Back to Journals » Drug Design, Development and Therapy » Volume 12
Identification of new inhibitors of Mdm2–p53 interaction via pharmacophore and structure-based virtual screening
Authors Atatreh N, Ghattas MA ![]() , Bardaweel SK, Al Rawashdeh S, Al Sorkhy M
, Bardaweel SK, Al Rawashdeh S, Al Sorkhy M ![]()
Received 3 August 2018
Accepted for publication 3 October 2018
Published 2 November 2018 Volume 2018:12 Pages 3741—3752
DOI https://doi.org/10.2147/DDDT.S182444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Noor Atatreh,1 Mohammad A Ghattas,1 Sanaa K Bardaweel,2 Sara Al Rawashdeh,1 Mohammad Al Sorkhy1
1Department of Pharmaceutical Sciences, College of Pharmacy, Al Ain University of Science and Technology, Al Ain, Abu Dhabi, United Arab Emirates; 2Department of Pharmaceutical Sciences, School of Pharmacy, The University of Jordan, Amman 11942, Jordan
Background: The tumor suppressor protein p53 plays an important role in preventing tumor formation and progression through its involvement in cell division control and initiation of apoptosis. Mdm2 protein controls the activity of p53 protein through working as ubiquitin E3 ligase promoting p53 degradation through the proteasome degradation pathway. Inhibitors for Mdm2-p53 interaction have restored the activity of p53 protein and induced cancer fighting properties in the cell.
Purpose: The objective of this study is to use computer-aided drug discovery techniques to search for new Mdm2-p53 interaction inhibitors.
Methods: A set of pharmacophoric features were created based on a standard Mdm2 inhibitor and this was used to screen a commercial drug-like ligand library; then potential inhibitors were docked and ranked in a multi-step protocol using GLIDE. Top ranked ligands from docking were evaluated for their inhibition activity of Mdm2-p53 interaction using ELISA testing.
Results: Several compounds showed inhibition activity at the submicromolar level, which is comparable to the standard inhibitor Nutlin-3a. Furthermore, the discovered inhibitors were evaluated for their anticancer activities against different breast cancer cell lines, and they showed an interesting inhibition pattern.
Conclusion: The reported inhibitors can represent a starting point for further SAR studies in the future and can help in the discovery of new anticancer agents.
Keywords: protein–protein interaction, virtual screening, Mdm2, p53, anticancer, docking, pharmacophore, ELISA
Introduction
Tumor suppressor protein p53 plays an important role in the survival of cells and control of cell division and growth.1 The importance of this protein comes from its vital role in the initiation of cell apoptosis,2 angiogenesis,3 autophagy,4 and cell cycle control.5 It is believed that impairment of its activity can lead to the development of cancer and tumor progression.6
Previous research showed that 50% of human cancers express mutated TP53 gene that would lead to damaged p53 functionality.7 This damaged activity will affect cell proliferation resulting in abrogated cell cycle control leading to cancer development.8 In addition to the mutation of p53 protein, activity of this protein is highly orchestrated by Mdm2 protein as well as ARF. The tumor suppresser ARF has been found to induce cell cycle arrest in a p53-dependent manner by binding to Mdm2 promoting its rapid degradation, leading to stabilization of p53.9–12
In normal unstressed cells, p53 is an unstable protein and is present at very low cellular levels, owing to continuous binding to its specific E3 ubiquitin ligase Mdm2, which controls its degradation through the proteasome pathway.13
It has been proven that overexpression of Mdm2 and subsequent deactivation of p53 protein have a major impact on different cell cycle checkpoints resulting in failure of apoptosis and cancer cell survival. It is well established that abrogating p53/Mdm2 protein binding would lead to the increase of p53 antitumor activities within the cell.14 In fact targeting p53–Mdm2 interaction received great attention in order to restore p53 protein activity and enhance its tumor suppression proprties.2,12,15–17
The p53–Mdm2 interaction surface was intensively investigated. Crystallization studies reported that the interaction surface is ~700 Å2 and this surface provides an excellent opportunity for small molecule inhibitors to disrupt p53–Mdm2 interaction. Fourteen amino acids form a deep hydrophobic cavity on the Mdm2 protein structure that can be occupied by small molecule inhibitors. These residues are Leu54, Leu57, Ile61, Met62, Tyr67, Gln72, Val75, Phe86, Phe91, Val93, His96, Ile99, Tyr100, and Ile101.18 On the other hand, three amino acids, namely Phe19, Trp23, and Leu26, from p53 protein are involved directly with Mdm2 interaction surface residues.
For more than 15 years, using several drug discovery techniques, several inhibitors for p53–Mdm2 interaction were reported. Interestingly, these inhibitors showed diversity in their chemical structures. Estrada-Ortiz et al19 have recently published a review listing p53–Mdm2 small molecule inhibitors and listed these molecules according to their chemical structure similarities to nutlins,20 imidazoles,21 imidazothiazoles,22 indoles,23 spirooxindoles,24 pyrrolidines,25 isoquinolines,26 piperidinones,27 morpholinones,28 and benzodiazepines.29 These inhibitors show diverse physiochemical properties that affect their potency, selectivity, and inhibitory effects. Several examples of inhibitors are shown in Figure 1 along with their interaction fingerprints with the Mdm2 residues. It can be clearly said that the most dominant interaction in this case is the van der Waal (vdW) contacts, where hydrophobic residues (Val93 and Leu54) are mostly involved in such interactions. Hence, the Mdm2 active site favors ligands with hydrophobic character with multiple aromatic systems.
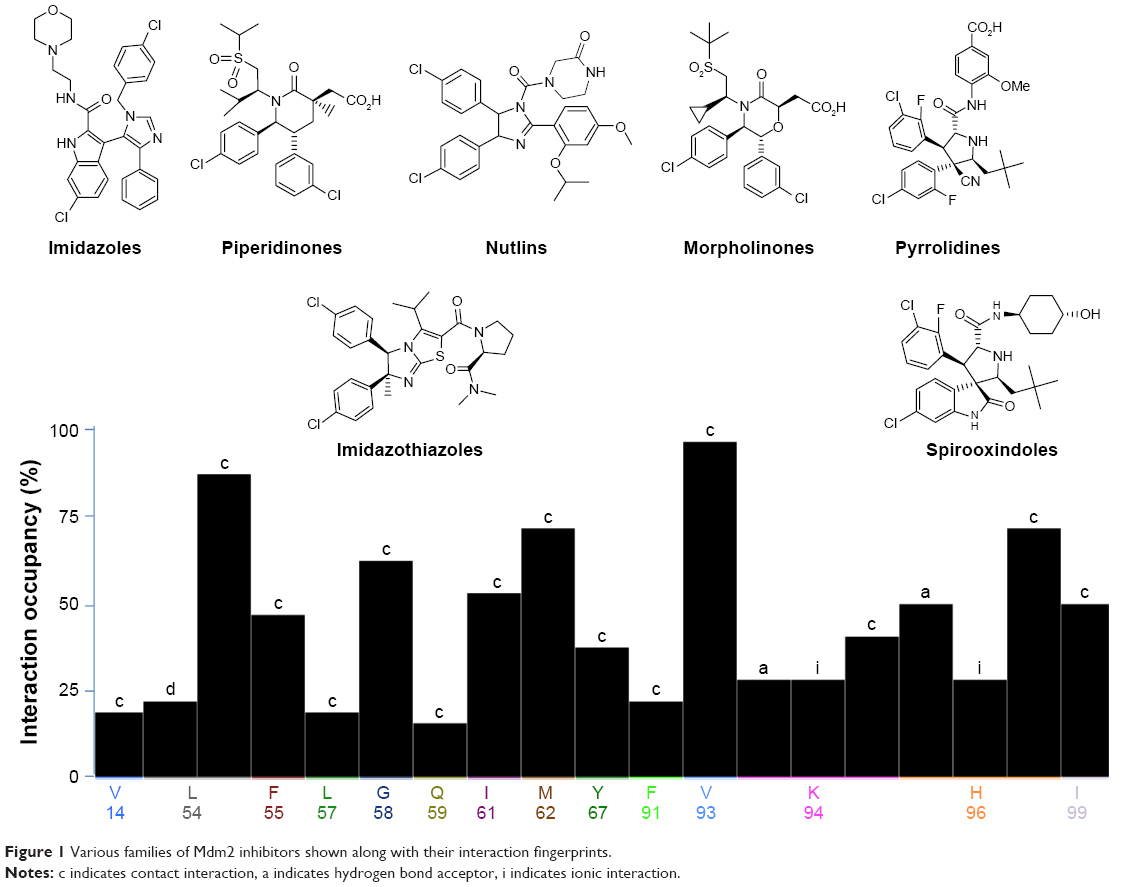 | Figure 1 Various families of Mdm2 inhibitors shown along with their interaction fingerprints. |
So far, none of these drugs had reached the clinics yet; some of them are in preclinical trials. Toxicity and drug resistance are still the major challenges of these drugs. Therefore, the need to discover new inhibitors that overcome these two obstacles is a crucial step toward cancer therapy and management. In this study we utilize several computer-aided drug discovery techniques to search for new Mdm2–p53 interaction inhibitors. First, we built a pharmacophore set in the druggable pocket of the Mdm2 protein (where p53 binds), which was used to filter out an initial drug-like ligand library, then the resultant set of potential inhibitors was screened against the targeted protein using docking, and finally, the activity of these inhibitors was evaluated in vitro.
Methods
Molecular modeling
Protein and ligand library preparation
The crystal structure of Mdm2 was obtained from the protein data bank (PDB; http://www.rcsb.org, 3JZK).30 All solvent molecules were eliminated prior to protein preparation. The MOE 3D protonate module31 was employed to add hydrogen atoms to the protein structure and to assign partial charges to each atom based on the MMFF94× force field.32
A ligand library of 582,474 compounds was obtained from TimTec Compound Libraries for screening (http://www.timtec.net/). The ligands were filtered based on the Veber’s rules33 and the Lipinski’s rule of five.34 These drug-likeness filters included: molecular weight ≤500 Da, hydrogen bond donor ≤5, hydrogen bond acceptor ≤10, logP ≤5, polar surface area ≤140 Å2, and rotatable bonds ≤10. Ligands were protonated/deprotonated based on the wash module in MOE31 and were then assigned partial charges based on the MMFF94× force field.
Virtual screening
Three pharmacophoric features were created using the cocrystallized Mdm2 inhibitor from the PDB entry 3JZK, including one aromatic, one hydrophobic, and one hydrophobic/aromatic. And as a validation step, a total number of 32 known Mdm2 inhibitors were screened against the pharmacophore, of which 31 were successfully capable of passing the pharmacophore. The parmacophore algorithm was then used to filter the ligand library. In the pharmacophore algorithm, each ligand in the ligand library was placed on the predefined pharmacophore and was then oriented to take the best fit. Only ligands that satisfied the three aforementioned pharmacophoric features were kept in the resulting ligand library.
Subsequently, the cocrystallized ligand was used to define the Mdm2 binding pocket. Then, the prepared and filtered drug-like ligand library was docked into the Mdm2 active site using the grid-based ligand docking with energetics (GLIDE) module of the Schrödinger molecular modeling suite.35 A two-step docking protocol was employed to allow gradual increase in docking precision: standard precision and then extra precision. The top-ranked ligands obtained from the extra-precision docking step were then visually inspected and the ligands that showed convenient binding modes and nice pocket filling were selected for in vitro testing.
Biological assessment of p53/Mdm2 complex-specific enzyme immunoassay
Capture antibody (100 μL) (from the 250× stock prepared in coating buffer) was dispensed into 96-well plate (96-well high-binding polystyrene microtiter plates, Cat. #80–1930) using a precision pipet. The plate was sealed and incubated overnight at room temperature (RT) in a microplate reader/incubator (Multiskan™ GO Microplate Spectrophotometer; Thermo Fisher Scientific, Waltham, MA, USA). Each well was aspirated to remove coating solution, and 200 μL of blocking buffer per well was immediately added, and the plate was sealed and incubated for at least 1 hour at RT, after which each well was aspirated to remove blocking solution.
p53 Standard, prepared in assay buffer from the 20× stock (1 μg/mL), was pipetted into the bottom of the appropriate wells (standards and samples) and 50 μL of assay buffer into the blank wells. The plate was then sealed and incubated on a plate shaker for 1 hour at RT.
Mdm2 standard, prepared in assay buffer from the 20× stock (0.32 μg/mL), was pipetted into the bottom of lidded polypropylene micro-vials (the same number as the wells of the plate) and then 5 μL of dimethyl sulfoxide (DMSO), samples, and nutlin (the control Mdm2 inhibitor) were added to the appropriate micro-vial; both samples and nutlin were dissolved in DMSO (20 μM). Then, the contents of each vial were mixed well using several pipetting and releasing motion.
After mixing, the samples were incubated for 1 hour at RT in an incubator (WiseCube incubator; witeg Labortechnik GmbH, Wertheim, Germany).
Subsequently, 50 μL of Mdm2 mixtures were added to the appropriate P35-containig wells and the plate was incubated on a plate shaker for 1 hour at RT.
The contents of the wells were emptied and washed by adding 400 μL of wash buffer to every well. This was repeated thrice for a total of four washes. After the final wash, the wells were aspirated and the plate was firmly tapped on a lint-free paper towel to remove any remaining wash buffer. Diluted Mdm2 detection antibody (from the 250× stock prepared in assay buffer) was added into each well, except the blank well. The plate was sealed and incubated on a plate shaker for 1 hour at RT. Then the wells were aspirated and washed by adding 400 μL of wash buffer to every well. This was repeated for a total of four washes. After the final wash, the wells were aspirated and the plate was firmly tapped on a paper towel to remove any remaining wash buffer.
Afterward, 100 μL of the diluted streptavidin-horseradish peroxidase conjugate was added into each well except the blank well. The plate was then sealed and incubated on a plate shaker for 30 minutes at RT, and the wells were aspirated and washed four times as previously mentioned. Then, 100 μL of 3,3′,5,5′-tetramethylbenzidine solution was added into each well, and the plate was sealed and incubated on a plate shaker for 30 minutes at RT. Finally, 100 μL 1N HCl was pipetted into each well.
After blanking the plate reader against the substrate, OD at 450 nm was measured. All the results were computed and expressed as mean±SD from three determinations performed in duplicates (n=6). Inhibitory concentration of 50% of the sample (IC50) values were derived from a sigmoidal dose–response (variable slope) curve using GraphPad Prism 7 software (GraphPad Software, Inc., La Jolla, CA, USA).
Cells and cell culture conditions
The human breast adenocarcinoma MCF-7, the human ductal carcinoma T47D, the human basal triple-negative breast cancer (mammary gland) MDA-231, and human skin fibroblast cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown and maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS and penicillin (100 U/mL) and were incubated at 37°C in a humidified atmosphere of 95% O2 and 5% CO2. The cells were seeded at a density of 6-8×103 cells/well in 96-well plates and incubated for 24 hours for adhesion.
Cell proliferation assay (MTT)
The MTT colorimetric assay was performed to assess cell proliferation as previously described.36 In short, test samples were prepared by dissolving the inhibitors in DMSO followed by further dilution with DMEM to reach the desired final concentration. The final DMSO concentration in the assay was kept as low as 1%. Test samples were applied to the wells and incubated for a period of 48 hours. MTT solution was added to each well and incubated for 4 hours at 37°C. DMSO was added to each well to solubilize the formed purple formazan crystals before absorbance is read using a microplate reader at 570 nm. Doxorubicin was used as the positive control and 1% DMSO in DMEM was used as the solvent control.
Assessing structural similarities with known Mdm2 inhibitors
To assess the structural similarities of the synthesized compounds with some known Mdm2 inhibitors, the 2D fingerprint calculation method from the MOE-Similarity function was used where a 2D fingerprint was generated for each ligand using the MACCS (structural keys) and Tanimoto coefficient calculation. It is a method that encodes the structural features of molecules as bit strings, where each bit indicates the presence or absence of predefined structural and chemical patterns such as atom sequences, electronic configurations, atom pairs, and ring systems. Ligands from all crystal structures that were used to generate the pharmacophore were examined as known Mdm2 inhibitors. All ligands were evaluated based on MACCS similarity with one inhibitor at a time as query source.
Results and discussion
Interaction between Mdm2 and p53 received huge attention from scientists in the past, and researchers are still looking for inhibitors for this interaction due to the important role it plays in the induction of apoptosis and cancer cell survival.
The focus of this study was on finding new inhibitors for this interaction using multiple drug discovery techniques. We started with generating a 3D pharmacophore using a cocrystallized Mdm2 inhibitor. The pharmacophore was created inside the p53 binding pocket on the Mdm2 protein, and it contained three pharmacophoric features (one aromatic, one hydrophobic, and one hydrophobic/aromatic) as shown in Figure 2. As a validation step, a total number of 32 known Mdm2 inhibitors were screened against the three-set pharmacophore of which 31 were successfully capable of passing the pharmacophore. Consequently, pharmacophore-based screening was conducted on a commercial drug-like ligand library (582,474 compounds) to filter out all compounds that did not match the required features.
 | Figure 2 The Mdm2 inhibitor 3D pharmacophore, used to filter out the initial drug-like library, is shown inside the pocket of Mdm2 protein (shown as surface). |
In the next step, receptor-based virtual screening was performed using GLIDE docking to screen the remaining ligand library (which passed the Mdm2 inhibitor pharmacophore) according to the docking protocol shown in Figure 3. The top-scoring 500 compounds were visually checked for their fitting into the p53 binding site of the Mdm2 protein, and 40 of which were selected for experimental testing.
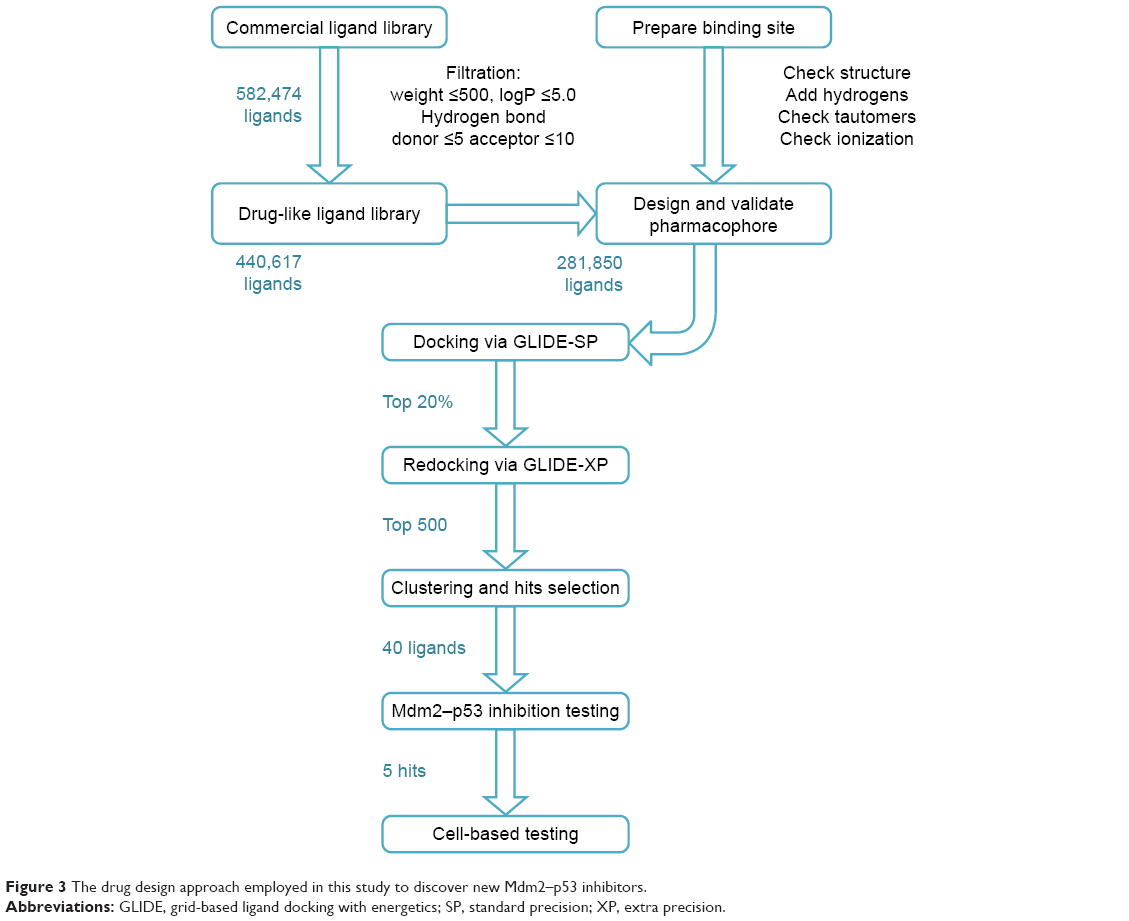 | Figure 3 The drug design approach employed in this study to discover new Mdm2–p53 inhibitors. |
Inhibitory activity of the tested compounds was measured by IC50. The outcomes of the in vitro biological testing showed that the examined compounds have strong inhibitory effect, interestingly, all of which showed IC50 values in the submicromolar level (Table 1). Furthermore, compound S02 exhibited potent inhibitory activity that is comparable to Nutlin 3a (IC50 was 0.42 and 0.30 μM, respectively). This ability to interfere with Mdm2–p53 interaction is mainly attributed to the presence of the previously mapped pharmacophoric features required for Mdm2 binding, indicating the importance of combining pharmacophore screening and docking in the virtual screening protocol to predict compounds with strong inhibitory effects.37,38
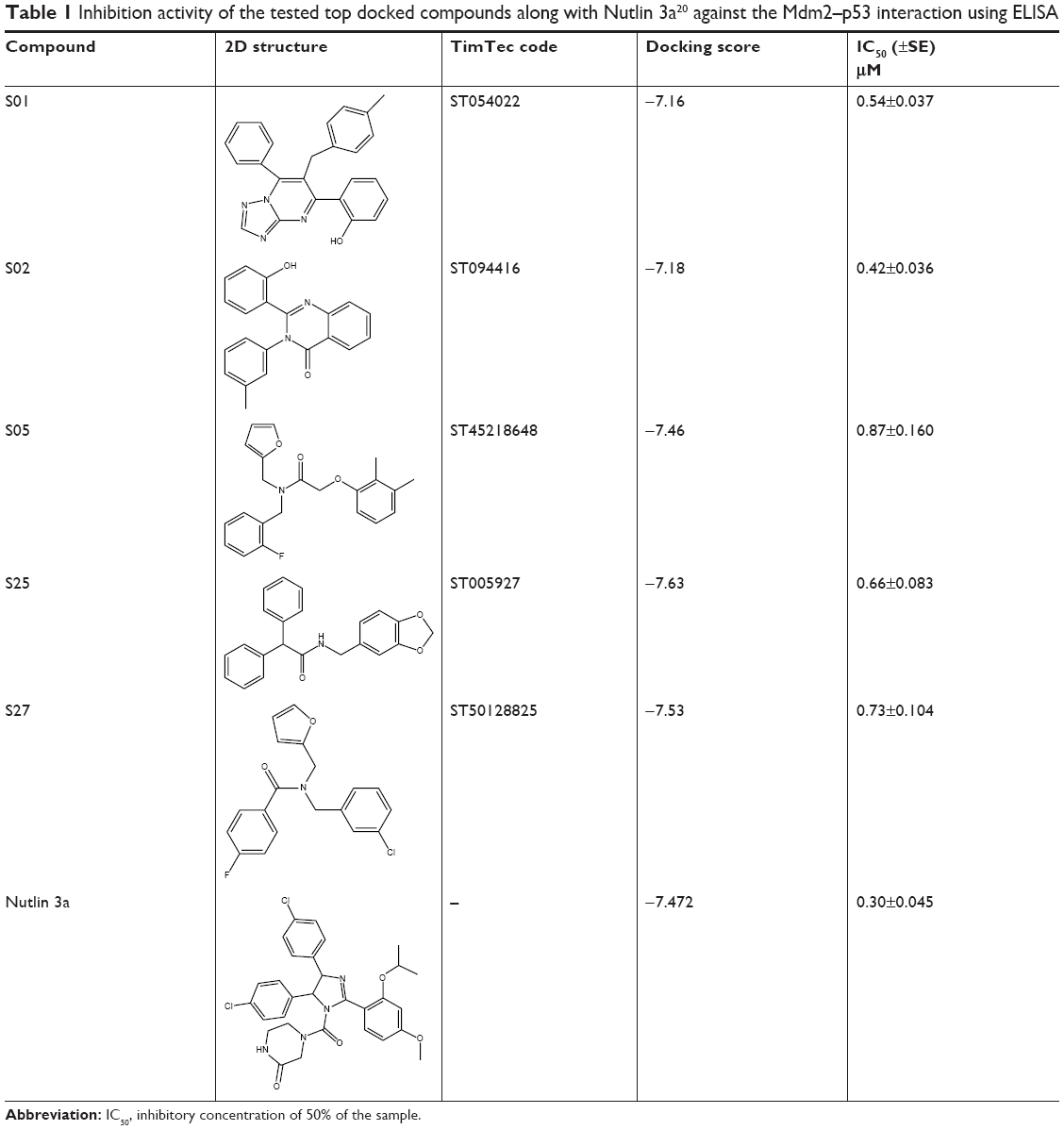 | Table 1 Inhibition activity of the tested top docked compounds along with Nutlin 3a20 against the Mdm2–p53 interaction using ELISA |
Docked compounds showed different binding modes in the active site. These compounds bind to the Mdm2 protein by interacting with key residues, such as Leu54, Leu57, Gly58, Ile61, Met62, Val93, His96, Ile99, and Tyr100. Interestingly, compound S01 interacts with Gly58 in Mdm2 active site (Figure 4), which is not a common interaction formed by Mdm2 cocrystallized inhibitors. S01 belongs to triazolopyrimidine structural scaffold, which, to our knowledge, has never been discovered as Mdm2–p53 interaction inhibitor.
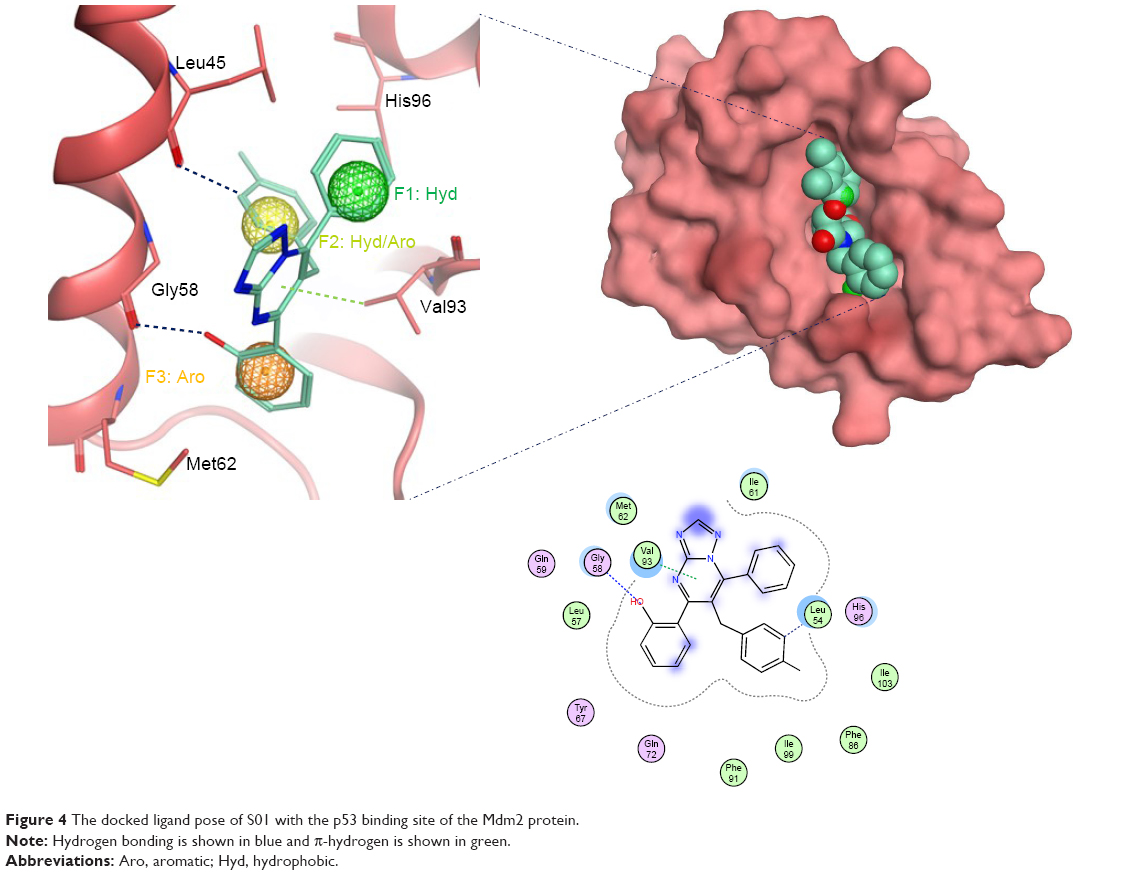 | Figure 4 The docked ligand pose of S01 with the p53 binding site of the Mdm2 protein. |
Compound S02 was the most potent compound from the docked ligands to the Mdm2 active site with IC50=0.42 μM. Visual examination of the docked position of the ligand showed several interactions between the ligand and Mdm2 protein (Figure 5). It forms hydrogen bond interaction with Leu54 and also produces Van der Waals interaction with Met62. Moreover, π-hydrogen interactions were formed between S02 and multiple residues such as His96, Val93, and Ile99. S02 belongs to the quinazolinone family that was previously reported to restore the p53 function via reacting with the p53 protein itself rather than binding to Mdm2, where S02 is suggested to bind.39
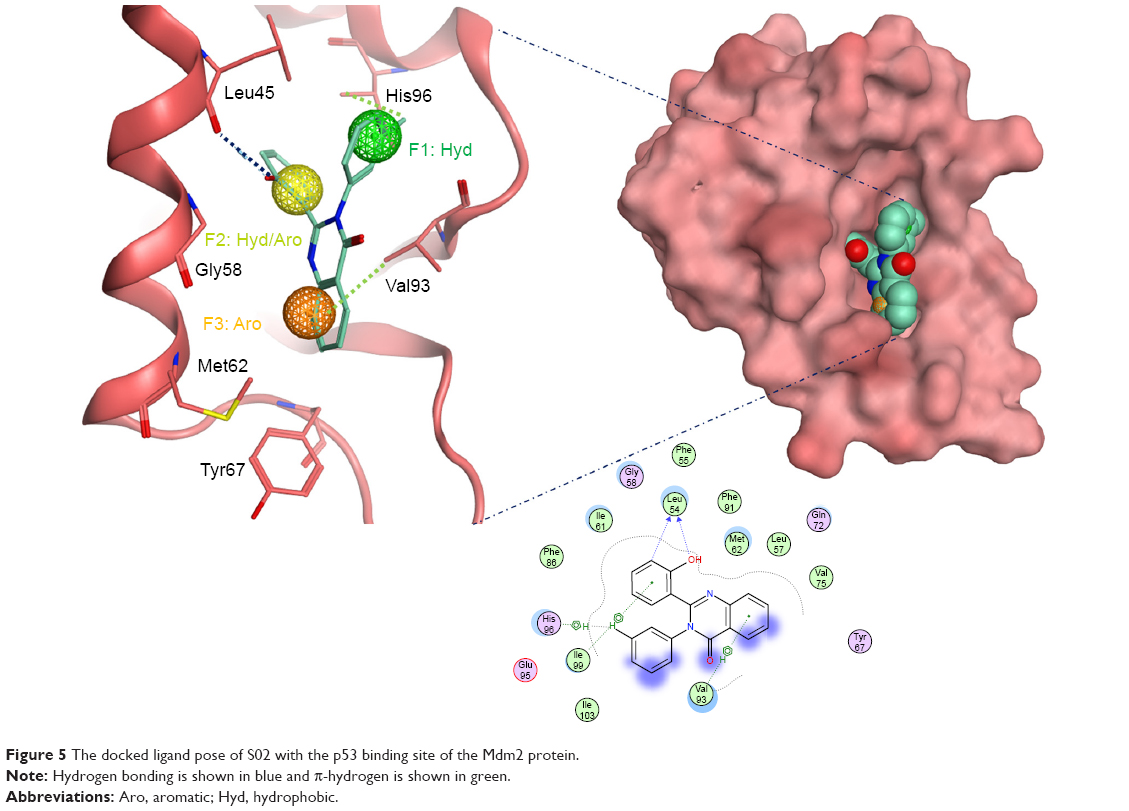 | Figure 5 The docked ligand pose of S02 with the p53 binding site of the Mdm2 protein. |
S05 is another interesting hit which exhibits the most potent anticancer activity (as shown in Figure 5). It is a furan derivative, and this class of compound has been reported before as Mdm2 inhibitor, however, the furan ring usually takes the center of the molecule where other aromatic/heteroaromatic ring (needed for binding) branches from. S05 has amide moiety in the middle where three aromatic rings branch and satisfy the required pharmacophore. This compound has got a nice filling to the p53 binding pocket in addition to several polar contacts with the surrounding residues, as shown in Figure 6. The ability to interfere with Mdm2–p53 interaction is mainly attributed to the presence of the previously mapped pharmacophore (not only in S05 but also in S01 and S02), indicating the importance of combining pharmacophore screening and docking in the same structure-based design protocol to predict compounds with strong inhibitory effects.37,38
 | Figure 6 The docked ligand pose of S05 with the p53 binding site of the Mdm2 protein. |
The last two discovered hits, S25 and S27, also exhibited convincing binding mode inside the Mdm2 pocket, having good shape complementarity with the binding site. S25 showed several hydrogen–arene interactions in addition to a hydrogen bond with the side chain of Leu54 (Figure 7A). S27 is an analog of S05 and another Mdm2 inhibitor that belongs to the furan ring family, with good binding mode (Figure 7B). Both S25 and S27 have three hydrophobic groups that satisfy the three general pharmacophoric features of Mdm2 inhibitors and hence exhibit interesting binding and activity against Mdm2 protein.
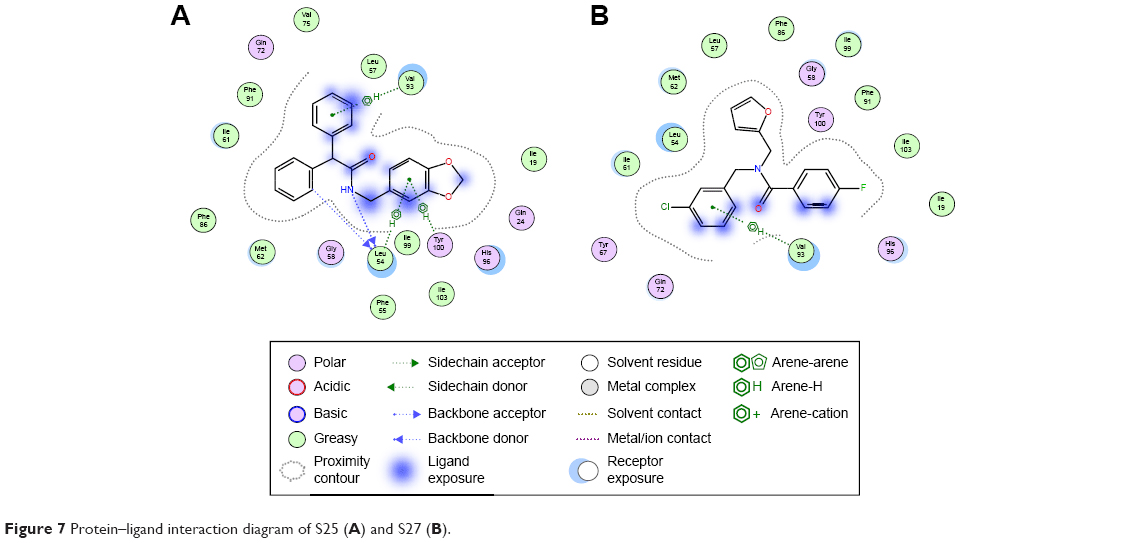 | Figure 7 Protein–ligand interaction diagram of S25 (A) and S27 (B). |
To further evaluate the activity of the presumed inhibitors, they were tested against the breast cancer cell lines MCF-7, T47D, and MDA231. Estrogen receptor a (ERa+) breast cancer cells, such as MCF-7 and T47D cell lines, are often reported to express wild-type p53.40,41 On the other hand, more aggressive breast cancers, represented by the estrogen receptor negative breast tumor cell line MDA231, are reported to have either a mutant or nonfunctional p53 protein with significantly reduced expression of Mdm2. In an attempt to study the differential effects of the presumed inhibitors on diverse subtypes of breast cancer, the ER+ and ER-, we studied the direct anticancer activity of these inhibitors on the model breast cancer cell lines. Interestingly, we show that the examined inhibitors demonstrated preferential antiproliferative activity on the ER+ cell lines, MCF-7 and T47D, as illustrated in Figure 8. The positive control nutlin confirmed this observation with the same trend of activity. The reduced activity in ER- cells can be explained in the light of reduced expression of both p53 and Mdm2 proteins in MDA231 cell line.40,41 Interestingly, there was no significant change to the human fibroblast cell proliferation, relative to the nontreated control, after exposure to any of the presumed inhibitors for 48 hours, suggesting a reasonable safety margin to normal human cells.
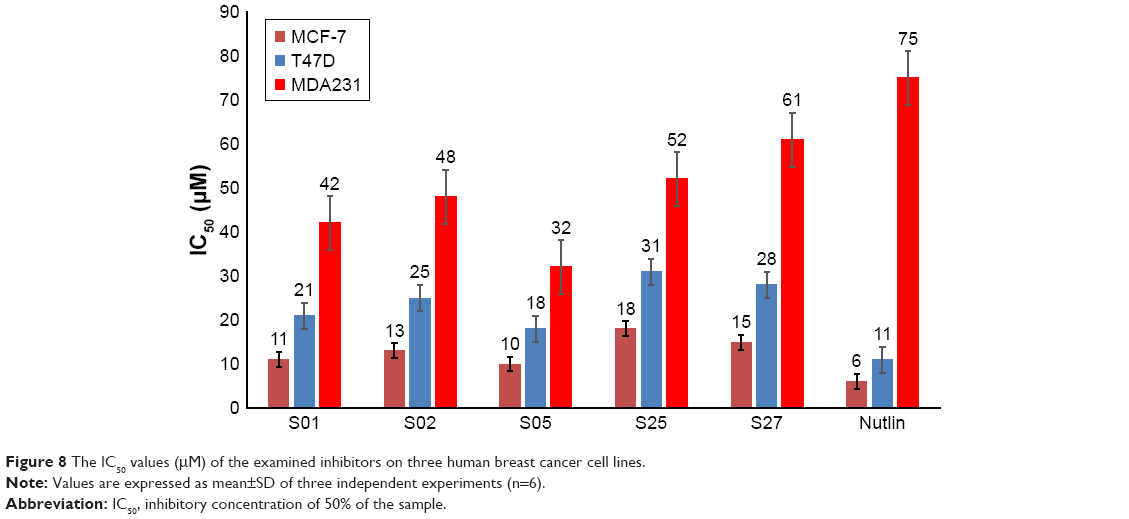 | Figure 8 The IC50 values (μM) of the examined inhibitors on three human breast cancer cell lines. |
To understand the structural novelty of these hit compounds, the similarity scores with previously reported inhibitors (from the database used in pharmacophore generation) were calculated. As shown in Table 2, compound S01 has shown the highest similarity with a chromenotriazolopyrimidine compound (PDB: 3JZK), obviously sharing the triazolopyrimidine ring in their structures. On the other hand, all other four hits showed less than 60% overlapping. Overall, this study generated novel hits with newly discovered structural cores that could act as starting point for clinically useful anticancer agents.
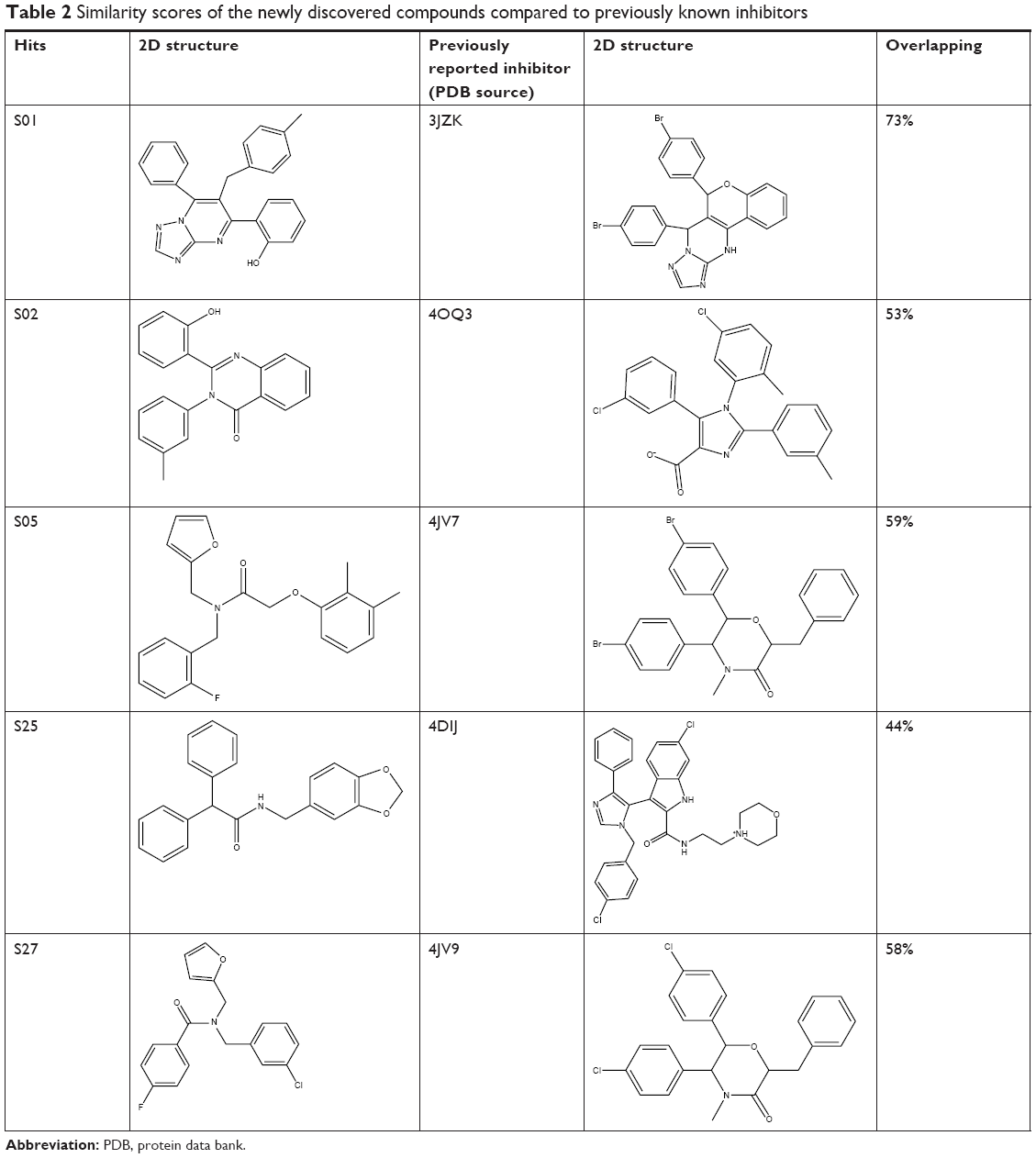 | Table 2 Similarity scores of the newly discovered compounds compared to previously known inhibitors |
Conclusion
Mdm2–p53 protein–protein interaction is an interesting model for scientists to target and discover new anticancer drugs. In this study, we tried to discover new potential inhibitors through the deployment of several molecular modeling tools such as pharmacophoric mapping and ligand-based protein docking. Biological evaluation of the top docked compounds was done by ELISA which provided a good validation technique for the activity of the potential inhibitors. Accordingly, selected compounds from the molecular docking were biologically tested against Mdm2–p53.
Many compounds exhibited potential anticancer activity when examined against different subtypes of breast cancer cell lines, mainly compound S02 showed interesting binding activity to Mdm2 protein, predicted by docking to pose several hydrogen bonds, Van der Waals forces, and π-hydrogen interactions. Another interesting compound is S01 that was found to bind Gly58 in Mdm2 active site, a noncanonical interaction formed by Mdm2 cocrystallized inhibitors. S01 belongs to triazolopyrimidine, which, to our knowledge, has never been reported to block Mdm2–p53 interaction.
These compounds can represent a starting point for further structure–activity relationship studies in the future and hopefully help find new potent inhibitors.
Acknowledgment
This work was funded by the Deanship of Scientific Research and Graduate Studies at Al Ain University of Science and Technology, Al Ain, UAE.
Disclosure
The authors report no conflicts of interest in this work.
References
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–16. | ||
Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer. 2004;91(8):1415–1419. | ||
Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8. | ||
Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. | ||
Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. | ||
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283. | ||
Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9(10):701–713. | ||
Vaughan C, Pearsall I, Yeudall A, Deb SP, Deb S. p53: its mutations and their impact on transcription. Subcell Biochem. 2014;85:71–90. | ||
Momand J, Zambetti GP. Mdm-2: “big brother” of p53. J Cell Biochem. 1997;64(3):343–352. | ||
Michael D, Oren M. The p53 and Mdm2 families in cancer. Curr Opin Genet Dev. 2002;12(1):53–59. | ||
Manfredi JJ. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24(15):1580–1589. | ||
Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. | ||
Lahav G, Rosenfeld N, Sigal A, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36(2):147–150. | ||
Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–155. | ||
Selivanova G. Therapeutic targeting of p53 by small molecules. Semin Cancer Biol. 2010;20(1):46–56. | ||
Stegh AH. Targeting the p53 signaling pathway in cancer therapy – the promises, challenges and perils. Expert Opin Ther Targets. 2012;16(1):67–83. | ||
Essmann F, Schulze-Osthoff K. Translational approaches targeting the p53 pathway for anti-cancer therapy. Br J Pharmacol. 2012;165(2):328–344. | ||
Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948–953. | ||
Estrada-Ortiz N, Neochoritis CG, Dömling A. How to design a successful p53-MDM2/X interaction inhibitor: a thorough overview based on crystal structures. ChemMedChem. 2016;11(8):757–772. | ||
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. | ||
Popowicz GM, Czarna A, Wolf S, et al. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle. 2010;9(6):1104–1111. | ||
Miyazaki M, Naito H, Sugimoto Y, et al. Synthesis and evaluation of novel orally active p53-MDM2 interaction inhibitors. Bioorg Med Chem. 2013;21(14):4319–4331. | ||
Huang Y, Wolf S, Koes D, et al. Exhaustive fluorine scanning toward potent p53-Mdm2 antagonists. ChemMedChem. 2012;7(1):49–52. | ||
Gonzalez-Lopez de Turiso F, Sun D, Rew Y, et al. Rational design and binding mode duality of MDM2-p53 inhibitors. J Med Chem. 2013;56(10):4053–4070. | ||
Ding Q, Zhang Z, Liu JJ, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979–5983. | ||
Gessier F, Kallen J, Jacoby E, et al. Discovery of dihydroisoquinolinone derivatives as novel inhibitors of the p53-MDM2 interaction with a distinct binding mode. Bioorg Med Chem Lett. 2015;25(17):3621–3625. | ||
Yu M, Wang Y, Zhu J, et al. Discovery of Potent and Simplified Piperidinone-Based Inhibitors of the MDM2-p53 Interaction. ACS Med Chem Lett. 2014;5(8):894–899. | ||
Gonzalez AZ, Eksterowicz J, Bartberger MD, et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J Med Chem. 2014;57(6):2472–2488. | ||
Yu Z, Zhuang C, Wu Y, et al. Design, synthesis and biological evaluation of sulfamide and triazole benzodiazepines as novel p53-MDM2 inhibitors. Int J Mol Sci. 2014;15(9):15741–15753. | ||
Allen JG, Bourbeau MP, Wohlhieter GE, et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J Med Chem. 2009;52(22):7044–7053. | ||
Chemical Computing Group Inc. Molecular Operating Environment (MOE, version 2013.08) Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015. | ||
Halgren TA. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J Comput Chem. 1996;17(5–6):616–641. | ||
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | ||
Friesner RA, Banks JL, Murphy RB, et al. GLIDE: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. | ||
ISO – International Organization for Standardization. ISO 10993-5 – Biological Evaluation of Medical Devices. Part 5: Testes for In Vitro Cytotoxicity. 3rd ed. Geneva: ISO; 2009. | ||
Ghattas MA, Atatreh N, Bichenkova EV, Bryce RA. Protein tyrosine phosphatases: ligand interaction analysis and optimisation of virtual screening. J Mol Graph Model. 2014;52:114–123. | ||
Ghattas MA, Mansour RA, Atatreh N, Bryce RA. Analysis of Enoyl-Acyl carrier protein reductase structure and interactions yields an efficient virtual screening approach and suggests a potential allosteric site. Chem Biol Drug Des. 2016;87(1):131–142. | ||
Zache N, Lambert JM, Rökaeus N, et al. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol Oncol. 2008;2(1):70–80. | ||
Gudas JM, Nguyen H, Klein RC, Katayose D, Seth P, Cowan KH. Differential expression of multiple MDM2 messenger RNAs and proteins in normal and tumorigenic breast epithelial cells. Clin Cancer Res. 1995;1(1):71–80. | ||
Kim K, Burghardt R, Barhoumi R, Lee SO, Liu X, Safe S. MDM2 regulates estrogen receptor α and estrogen responsiveness in breast cancer cells. J Mol Endocrinol. 2011;46(2):67–79. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.