Back to Journals » Drug Design, Development and Therapy » Volume 12
Identification of Mycobacterium tuberculosis BioA inhibitors by using structure-based virtual screening
Authors Singh S, Khare G, Bahal RK, Ghosh PC, Tyagi AK ![]()
Received 17 June 2017
Accepted for publication 28 September 2017
Published 1 May 2018 Volume 2018:12 Pages 1065—1079
DOI https://doi.org/10.2147/DDDT.S144240
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sukesh Voruganti
Video abstract presented by Dr Garima Khare.
Views: 2461
Swati Singh,1 Garima Khare,1 Ritika Kar Bahal,1 Prahlad C Ghosh,1 Anil K Tyagi1,2
1Department of Biochemistry, University of Delhi South Campus, New Delhi, India; 2Guru Gobind Singh Indraprastha University, New Delhi, India
Background: 7,8-Diaminopelargonic acid synthase (BioA), an enzyme of biotin biosynthesis pathway, is a well-known promising target for anti-tubercular drug development.
Methods: In this study, structure-based virtual screening was employed against the active site of BioA to identify new chemical entities for BioA inhibition and top ranking compounds were evaluated for their ability to inhibit BioA enzymatic activity.
Results: Seven compounds inhibited BioA enzymatic activity by greater than 60% at 100 µg/mL with most potent compounds being A36, A35 and A65, displaying IC50 values of 10.48 µg/mL (28.94 µM), 33.36 µg/mL (88.16 µM) and 39.17 µg/mL (114.42 µM), respectively. Compounds A65 and A35 inhibited Mycobacterium tuberculosis (M. tuberculosis) growth with MIC90 of 20 µg/mL and 80 µg/mL, respectively, whereas compound A36 exhibited relatively weak inhibition of M. tuberculosis growth (83% inhibition at 200 µg/mL). Compound A65 emerged as the most potent compound identified in our study that inhibited BioA enzymatic activity and growth of the pathogen and possessed drug-like properties.
Conclusion: Our study has identified a few hit molecules against M. tuberculosis BioA that can act as potential candidates for further development of potent anti-tubercular therapeutic agents.
Keywords: Mycobacterium tuberculosis, BioA, virtual screening, drug discovery
Introduction
Mycobacterium tuberculosis (M. tuberculosis), the etiological agent of tuberculosis (TB), has been threatening mankind for millennia and is amongst the deadliest diseases in the world.1 The remarkable capacity of this pathogen to evade the host immune responses makes it a successful and difficult pathogen. The current chemotherapy is a multidrug regimen consisting of rifampicin, isoniazid, pyrazinamide and ethambutol that requires 6–9 months to achieve high cure rates. Currently available standard treatment of TB has a poor compliance due to prolonged treatment duration resulting in the emergence of multidrug-resistant and extremely drug-resistant strains.2,3 Hence, there is an urgent need to improve the treatment by identifying new chemical entities with potent activities against M. tuberculosis.
Biotin is an essential cofactor required for fatty acid metabolism, amino acid biosynthesis and gluconeogenesis.4 M. tuberculosis harbors four necessary genes, namely, bioF, bioA, bioD and bioB, which encode enzymes required for the biosynthesis of biotin from pimeloyl-CoA.5,6 The second step of biotin biosynthesis pathway is catalyzed by 7,8-diaminopelargonic acid synthase (BioA), a pyridoxal-5′-phosphate (PLP)-dependent aminotransferase, which is a crucial enzyme involved in the transamination of 7-keto-8-aminopelargonic acid (KAPA) into 7,8-diaminopelargonic acid (DAPA) uniquely utilizing S-adenosylmethionine (SAM) as an amino donor.7
The importance of biotin biosynthesis in mycobacteria was first demonstrated in a study by Sassetti and Rubin who showed by employing transposon mutagenesis that the genes involved in the biotin biosynthesis are essential for M. tuberculosis growth in vivo.8 Later, Park et al had also demonstrated an essential role of bioA in M. tuberculosis by using conditionally regulated gene expression system wherein the M. tuberculosis mutant lacking bioA displayed an in vitro growth defect under biotin deprivation as well as was unable to cause infection in mice, thus establishing the role of bioA in the persistence of M. tuberculosis in mice.9 Moreover, there is no homolog of BioA in humans as they lack the de novo biotin biosynthesis pathway. Based on these features, BioA appears to be an extremely promising target for anti-mycobacterial drug development.
In the past few years, several efforts have been made toward the identification of potential and selective inhibitors of M. tuberculosis BioA. Amiclenomycin (ACM) was a potent inhibitor of mycobacterial BioA but it failed in animal models due to its low chemical stability.10–13 Following this, many derivatives of ACM have been tried but the stability could be achieved only at the expense of potency.14,15 Further, several other approaches have also been tried for the identification of BioA inhibitors that include biochemical screening,16 mechanism-based inhibitors,14 reversible covalent hydrazines by fragment-based screening,17 target-based whole-cell screening approach,18 fragment library screening using differential scanning fluorimetry and crystallography,19 and structure-based pharmacophore screening.20,21
Here, we present the identification of new BioA inhibitors by employing structure-based virtual screening against the substrate binding site of BioA. A filtered National Cancer Institute (NCI) library was screened to identify the compounds with the highest binding energy and the procured compounds were tested for their inhibitory potential against BioA. Seven compounds displayed greater than 60% inhibition of BioA activity at 100 μg/mL; three of these compounds inhibited greater than 80% of enzymatic activity of BioA at 100 μg/mL. The most potent compound exhibited an IC50 of 10.48 μg/mL (28.94 μM), followed by two others with IC50 values of 33.36 μg/mL (88.16 μM) and 39.17 μg/mL (114.42 μM), respectively. These hits were further evaluated for their whole-cell inhibitory potential against M. tuberculosis in broth culture. Potential molecules were further employed for their evaluation for drug-likeness to provide a foundation for the lead optimization for future drug design studies. Our study has identified few molecules that can be further optimized for drug designing against M. tuberculosis.
Materials and methods
Oligos for polymerase chain reaction, aspartate transaminase (AST) activity kit, SAM, PLP, TAPS [N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid] and CHM-1 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). KAPA and orthophthaldehyde (OPA) were supplied by Cayman Chemical Co. (Ann Arbor, MI, USA), and DAPA was from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Nickel nitrilotriacetic acid (Ni-NTA) was obtained from IBA Lifesciences (Goettingen, Germany). DMEM medium, antibiotic-antimycotic solution and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Difco middlebrook 7H9 media and albumin-dextrose-catalase (ADC) were obtained from Becton, Dickinson and Company (Franklin Lakes, NJ, USA). The compounds were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program (DTP), Division of Cancer Treatment and Diagnosis, NCI, National Institutes of Health.
Cloning and expression of bioA
The gene bioA was PCR amplified from M. tuberculosis H37Rv genomic DNA by using the primers 5′-GATTATCATATGGGATCCATGGCTGCGGCGACTGGC-3′ containing NdeI, BamHI sites (forward primer) and 5′-CTTATATCCTCGAGTCA TGGCAGTGAGCCTAC-3′ containing XhoI, HindIII sites (reverse primer). The amplified PCR product after digestion with NdeI and XhoI was cloned into pET28c vector digested with the same enzymes to create pET28c-bioA for the synthesis of N-terminal His tagged BioA. For expression, E. coli BL21 (λDE3) cells transformed with pET28c-bioA were grown at 37°C in Luria Bertani media containing 25 μg/mL kanamycin till the A600nm of 0.8. The culture was then induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside and was allowed to grow for 16 hours at 25°C. The cells were harvested by centrifugation at 4°C, 6,000× g for 10 minutes.
Purification of BioA
For purification, the cells from the induced culture were harvested and resuspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 10 mM imidazole, 500 mM NaCl, 5 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride and 100 μM PLP and lysed by sonication followed by centrifugation to remove cell debris (15,000× g, 45 minutes, 4°C). Clarified lysate was loaded onto a Ni-NTA metal affinity column pre-equilibrated with lysis buffer. The column was washed initially with buffer containing 20 mM Tris-HCl (pH 8.0), 10 mM imidazole, 500 mM NaCl and later with same buffer containing 20 mM and 50 mM imidazole. The protein was eluted with buffer containing 250 mM imidazole, and the purity of the protein was analyzed by SDS-PAGE by using a 12.5% polyacrylamide gel. The fractions containing BioA were pooled, concentrated by Amicon Ultra Protein Concentrator (EMD Millipore) and loaded onto a Sephadex G-10 column equilibrated with 20 mM Tris-HCl (pH 8.0), 100 mM NaCl and 10 μM PLP. The purified protein was stored at −80°C.
BioA enzyme assay
Enzyme assays were performed as described by Mann et al22 with some modifications. Briefly, the enzymatic reaction mixture comprised 100 mM TAPS buffer (pH 8.6), 100 μM PLP, 20 μM KAPA, 1 mM SAM and 2 μM BioA. The reaction was initiated by the addition of KAPA after 10 minutes of pre-incubation of the enzyme with all other components at 37°C. Enzymatic reaction was terminated by heating at 100°C for 10 minutes and centrifuged to obtain the supernatant, which was then transferred to a 96-well plate. To the supernatant, 200 μL of a freshly made derivatizing solution composed of OPA–2ME reagent (20 μL), 0.26 M sodium borate buffer (pH 9.4, 135 μL) and ethanol (45 μL) was added. The reaction was incubated for 2 hours at room temperature and then fluorescence was measured at an emission wavelength of 470 nm with an excitation wavelength of 410 nm using Cary Varian fluorescence detector. The concentrations of DAPA were calculated from a standard curve of pure DAPA derivatized in the enzymatic assay conditions without BioA.
Virtual screening
The crystal structure of M. tuberculosis BioA in complex with sinefungin, an analog of SAM (PDB ID-3LV2),23 was downloaded from the RCSB Protein Data Bank and the active site was selected for virtual screening. The docking parameters of Autodock4.2 utilized in the study included genetic algorithm with default parameters, 1,750,000 energy evaluations and 20 runs. Virtual screening was performed by using small molecule library comprising 260,071 compounds from NCI Open Database. These compounds were filtered by using FAF server program, which resulted in 95,748 compounds24 that were used for screening against the active site of BioA by using Autodock4.2.25,26 Based on the availability, top 81 high scoring molecules were procured from NCI-DTP for inhibition studies. Compounds obtained were denoted as A1-A81. One compound (A38) could not be dissolved in DMSO; hence, 80 compounds were evaluated experimentally. Subsequently, re-ranking of the docked ligands was carried out by software X-Score.27 Another top new 80 compounds based on X-Score were procured from NCI-DTP depending upon their availability for conducting inhibition studies. Compounds obtained on the basis of X-Score were denoted as X1-X80. Amongst the 80 compounds from X-Score, two compounds were not proceeded with because of their limited solubility (X49) and high toxicity (X80). Hence, 78 compounds from X-Score were experimentally evaluated for inhibition studies.
In vitro enzymatic assay for inhibition study
For BioA inhibition study, all the compounds were dissolved in DMSO to prepare a stock solution of 4 mg/mL and were screened for their ability to inhibit BioA activity at a concentration of 100 μg/mL in a reaction volume of 40 μL. The inhibitory potential of the compounds was evaluated by employing an assay where the product of the BioA-catalyzed reaction was fluorescently quantitated by adding OPA derivatizing agent (Figure 1). The enzymatic reaction mixture was composed of 100 mM TAPS buffer, 2 μM BioA and each compound at 100 μg/mL in a volume of 40 μL, which was then incubated at 37°C for 10 minutes. After 10 minutes, 100 μM PLP, 20 μM KAPA and 1 mM SAM were added and reaction was proceeded for further 20 minutes followed by heating at 100°C for 10 minutes. Rest of the steps was same as already mentioned. Sample containing CHM-1 (the known inhibitor of mycobacterial BioA, identified in biochemical screening of LOPAC library16) was treated as positive control, whereas samples with no compound and sample containing DMSO were used as a negative control for the assay. The percent inhibition of compounds and their IC50 values were calculated by subtracting the inhibition displayed by DMSO in the enzymatic assay. IC50 is defined as the concentration of the compound at which 50% inhibition of enzymatic activity was observed.
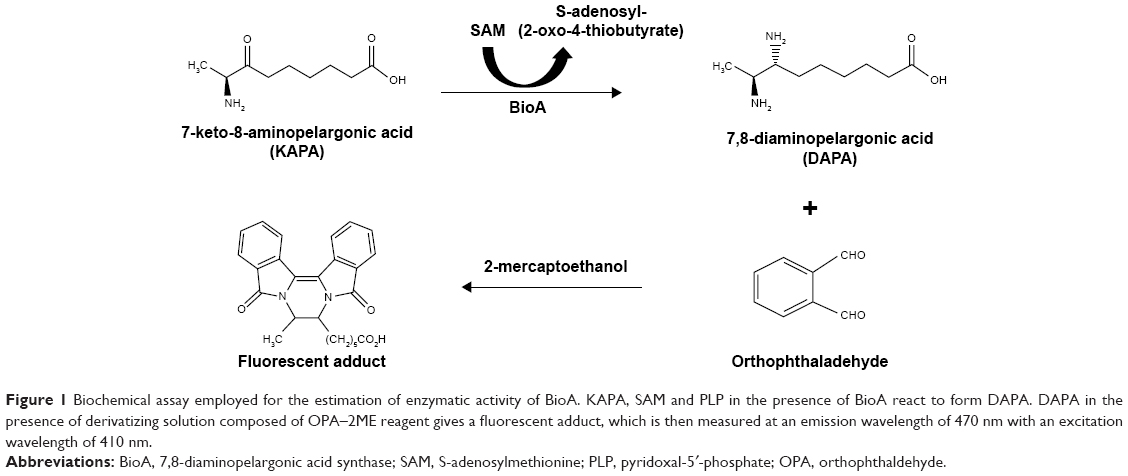 | Figure 1 Biochemical assay employed for the estimation of enzymatic activity of BioA. KAPA, SAM and PLP in the presence of BioA react to form DAPA. DAPA in the presence of derivatizing solution composed of OPA–2ME reagent gives a fluorescent adduct, which is then measured at an emission wavelength of 470 nm with an excitation wavelength of 410 nm. |
Aspartate transaminase enzymatic assay
Inhibition of AST was checked by AST kit (Sigma-Aldrich Co.) according to manufacturer’s instructions. Each compound was checked at a concentration of 100 μg/mL to evaluate its inhibitory potential against AST. Assay was carried out with the positive control provided with the kit. Briefly, in the 96-well flat bottom plate, AST positive control (0.5 μL) and compounds were incubated in AST buffer for 10 minutes at 37°C followed by the addition of AST substrate, AST enzyme mix and AST developer and incubated at 37°C for further 30 minutes. Wells with no compound and containing DMSO were used as a negative control. The plate was read at 450 nm by using Elisa plate reader at the beginning and after every 5 minutes from the commencement of the assay till 30 minutes. For each sample, the inhibition was calculated by subtracting the absorbance at initial time (t=0) from the absorbance at the final time.
Whole-cell growth assay
M. tuberculosis H37Rv was grown in Difco Middlebrook 7H9 (MB-7H9) with 0.5% glycerol supplemented with 0.2% tween 80 and 1X ADC for media enrichment till logarithmic phase. M. tuberculosis H37Rv cells with A600nm of 0.02 (~2×106 cfu/mL) were incubated with varying concentrations of compounds for 7 days at 37°C in 96-well U-bottom microtiter plates followed by the addition of 30 μL of 0.01% resazurin reagent. For evaluation of biotin-dependent growth inhibition of M. tuberculosis, the cells were grown in Sauton’s medium till logarithmic phase and diluted to an A600nm of 0.02 (~2×106 cfu/mL) with varying concentrations of compounds for 7 days at 37°C in 96-well U-bottom microtiter plates followed by the addition of 30 μL of 0.01% resazurin reagent. In total, 1 μM biotin was supplemented to the media wherever needed. Blue color depicts no growth, while conversion to pink color depicts viable cells. Fluorescence was measured at an emission wavelength of 590 nm with an excitation wavelength of 530 nm after 24 hours and percent inhibition was calculated. Rifampicin was employed as a positive control at concentrations of 2 μg/mL, 1 μg/mL, 0.5 μg/mL and 0.25 μg/mL, whereas wells with DMSO were used as a negative control. MIC90 was defined as the concentration of compound at which growth of the pathogen was inhibited by 90%.
Cytotoxicity assay
The compounds were evaluated for their cytotoxicity against Chinese hamster ovary cell line (CHO). Cells were grown in DMEM media with 10% FBS and 1X antibiotic-antimycotic solution. Cells were seeded at a density of 1×104 cells per well in 96-well flat bottom plate and incubated with compounds at varying concentrations ranging from 1 μg/mL to 200 μg/mL for 48 hours in a volume of 200 μL followed by addition of 30 μL of 0.01% resazurin dye to measure cell viability.
Results
Cloning, expression and purification
The bioA gene from M. tuberculosis was cloned in pET28c vector and expressed as described in “Materials and methods” section. Positive clones were selected by using restriction digestion resulting in a fallout of 1.3 kb corresponding to the size of bioA gene and further confirmed by DNA sequencing (data not shown). The selected recombinant clone was subjected to expression and localization studies. The protein was found to be localized predominantly in the insoluble fraction of the cell. However, soluble fraction containing a small fraction of the synthesized protein was employed for purification by nickel-NTA affinity chromatography (Figure S1).
Virtual screening against the active site of BioA
The crystal structure of M. tuberculosis BioA (PDB ID-3LV2) was selected for virtual screening.23 A library of NCI Open Database containing 260,071 compounds was filtered on the basis of Lipinski guidelines for drug-likeness, which resulted in 95,748 compounds.24 These compounds were virtually screened against the substrate binding site of BioA by employing Autodock4.2.25,26 Grid was prepared surrounding the active site residues, and docking of the ligands was carried out by using the parameters defined by this grid box (Figure 2A and B). The selection of this grid was based on the area where sinefungin (an analog of SAM) binds in the crystal of M. tuberculosis BioA (Figure 2C). The ligand sinefungin was extracted from the crystal structure and was docked again at the active site of BioA structure by using same parameters as employed in the virtual screening (Figure 2D). The crystallographic and docked modes of sinefungin were superimposed and they displayed almost the same pose with a root mean square deviation (RMSD) value of 1.88 Å, which is in the acceptable range (Figure 2E). To validate the strategy used for the docking protocol, we have separately docked CHM-1 (the known inhibitor of mycobacterial BioA, identified in biochemical screening of LOPAC library16) at the active of BioA by Autodock4.2 using the same grid co-ordinates as used in our docking studies. It was observed that the docked complex of BioA-CHM-1 was found to be in vicinity of Tyr25, Trp64, Trp65, Val383, Leu385, Arg400, Phe402, Asp160 and Tyr157, which were in accordance with earlier reported study (Figure S2A).19 We superimposed the crystallographic and docked mode of CHM-1, which gave an RMSD of 0.00 as shown in Figure S2B, thereby validating the docking protocol. As a reference molecule, sinefungin was docked at the active site by using same grid parameters and it was found to exhibit a docking score of −7.14. It is to be noted that the compounds obtained in our virtual screening exercise had the highest docking score of −12.52 and the compounds that could be procured for inhibition assays were having docking scores in the range of −11 to −10. Based on the scores generated by Autodock4.2, which is dependent on the binding energy of the docked ligand, top 81 compounds were procured from NCI-DTP depending upon the availability of the compounds and 80 compounds were proceeded with for further experiments. One of the docked compounds, ie, compound A36 and its zoomed view of docked state, is shown in Figure 2F. Docking results obtained by employing Autodock4.2 were further rescored by using software X-Score, which is a scoring function that computes the binding score of a ligand molecule to a target protein.27 Based on re-ranking of the docked ligands by X-Score, another 80 compounds were requested from NCI-DTP as per their availability and 78 compounds were used for further experiments. In total, 158 compounds obtained from NCI-DTP were experimentally screened for their inhibitory potential against BioA enzymatic activity.
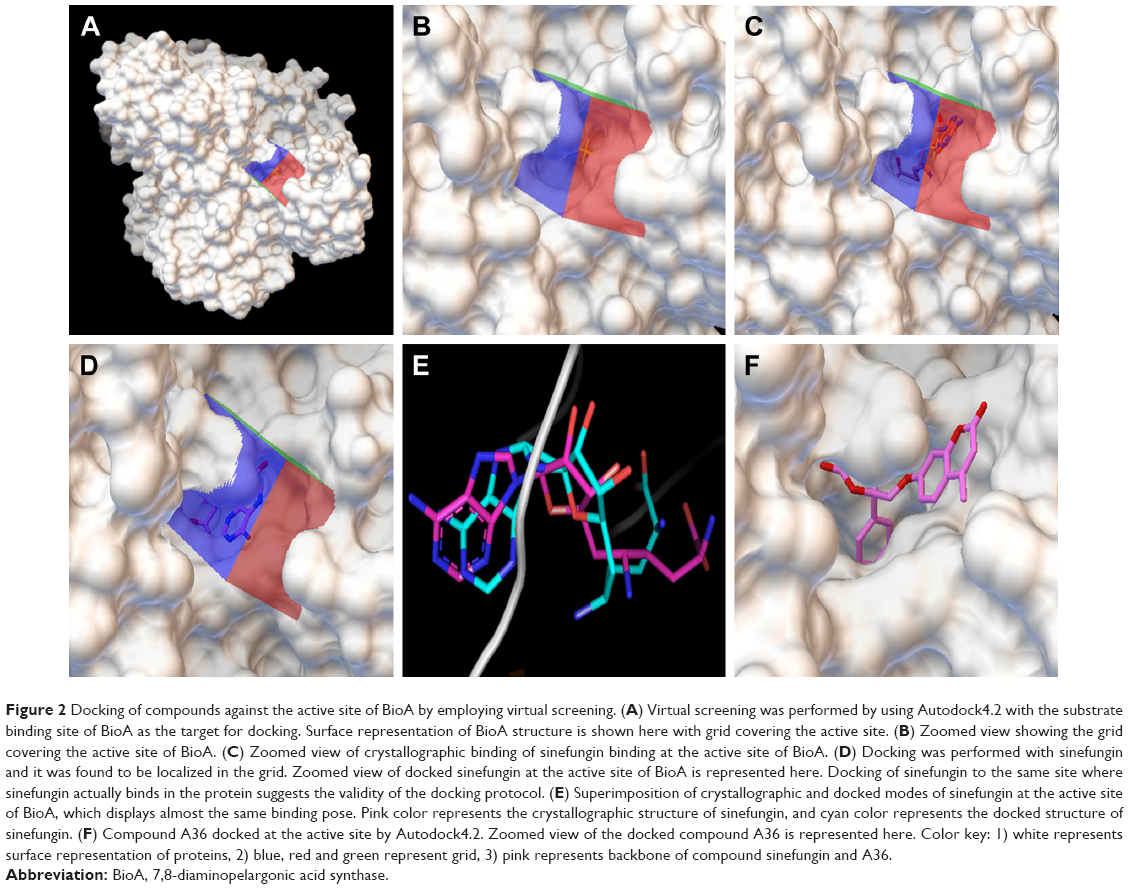 | Figure 2 Docking of compounds against the active site of BioA by employing virtual screening. (A) Virtual screening was performed by using Autodock4.2 with the substrate binding site of BioA as the target for docking. Surface representation of BioA structure is shown here with grid covering the active site. (B) Zoomed view showing the grid covering the active site of BioA. (C) Zoomed view of crystallographic binding of sinefungin binding at the active site of BioA. (D) Docking was performed with sinefungin and it was found to be localized in the grid. Zoomed view of docked sinefungin at the active site of BioA is represented here. Docking of sinefungin to the same site where sinefungin actually binds in the protein suggests the validity of the docking protocol. (E) Superimposition of crystallographic and docked modes of sinefungin at the active site of BioA, which displays almost the same binding pose. Pink color represents the crystallographic structure of sinefungin, and cyan color represents the docked structure of sinefungin. (F) Compound A36 docked at the active site by Autodock4.2. Zoomed view of the docked compound A36 is represented here. Color key: 1) white represents surface representation of proteins, 2) blue, red and green represent grid, 3) pink represents backbone of compound sinefungin and A36. |
Inhibitory potential of the compounds against BioA activity
Compounds were evaluated for their potential to inhibit BioA activity at a concentration of 100 μg/mL. Out of the compounds obtained after screening through Autodock4.2, 39 compounds exhibited greater than 20% inhibition of BioA activity at 100 μg/mL (Figure 3A, Table S1). Compounds procured on the basis of new scores generated by X-Score were also evaluated for their inhibitory potential against BioA at 100 μg/mL. Thirty-seven compounds displayed inhibition of BioA activity with more than 20% inhibition at 100 μg/mL (Figure 3B, Table S2). Moreover, seven compounds (A5, A26, A35, A36, A48, A65 and A70) were active with more than 60% inhibition and three of these compounds, ie, compound A35, compound A36 and compound A65, exhibited greater than 80% inhibition at 100 μg/mL (Figure 3A and Table 1). Compounds A5 and A70 exhibited greater than 70% inhibition of BioA activity (Figure 3A and Table 1). CHM-1 (an inhibitor of BioA that was identified in biochemical screening of LOPAC library16) was utilized in the assay as a positive control and displayed 78.73% inhibition of BioA activity at 100 μg/mL and exhibited an IC50 value of 0.68 μg/mL or 2.42 μM.
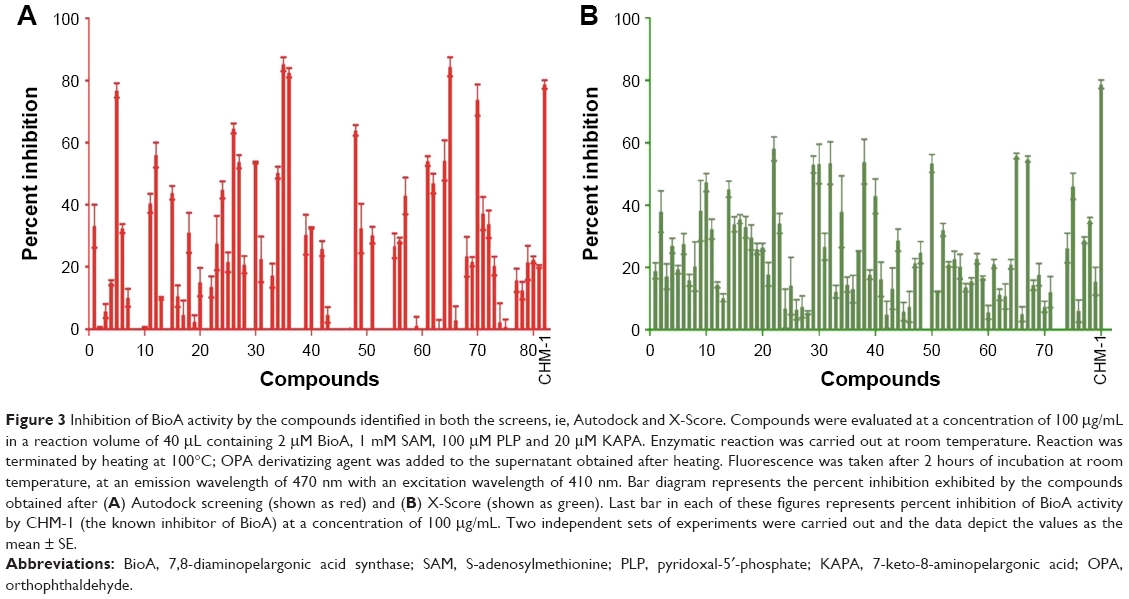 | Figure 3 Inhibition of BioA activity by the compounds identified in both the screens, ie, Autodock and X-Score. Compounds were evaluated at a concentration of 100 μg/mL in a reaction volume of 40 μL containing 2 μM BioA, 1 mM SAM, 100 μM PLP and 20 μM KAPA. Enzymatic reaction was carried out at room temperature. Reaction was terminated by heating at 100°C; OPA derivatizing agent was added to the supernatant obtained after heating. Fluorescence was taken after 2 hours of incubation at room temperature, at an emission wavelength of 470 nm with an excitation wavelength of 410 nm. Bar diagram represents the percent inhibition exhibited by the compounds obtained after (A) Autodock screening (shown as red) and (B) X-Score (shown as green). Last bar in each of these figures represents percent inhibition of BioA activity by CHM-1 (the known inhibitor of BioA) at a concentration of 100 μg/mL. Two independent sets of experiments were carried out and the data depict the values as the mean ± SE. |
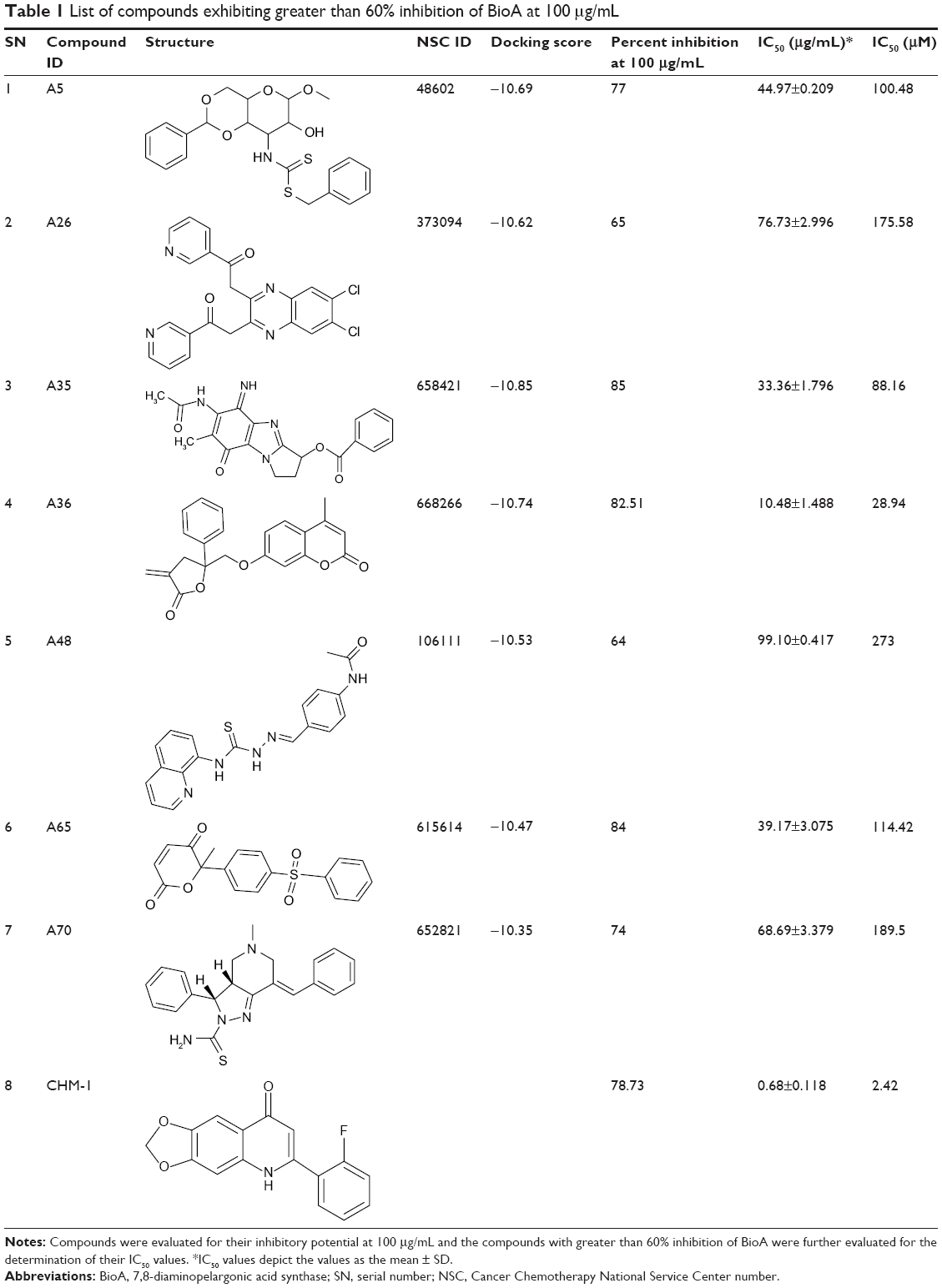 | Table 1 List of compounds exhibiting greater than 60% inhibition of BioA at 100 μg/mL |
All the compounds that exhibited greater than 60% inhibition at a concentration of 100 μg/mL were subjected to dose-response studies for the determination of their IC50 values (Table 1). Compound A36 with an IC50 value of 10.48 μg/mL or 28.94 μM was found to be the most potent one followed by compounds A35 and A65 with IC50 values of 33.36 μg/mL or 88.16 μM and 39.17 μg/mL or 114.42 μM, respectively. Rest of the compounds showed an IC50 value in the range of 45–99 μg/mL (Table 1).
To negate the possibility of the inhibition exerted by the compounds as a result of pre-incubation with the protein, the effect of incubation was evaluated for the compounds with greater than 60% inhibition at a concentration of 100 μg/mL. None of the compounds displayed any significant variation in the percent inhibition exhibited by them at 100 μg/mL with or without pre-incubation conditions (data not shown). These compounds were further evaluated to rule out the possibility of their reactivity with OPA. OPA alone exhibited very little fluorescence, and these compounds in the presence of only OPA showed insignificant variation in the fluorescence intensity when compared to OPA alone. However, very little fluorescence in the absence of enzyme and increased fluorescence intensity of compounds in the presence of enzyme indicated that these compounds did not react with OPA (data not shown). Moreover, to rule out the possibility of compound aggregation in inhibition studies, the enzymatic assay was also performed in the presence of 0.1% Tween 20. All the compounds except A26 displayed no apparent changes in inhibition exhibited by them at 100 μg/mL (data not shown), suggesting that the inhibition displayed by them was because of the enzyme inhibition and not due to any non-specific aggregation of compound itself leading to enzyme inhibition.
AST activity assay
Compounds with greater than 60% inhibition of BioA activity at 100 μg/mL in biochemical assay were further evaluated for their inhibitory potential against another PLP-dependent enzyme AST, an ubiquitous enzyme that catalyzes the conversion of aspartate and α-keto-glutarate to oxaloacetate and glutamate, to rule out their non-specific inhibition of PLP-dependent enzymes. The inhibition assay for AST was carried out by employing the AST activity assay kit by Sigma-Aldrich Co. as described in “Materials and methods” section. None of the compounds showed any inhibition against the enzymatic activity of AST, thereby suggesting their selectivity toward BioA (Figure S3).
Evaluation of the anti-mycobacterial activity of the compounds
In search for the potent inhibitors that target biotin biosynthesis pathway in M. tuberculosis, we combined target-based approach with whole-cell screening approach by employing Resazurin Microtiter Assay (REMA)28–30 to identify the compounds active against M. tuberculosis growth in broth culture. The whole-cell inhibitory potential of the compounds was evaluated against M. tuberculosis. Rifampicin was used as a positive control in the assay, which displayed MIC90 of 0.25 μg/mL or 0.30 μM. Twelve compounds inhibited M. tuberculosis growth in vitro at varying concentration range. The most potent inhibition against M. tuberculosis was exhibited by compound X21 (NSC_115985, 4-{[2-(napthalen-1-ylcarbonyl)hydrazinylidene]methyl}benzoic acid), which displayed complete inhibition of growth at 2.5 μg/mL or 7.86 μM (Table 2 and Figure S4A). Besides, compounds A66 and X58 also exhibited inhibitory potential with MIC90 value of 5 μg/mL (14.2 μM and 14.7 μM, respectively) followed by compound A6 and X50 that showed no growth at 10 μg/mL (30.67 μM and 23.41 μM). Most of the other compounds showed modest activity against M. tuberculosis with MIC90 value of 20 μg/mL (Table 2 and Figure S4A).
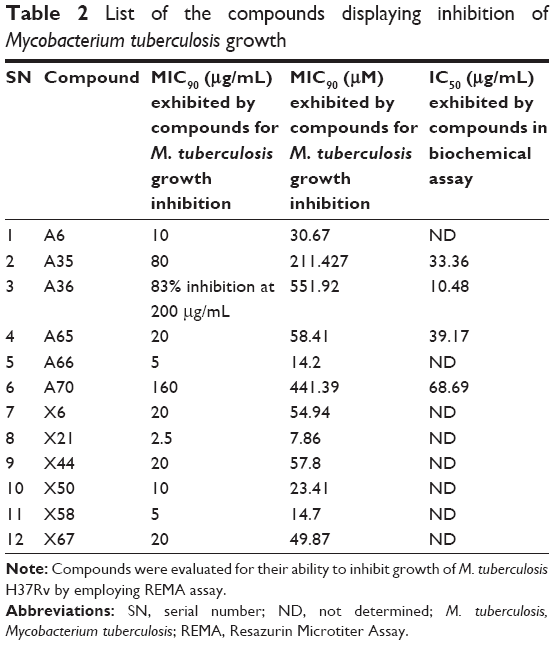 | Table 2 List of the compounds displaying inhibition of Mycobacterium tuberculosis growth |
Compounds A66 and X21 displayed high potency of mycobacterial growth inhibition in spite of showing weak inhibition of BioA in in vitro assays, suggesting their probable off target action. However, compound X50 with MIC90 of 10 μg/mL or 23.41 μM inhibited BioA by 53% at 100 μg/mL, indicating that it could be a BioA-dependent inhibition of mycobacterial growth. Amongst the compounds possessing anti-mycobacterial activity, compound A65 with MIC90 of 20 μg/mL or 58.41 μM also displayed inhibition of BioA activity with IC50 of 39.17 μg/mL, emphasizing its probable BioA-dependent inhibition of mycobacterial broth culture (Table 2).
Compounds that exhibited potent inhibition of BioA activity but failed to inhibit mycobacterial growth till a concentration of 25 μg/mL were evaluated again by using a higher concentration range of 12.5–200 μg/mL. Compound A36 with the most potent activity against BioA with an IC50 of 10.48 μg/mL displayed weak anti-mycobacterial activity with 83% inhibition at 200 μg/mL or 551.92 μM (Table 2 and Figure S4B). Compound A35 with IC50 of 33.36 μg/mL displayed MIC90 of 80 μg/mL or 211.42 μM. Similarly, compound A70 with IC50 of 68.69 μg/mL exhibited MIC90 of 160 μg/mL or 441.39 μM (Table 2 and Figure S4B). Apart from these, the remaining three compounds, ie, A5, A26, A48, which inhibited BioA enzymatic activity by greater than 60% at 100 μg/mL, did not display any anti-mycobacterial activity till 200 μg/mL.
To summarize, we identified four molecules, ie, compounds A65, A35, A70 and A36, that exhibited growth inhibition against M. tuberculosis as well as BioA activity inhibition in biochemical assays.
To evaluate whether compounds showing growth inhibition of M. tuberculosis exert their effect through inhibition of BioA, we determined the inhibitory potential of compounds against growth of M. tuberculosis in Sauton’s medium supplemented with or without biotin (as also reported in earlier studies).18,19 The compounds A35, A65 and A70 that exhibited potent inhibition of BioA and also displayed growth inhibition of M. tuberculosis were evaluated to check biotin-dependent growth inhibition of M. tuberculosis (Figure 4). Compound A36 displayed a poor antibacterial activity and thus it was not determined for its on-target effect inside bacterial cells. Compound A35 displayed MIC90 of 80 μg/mL in Sauton’s medium; however, its MIC90 was shifted to 160 μg/mL on supplementation of media with 1 μM of biotin. Similarly, compound A70 displayed MIC90 of 160 μg/mL, whereas supplementation of biotin showed no growth inhibition of M. tuberculosis till 200 μg/mL. A shift in the MIC90 was also observed in the case of compound A65 that displayed MIC90 of 80 μg/mL, which shifted to 100 μg/mL on supplementation. However, compound A65 displayed MIC90 of 20 μg/mL in 7H9 medium but its MIC90 got shifted to 80 μg/mL in Sauton’s medium; the reason for this change is yet unknown. Thus, these changes in MIC90 of compounds in the presence of biotin indicated that the observed growth inhibition of M. tuberculosis could be due to the inhibition of BioA inside the cells of the pathogen.
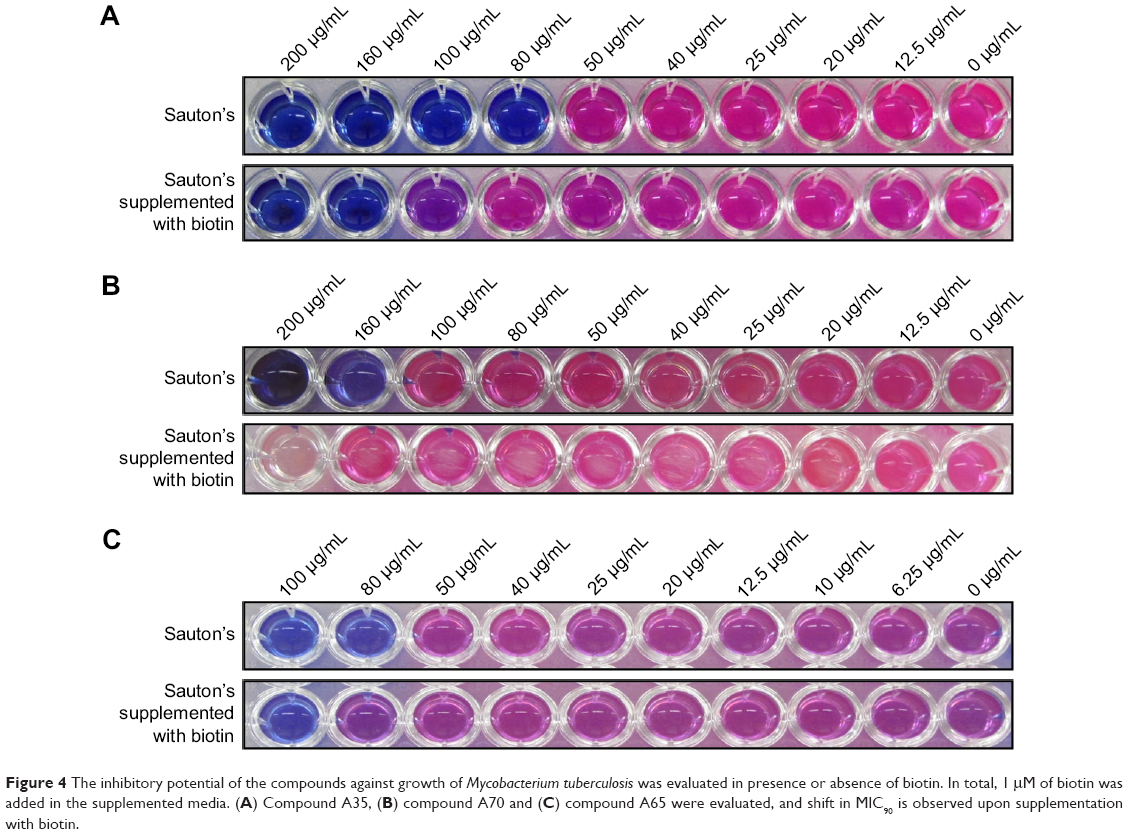 | Figure 4 The inhibitory potential of the compounds against growth of Mycobacterium tuberculosis was evaluated in presence or absence of biotin. In total, 1 μM of biotin was added in the supplemented media. (A) Compound A35, (B) compound A70 and (C) compound A65 were evaluated, and shift in MIC90 is observed upon supplementation with biotin. |
Drug-likeness prediction and evaluation of the cytotoxicity
Seven compounds that exhibited potent inhibition of BioA in in vitro assays were further assessed for their drug-likeness properties. Most of these inhibitors satisfied the rules for drug-likeness. Lipophilicity and aqueous solubility of the compounds were predicted by online software AlogPs.31,32 The molecular weights of all the inhibitors were found to be less than 500 Da, the number of hydrogen bond donors was fewer than 5 and the number of hydrogen bond acceptors was fewer than 10. The predicted logP and logS values were found to be in the acceptable range (Table S3). To support these results, cytotoxicity of these top hits was evaluated in CHO cell line at a concentration range from 1 μg/mL to 200 μg/mL. Compound A5, A26, A48 and A70 were not cytotoxic till 200 μg/mL (Table S4). Compound A35 displayed IC50 of 150 μg/mL, whereas compound A65 displayed moderate toxicity at 50 μg/mL (Table S4). Compound A36 was not evaluated for its cytotoxicity because of the limited quantity provided by NCI. All these results suggested that most of the inhibitors identified in this study possessed valuable potential.
Discussion
Biotin is an essential micronutrient as it serves as an enzyme cofactor in various metabolic processes. In M. tuberculosis, biotin is an essential cofactor for two important enzymes namely pyruvate carboxylase, an enzyme involved in carbon metabolism, and acyl-CoA carboxylase, an enzyme required for carboxylation of various acyl CoA substrates. Hence, the metabolic functions of these essential enzymes are dependent on the presence of biotin, thus making it a crucial cofactor.5,33–35 Several studies in literature reflect the importance of biotin synthesizing genes in M. tuberculosis. bioF and bioB mutants showed an attenuation of M. tuberculosis growth in macrophages.36 In addition, the mutant strains of bioF, bioA and bioB also reduced pathogen’s growth rate in murine model of infection.8 In the case of M. smegmatis also, disruption of bioA resulted in the attenuation of its growth in carbon-depleted media.37 All these studies emphasize the importance of biotin biosynthesis pathway in mycobacteria. BioA or 7,8-diaminopelargonic acid synthase catalyzes the antepenultimate step of biotin biosynthesis pathway in M. tuberculosis and plays an important role by transferring amino group from SAM to KAPA.
In the recent years, biotin biosynthesis pathway has emerged as an attractive target for the discovery of new inhibitors against M. tuberculosis.14–19 Hence, in this study, we have focused our efforts on identifying small molecule inhibitors against BioA, which may serve as lead molecules for the development of novel anti-tubercular drugs. The various approaches for drug discovery majorly include high-throughput phenotypic screening, structure-based virtual screening or target-based screening assays. Although the use of phenotypic screening results in the identification of the molecules directly acting on the target organism, it suffers from the problem of screening of a very large number of molecules and often selection of non-specific cytotoxic compounds. However, in contrast to these conventional high-throughput screening methods, which are expensive and time consuming, virtual screening is inexpensive and provides an advantage of rapid in silico screening of large number of molecules and decreases the number of compounds to be screened in vitro. Hence, this technique has gained importance in early stages of drug discovery. Moreover, virtual screening has not been attempted in the previous studies pertaining to the identification of inhibitors against BioA. Hence, we made efforts to identify the inhibitory molecules against substrate binding site of BioA by employing structure-based virtual screening.
With this aim, the active site of BioA was employed for structure-based virtual screening. The active site of BioA is shaped like a funnel that opens into a deep pocket providing sites for binding of substrates. Figure 5 depicts the strategy employed in this study. Structure-based virtual screening was carried out against the active site of BioA by using NCI library containing diverse compounds by employing software Autodock4.2; subsequently, re-ranking of the docked ligands was performed by using X-Score. Top hits were procured from NCI-DTP and evaluated for their inhibitory potential against BioA enzymatic activity. It was observed that there is a difference in both A-list and X-list and only a few compounds were found to be overlapping between both the series. Although the precise reason for such an observation cannot be assessed, it could be possible due to the use of different algorithms for these software. Autodock is based on force field-based scoring function, whereas X-Score utilizes empirical scoring function. Force field-based scoring function computes binding free energy as the non-covalent ligand–target interactions, which is calculated by addition of strength of van der Waals, electrostatic interaction and hydrogen bonding between ligand and target, whereas empirical scoring function is calculated by counting the number of various types of interaction such as hydrophobic contacts, number of rotatable bonds and number of hydrogen bonds in ligand–protein complex. Therefore, it is indeed possible to have different results by employing these two different scoring functions. Also, empirical scoring functions include common protein–ligand interactions and usually ignore less common interactions such as cation–π interactions. This could be another reason for the variation in the compounds in A-list and X-list. Also, our in vitro data of BioA inhibition assays suggest the accuracy of scoring of Autodock4.2 is higher than the accuracy of scoring of X-Score.
 | Figure 5 Strategy employed for the identification of inhibitors against BioA. |
Three compounds, ie, A35 (syn/anti-6-acetamido-5-imino-7-methyl-2,3-dihydro-1H-pyrrolo[1,2-a]benzamidazole-8-one-3-benzoate), A36 (4-methyl-7-[(4-methylidiene-5-oxo-2-phenyltetrahydrofuran-2-yl)methoxy]-2H-chromen-2-one) and A65 (6-methyl-6-[4-(phenylsulfonyl)phenyl]-2H-pyran-2,5(6H)-dione), were identified as potential inhibitors of M. tuberculosis BioA. On superimposing the structures of these compounds, the most common feature that emerged was that the ring present in each of the compounds was found in the vicinity of the residues Trp64, Trp65 and Phe402 (Figure S5). Although compound A36 was found to be the most potent one with an IC50 value of 10.48 μg/mL or 28.94 μM, it displayed a relatively weak inhibition of M. tuberculosis growth (83% at 200 μg/mL or 551.92 μM) than compound A35, which exhibited an IC50 value of 33.36 μg/mL and MIC90 value of 80 μg/mL or 211.427 μM. Compound A65 was found to be the most promising amongst all the three shortlisted compounds, which displayed an IC50 value of 39.17 μg/mL or 114.42 μM with MIC90 value of 20 μg/mL or 58.41 μM. Similarly, we found another set of compounds, ie, compound A6, A66, X6, X21, X44, X50, X58 and X67, that exhibited inhibition of M. tuberculosis growth in broth culture but showed poor inhibition of BioA activity. These observations suggest that we cannot discern any direct correlation between the IC50 and MIC values, and one of the probable reasons for such an observation could be related to the inability of potent enzyme inhibitors to permeate inside the cells due to their chemical nature and thus displaying poor MIC values. Compound A35, A65 and A70 were found to exert the inhibition of M. tuberculosis growth in a biotin-dependent manner, suggesting BioA to be their target inside the cells. We performed the multiple sequence alignment of M. tuberculosis BioA with its homologs from the other organisms such as Saccharomyces cerevisiae, Haemophilus influenzae, Serratia marcescens, Escherichia coli, Erwinia herbicola, Brevibacterium flavum, Mycobacterium leprae, Bacillus subtilis, Methanococcus jannaschii, Bacillus sphaericus and Helicobacter pylori (Figure S6).38 It was found that residues Trp65, Tyr157, Lys283 and Arg400 were conserved in all species and the residues Tyr25, Trp64 and Phe402 were found to be present in most of the species. These residues were found to be in accordance with the previously published results.38 To establish the significance of the interactions of these compounds with the active site residues, we compared the interactions of the substrate with the active site residues of M. tuberculosis BioA. In M. tuberculosis BioA structure, the aliphatic carbon atoms of the KAPA molecule form van der Waals interactions with Tyr25, Trp64, Tyr157 and Phe402. The aliphatic chain of sinefungin also forms van der Waals contacts with the side chains of Trp64, Trp65 and Phe402 in M. tuberculosis BioA.23 The carboxy oxygen atom of sinefungin also forms hydrogen bonding with Tyr157 via a water molecule in the active site. Residue Arg400 in M. tuberculosis is involved in the recognition of KAPA by formation of salt bridges. The residue Lys283 forms covalent bond with the cofactor PLP.23
We carried out in silico prediction of the mode of binding of the docked compounds A35, A36 and A65 by employing the BioA structure complexed with sinefungin (BioA-SFG). We initiated this work much before the crystal structure of BioA in complex with KAPA (PDB id 4CXQ, BioA-KAPA) was available. Moreover, the BioA structure employed in our study (PDB id 3LV2, BioA-SFG) exhibited a different conformation of Tyr25 in comparison to other reported structures of BioA and also has H315R mutation. Thus, we aligned the crystal structures of BioA in complex with sinefungin (PDB ID-3LV2) and BioA in complex with KAPA (PDB ID-4CXQ), which displayed the RMSD value of 0.205 Å (Figure S7A). We also aligned the active site residues of BioA complexed with sinefungin (PDB ID-3LV2) with the structure of BioA with KAPA (PDB ID-4CXQ), which resulted in an RMSD value of 0.191 Å. It is evident from Figure S7B that most of the residues are in similar conformation except Tyr25 (as also reported in earlier studies).17 To verify the binding mode of compounds, we re-docked these compounds to the crystal structure of BioA-KAPA to predict their binding mode to the wild-type protein. Compound A35 was found to be docked into the deep pocket of BioA-SFG and was surrounded by residues Trp65, Phe402, Tyr25, Trp64, Arg400, Arg403 and Leu385 near to the active site (Figure 6A). Compound A35 was then docked at the active site of BioA-KAPA and the superimposition of the docked conformations of compound A35 docked at the active site of BioA-SFG and BioA-KAPA gave an RMSD value of 0.083, suggesting that the compound docked in a similar manner to both the structures (Figure 6B). Ligand interaction diagram of the compound A35 with BioA-KAPA shows the involvement of many active site residues. Compound A35 exhibited hydrophobic interactions with Ala226, Arg400, Phe402 at a distance of 4.49 Å and with Trp65 (Figure 6C). Compound A35 also showed hydrogen bond formation with the residues Tyr407, Pro401, Tyr157 and Tyr25. In addition, compound A35 displayed pi-donor hydrogen bond formation with Arg400 and Trp64 and pi-sulfur bond formation with Met174 (Figure 6C).
 | Figure 6 Binding mode of compounds. (A) Zoomed view of compound A35 at the active site of BioA-SFG showing its interaction with the neighboring active site residues. (B) Superimposition of compound A35 docked at the active sites of BioA-SFG and BioA-KAPA. (C) Ligand interaction diagram showing the probable interactions of compound A35 with residues of BioA-KAPA. (D) Zoomed view of compound A36 at the active site of BioA-SFG showing its interaction with the neighboring active site residues of BioA. (E) Superimposition of compound A36 docked at the active sites of BioA-SFG and BioA-KAPA. (F) Ligand interaction diagram showing the probable interactions of compound A36 with residues of BioA. (G) Zoomed view of compound A65 at the active site showing its interaction with the neighboring active site residues of BioA. (H) Superimposition of compound A65 docked at the active sites of BioA-SFG and BioA-KAPA. (I) Ligand interaction diagram showing the probable interactions of compound A65 with residues of BioA. Figure 6A, D and G were generated by software Pymol.39 Ligand interaction diagrams were generated by using software Accelrys Discovery Studio.40 |
Compound A36 was also found to be docked at the active site of BioA-SFG (Figure 6D) and was observed to be surrounded with residues Tyr25, Leu385, Arg400, Asp384, Phe402, Trp64 and Trp65. Coumarin ring of the compound A36 was present in close proximity to Tyr25, Leu385 and Arg400. Benzene ring was found close to Phe402, whereas oxolan-2-one was found close to residues Trp64 and Tyr25 (Figure 6D). As observed in the case of compound A35, the docked conformations of compound A36 at BioA-SFG and BioA-KAPA also superimposed well with an RMSD value of 0.713 (Figure 6E). Ligand interaction diagram of the compound A36 showed the involvement of hydrophobic interactions with Arg400 and Phe402 (Figure 6F). Compound A36 also displayed pi-donor hydrogen bond formation with Trp64, Tyr25 and Tyr157 and pi-cation bond formation with Lys283.
Compound A65 was also found to be docked in the binding groove of BioA-SFG and was surrounded by the residues Phe402, Arg400, Arg403, Tyr25, Trp65, Trp64, Val383 and Leu385. 6H-pyran-2,5-dione moiety was found to be in close proximity to Val383 and Leu385 (Figure 6G). The superimposition of the compound A65 docked at the active sites of BioA-SFG and BioA-KAPA gave an RMSD value of 0.701, again suggesting that the compounds docked in a similar manner to both the structures (Figure 6H). Ligand interaction diagram of compound A65 displayed hydrogen bonding with Lys283 (Figure 6I). Compound A65 also displayed pi-donor hydrogen bond formation with Trp64, pi-sulfur bond formation with Phe402 and hydrophobic interactions with Arg400, Trp64, Trp65 and Ala226 (Figure 6I).
The compounds were also evaluated for their drug-like properties. All the compounds that displayed greater than 60% inhibition at 100 μg/mL were evaluated and were found to adhere to the Lipinski guidelines for drug-likeness. Solubility of compounds presents another challenge in drug discovery; hence, determination of aqueous solubility of a candidate molecule is important as it reflects on the bioavailability of the compound. Similarly, lipophilicity of compounds is another important analysis as lipophilicity is required for the permeation of compounds through various biological membranes. All the compounds exhibited logS and logP in the acceptable range, indicating that these compounds fulfilled these important criteria in addition to Lipinski guidelines, and this was further confirmed by evaluating the cytotoxicity of the molecules in CHO cell line.
Thus, we have been able to identify a few new moieties that inhibit BioA enzyme activity as well as M. tuberculosis growth, and the most potent compound identified in the study is compound A65.
Conclusion
Structure-based virtual screening was carried out to identify new inhibitors against BioA of M. tuberculosis. We have identified a promising lead molecule (compound A65) that is a potent inhibitor of BioA activity in vitro, exhibits inhibition against M. tuberculosis growth in broth culture and possesses drug-like properties. Improving this compound without disturbing its core scaffold could lead to a molecule with superior potential for inhibition of M. tuberculosis. The other two compounds, ie, A35 and A36, were also identified in the screening that showed both inhibition of enzymatic activity and growth of the pathogen. Docking and structural information of all these compounds could also be useful for the medicinal chemists in designing new moieties for inhibition of BioA.
Acknowledgments
The Drug Synthesis and Chemistry Branch, DTP, Division of Cancer Treatment and Diagnosis, NCI, National Institutes of Health, Bethesda, MD, USA, is highly acknowledged for providing us the compounds. The authors thank the Computer Centre, Central Instrumentation Facility, University of Delhi South Campus (UDSC), for providing the computational help and facilities. Akshay Rohilla (UDSC) is acknowledged for generating filtered NCI library and his help in carrying out virtual screening. Priti Singh (UDSC) and Tanu Priya Gosain are acknowledged for their excellent technical help. The DNA Sequencing Facility, UDSC, New Delhi, is acknowledged for sequencing service. The Distributed Information Subcenter, DBT, is acknowledged for providing computer and Internet services. Swati Singh is thankful to the Indian Council of Medical Research, India, for providing fellowship. Ritika Kar Bahal is grateful to the Council of Scientific & Industrial Research, India, for providing fellowship. This work is supported by a financial grant from the Department of Biotechnology, Government of India (grant number – BT/01/COE/05/06-II).
Disclosure
The authors report no conflicts of interest in this work.
References
WHO [webpage on the Internet]. Global tuberculosis report 2017. Geneva: WHO; 2017. Available from: http://www.who.int/tb/publications/global_report/en/. Accessed March 24, 2018. | ||
Nachega JB, Chaisson RE. Tuberculosis drug resistance: a global threat. Clin Infect Dis. 2003;36:24–30. | ||
Raviglione MC, Smith IM. XDR tuberculosis–implications for global public health. N Engl J Med. 2007;356(7):656–659. | ||
Knowles JR. The mechanism of biotin-dependent enzymes. Annu Rev Biochem. 1989;58:195–221. | ||
Salaemae W, Azhar A, Booker GW, Polyak SW. Biotin biosynthesis in Mycobacterium tuberculosis: physiology, biochemistry and molecular intervention. Protein Cell. 2011;2(9):691–695. | ||
Salaemae W, Booker GW, Polyak SW. The role of biotin in bacterial physiology and virulence: a novel antibiotic target for Mycobacterium tuberculosis. Microbiol Spectr. 2016;4(2). | ||
Mann S, Ploux O. 7,8-Diaminoperlargonic acid aminotransferase from Mycobacterium tuberculosis, a potential therapeutic target. Characterization and inhibition studies. FEBS J. 2006;273(20):4778–4789. | ||
Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100(22):12989–12994. | ||
Park SW, Klotzsche M, Wilson DJ, et al. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 2011;7(9):e1002264. | ||
Kitahara T, Hotta K, Yoshida M, Okami Y. Biological studies of amiclenomycin. J Antibiot (Tokyo). 1975;28(3):215–221. | ||
Sandmark J, Mann S, Marquet A, Schneider G. Structural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycin. J Biol Chem. 2002;277(45):43352–43358. | ||
Okami Y, Kitahara T, Hamada M, Naganawa H, Kondo S. Studies on a new amino acid antibiotic, amiclenomycin. J Antibiot (Tokyo). 1974;27(9):656–664. | ||
Hotta K, Kitahara T, Okami Y. Studies of the mode of action of amiclenomycin. J Antibiot (Tokyo). 1975;28(3):222–228. | ||
Shi C, Geders TW, Park SW, et al. Mechanism-based inactivation by aromatization of the transaminase BioA involved in biotin biosynthesis in Mycobaterium tuberculosis. J Am Chem Soc. 2011;133(45):18194–18201. | ||
Shi C, Aldrich CC. Design and synthesis of potential mechanism-based inhibitors of the aminotransferase BioA involved in biotin biosynthesis. J Org Chem. 2012;77(14):6051–6058. | ||
Wilson DJ, Shi C, Duckworth BP, et al. A continuous fluorescence displacement assay for BioA: an enzyme involved in biotin biosynthesis. Anal Biochem. 2011;416(1):27–38. | ||
Dai R, Wilson DJ, Geders TW, Aldrich CC, Finzel BC. Inhibition of Mycobacterium tuberculosis transaminase BioA by aryl hydrazines and hydrazides. Chembiochem. 2014;15(4):575–586. | ||
Park SW, Casalena DE, Wilson DJ, et al. Target-based identification of whole-cell active inhibitors of biotin biosynthesis in Mycobacterium tuberculosis. Chem Biol. 2015;22(1):76–86. | ||
Dai R, Geders TW, Liu F, et al. Fragment-based exploration of binding site flexibility in Mycobacterium tuberculosis BioA. J Med Chem. 2015;58(13):5208–5217. | ||
Billones JB, Carrillo MC, Organo VG, Macalino SJ, Emnacen IA, Sy JB. Virtual screening against M. tuberculosis 7,8-diaminopelargonic acid synthase (MtbBioA) and in silico toxicity evaluation of top hits. Curr Enzym Inhib. 2014;10(2):105–112. | ||
Billones JB, Carrillo MC, Organo VG, et al. In silico discovery and in vitro activity of inhibitors against Mycobacterium tuberculosis 7,8-diaminopelargonic acid synthase (Mtb BioA). Drug Des Devel Ther. 2017;11:563–574. | ||
Mann S, Eveleigh L, Lequin O, Ploux O. A microplate fluorescence assay for DAPA aminotransferase by detection of the vicinal diamine 7,8-diaminopelargonic acid. Anal Biochem. 2013;432(2):90–96. | ||
Dey S, Lane JM, Lee RE, Rubin EJ, Sacchettini JC. Structural characterization of the Mycobacterium tuberculosis biotin biosynthesis enzymes 7,8-diaminopelargonic acid synthase and dethiobiotin synthetase. Biochemistry. 2010;49(31):6746–6760. | ||
Rohilla A, Khare G, Tyagi AK. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci Rep. 2017;7(1):4653. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem. 2009;30(16):2785–2791. | ||
AutoDock website. http://autodock.scripps.edu/downloads/autodock-4.0-registration/autodock-4-2-download-page. Accessed March 24, 2018. | ||
Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des. 2002;16(1):11–26. | ||
Palomino JC, Martin A, Camacho M, Guerra H, Swings J, Portaels F. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46(8):2720–2722. | ||
Martin A, Camacho M, Portaels F, Palomino JC. Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob Agents Chemother. 2003;47(11):3616–3619. | ||
Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41(5):1004–1009. | ||
Tetko IV, Gasteiger J, Todeschini R, et al. Virtual computational chemistry laboratory–design and description. J Comput Aided Mol Des. 2005;19(6):453–463. | ||
VCCLAB. Virtual Computational Chemistry Laboratory. Available from: http://www.vcclab.org, 2005. Accessed March 24, 2018. | ||
Eisenreich W, Dandekar T, Heesemann J, Goebel W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol. 2010;8(6):401–412. | ||
Gago G, Diacovich L, Arabolaza A, Tsai SC, Gramajo H. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol Rev. 2011;35(3):475–497. | ||
Arabolaza A, Shillito ME, Lin TW, et al. Crystal structures and mutational analyses of acyl-CoA carboxylase beta subunit of Streptomyces coelicolor. Biochemistry. 2010;49(34):7367–7376. | ||
Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A. 2005;102(23):8327–8332. | ||
Keer J, Smeulders MJ, Gray KM, Williams HD. Mutants of Mycobacterium smegmatis impaired in stationary-phase survival. Microbiology. 2000;146(Pt 9):2209–2217. | ||
Käck H, Sandmark J, Gibson K, Schneider G, Lindqvist Y. Crystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymes. J Mol Biol. 1999;291(4):857–876. | ||
The PyMol Molecular Graphics System, Version 1.1. eval. Schrodinger, LLC. | ||
Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment. Release 2017. San Diego, CA: Dassault Systèmes; 2016. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.