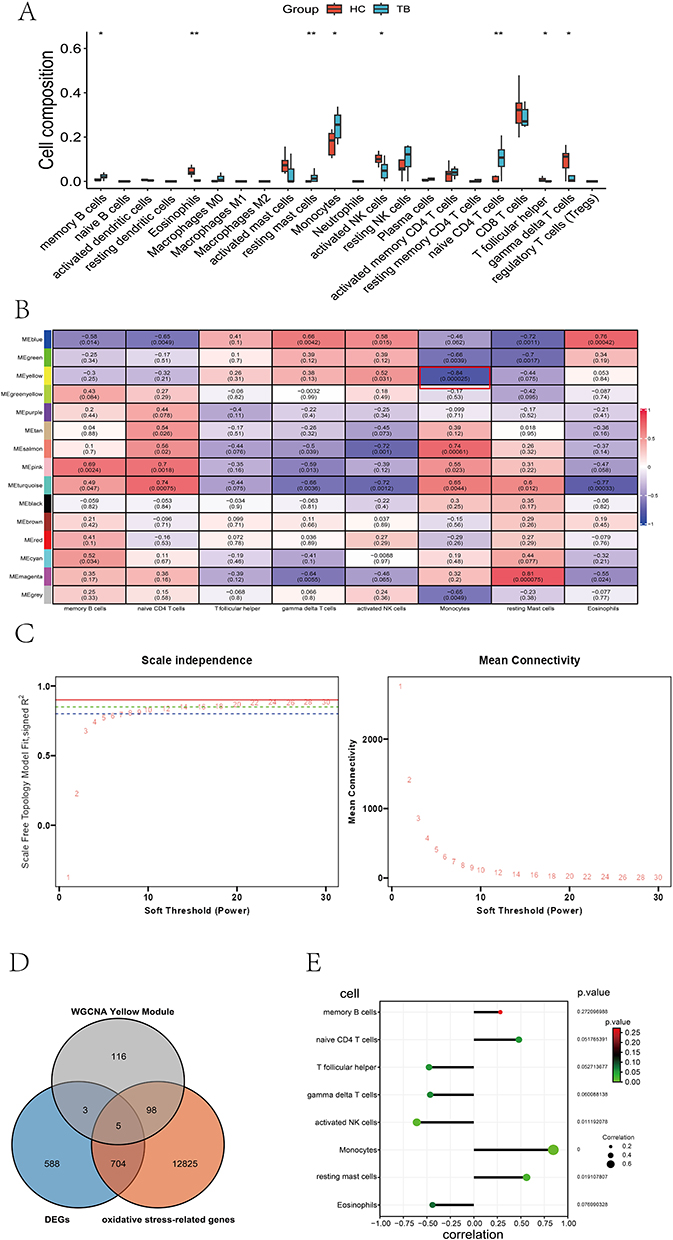 |
Figure 3 Immune infiltration analysis and construction of WGCNA. (A) The proportion of 22 kinds of immune cells in samples from HCs and TB patients. (B) Immune infiltration analysis and construction of weighted gene co-expression networks. The Yellow module, highlighted by a red frame, exhibited a strong negative correlation with monocyte subsets and was consequently selected for further investigation. (C) Determination of the scale-free fit index by soft-thresholding power analysis. The solid red line indicates R2 = 0.90 (standard threshold), the dotted green line indicates R2 = 0.85 (our selected cutoff), and the dotted blue line indicates R2 = 0.80 (minimum reference limit). The red numbers represent soft threshold powers (1–30). We selected power 18 as the optimal threshold, where R2 first exceeds 0.85. (D) Venn diagrams of DEIOSGs. (E) The lollipop diagram of the correlation between TNFSF13B and immune cells. * p < 0.05; ** p < 0.01. |
We employed WGCNA using the total expression profiles in microarray to identify the key modules related to TB immune response. A soft-threshold power of 18 was calibrated in our research (scale-free R2 = 0.85) (Figure 3B), and a total of 15 modules was identified by the WGCNA analysis (Figure 3C). The yellow module showed a strong negative correlation with monocyte subsets and was selected for further investigation.
Identification of DEOSGs Associated with Monocytes
In the yellow module, 222 hub genes were identified with gene significance (GS) > 0.5 and module membership (MM) > 0.5. We further selected the differentially expressed genes related to oxidative stress in monocytes through the triple intersection of the DEGs, the genes in the WGCNA-filtered yellow module, and the ROS-related genes from the Genecard database. A total of five genes (TNFSF13B, GNAL, SH2D2A, KIR2DL4, KIR2DL3) were identified, suggesting their potential involvement in oxidative stress response pathways within monocytes (Figure 3D). Correlation analysis between TNFSF13B and various immune cell subsets revealed that TNFSF13B exhibits the strongest association with monocytes (Figure 3E).
Validation in the External Databases
We employed two external datasets (GSE83456 and GSE19439) to confirm the expression profiles of the five genes associated with oxidative stress in monocytes. Among them, TNFSF13B exhibited consistent expression patterns and exhibited significant disparities between the TB and HCs groups in these external datasets (Figure 4). Furthermore, the other four genes also showed consistent expression patterns between the external dataset (GSE83456) and our microarray results, although only SH2D2A and KIR2DL3 differed significantly between the TB and HC groups. KIR2DL4 in the dataset GSE83456 and SH2D2A in the dataset GSE19439 also showed consistent expression patterns with our microarray results. These results verified the feasibility of our microarray data.
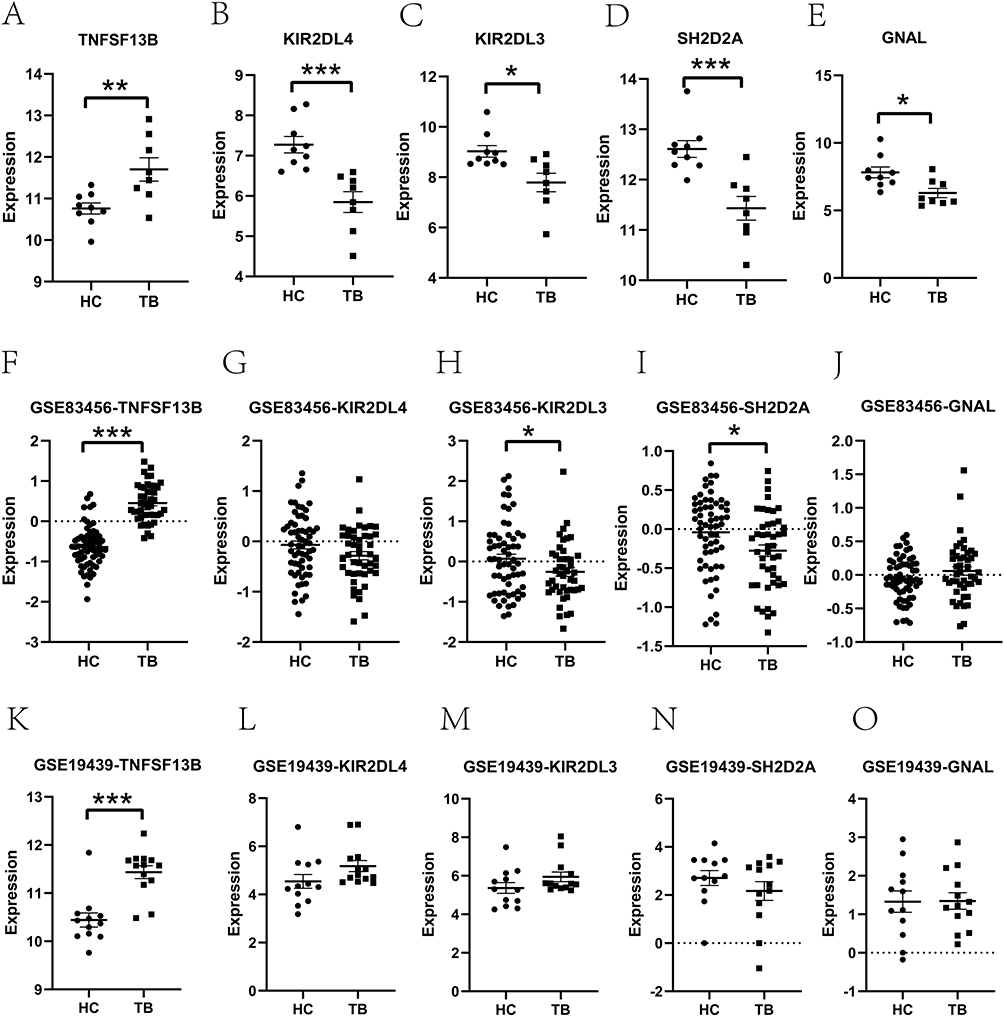 |
Figure 4 Verification of the hub genes. Each column represents a gene; Rows represent different groups. Expression of the five hub genes in our microarray data (A–E), the dataset GSE83456 (F–J), and GSE19439 (K–O). The dotted lines indicate the zero expression baseline after normalization, used as a reference level for comparing gene expression between groups. * p < 0.05; ** p < 0.01; *** p < 0.001. |
Regulatory Network Construction
Using the TRRUST database, 2 predicted TFs (STAT1, NFKB1) of TNFSF13B were finally obtained. In addition, potential miRNA-target interactions were predicted by Mirwalk, TargetScan, and miRDB databases. Among these, three microRNAs (miRNAs), including hsa-miR-6838-5p, hsa-miR-452-5p, and hsa-miR-892c-3p, were identified as the upstream regulators targeting TNFSF13B (Figure 5A).
 |
Figure 5 In-Depth Analysis of TNFSF13B. (A) The TF-mRNA-miRNA regulatory network analysis. Yellow represents transcription factors regulating TNFSF13B; purple represents the hub gene TNFSF13B; red represents miRNAs potentially regulated by TNFSF13B. (B) The sgGSEA analysis of TNFSF13B. (C) The expression level of TNFSF13B in THP-1 cells infected with Mtb. ** p < 0.01; *** p < 0.001. |
Single-Gene GSEA
Single-gene GSEA was performed, and the results showed that TNFSF13B was mainly enriched in pathways related to NAD⁺ homeostasis and mitochondrial complex I function (Figure 5B), which illustrated that TNFSF13B may be critically involved in the regulation of energy metabolism processes within the host immune response to TB infection, highlighting its potential role in modulating energetic metabolism essential for host anti-TB immunity.
Validation of TNFSF13B in Mtb-Infected Macrophages
Since TNFSF13B was selected as a ROS-related gene associated with monocytes, we further validated the expression in Mtb-infected THP-1 cells. Consistent with our microarray results from the patients with active TB and HCs, the expression level of TNFSF13B was also significantly upregulated in the Mtb-infected THP-1 cells than that in the uninfected THP-1 cells, with the most significant upregulation observed at 24h post-infection (FC = 5.98, p < 0.001). These results suggested that TNFSF13B may play a crucial role in the host immune response to Mtb infection and highlight its potential as a promising diagnostic biomarker for TB, warranting further functional studies to elucidate its mechanistic involvement. (Figure 5C).
ROC Curve Analysis
To explore the diagnostic performance of TNFSF13B, the ROC curve analysis was implemented (Figures 6A–C). In our microarray data, the AUC value was 0.861 (95% CI: 0.659–1.000) in discriminating TB from HCs. Notably, the AUC value was 0.939 (95% CI: 0.896–0.982) in the GSE83456 dataset, demonstrating excellent diagnostic performance. Furthermore, the AUC value was 0.891 (95% CI: 0.735–1.000) in the GSE19439 dataset, providing preliminary support for the diagnostic relevance of TNFSF13B across multiple independent cohorts.
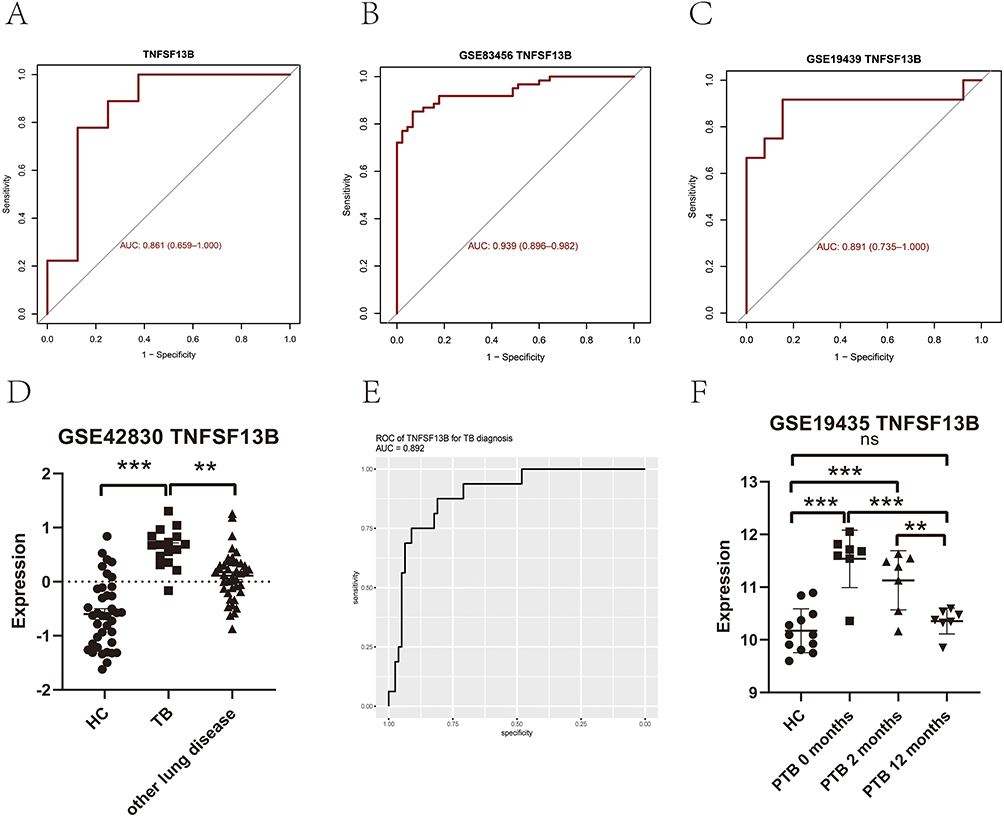 |
Figure 6 Diagnostic performance of TNFSF13B. (A) TNFSF13B in our microarray data. (B and C) TNFSF13B in GSE83456 and GSE19435 external datasets. (D) The expression levels of TNFSF13B in the peripheral blood derived from TB patients and other non-TB individuals in the GSE42830 dataset. (E) ROC curves of TNFSF13B were generated based on the GSE42830 dataset, using TB patients as the disease cohort and the other non-TB individuals as controls. (F) The expression levels of TNFSF13B in the peripheral blood of patients before and after anti-TB treatment (GSE19435). ** p < 0.01; *** p < 0.001. Abbreviations: ns, no significant; HC, healthy donor; TB, tuberculosis; ROC, Receiver operating characteristic; AUC, area under the ROC curve. |
Utility of TNFSF13B in Discriminating TB from Other Non-TB Patients
To further validate the value of TNFSF13B in discriminating TB patients from other non-TB patients, we retrieved dataset GSE42830, which enrolled patients with four major pulmonary diseases—active TB, sarcoidosis, lung cancer, and pneumonia. Comparative analysis of TNFSF13B expression was conducted between TB patients and non-TB controls. The results demonstrated that TB cases displayed a significantly higher expression level of TNFSF13B than that of other pulmonary diseases (p < 0.01) (Figure 6D). To further assess the discriminatory performance of TNFSF13B, the ROC analysis was performed, and the AUC value reached 0.89 (95% CI: 0.81–0.97) in discriminating TB patients from non-TB individuals (Figure 6E). These analyses preliminarily identified TNFSF13B as a potentially diagnostically informative biomarker for active TB, pending further validation.
The Decline of TNFSF13B Expression Following Anti-TB Therapy
Additionally, we further downloaded the GSE19435 dataset and analyzed the variations in the expression level of TNFSF13B before and after anti-TB treatment (Figure 6F). The results showed that TNFSF13B levels were markedly upregulated in the TB cohort compared with HCs. Notably, a significant reduction in TNFSF13B expression (p = 0.0001) was observed post-treatment, and this decrease displayed a treatment duration-dependent pattern, indicating a progressive decline with ongoing therapy. These results support the candidacy of TNFSF13B as a potential biomarker for TB diagnosis and therapeutic monitoring, suggesting its possible utility in future clinical diagnostic strategies pending further validation.
Discussion
Oxidative stress is a functional hallmark of Mtb infection, rendering the genes involved in this process as promising diagnostic biomarkers.15 Monocyte-derived macrophages, which were the key effectors of innate immunity, serve as the primary cellular niche for Mtb survival. Upon infection, they could trigger a robust intracellular oxidative burst that contributes to anti-mycobacterial defense.16 Here, an integrative bioinformatics approach was employed to identify TNFSF13B as a monocyte-derived oxidative-stress-related gene that can be used to diagnose active TB. Finally, independent datasets from public TB-cohort transcriptomes and further validation in Mtb-infected macrophages suggest TNFSF13B as a potential cell-type-specific diagnostic indicator of active TB.
The limitations of current microbiological and rapid molecular diagnostics, which can yield false-negative results, underscore the need for developing novel biomarkers derived from blood or other non-sputum fluids. Here, we integrated transcriptomics to discover robust, host-derived biomarkers of active TB. Given that the host response to Mtb infection and TB pathogenesis involves an oxidative stress response to limit bacterial infection and disease progression, we first mapped ROS-related transcriptional signatures that distinguished patients with TB from HCs. Through WGCNA, we identified immune-related co-expression modules and found that the gene module was most strongly associated with monocytes. Within this module, TNFSF13B exhibited potential for the diagnosis of TB and was subsequently validated in independent cohorts from public databases.
TNFSF13B (also known as BAFF), a member of the TNF superfamily, is a cytokine mainly secreted by myeloid cells, such as monocytes, macrophages, and dendritic cells. Conventionally, TNFSF13B plays a critical role in the regulation of B-cell survival, maturation, and antibody production, thereby enhancing adaptive immune response.17,18 However, a growing body of research has revealed that TNFSF13B engages in immune and inflammatory responses through a spectrum of molecular mechanisms, including its significant functions in innate immunity. It was reported that ROS can raise the expression level of TNFSF13B via the ROS–Epac1–Rap1–NF-κB axis.19,20 The JAK/STAT21 and NF-κB22 signaling pathways are both centrally involved during Mtb infection, further suggesting that TNFSF13B may exert its immunological effects within macrophages via one or more of these key signaling cascades. Studies in THP-1 cells have shown that reactive oxygen species (ROS) recruit the transcription factor c-Fos to the TNFSF13B promoter, thereby boosting promoter activity and elevating TNFSF13B expression.23 TNFSF13B also elicits the expression of inflammatory mediators in macrophages while suppressing cytoskeletal dynamics required for phagocytosis and trogocytosis.24,25 Single-cell RNA sequencing (scRNA-seq) data revealed a significant upregulation of TNFSF13B expression in patients with severe TB, and monocyte-derived TNFSF13B may play a pivotal role in driving cytokine storms in individuals suffering from severe TB, indicating its involvement in the progression of TB disease.26 Furthermore, in other infectious diseases, plasma TNFSF13B is markedly elevated in children with severe adenovirus pneumonia and tracks closely with the intensity of the cytokine storm.27 Studies also show that the absence of TNFSF13B exacerbates pseudomonas aeruginosa infection.28
In this study, we demonstrated that the expression level of TNFSF13B was significantly up-regulated in response to Mtb infection. Immune-infiltration deconvolution of bulk transcriptomes revealed the elevated expression of TNFSF13B in monocytes/macrophages from patients with TB, suggesting a critical role for this cytokine in macrophage-mediated anti-mycobacterial immunity. Moreover, TNFSF13B was significantly elevated in active TB and declined after anti-tuberculous therapy, supporting its potential as a diagnostic biomarker and a tool for treatment monitoring. These findings were consistent with earlier investigations, which also showed elevated TNFSF13B in active TB patients compared to individuals with latent TB infection and HCs.29,30
It has been established that IFN-γ stimulation can induce TNFSF13B release in monocytes, and the upregulation of TNFSF13B following viral infection is also dependent on IFN-γ release, suggesting the positive correlation between TNFSF13B and IFN-γ.31 It is well documented that lymphocytes from TB patients can release large amounts of IFN-γ following Mtb infection.32 It is plausible that the elevated TNFSF13B expression observed in our data is similarly mediated by IFN-γ signaling. Furthermore, TNFSF13B contributes to macrophage polarization by promoting M1 differentiation while suppressing M2 phenotypic maturation.33 Given that M1 polarization is a hallmark of active TB pathogenesis, the upregulation of TNFSF13B likely facilitates the skewing of macrophages toward an M1 phenotype during disease progression.
Therefore, the TNFSF13B gene represents a novel and promising transcriptome marker with greater potential compared to other mRNA diagnostic markers9 and lncRNA.10 Although we identified TNFSF13B as a potential modulator of host protective immunity against Mtb, as well as a potential biomarker that can be exploited for both diagnosis and treatment-response assessment for TB, by integrating bulk transcriptome analysis. Several limitations remain inherent to this investigation. Firstly, the molecular mechanisms of TNFSF13B in TB pathogenesis remain unclear; therefore, future studies are warranted to elucidate how it contributes to disease occurrence and development. In addition, given the limitations of the small primary cohort sample size, potential selection bias from the single-center design, limited validation cohort sizes, and the inherent inability to randomize in observational studies, larger-scale, multicenter clinical datasets are required to validate the predictive value of TNFSF13B in TB treatment. Moreover, a single gene often has limited diagnostic power. In-depth analysis suggests that multi-gene panels, including TNFSF13B, may offer promising potential for clinical translation. The cost-effectiveness and accessibility of TNFSF13B testing relative to existing diagnostics (eg, IGRA, Xpert MTB/RIF) remain to be evaluated. Prospective studies are further needed to determine whether TNFSF13B expression dynamics can reliably guide treatment duration and predict relapse, extending beyond mere diagnostic discrimination. Addressing these hurdles will be essential to realize the clinical potential of our findings.
Conclusion
By intersecting ROS-related DEGs and the WGCNA-identified monocyte module, coupled with further validation in public TB cohort databases, we identified TNFSF13B as a promising candidate biomarker with preliminary diagnostic and treatment monitoring potential for active TB. These findings refine the mechanistic understanding of oxidative stress in active TB and suggest a potential target for future non-sputum diagnostic strategies. However, given the exploratory, transcriptomic-driven nature of this study and the limited sample size of the discovery cohort, these conclusions should be validated in larger, multicenter cohorts to confirm the diagnostic utility and translational feasibility of TNFSF13B before further clinical application.
Abbreviation
TB, tuberculosis; Mtb, Mycobacterium tuberculosis; HCs, healthy donors; DEGs, differentially expressed genes; ROS, reactive oxygen species; MDR-TB, multidrug-resistant TB; XDR-TB, extensively drug-resistant TB; HDTs, host-directed therapies; NAC, N-acetylcysteine; PBMCs, peripheral blood mononuclear cells; WGCNA, weighted gene co-expression network analysis; IGRA, interferon-γ release assay; RIN, RNA integrity number; DEOSGs, differentially expressed genes related to oxidative stress; MAD, median absolute deviation; MEs, module eigengenes; DEIOSGs, differentially expressed immune-related oxidative stress genes; GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes Genomes; sgGSEA, Single-gene GSEA; TF, Transcription factor; TRRUST, Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining; ROC, Receiver operating characteristic; AUC, area under the ROC curve; SD, standard deviation; PMA, phorbol 12-myristate 13-acetate; GS, gene significance; MM, module membership; scRNA-seq, Single-cell RNA sequencing; miRNAs, microRNAs.
Data Sharing Statement
The datasets used and analyzed in the current study are available from the corresponding author on reasonable request. The microarray data has been uploaded to the Gene Expression Omnibus (GEO) database. GEO Series accession number is GSE313408.
Ethics Approval and Consent to Participate
This retrospective investigation adhered to the ethical guidelines established by the Declaration of Helsinki and received approval from the Ethics Committee of Beijing Chest Hospital, Capital Medical University (approval number: BJXK-2017-40-01). Given the retrospective nature of the study design, obtaining informed consent from participants was not feasible.
Acknowledgments
We acknowledge with deep thanks the commitment of all volunteers who engaged in this project.
Funding
Funding for this project was secured from the Prevention and Control of Emerging and Major Infectious Disease-National Science and Technology Major Project (2025ZD01907200), National Natural Science Foundation (82172279), the Beijing Natural Science Foundation (L234055), Beijing High-level Public Health Project (G2024-2-007).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. World Health Organization. Global tuberculosis report 2025.
2. World Health Organization. High-priority target product profiles for new tuberculosis diagnostics: report of a consensus meeting. WHO Meet Rep. 2014;1–14.
3. Tripathi A, Kalita J, Kant S, Misra UK. Oxidative stress and ER stress are related to severity of tubercular infection. Microb Pathogenesis. 2022;172:105764. doi:10.1016/j.micpath.2022.105764
4. Kim TS, Jin YB, Kim YS, et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy. 2019;15(8):1356–1375. doi:10.1080/15548627.2019.1582743
5. Borbora SM, Satish BA, Sundar S, B M, Bhatt S, Balaji KN. Mycobacterium tuberculosis elevates SLIT2 expression within the host and contributes to oxidative stress responses during infection. J Infect Dis. 2023. doi:10.1093/infdis/jiad126
6. Behera A, Jain P, Ganguli G, et al. Mycobacterium tuberculosis acetyltransferase suppresses oxidative stress by inducing peroxisome formation in macrophages. Int J Mol Sci. 2022;23(5):2584. doi:10.3390/ijms23052584
7. Zhang LY, Yin H, Wang YC, et al. The acetyltransferase CysE modulates virulence and drug resistance of Mycobacterium tuberculosis by interfering with oxidative stress responses. Commun. Biol. 2025;8(1):1425. doi:10.1038/s42003-025-08670-z
8. Araújo-Pereira M, Andrade BB. Oxidative battles in tuberculosis: walking the ferroptotic tightrope. Trends Immunol. 2025;46(4):338–351. doi:10.1016/j.it.2025.02.009
9. Mapamba DA, Sabi I, Lalashowi J, et al; TB SEQUEL consortium. N-acetylcysteine modulates markers of oxidation, inflammation and infection in tuberculosis. J Infect. 2025;90(2):106379. doi:10.1016/j.jinf.2024.106379
10. Pan L, Huang M, Jia H, et al. Diagnostic performance of a novel CXCL10 mRNA release assay for Mycobacterium tuberculosis infection. Front Microbiol. 2022;13:825413. doi:10.3389/fmicb.2022.825413
11. Dong J, Song R, Shang X, et al. Identification of important modules and biomarkers in tuberculosis based on WGCNA. Front Microbiol. 2024;15:1354190. doi:10.3389/fmicb.2024.1354190
12. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods. 2015;12(5):453–457. doi:10.1038/nmeth.3337
13. Tang D, Chen M, Huang X, et al. SRplot: a free online platform for data visualization and graphing. PLoS One. 2023;18(11):e0294236. doi:10.1371/journal.pone.0294236
14. Chen C, Wu Y, Li J, et al. TBtools-II: a “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant. 2023;16(11):1733–1742. doi:10.1016/j.molp.2023.09.010
15. Ganguli G, Mukherjee U, Sonawane A. Peroxisomes and oxidative stress: their implications in the modulation of cellular immunity during mycobacterial infection. Front Microbiol. 2019;10:1121. doi:10.3389/fmicb.2019.01121
16. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264(1):182–203. doi:10.1111/imr.12266
17. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND April: a tutorial on B cell survival. Ann Rev Immunol. 2003;21:231–264. doi:10.1146/annurev.immunol.21.120601.141152
18. Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. 2002;2(7):465–475. doi:10.1038/nri844
19. Moon EY, Lee JH, Oh SY, et al. Reactive oxygen species augment B-cell-activating factor expression. Free Radic Biol Med. 2006;40(12):2103–2111. doi:10.1016/j.freeradbiomed.2006.02.007
20. Moon EY, Lee JH, Lee JW, Song JH, Pyo S. ROS/Epac1-mediated Rap1/NF-kappaB activation is required for the expression of BAFF in Raw264.7 murine macrophages. Cell. Signalling. 2011;23(9):1479–1488. doi:10.1016/j.cellsig.2011.05.001
21. Harling K, Adankwah E, Güler A, et al. Constitutive STAT3 phosphorylation and IL-6/IL-10 co-expression are associated with impaired T-cell function in tuberculosis patients. Cell. Mol. Immunol. 2019;16(3):275–287. doi:10.1038/cmi.2018.5
22. Jiang J, Cao Z, Li B, et al. Disseminated tuberculosis is associated with impaired T cell immunity mediated by non-canonical NF-κB pathway. J Infect. 2024;89(3):106231. doi:10.1016/j.jinf.2024.106231
23. Lee GH, Lee MH, Yoon YD, Kang JS, Pyo S, Moon EY. Protein kinase C stimulates human B cell activating factor gene expression through reactive oxygen species-dependent c-Fos in THP-1 pro-monocytic cells. Cytokine. 2012;59(1):115–123. doi:10.1016/j.cyto.2012.03.017
24. Jeon ST, Kim WJ, Lee SM, et al. Reverse signaling through BAFF differentially regulates the expression of inflammatory mediators and cytoskeletal movements in THP-1 cells. Immunol cell biol. 2010;88(2):148–156. doi:10.1038/icb.2009.75
25. Lee SM, Kim EJ, Suk K, Lee WH. BAFF and April induce inflammatory activation of THP-1 cells through interaction with their conventional receptors and activation of MAPK and NF-κB. Inflammation Res. 2011;60(9):807–815. doi:10.1007/s00011-011-0336-3
26. Wang Y, Sun Q, Zhang Y, et al. Systemic immune dysregulation in severe tuberculosis patients revealed by a single-cell transcriptome atlas. J Infect. 2023;86(5):421–438. doi:10.1016/j.jinf.2023.03.020
27. Fan H, Lu B, Cao C, et al. Plasma TNFSF13B and TNFSF14 function as inflammatory indicators of severe adenovirus pneumonia in pediatric patients. Front Immunol. 2021;11:614781. doi:10.3389/fimmu.2020.614781
28. Garić D, Tao S, Ahmed E, et al. Depletion of BAFF cytokine exacerbates infection in Pseudomonas aeruginosa infected mice. J Cyst Fibros. 2019;18(3):349–356. doi:10.1016/j.jcf.2018.11.015
29. Liu K, Zhang Y, Hu S, et al. Increased levels of BAFF and April related to human active pulmonary tuberculosis. PLoS One. 2012;7(6):e38429. doi:10.1371/journal.pone.0038429
30. Kathamuthu GR, Moideen K, Banurekha VV, et al. Altered circulating levels of B cell growth factors and their modulation upon anti-tuberculosis treatment in pulmonary tuberculosis and tuberculous lymphadenitis. PLoS One. 2018;13(11):e0207404. doi:10.1371/journal.pone.0207404
31. Nardelli B, Belvedere O, Roschke V, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97(1):198–204. doi:10.1182/blood.v97.1.198
32. Mthembu M, Bowman KA, Davies LRL, et al. Discrepancy between Mtb-specific IFN-γ and IgG responses in HIV-positive people with low CD4 counts. EBioMedicine. 2023;90:104504. doi:10.1016/j.ebiom.2023.104504
33. Allman WR, Dey R, Liu L, et al. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc Natl Acad Sci USA. 2015;112(30):E4094–E4103. doi:10.1073/pnas.1421580112
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Omics Biomarkers for Monitoring Tuberculosis Treatment: A Mini-Review of Recent Insights and Future Approaches

Pitaloka DAE, Syamsunarno MRAA, Abdulah R, Chaidir L
Infection and Drug Resistance 2022, 15:2703-2711
Published Date: 28 May 2022
Association Between Serum Uric Acid Levels and Suicide Attempts in Adolescents and Young Adults with Major Depressive Disorder: A Retrospective Study
Kong Y, Liu C, Zhang C, Wang W, Li Y, Qiu H, Wang G, Li D, Chen X, Lv Z, Zhou D, Wan L, Ai M, Chen J, Ran L, Kuang L
Neuropsychiatric Disease and Treatment 2022, 18:1469-1477
Published Date: 20 July 2022