Back to Journals » Drug Design, Development and Therapy » Volume 8
Identification of novel multitargeted PPARα/γ/δ pan agonists by core hopping of rosiglitazone
Authors Wang X, Zhang J, Wang S, Xu W, Cheng X, Wang R
Received 1 July 2014
Accepted for publication 27 August 2014
Published 7 November 2014 Volume 2014:8 Pages 2255—2262
DOI https://doi.org/10.2147/DDDT.S70383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Xue-Jiao Wang,1 Jun Zhang,1 Shu-Qing Wang,1 Wei-Ren Xu,2 Xian-Chao Cheng,1 Run-Ling Wang1
1Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics (Theranostics), School of Pharmacy, Tianjin Medical University, Tianjin, People’s Republic of China; 2Tianjin Key Laboratory of Molecular Design and Drug Discovery, Tianjin Institute of Pharmaceutical Research, Tianjin, People’s Republic of China
Abstract: The thiazolidinedione class peroxisome proliferator-activated receptor gamma (PPARγ) agonists are restricted in clinical use as antidiabetic agents because of side effects such as edema, weight gain, and heart failure. The single and selective agonism of PPARγ is the main cause of these side effects. Multitargeted PPARα/γ/δ pan agonist development is the hot topic in the antidiabetic drug research field. In order to identify PPARα/γ/δ pan agonists, a compound database was established by core hopping of rosiglitazone, which was then docked into a PPARα/γ/δ active site to screen out a number of candidate compounds with a higher docking score and better interaction with the active site. Further, absorption, distribution, metabolism, excretion, and toxicity prediction was done to give eight compounds. Molecular dynamics simulation of the representative Cpd#1 showed more favorable binding conformation for PPARs receptor than the original ligand. Cpd#1 could act as a PPARα/γ/δ pan agonist for novel antidiabetic drug research.
Keywords: PPARs, diabetes, docking, molecular dynamics simulation, ADMET
Introduction
Peroxisome proliferator-activated receptors (PPARs) are nuclear ligand-activated transcription factors and include three subtypes, namely PPARα, PPARγ, and PPARδ.1–3 The drugs targeting PPARs mainly include: 1) PPARγ agonists4 such as rosiglitazone and pioglitazone, which are used as antidiabetic drugs and also possess anti-inflammatory or antineoplastic activities,5,6 and 2) PPARα agonists such as fenofibrate and bezafibrate, which are used as antilipemic drugs (Figure 1).7,8 Rosiglitazone and pioglitazone have shown side effects in clinical use, such as liver function abnormity, edema, and weight gain.9 Especially in 2007, Nissen and Wolski10 reported the cardiac safety of rosiglitazone, which showed that singly selective agonism of PPARγ not only enhanced insulin sensitivity and the therapeutic effect of insulin metabolism but also caused edema, weight gain, and the potential risk of heart failure.
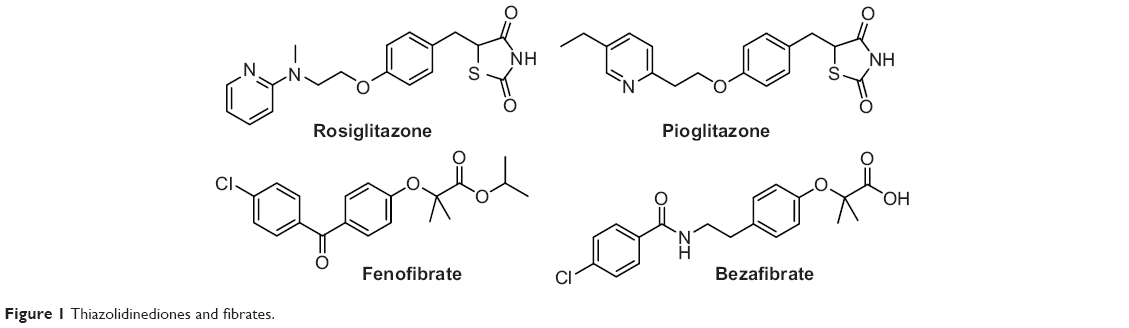 | Figure 1 Thiazolidinediones and fibrates. |
In recent years, some novel PPARs concepts appeared in the antidiabetic drug research area, such as multitargeted cooperative PPARα/γ dual agonists and PPARα/γ/δ pan agonists. These multitargeted agonists could cooperatively improve glucose and lipid metabolism. They could not only effectively control blood sugar but also reduce the content of triglyceride, free fatty acid, and low-density lipoprotein, as well as increase high-density lipoprotein concentration, thus having a preventive effect on cardiovascular complications of type 2 diabetes patients. Some of these multitargeted PPARs agonists have entered clinical trials and represent promising PPARs drug research.11–13
The pharmacophore of PPARs agonists consists of the polar head, linker, and hydrophobic tail. The polar head of PPARs agonists could form a hydrogen bond with Tyr residue at the AF-2 region, producing a transactivation effect. The hydrophobic tail of PPARs agonists could bind to the residues at the active site entrance, affecting subtype selectivity. This indicates that by modification of the polar head and hydrophobic tail, various pharmacological effects can be produced, such as PPARα/γ dual agonistic activity and PPARα/γ/δ pan agonistic activity.
In our previous research, using GW409544 as the starting point, by means of “core hopping” and “glide docking” techniques, a novel class of PPARα/γ dual agonists was discovered.14 In this paper, starting from rosiglitazone as the lead compound, using a core hopping approach, the polar head, linker, and hydrophobic tail of rosiglitazone were modified to produce various compounds. These compounds were then screened by docking and absorption, distribution, metabolism, excretion, and toxicity (ADMET) prediction to discover some excellent PPARα/γ/δ pan agonists. Molecular dynamics simulations of the representative Cpd#1 with PPARα/γ/δ were also done to study the binding details (Figure 2).
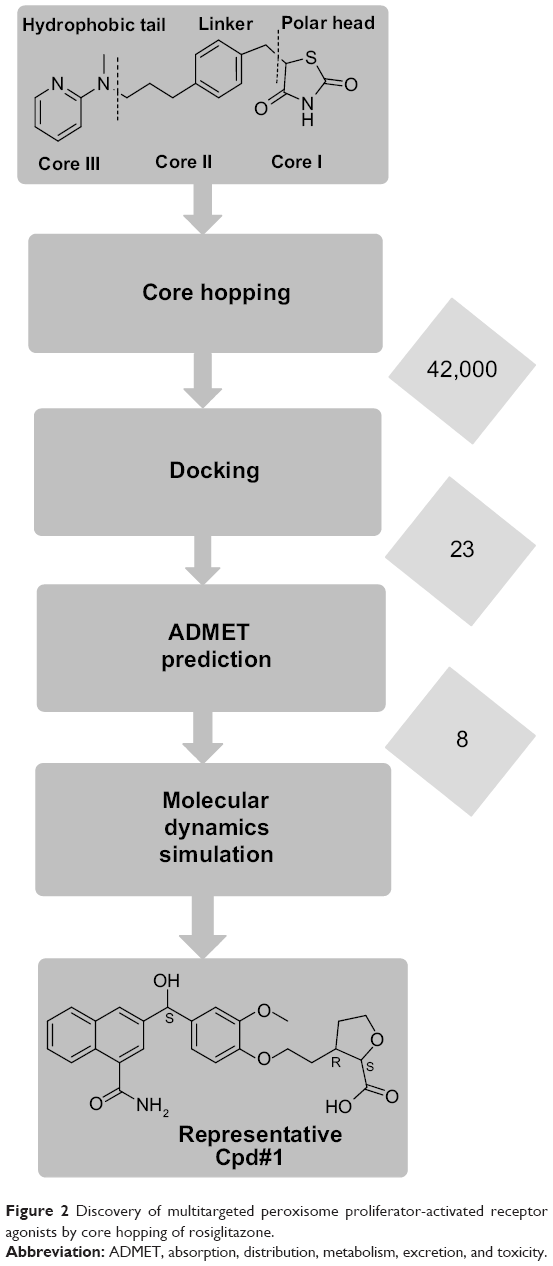 | Figure 2 Discovery of multitargeted peroxisome proliferator-activated receptor agonists by core hopping of rosiglitazone. |
Materials and methods
Preparation of PPAR receptors structures
The crystal structures of PPARα, PPARγ, and PPARδ receptors were downloaded from the Protein Data Bank (PDB) with PDB identification numbers 1I7G, 2PRG, and 2ZNP, respectively.15–17 The preparation of these receptors was performed on the Protein Preparation Wizard embedded in Schrodinger 2009. The process of preparing receptors included assigning bond orders, adding hydrogen, treating metals, treating disulfides, deleting waters, alleviating potential steric clashes, adjusting formal charges, minimizing proteins with the OPLS (Optimized Potentials for Liquid Simulation) 2005 force field,18 and refining the protein by limiting value of root mean square deviation (RMSD) to 0.50 Å as the constraint. Then, the original ligand was centered and redocked into the binding site to generate a docking box for molecular docking.
Core hopping and docking
The Core Hopping module in Schrodinger 2009 software was used to modify the polar head, linker, and hydrophobic tail of rosiglitazone (Figure 2).19 Core hopping is a docking algorithm that has the functions of fragment-based replacing and molecular docking.20–22 The first step of core hopping was to define the points at which the cores were attached to the scaffold. It was performed in the Define Combinations Step from the Combinatorial Screening panel. The second step was to define “the receptor grid file”, which was done in the Receptor Preparation panel. The third step was to prepare the cores attached to the scaffold using fragment database derived from ZINC.23,24 The fourth step was to align and dock the entire molecular structure built up by the core and scaffold. The cores were sorted and filtered by goodness of alignment and then redocked into the receptor after attaching the scaffold, followed by using the docking scores to sort the final molecules.25–27 The original ligand AZ242, rosiglitazone, and TIPP204 were used as positive control compounds.
ADMET prediction
The ADMET module of Discovery Studio 3.1 was used to predict pharmacokinetics and toxicity of the compounds (Figure 2). Taking rosiglitazone as control, the compounds as a mol2 file were imported into the ADMET Descriptors module and the Toxicity Prediction Extensible and Toxicity Prediction TOPKAT modules, respectively, obtaining pharmacokinetics and toxicity parameters.
Molecular dynamics simulation
In order to study the binding stability of compounds with PPARs active site, the 10ns molecular dynamics simulations were performed using the open GROMACS 4.0 package for Linux (Figure 2).28 Before the simulations, the coordinate file and topology file were prepared29 and the water box was constructed and filled with simple point charge water solution, which was then neutralized by sodium ions or chloride ions.30,31 The 1,000-step energy minimization of the system was performed using the steepest descending method. The NVT (constant number, volume and temperature) ensembles were used with temperature being maintained at 300 K. The cutoff radius of van der Waals interaction was 1.4 nm, and particle mesh Ewald algorithm was used for the electrostatic interaction.32,33 The Linear Constraint Solver algorithm was used for all of the bond restriction.34–36
Results and discussion
Ligand binding domains of the PPAR receptors
The X-ray crystallography studies showed that the ligand binding domain of the PPARs was composed of 12α-helix and 4 antiparalleled β-sheet. The three subtypes of PPARs were 60%–70% sequence similarities with RMSD between Cα atoms <1 Å. In addition, the ligand binding domain of the PPARα/γ/δ formed a Y-shaped hydrophobic pocket with a volume of about 1,300 Å3. The AF-2 region of H12 helix played an important role in the process of the activation of PPARs. As for PPARδ, the AF-2 region was significantly narrower, which was not suitable for binding ligands with a larger polar head.37
Core hopping and docking
A total of 42,000 compounds were obtained by core hopping of rosiglitazone. These compounds were docked into PPARα (pdb 1I7G), PPARγ (pdb 2PRG), and PPARδ (pdb 2ZNP), respectively, screening out 23 compounds with higher docking scores and better binding poses than the original ligands. Further ADMET prediction studies produced the top eight compounds (Table 1). The docking scores of these compounds with PPARα and PPARγ were higher than the original ligand AZ242 and rosiglitazone, respectively. The docking scores of these compounds with PPARδ were somewhat lower than the original ligand TIPP204. In addition, the hydrogen bond distances between compounds and PPARs were <3.20 Å,38,39 and the values were equal to the original ligand.
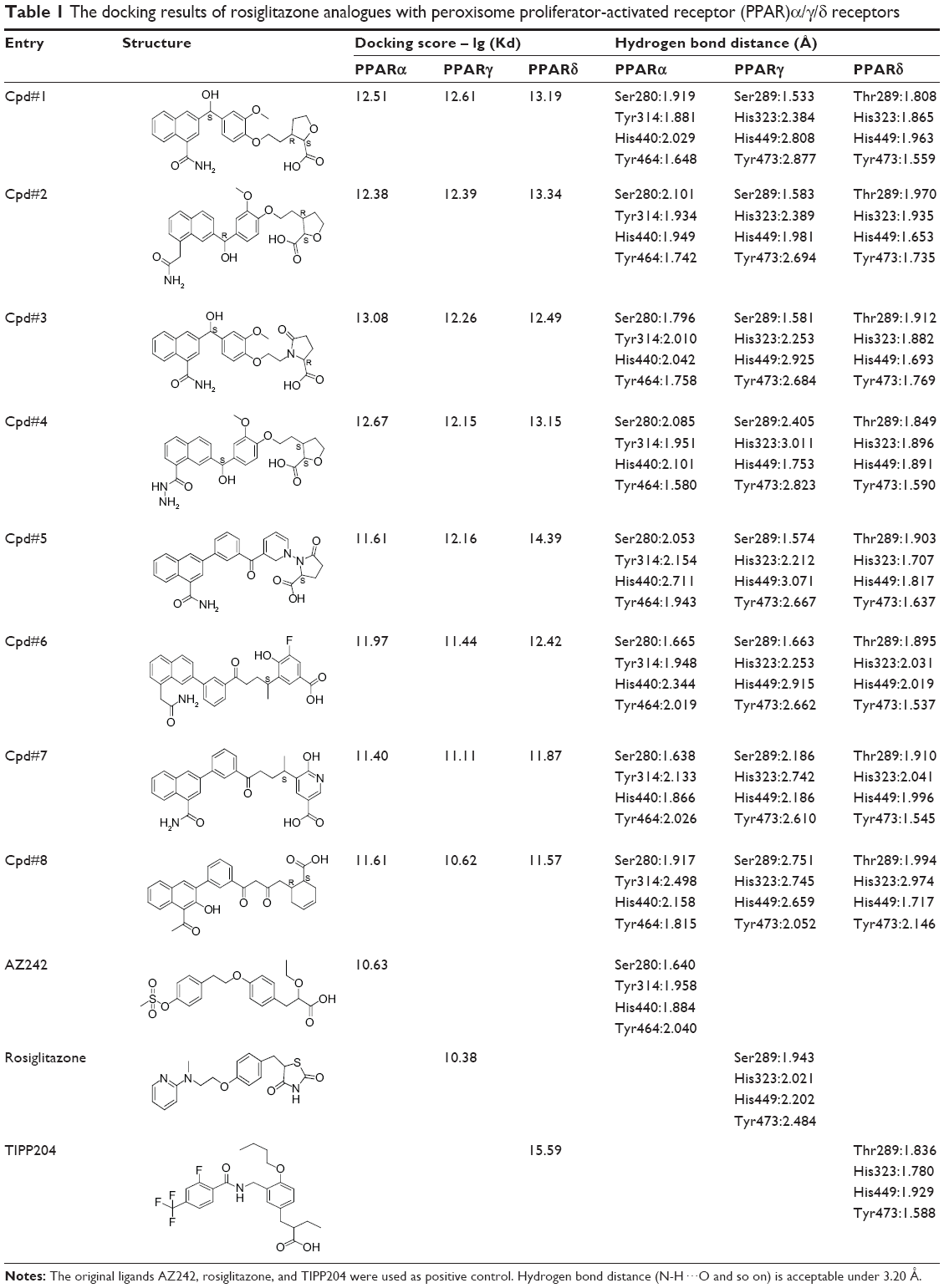 | Table 1 The docking results of rosiglitazone analogues with peroxisome proliferator-activated receptor (PPAR)α/γ/δ receptors |
The docking mode of the representative Cpd#1 with the active site of PPARα/γ/δ receptors is shown in Figure 3. The carboxyl acidic head of Cpd#1 formed hydrogen bonds with the key residues of PPARα (Ser280, Tyr314, Tyr464, and His440), PPARγ (Ser289, His323, Tyr473, and His449), and PPARδ (His323, His449, and Tyr473) receptors, respectively. The aromatic hydrophobic tail and the linker of Cpd#1 bound to PPARα/γ/δ with similar conformations to the original ligand AZ242, rosiglitazone, and TIPP204, respectively.
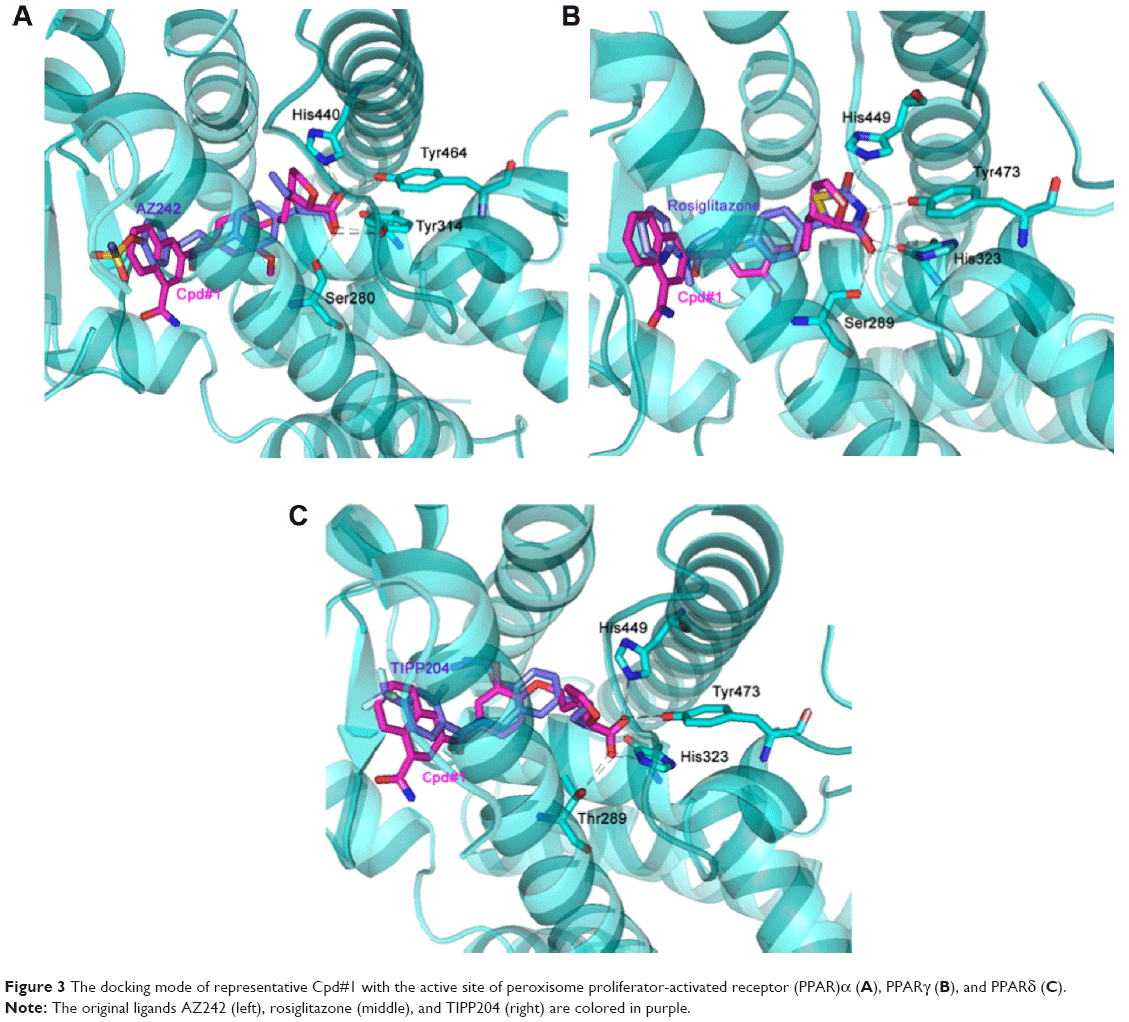 | Figure 3 The docking mode of representative Cpd#1 with the active site of peroxisome proliferator-activated receptor (PPAR)α (A), PPARγ (B), and PPARδ (C). |
ADMET prediction
The development of the PPARα/γ dual agonist muraglitazar has been discontinued during clinical trials because of danger and mortality rate of cardiovascular events.40 Thus, the prediction of drug ADME/Tox was crucial and could reduce the risk of the drug development. The pharmacokinetics and toxicity of the top eight compounds were predicted using the ADMET module of Discovery Studio 3.1. The molecular weight (MW), octanol–water partition coefficient (AlogP98), polar surface area (PSA-2D), aqueous solubility (QplogS), number of hydrogen bond acceptors (nON), number of hydrogen bond donors (nOHNH), and mutagenicity of rosiglitazone analogues are listed in Table 2, respectively. These compounds accorded with Lipinski’s rule of five (Mol_MW<500, 0.4<AlogP98<5.6, nOHNH<5, nON<10, 7<PSA-2D<200, 0.5<QplogS<6.5),41,42 and the values were equal to the positive control AZ242, rosiglitazone, and TIPP204. The probabilities of mutagenicity of these compounds were also lower than for rosiglitazone.
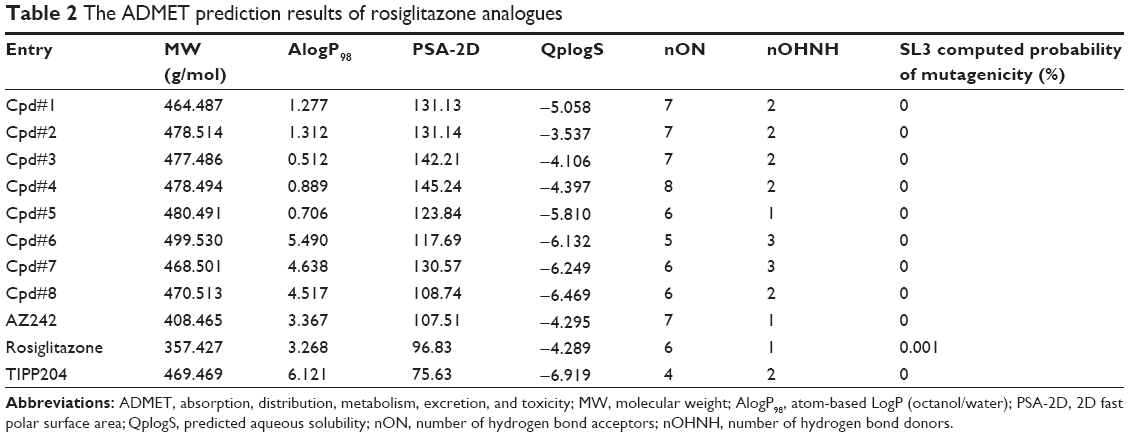 | Table 2 The ADMET prediction results of rosiglitazone analogues |
Molecular dynamics trajectory analysis
In order to study the dynamics behaviors and the binding stability of the PPARs–Cpd#1 complex, the 10ns molecular dynamics simulations were performed on PPARs–apo, PPARs–original ligand complex, and PPARs–Cpd#1 complex.
The RMSD versus the simulation time was considered as a significant criterion to evaluate the stability of dynamic behavior. The final RMSD values for all the simulation systems were <0.8 nm (Figure 4). After 3ns, the RMSD values for Cpd#1–PPARs system (red) was the lowest one among these three simulation trajectories.
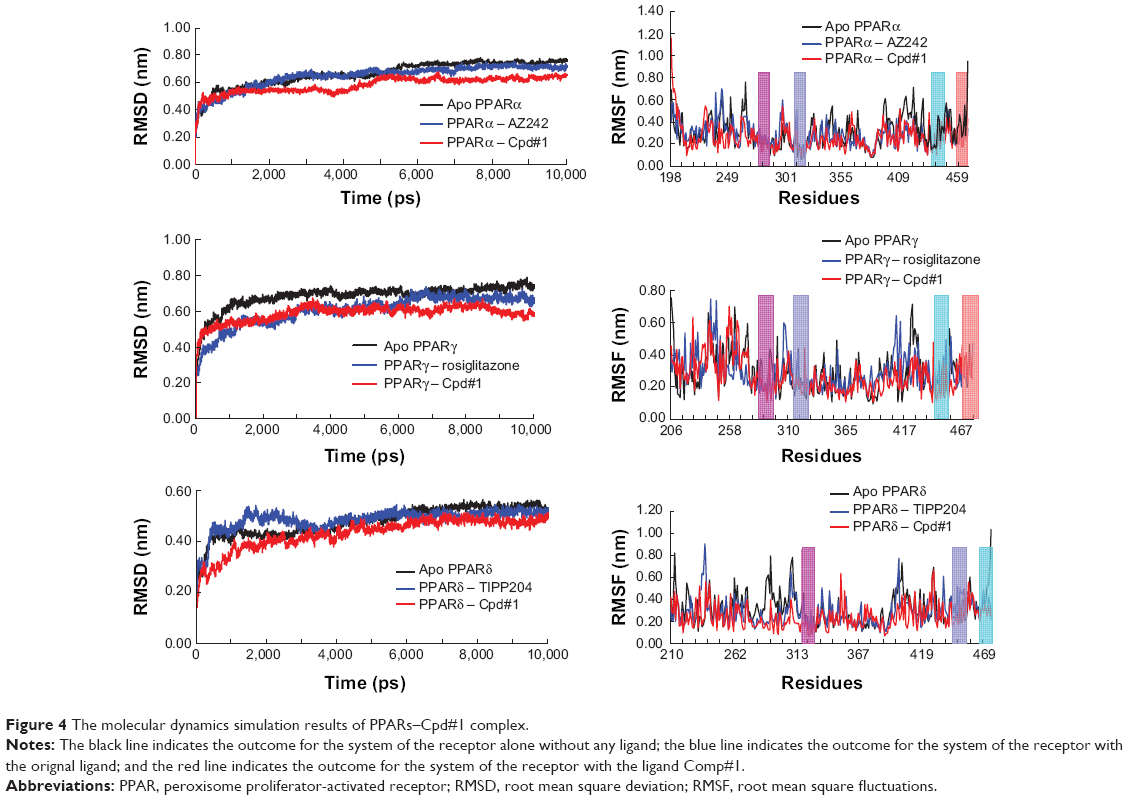 | Figure 4 The molecular dynamics simulation results of PPARs–Cpd#1 complex. |
In order to study the dynamic details of key residues interacted with the ligand, the root mean square fluctuations (RMSF) of all the side chain residues were obtained. The RMSF curve of PPARs–Cpd#1 complex was similar to that of PPARs–original ligand complex (Figure 4). At the key residues of PPARα such as Ser280 (the pink area), Tyr314 (the conch area), Tyr464 (the cyan area), and His440 (the coral area), the RMSF values of the PPARα–Cpd#1 complex were somewhat lower than those of the PPARα–original ligand complex and PPARα–apo form. As for PPARγ/δ, similar circumstances existed just as with PPARα. These molecular dynamics simulation trajectories indicated that PPARs became more stable after binding Cpd#1.
Conclusion
In this study, rosiglitazone was modified by core hopping strategy to produce various analogues. Using docking and ADMET prediction technique, eight novel compounds were identified as multitargeted PPARα/γ/δ pan agonists with excellent pharmacokinetic properties. Molecular dynamics simulations of the representative Cpd#1 showed that Cpd#1 bound steadily to PPARα/γ/δ active site and restricted the target movement. These compounds have not been reported in the literature and could act as novel PPARs multitargeted agonists for antidiabetic drug research.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21202120), a China Postdoctoral Science Foundation funded project (2012T50237). The authors wish to thank the anonymous reviewers for their valuable suggestions, which were very helpful for strengthening the presentation of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Monsalve FA, Pyarasani RD, Delgado-Lopez F, Moore-Carrasco R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediators Inflamm. 2013;2013:549627. | ||
Aleshin S, Strokin M, Sergeeva M, Reiser G. Peroxisome proliferator-activated receptor (PPAR) beta/delta, a possible nexus of PPARalpha- and PPARgamma-dependent molecular pathways in neurodegenerative diseases: review and novel hypotheses. Neurochem Int. 2013;63(4):322–330. | ||
Seok H, Cha BS. Refocusing peroxisome proliferator activated receptor-alpha: a new insight for therapeutic roles in diabetes. Diabetes Metab. 2013;37(5):326–332. | ||
Markt P, Petersen RK, Flindt EN, et al. Discovery of novel PPAR ligands by a virtual screening approach based on pharmacophore modeling, 3D shape, and electrostatic similarity screening. J Med Chem. 2008;51(20):6303–6317. | ||
Antonelli A, Ferrari SM, Fallahi P, et al. Monokine induced by interferon gamma (IFNgamma) (CXCL9) and IFNgamma inducible T-cell alpha-chemoattractant (CXCL11) involvement in Graves’ disease and ophthalmopathy: modulation by peroxisome proliferator-activated receptor-gamma agonists. J Clin Endocrinol Metab. 2009;94(5):1803–1809. | ||
Antonelli A, Fallahi P, Ferrari SM, et al. Dedifferentiated thyroid cancer: a therapeutic challenge. Biomed Pharmacother. 2008;62(8):559–563. | ||
Eldor R, DeFronzo RA, Abdul-Ghani M. In vivo actions of peroxisome proliferator-activated receptors: glycemic control, insulin sensitivity, and insulin secretion. Diabetes Care. 2013;36 Suppl 2:162–174. | ||
Kostapanos MS, Kei A, Elisaf MS. Current role of fenofibrate in the prevention and management of non-alcoholic fatty liver disease. World J Gastroentero. 2013;5(9):470–478. | ||
Lewis SN, Brannan L, Guri AJ, et al. Dietary alpha-eleostearic acid ameliorates experimental inflammatory bowel disease in mice by activating peroxisome proliferator-activated receptor-gamma. PLoS One. 2011;6(8):e24031. | ||
Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356(24):2457–2471. | ||
Wickens P, Zhang C, Ma X, et al. Indanylacetic acids as PPAR-delta activator insulin sensitizers. Bioorg Med Chem Lett. 2007;17(15):4369–4373. | ||
Batista FA, Trivella DB, Bernardes A, et al. Structural insights into human peroxisome proliferator activated receptor delta (PPAR-delta) selective ligand binding. PloS One. 2012;7(5):e33643. | ||
Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, Staels B. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta. 2007;1771(8):1065–1081. | ||
Ma Y, Wang SQ, Xu WR, Wang RL, Chou KC. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PloS One. 2012;7(6):e38546. | ||
Cronet P, Petersen JF, Folmer R, et al. Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ242; ligand selectivity and agonist activation in the PPAR family. Structure. 2001;9(8):699–706. | ||
Nolte RT, Wisely GB, Westin S, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395(6698):137–143. | ||
Oyama T, Toyota K, Waku T, et al. Adaptability and selectivity of human peroxisome proliferator-activated receptor (PPAR) pan agonists revealed from crystal structures. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 8):786–795. | ||
Banks JL, Beard HS, Cao Y, et al. Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem. 2005;26(16):1752–1780. | ||
Zapata-Sudo G, Lima LM, Pereira SL, et al. Docking, synthesis and anti-diabetic activity of novel sulfonylhydrazone derivatives designed as PPAR-gamma agonists. Curr Top Med Chem. 2012;12(19):2037–2048. | ||
Cai L, Wang Y, Wang JF, Chou KC. Identification of proteins interacting with human SP110 during the process of viral infections. Med Chem. 2011;7(2):121–126. | ||
Chou KC, Wei DQ, Zhong WZ. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem Bioph Res Co. 2003;308(1):148–151. | ||
Liao QH, Gao QZ, Wei J, Chou KC. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR). Med Chem. 2011;7(1):24–31. | ||
Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. | ||
da Silva FM, dos Santos JC, Campos JL, et al. Structure-based identification of novel PPAR gamma ligands. Bioorg Med Chem Lett. 2013;23(21):5795–5802. | ||
Nevin DK, Peters MB, Carta G, Fayne D, Lloyd DG. Integrated virtual screening for the identification of novel and selective peroxisome proliferator-activated receptor (PPAR) scaffolds. J Med Chem. 2012;55(11):4978–4989. | ||
Guasch L, Sala E, Mulero M, et al. Identification of PPARgamma partial agonists of natural origin (II): in silico prediction in natural extracts with known antidiabetic activity. PloS One. 2013;8(2):e55889. | ||
Guasch L, Sala E, Castell-Auvi A, et al. Identification of PPARgamma partial agonists of natural origin (I): development of a virtual screening procedure and in vitro validation. PloS One. 2012;7(11):e50816. | ||
Fruchart JC. Selective peroxisome proliferator-activated receptor alpha modulators (SPPARMalpha): the next generation of peroxisome proliferator-activated receptor alpha agonists. Cardiovasc Diabetol. 2013;12:82. | ||
Liu L, Ma Y, Wang RL, Xu WR, Wang SQ, Chou KC. Find novel dual-agonist drugs for treating type 2 diabetes by means of cheminformatics. Drug Des Dev Ther. 2013;7:279–288. | ||
Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363. | ||
Bernardes A, Souza PC, Muniz JR, et al. Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: a new mode of ligand recognition and receptor stabilization. J Mol Biol. 2013;425(16):2878–2893. | ||
Puhl AC, Bernardes A, Silveira RL, et al. Mode of peroxisome proliferator-activated receptor gamma activation by luteolin. Mol Pharmacol. 2012;81(6):788–799. | ||
Kouskoumvekaki I, Petersen RK, Fratev F, et al. Discovery of a novel selective PPARgamma ligand with partial agonist binding properties by integrated in silico/in vitro work flow. J Chem Inf Model. 2013;53(4):923–937. | ||
Lian P, Wei DQ, Wang JF, Chou KC. An allosteric mechanism inferred from molecular dynamics simulations on phospholamban pentamer in lipid membranes. PloS One. 2011;6(4):e18587. | ||
Wang YJ, Wang JF, Ping J, et al. Computational studies on the substrate interactions of influenza A virus PB2 subunit. PloS One. 2012;7(9):e44079. | ||
Li XB, Wang SQ, Xu WR, Wang RL, Chou KC. Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PloS One. 2011;6(11):e28111. | ||
Issemann I, Prince RA, Tugwood JD, Green S. The peroxisome proliferator-activated receptor: retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J Mol Endocrinol. 1993;11(1):37–47. | ||
Feng G, Evangelisti L, Favero LB, Grabow JU, Xia Z, Caminati W. On the weak O-H···halogen hydrogen bond: a rotational study of CH3CHClF···H2O. Phys Chem Chem Phys. 2011;13(31):14092–14096. | ||
Taylor R. Life-science applications of the cambridge structural database. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 6 No 1):879–888. | ||
Eghdamian B, Ghose K. Mode of action and adverse effects of lipid lowering drugs. Drug Today. 1998;34(11):943–956. | ||
Lipinski CA. Chris lipinski discusses life and chemistry after the rule of five. Drug Discov Today. 2003;8(1):12–16. | ||
Egan WJ, Lauri G. Prediction of intestinal permeability. Adv Drug Deliv Rev. 2002;54(3):273–289. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.