Back to Journals » Cancer Management and Research » Volume 12
Identification of Potential Prognostic Long Non-Coding RNA Biomarkers for Predicting Recurrence in Patients with Cervical Cancer
Authors Zhang Y, Zhang X, Zhu H, Liu Y, Cao J, Li D, Ding B, Yan W, Jin H, Wang S ![]()
Received 20 September 2019
Accepted for publication 19 December 2019
Published 31 January 2020 Volume 2020:12 Pages 719—730
DOI https://doi.org/10.2147/CMAR.S231796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Seema Singh
Yan Zhang,1 Xing Zhang,1 Haixia Zhu,2 Yang Liu,3 Jian Cao,3 Dake Li,3 Bo Ding,4 Wenjing Yan,1 Hua Jin,2,* Shizhi Wang1,*
1Key Laboratory of Environmental Medicine Engineering, Ministry of Education, School of Public Health, Southeast University, Nanjing, People’s Republic of China; 2Clinical Laboratory, Affiliated Tumor Hospital of Nantong University (Nantong Tumor Hospital), Nantong, People’s Republic of China; 3Department of Gynecology, Women’s Hospital of Nanjing Medical University, Nanjing Maternity and Child Health Care Hospital, Nanjing, People’s Republic of China; 4Department of Gynecology and Obstetrics, Zhongda Hospital, School of Medicine, Southeast University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shizhi Wang
Key Laboratory of Environmental Medicine Engineering, Ministry of Education, School of Public Health, Southeast University, 87 Dingjiaqiao, Gulou District, Nanjing 210009, People’s Republic of China
Tel +86 25 83272566
Fax +86 25 83324322
Email [email protected]
Hua Jin
Clinical Laboratory, Affiliated Tumor Hospital of Nantong University (Nantong Tumor Hospital), No. 30, North Tongyang Road, Tongzhou District, Nantong 226361, People’s Republic of China
Tel +86 513 89002658
Fax +86 513 85169100
Email [email protected]
Background: Cervical cancer (CC) is one of the most common malignant tumors in women, and its treatment is often accompanied by high recurrence. We aimed to identify the long non-coding RNAs (lncRNAs) associated with CC recurrence.
Methods: We downloaded lncRNAs expression data of CC patients from The Cancer Genome Atlas (TCGA) dataset and used Cox regression models to analyze the lncRNAs relationship with CC recurrence. The significantly associated lncRNAs were utilized to construct a recurrence risk score (RRS) model. Bioinformatics analyses were used to assess the potential role of the critical lncRNAs in CC recurrence. The effect of critical lncRNAs on CC phenotype was determined by in vitro experiments.
Results: Using Cox regression analysis, four lncRNAs, ie, HCG11, CASC15, LINC00189, and LINC00905, were markedly associated with worse recurrence-free survival (RFS) of CC, whereas three lncRNAs, including HULC, LINC00173, and MIR22HG, were the opposite. After constructing the RRS model, Kaplan-Meier analysis revealed that patients with high RRS had significantly increased risk of recurrence. Among the 20 types of tumors in the TCGA database which all had adjacent normal tissues, MIR22HG and HCG11were significantly downregulated in 18 and 10 types of tumors including CC, respectively. Increased MIR22HG was significantly relevant to decreased risks of recurrence among the subgroups of age at diagnosis < 45 (Hazard Ratio (HR) = 0.26), stage I/II (HR = 0.33), T stage I/II (HR = 0.30), chemotherapy (HR = 0.18), and molecular therapy (HR = 0.16). Functionally, elevated MIR22HG expression could suppress CC cell proliferation, migration and invasion.
Conclusion: MIR22HG has a fundamental role in CC recurrence and could be served as a potential prognostic biomarker.
Keywords: TCGA, lncRNAs, cervical cancer, recurrence, biomarker
Introduction
Cervical cancer (CC) is one of the most common malignant tumors in women. The global cancer statistics demonstrate that CC is the fourth most common cancer in females, with an annual incidence of about 530,000 new cases and a death toll of 270,000.1 With the development of the prophylactic HPV vaccination and CC screening, the 5-year overall survival (OS) rate of CC has been prolonged.2 However, in many developing countries, the 3- to 5-year OS rate of CC is less than 50%; especially in patients with advanced disease, the recurrence rate of CC is as high as 70%.3,4 The cure rate for recurrent CC is very low, and the 5-year survival rate for relapsed patients is less than 5%. According to the International Federation of Obstetrics and Gynecology (FIGO), the recurrence rate of CC is 11% to 22% and 28% to 64% for the FIGO stage IB-IIA and IIB-IVA, respectively.5 The prognosis of recurrent CC is poor, the treatment time is limited and the quality of life deteriorates rapidly. The treatment of recurrent CC remains challenging.6 Therefore, it is necessary to find new biomarkers related to CC recurrence and provide evidence for prevention and personalized treatment of CC recurrence.
Long non-coding RNAs (lncRNAs) are a kind of long RNA transcripts, which are longer than 200 nucleotides and unable to encode proteins.7 With the development of bioinformatics and the application of a series of new experimental methods such as high throughput sequencing, gene chip, and genome enrichment analysis, more and more lncRNAs have been found.8 Some of the dysregulated lncRNAs, involving the regulation of multiple cellular pathways, have revealed oncogenic and tumor-suppressive roles in cancer development, progression, and metastasis.9 Accumulating evidence has demonstrated that some specific lncRNAs are abnormally expressed in CC cells and participate in gene regulation, such as reducing or enhancing the expression of target genes in the process of carcinogenesis at the transcriptional level, and blocking or promoting the development of cancer.10 Much work so far had reported that lncRNAs can be used as biomarkers for CC invasion, metastasis and prognosis.11 Some lncRNAs were significantly associated with CC recurrence, such as HOTAIR, snaR and MEG3.12–14 LncRNAs provide new insights into the molecular mechanisms and treatments of cancer, and thus lay the ground work for the clinic.
In this study, we used The Cancer Genome Atlas (TCGA) data to identify lncRNAs associated with CC recurrence by a variety of data mining and bioinformatics methods. The identified lncRNAs have the potential to be biomarkers for the prediction of CC recurrence.
Materials and Methods
TCGA Database and Patient Information
The expression and clinical data of 310 CC patients were downloaded from TCGA database (up to December 18, 2018). The inclusion criteria were as follows: 1) patients with pathological diagnosis of primary CC; 2) patients with gene expression and clinical features; 3) patients with information of recurrence outcomes. Overall, a total of 211 CC patients with corresponding clinical features such as diagnosing age, metastasis, lymph node status, stage, T stage, and histological type were enrolled in this study. Data processing procedures also complied with TCGA data and human subject protection policies (www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Differential Analysis of Expressed lncRNAs
To investigate the role of lncRNAs in CC recurrence, we searched the annotated lncRNAs on the website of HUGO gene nomenclature committee (HGNC), and a total of 4072 lncRNAs were obtained from the HGNC database (http://www.genenames.org/cgi-bin/statistics, up to February 19, 2019). The 4072 lncRNAs were searched for expression data in cervical squamous cell carcinoma (CSCC) (TCGA provisional) using the geno/transcriptomic analyses tool of the online bioinformatics website the cBioPortal for Cancer Genomics (hereinafter referred to as cBioPortal; http://www.cbioportal.org/);15 finally, there were 471 known lncRNAs with copy number variations in more than 1% of queried patients. Of the 471 lncRNAs, 248 lncRNAs met the criterion of Z-score with a threshold of ±2 and expression alteration in more than 1% CC patients. Genome-wide lncRNAs expression profiles were obtained from the RNA sequencing (RNA-seq) dataset of TCGA. The differentially expressed lncRNAs (DELs) were analyzed by R program (https://www.r-project.org/) using the limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html. org/packages/release/bioc/html/limma.html website). The fold change (FC) in the expression of individual lncRNAs was calculated and DELs with log2|FC| > 1.0 and P < 0.05 were selected.
Identifying lncRNAs Predictive of Recurrence-Free Survival (RFS)
To identify lncRNAs predictive of RFS, univariate and multivariate Cox regression analyses were performed to assess the relationship between expression of lncRNAs level and RFS. Then, we constructed a prognostic risk score, ie, recurrence risk score (RRS), for predicting RFS by using the regression coefficient (β) from the multivariate Cox regression model as following:
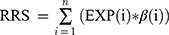
where n is the number of selected lncRNAs and EXP is the expression level of individual lncRNAs.16
According to the above formula, RRS of each patient was calculated, and patients were divided into high- and low-risk groups based on the median of RRS. Kaplan-Meier (K-M) analysis was applied to evaluate the different survival rates between patients with low- and high-RRSs, and Log rank test was used to evaluate the differences between them. The sensitivity and specificity of the RRS were estimated by time-dependent receiver operating characteristic (ROC) curves. To further analyze the association between RFS and clinical features, we carried out subgroup survival analyses to estimate the influence of clinical parameters on the RFS of CC patients.
Functional Enrichment Analysis
To investigate the biological roles of the candidate lncRNAs signature in CC, we obtained potential co-expressed genes of selected lncRNAs using circlncRNAnet (http://app.cgu.edu.tw/circlnc/).17 Gene ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of co-expressed genes were analyzed by STRING (https://string-db.org/cgi/input.pl). The P value < 0.05 and gene count 3 were set as the cut-off criteria.
3 were set as the cut-off criteria.
Plasmid Transient Transfection, Quantitative Real-Time PCR (qRT-PCR), Cell Counting Kit-8 (CCK-8) Assay, and Transwell Assay
These methods were performed as described previously and are detailed in Supplementary Material (Table S7).
Statistical Analysis
All the analyses, including the heatmap, Cox regression analyses, K–M survival curves, and ROC curves were performed by the R statistical software and SPSS 24.0 software. The P-value less than 0.05 was considered significant. Values are expressed as the mean ± standard error of the mean (SEM) from three separate experiments, except for a special annotation.
Results
Association of Demographic and Clinical Characteristics with RFS
Based on the inclusion criteria, we screened 210 patients from the TCGA database at an age of 47.175 ± 13.209 years. Among them, there were 167 CC subjects (79.1%) with tumor stage I and II, and 39 CC subjects (18.5%) with tumor stage III and IV. Meanwhile, we examined the association of RFS and clinical features in CC patients by the univariate Cox regression analysis. The results suggested that lymph node metastasis (Hazard Ratio (HR) = 3.47, P = 0.012) was associated with RFS of CC patients (Table 1).
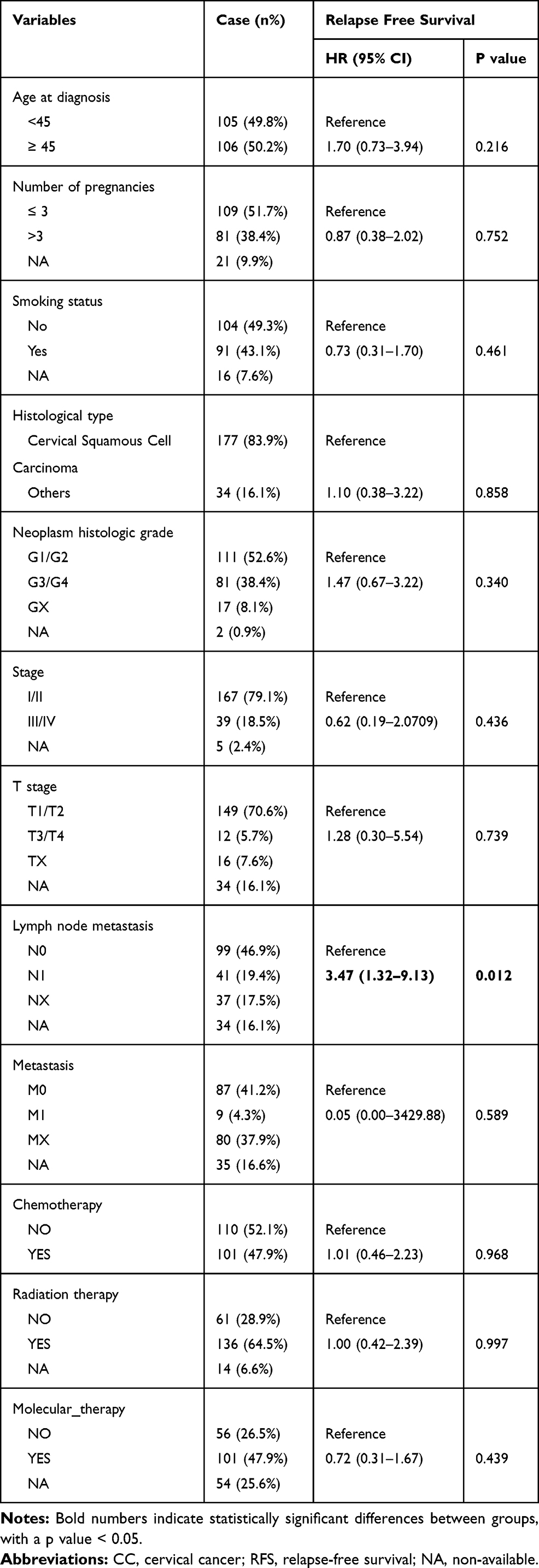 |
Table 1 Demographic and Clinical Characteristics of CC Patients and Their Relationship with RFS of CC |
Identification of DELs Associated with CC Recurrence
A total of 4072 lncRNAs were extracted from the HGNC database (Table S1). The OncoPrint from the cBioPortal showed 248 lncRNAs with expression alteration in more than 1% CC patients based on Z-score with a threshold of ± 2 (Table S2). Moreover, the expression alteration frequency of the 248 lncRNAs ranged from 1.4% to 16% in the CC patients with RNA-seq data (Figure 1).
 |
Figure 1 Differentially expressed lncRNAs in CC. The rows show 248 DELs, while the columns show 211 samples. The red dot represents up-regulated lncRNAs, and blue dot represents down-regulated lncRNAs. |
Univariate Cox regression analysis revealed that 14 DELs, ie, LINC00905, LINC00173, LINC00189, HULC, CASC15, MIR22HG, HCG11, MIR924HG, LINC00852, FAM95B1, SNHG9, MALAT1, H19, and ARRDC1-AS1, among the 284 DELs had prominent prognostic values for CC recurrence (Table 2). Afterward, the relationship between the 14 DELs and RFS was calculated by multivariate Cox regression analysis, and the results showed that four lncRNAs, ie, HCG11, CASC15, LINC00189, and LINC00905, were markedly associated with worse RFS of CC, whereas three lncRNAs, including HULC, LINC00173, and MIR22HG, were the opposite (Table 2 and Figure 2A–G). In addition, according to the cBioPortal, there were 61 (29%) patients with altered expression of any one of the 7 DELs, including amplification, deep deletion, mRNA up-regulation and mRNA down-regulation (Figure 3A). The genomic information of the 7 DELs is shown in Figure 3B. When all the 7 DELs were combined, their changes were also significantly associated with disease/progression-free survival and OS, respectively (P = 0.040 and P = 0.007; Figure 3C and D).
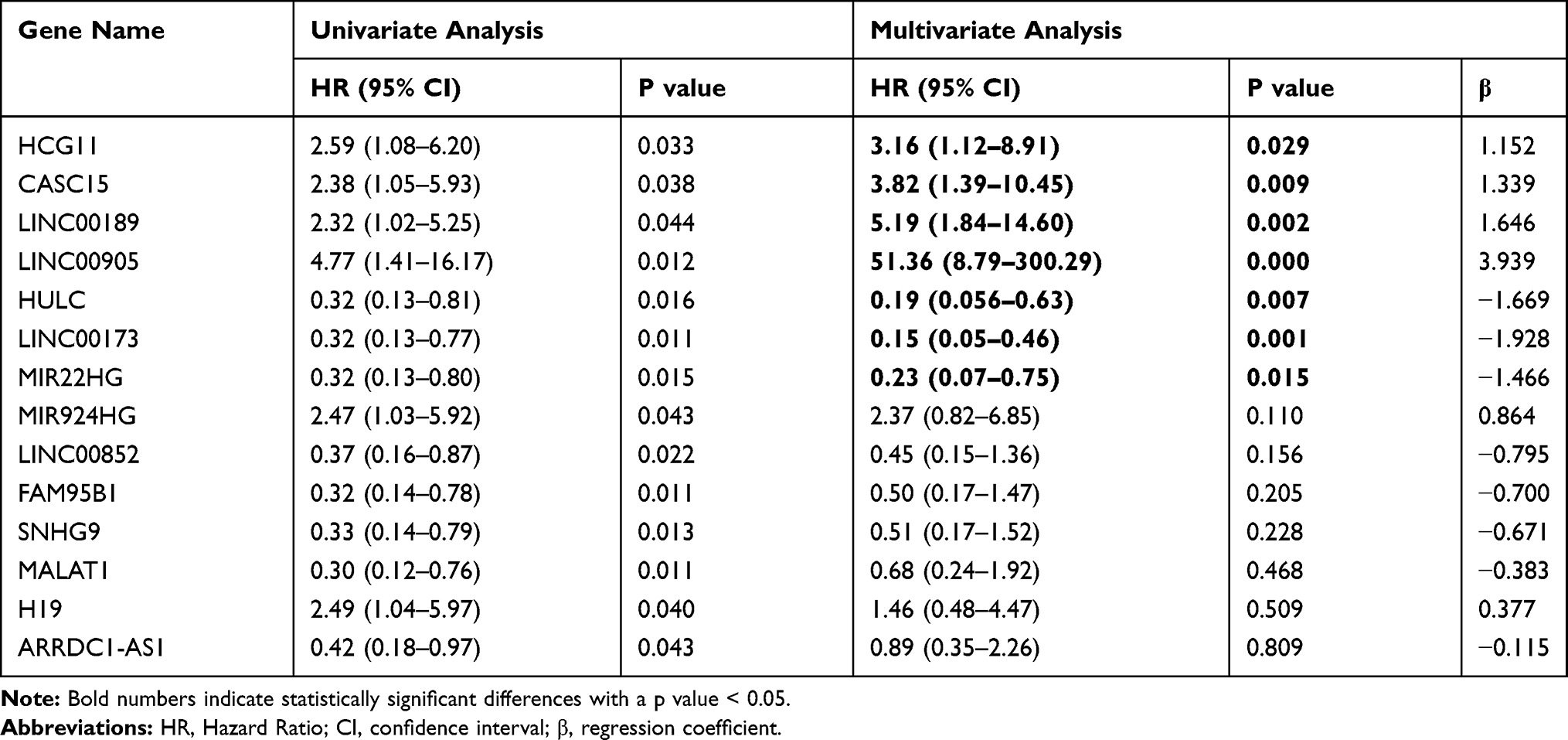 |
Table 2 Prognostic Significance of DELs from Univariate and Multivariate Cox Regression Analysis |
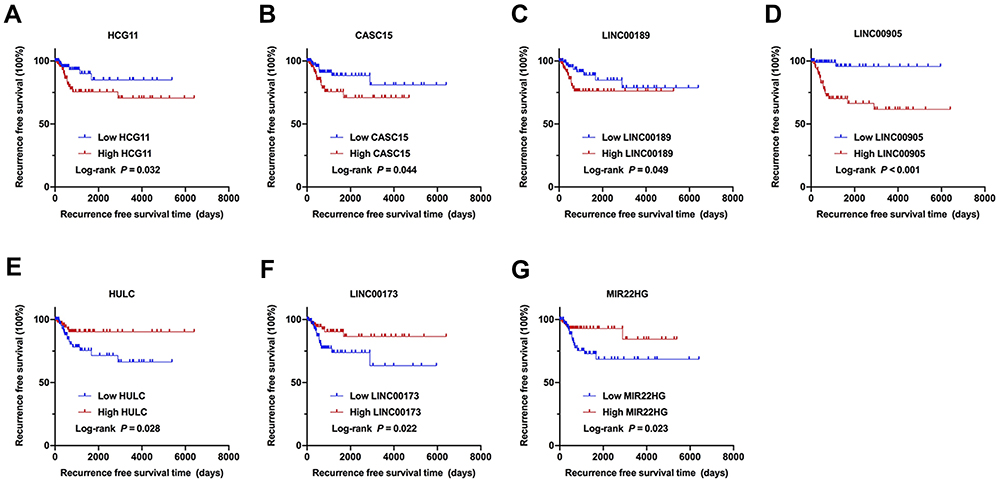 |
Figure 2 Identification of the 7 DELs associated with the recurrence of CC. (A–G) Kaplan–Meier survival curves of RFS in the TCGA cohort are shown according to HCG11, CASC15, LINC00189, LINC00905, HULC, LINC00173, and MIR22HG expression, respectively. |
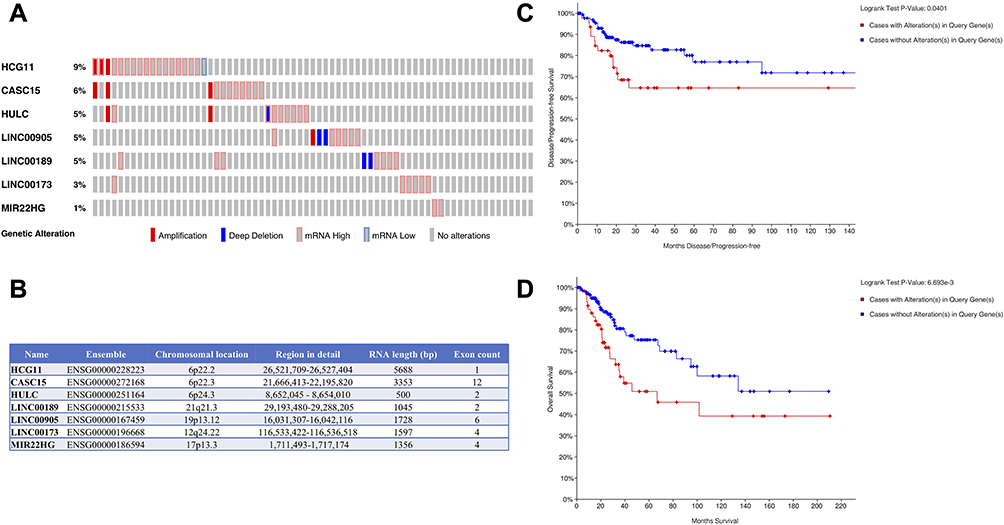 |
Figure 3 Identification of the 7 DELs signature associated with survival by cBioPortal. (A) The genetic expression alterations of the lncRNAs in CC. (B) Organizations of these 7 DELs and chromosome locations. (C) Kaplan–Meier curve for disease/progression-free survival based on alterations of the 7 DELs. (D) Kaplan–Meier curve for OS based on alterations of the 7 DELs. |
Prediction of CC Recurrence Risk by the RRS Based on DELs
RRS was established to predict the recurrence risk of CC patients and defined as: RRS = (1.152*EXPHCG11) + (1.339*EXPCASC15) + (1.646*EXPLINC00189) + (3.939*EXPLINC00905) + (−1.669*EXPHULC) + (−1.982*EXPLINC00173) + (−1.446*EXPMIR22HG). To evaluate the predictive power of the risk model, CC patients were classified into the low and high RRS groups based on the median RRS value (Figure 4A). Figure 4B shows the RFS time and disease prognosis for different RRS groups in CC patients. Furthermore, the expression pattern of the 7 DELs between the low and high RRS groups is shown in Figure 4C. The levels of HCG11, CASC15, LINC00189, and LINC00905 were remarkably higher in the high RRS group than that in the low RRS group, whereas three DELs, including HULC, LINC00173, and MIR22HG, were the opposite. K-M analysis revealed that the RFS time of the low RRS group was predominantly longer than that of the high RRS group (P < 0.001; Figure 4D). Meanwhile, the 7 DELs exhibited a well-predicted power of RFS for the CC patients, with an AUC value of 0.739 (P < 0.001; Figure 4E).
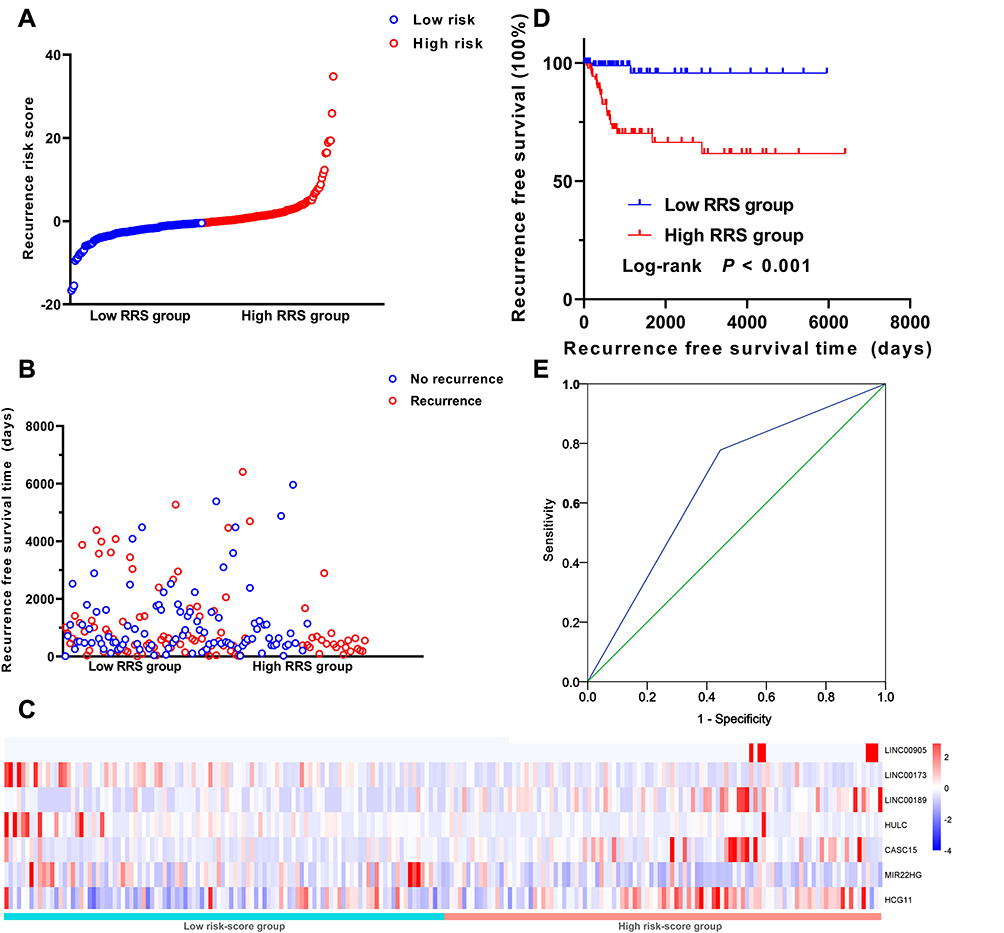 |
Figure 4 Prognostic value of the RRS constructed from 7 DELs in CC. (A) 7 DELs expression and RRS distribution by z-score. (B) Patients’ recurrence status and RFS time; (C) Heatmap of the lncRNA expression profiles. Rows represent lncRNAs, and columns represent patients. The red dot represents up-regulated lncRNA, and blue dot represents down-regulated lncRNA. (D) Kaplan-Meier analysis of the RFS between high/low RRS groups. (E) Prognostic value of the RRS displayed as a time-dependent ROC curve for predicting the survival status. AUC, area under curve; CI, confidence interval. |
MIR22HG as a Key Recurrence-Related lncRNAs in CC
We further investigated the difference of each lncRNA expression between the CC and matched normal tissues using the circlncRNAnet website, and found only the difference in HCG11 and MIR22HG expression between the tumors and controls was statistically significant, respectively (P = 0.028 and P = 0.029; Figure 5A and Table S3). To gain more insight, we analyzed the expression of the HCG11 and MIR22HG in 20 cancers which had adjacent normal tissues (Figure 5B and C and Table S4). The results showed that MIR22HG was significantly downregulated in 18 types of tumors while HCG11 downregulated in 10 and upregulated in one type of tumors. Therefore, we mainly focus on the fundamental role of MIR22HG.
 |
Figure 5 Relative expression of candidate lncRNAs from circlncRNAnet data. (A) The expression of DELs in tumor and adjacent tissues in CC. (B) Expression levels of HCG11 in tumor and adjacent tissues in 20 cancers. (C) Expression levels of MIR22HG in tumor and adjacent tissues in 20 cancers. ***<0.001; **<0,01; *<0.05. Abbreviations: BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma. |
To examine the association of the HCG11 and MIR22HG signature with clinical parameters, CC patients were divided into low- and the high-expression group using the median of HCG11 or MIR22HG expression level as the cutoff value. Subgroup analysis demonstrated that increased MIR22HG was significantly relevant to decreased risks of recurrence among the subgroups of age at diagnosis < 45 (HR = 0.26, P = 0.044), stage I/II (HR = 0.33, P = 0.028), T stage I/II (HR = 0.30, P = 0.032), chemotherapy (HR = 0.18, P = 0.024), and molecular therapy (HR =0.16, P = 0.018), suggesting it as a potential prognostic indicator of CC (Table 3). However, there was no marked correlation between HCG11 and clinical features (Table S5).
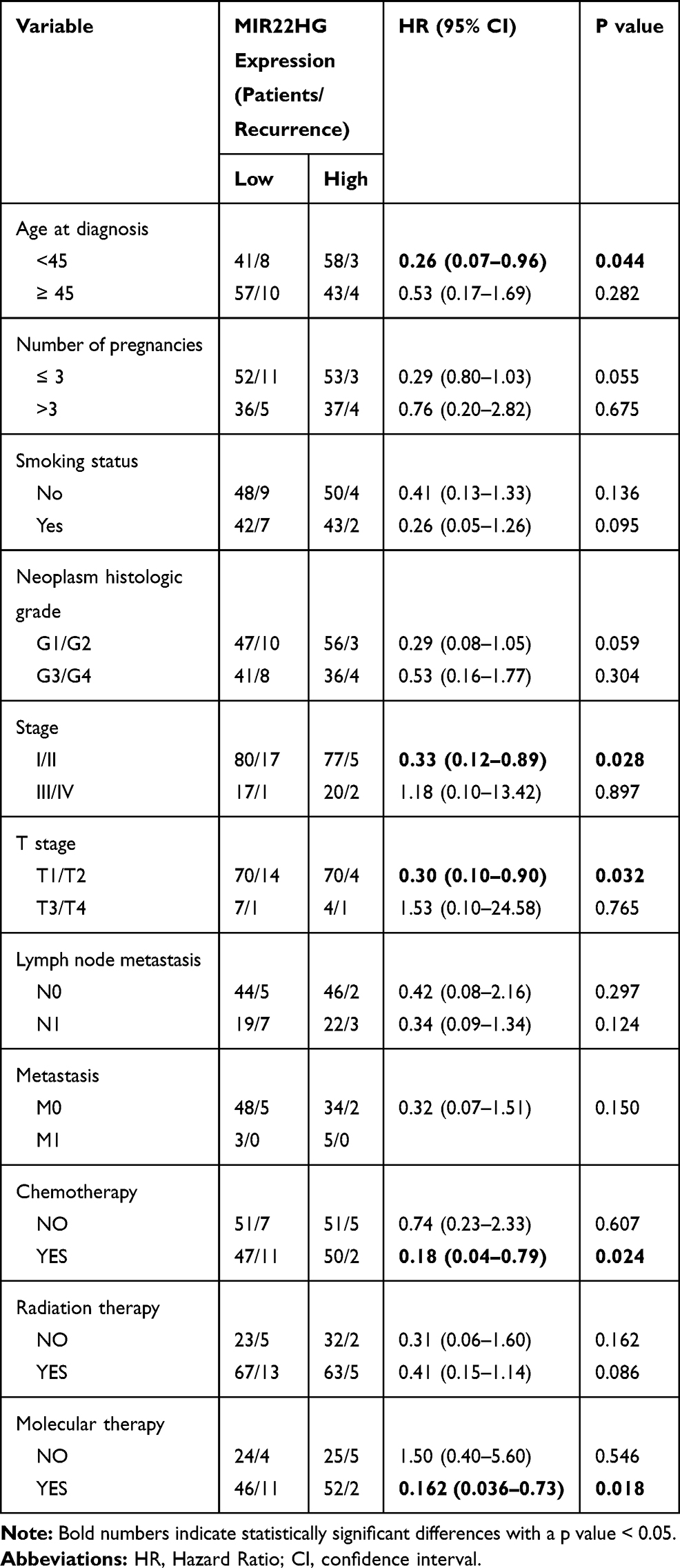 |
Table 3 Subgroup Survival Analyses for Correlation of Clinicopathological Characteristics and MIR22HG Expression in CC Patients |
Functional Enrichment Analysis of MIR22HG
To acquire more information about the molecular functions and signal pathways of the lncRNAs that had been selected, the co-expressed genes of MIR22HG were predicted by circlncRNAnet. Online website STRING was used to analyze the GO term and KEGG pathway enrichment of the co-expressed genes. We selected the top 5 most significantly enriched GO terms (biological process (BP), cellular component, and molecular function) (according to the P-value) and the top 15 most significantly enriched KEGG pathways (Figure 6 and Table S6). The enrichment results indicated the involvement of MIR22HG in the regulation of cell cycle since the most significant BP term and KEGG pathway were “cell cycle G1/S phase transition” and “Cell cycle”, respectively.
 |
Figure 6 Enrichment of GO terms and KEGG pathways for co-expressed mRNAs of MIR22HG. (A and B) KEGG and GO analysis of the related genes. Abbreviations: GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes; BP, biological process; CC, cellular component; MF, molecular function. |
Effects of MIR22HG on CC Cell Phenotypes
To further investigate the biological function of MIR22HG in CC, we transfected the GV146/MIR22HG vector into HeLa and SiHa cells, respectively (Figure 7A). Compared with the negative control (NC), HeLa and SiHa cells overexpressing MIR22HG had the lower proliferation (P < 0.05; Figure 7B and C). Transwell assays revealed that migration and invasion ability were suppressed in CC cells transfected with MIR22HG (P < 0.05; Figure 7D and E). In addition, to rule out the possibility that the inhibitory effect of MIR22HG on CC cell phenotype is dependent on HPV infection, similar experiments with C-33 A cell, an HPV-cervical cancer cell line, were performed and led to similar conclusions (Figure S1A–D).
 |
Figure 7 MIR22HG overexpression inhibited CC cells proliferation. (A) qRT-PCR determined expression efficiency of transfection with MIR22HG in HeLa and SiHa cells. Quantitative normalization of MIR22HG was performed in each sample using GAPDH expression as an internal control. (B and C) The CCK-8 assay was conducted to measure cell proliferation in HeLa and SiHa cells after transfection with MIR22HG. (D and E) Transwell assays were used for testing the migration and invasion ability of HeLa and SiHa cells after transfection with MIR22HG (10X). ***P<0.001, **P<0.01, *P<0.05. |
Discussion
Recurrent CC is almost always incurable and has no typical symptoms.18 Considering the high mortality rate among recurrent CC patients, it is necessary to develop suitable prognosis biomarkers with the potential to predict tumor progression. The ability to predict which patients have a high risk of recurrence would empower clinicians to better patient therapy.19 LncRNAs have been reported as key regulators in the progression and metastasis of cancer.20 Compared with mutations or aberrant expression in protein-coding genes, the expression of lncRNAs was more tissue-specific.20,21 Given that many lncRNAs were critical for cancer progression and prognostic, we needed to deeply investigate that lncRNAs may possess an unknown function in cancer.22
In this study, we analyzed the lncRNAs expression profiles of patients with CC downloaded from TCGA and identified seven DELs (HCG11, CASC15, LINC00189, LINC00905, HULC, LINC00173, and MIR22HG) signature associated with RFS. Among the 7 DELs, the potential of some lncRNAs as prognostic-associated biomarkers for cancer has been demonstrated in many cancers. CASC15 was up-regulated in CC, and its overexpression associated with lymph node metastasis and FIGO stage, indicating a poor prognosis for CC.23 HULC was abnormally upregulated in a variety of cancers and meta-analysis indicated that overexpression of HULC was associated with metastasis and a poor OS in cancer.24 Zhang et al25 presented that low expression HCG11 in tissues was associated with poor survival of prostate cancer patients. Likewise, in our study, downregulation of HCG11 expression was associated with shorter RFS of CC patients. However, HCG11 was down-regulated in CC compared to adjacent normal tissues, which meant that our findings of HCG11 needed further study.
In addition to HCG11, the comparison of the 7 DELs expression between tumor and adjacent normal tissues in TCGA suggested that only MIR22HG could act as a suppressor gene in CC. Decreased MIR22HG expression also significantly increased the risk of recurrence among the subgroups of age at diagnosis < 45, stage I/II, T stage I/II, chemotherapy, and molecular therapy while HCG11 failed to correlate with these clinicopathological features of CC accordantly. Emerging evidence suggested that MIR22HG was a tumor suppressor gene, which contributed to the initiation and progression of many cancers. Highly expressed MIR22HG inhibited cancer cell growth, migration and invasion, and overexpression MIR22HG had a better prognosis in HCC patients.26 The pathways that MIR22HG may mediate in CC remain unclear, so we performed functional enrichment analysis to identify MIR22HG-associated biological signaling pathways. The GO BP term and KEGG pathway most significantly associated with MIR22HG were “cell cycle G1/S phase transition” and “Cell cycle”, respectively. Functionally, elevated MIR22HG expression could suppress CC cell proliferation, migration and invasion. Those data indicated that MIR22HG could play important roles in CC recurrence. Cell-cycle dysregulation was an important indicator of tumor development. Cui et al27 reported that MIR22HG was downregulated in endometrial cancer, which inhibited endometrial cancer cell proliferation and arrested cells in G0/G1 phase to promote apoptosis by regulating miR-141-3p/DAPK1 axis. The role of MIR22HG in the recurrence of CC warrants further investigation. Except for those lncRNAs mentioned above, LINC00189, LINC00905 and LINC00173 are currently less studied, which imply that the biological functions of these lncRNAs remain to be explored.
Thus far, the studies on the role of lncRNAs in the recurrence of CC were limited and our findings may have some clinical implications. However, there were some limitations in this study. First, only a few lncRNAs (4072 lncRNAs extracted from the HGNC database) were investigated this time, so many lncRNAs which are not received in the HGNC database are not determined in the present study; second, the demographic data of patients in the TCGA may not represent those of other patient populations; third, we were not capable to validate the findings using our patients' cohort due to insufficient follow-up information; finally, the molecular mechanisms of the 7 DELs were unclear and further functionality studies were needed to better understand the roles of 7 DELs in the recurrence of CC.
In summary, this study reported a seven-lncRNA signature to predict recurrence in CC patients by comprehensive analysis of lncRNAs expression profiles in the TCGA database. Especially, MIR22HG might be a potential prognostic indicator for CC. We have shown that MIR22HG could suppress CC cells proliferation, metastasis and invasion. However, future functional investigations are required to further explore the mechanisms underlying the roles of these lncRNAs in CC recurrence.
Acknowledgments
This study was supported by Natural Science Foundation of China (81872684), Natural Science Foundation of Jiangsu Province (BK20171367), the Six Talent Peaks Project in Jiangsu Province (WSW-201), the “SIX ONE” Talent Research Project for the High-level Health Personnel of Jiangsu Province (LGY2018037), Nantong Municipal Science and Technology Plan (JC2018053 and YYZ17072), Nantong Municipal Key Talent Project (No. 9), and funded by Southeast University “Zhongying Young Scholars” Project.
Disclosure
All authors declare that they have no conflicts of interest in this work.
References
1. Small WJ, Bacon MA, Bajaj A, et al. Cervical cancer: a global health crisis. Cancer. 2017;123(13):2404–2412. doi:10.1002/cncr.v123.13
2. Zhu FC, Hu SY, Hong Y, et al. Efficacy, immunogenicity and safety of the AS04-HPV-16/18 vaccine in Chinese women aged 18-25 years: end-of-study results from a Phase II/III, randomised, controlled trial. Cancer Med. 2019:8(14):6195–6211.
3. Sankaranarayanan R, Swaminathan R, Brenner H, et al. Cancer survival in Africa, Asia, and Central America: a population-based study. Lancet Oncol. 2010;11(2):165–173. doi:10.1016/S1470-2045(09)70335-3
4. Friedlander M. Guidelines for the treatment of recurrent and metastatic cervical cancer. Oncologist. 2002;7(4):342–347.
5. Quinn MA, Benedet JL, Odicino F, et al. Carcinoma of the cervix uteri. FIGO 26th annual report on the results of treatment in gynecological cancer. Int J Gynaecol Obstet. 2006;95(Suppl 1):S43–S103. doi:10.1016/S0020-7292(06)60030-1
6. Petignat P, Roy M. Diagnosis and management of cervical cancer. BMJ. 2007;335(7623):765–768. doi:10.1136/bmj.39337.615197.80
7. Quinn JJCH, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;47–62. doi:10.1038/nrg.2015.10
8. Gao YF, Wang ZB, Zhu T, et al. A critical overview of long non-coding RNA in glioma etiology 2016: an update. Tumour Biol. 2016;37(11):14403–14413. doi:10.1007/s13277-016-5307-4
9. Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013;108(12):2419–2425. doi:10.1038/bjc.2013.233
10. Hoppe-Seyler F, Hoppe-Seyler K. Emerging topics in human tumor virology. Int J Cancer. 2011;129(6):1289–1299. doi:10.1002/ijc.26087
11. Peng L, Yuan X, Jiang B, Tang Z, Li GC. LncRNAs: key players and novel insights into cervical cancer. Tumour Biol. 2016;37(3):2779–2788. doi:10.1007/s13277-015-4663-9
12. Li J, Wang Y, Yu J, Dong R, Qiu H. A high level of circulating HOTAIR is associated with progression and poor prognosis of cervical cancer. Tumour Biol. 2015;36(3):1661–1665. doi:10.1007/s13277-014-2765-4
13. Zheng Z, Gao Y. Down-regulation of lncRNA snaR is correlated with postoperative distant recurrence of HPV-negative cervical squamous cell carcinoma. Biosci Rep. 2018;38(6). doi:10.1042/BSR20181213
14. Zhang J, Lin Z, Gao Y, Yao T. Downregulation of long noncoding RNA MEG3 is associated with poor prognosis and promoter hypermethylation in cervical cancer. J Exp Clin Cancer Res. 2017;36(1):5. doi:10.1186/s13046-016-0472-2
15. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
16. Wang K, Li J, Xiong YF, Zeng Z, Zhang X, Li HY. A potential prognostic long noncoding RNA signature to predict recurrence among ER-positive breast cancer patients treated with tamoxifen. Sci Rep. 2018;8(1):3179. doi:10.1038/s41598-018-21581-w
17. Wu SM, Liu H, Huang PJ, et al. circlncRNAnet: an integrated web-based resource for mapping functional networks of long or circular forms of noncoding RNAs. GigaScience. 2018;7(1):1–10.
18. Tempfer CB, Beckmann MW. State-of-the-art treatment and novel agents in local and distant recurrences of cervical cancer. Oncol Res Clin Cancer Res. 2016;39(9):525–533. doi:10.1159/000448529
19. Bodurka-Bevers D, Morris M, Eifel PJ, et al. Posttherapy surveillance of women with cervical cancer: an outcomes analysis. Gynecol Oncol. 2000;78(2):187–193. doi:10.1006/gyno.2000.5860
20. Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–3981. doi:10.1158/0008-5472.CAN-16-2634
21. Fatima R, Akhade VS, Pal D, Rao SM. Long noncoding RNAs in development and cancer: potential biomarkers and therapeutic targets. Mol Cell Ther. 2015;3:5.
22. Gao P, Wei GH. Genomic insight into the role of lncRNA in cancer susceptibility. Int J Mol Sci. 2017;18(6):1239.
23. Shan S, Li HF, Yang XY, et al. Higher lncRNA CASC15 expression predicts poor prognosis and associates with tumor growth in cervical cancer. Eur Rev Med Pharmacol Sci. 2019;23(2):507–512. doi:10.26355/eurrev_201901_16862
24. Ding Y, Sun C, Li J, et al. The significance of long non-coding RNA HULC in predicting prognosis and metastasis of cancers: a meta-analysis. Pathol Oncol Res. 2019;25(1):311–318. doi:10.1007/s12253-017-0351-y
25. Zhang Y, Zhang P, Wan X, et al. Downregulation of long non-coding RNA HCG11 predicts a poor prognosis in prostate cancer. Biomed Pharmacother. 2016;83:936–941. doi:10.1016/j.biopha.2016.08.013
26. Wu Y, Zhou Y, Huan L, et al. LncRNA MIR22HG inhibits growth, migration and invasion through regulating the miR-10a-5p/NCOR2 axis in hepatocellular carcinoma cells. Cancer Sci. 2019;110(3):973–984. doi:10.1111/cas.13950
27. Cui Z, An X, Li J, Liu Q, Liu W. LncRNA MIR22HG negatively regulates miR-141-3p to enhance DAPK1 expression and inhibits endometrial carcinoma cells proliferation. Biomed Pharmacother. 2018;104:223–228. doi:10.1016/j.biopha.2018.05.046
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.