Back to Journals » OncoTargets and Therapy » Volume 14
Identification of Metabolic-Associated Genes for the Prediction of Colon and Rectal Adenocarcinoma
Authors Cui Y ![]() , Han B, Zhang H, Liu H, Zhang F, Niu R
, Han B, Zhang H, Liu H, Zhang F, Niu R
Received 16 December 2020
Accepted for publication 5 March 2021
Published 31 March 2021 Volume 2021:14 Pages 2259—2277
DOI https://doi.org/10.2147/OTT.S297134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Yanfen Cui, Baoai Han, He Zhang, Hui Liu, Fei Zhang, Ruifang Niu
Public Laboratory, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin’s Clinical Research Center for Cancer, Key Laboratory of Breast Cancer Prevention and Therapy, Ministry of Education, Tianjin, 300060, People’s Republic of China
Correspondence: Ruifang Niu; Fei Zhang Tel +86 13752668992
; +86 18622221365
Email [email protected]; [email protected]
Background and Aim: Uncontrolled proliferation is the most prominent biological feature of tumors. In order to rapidly proliferate, tumor cells regulate their metabolic behavior by controlling the expression of metabolism-related genes (MRGs) to maximize the utilization of available nutrients. In this study, we aimed to construct prognosis models for colorectal adenocarcinoma (COAD) and rectum adenocarcinoma (READ) using MRGs to predict the prognoses of patients.
Methods: We first acquired the gene expression profiles of COAD and READ from the TCGA database, and then utilized univariate Cox analysis, Lasso regression, and multivariable Cox analysis to identify the MRGs for risk models.
Results: Eight genes (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR) in the colon cancer risk model and six genes (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) in the rectal cancer risk model were identified successfully. Multivariate Cox analysis indicated that these two models could accurately and independently predict overall survival (OS) for patients with COAD or READ. Furthermore, functional enrichment analysis was used to identify the metabolism pathway of MRGs in the risk models and analyzed these genes comprehensively. Then, we verified the prognosis model in independent COAD cohorts (GSE17538) and detected the correlations of the protein expression levels of GSR and ENPP2 with prognosis for COAD or READ.
Conclusion: In this study, 14 MRGs were identified as potential prognostic biomarkers and therapeutic targets for colorectal cancer.
Keywords: metabolism-related gene, colon adenocarcinoma, rectum adenocarcinoma, prognosis, ENPP2, GSR
Introduction
Colorectal cancer is a common malignant tumor of the digestive tract, which is a serious threat to human health.1 Given that the early symptoms of colorectal cancer are atypical, diagnosed cases are often in the middle and late stages of the disease. The development of surgical techniques and adjuvant chemotherapy and radiotherapy chemoradiotherapy has improved the survival time of colorectal cancer patients to a certain extent, but the 5-year survival rate of patients with colorectal cancer remains extremely low because of chemical drug resistance and other reasons.2 Therefore, biomarkers that can provide accurate prognosis and predict therapeutic effects are beneficial for patients with colorectal cancer. The prognoses of patients with colorectal cancer considerably vary, and thus establishing an accurate and effective prognosis model is necessary. Such model can be useful in assessing patients’ risk and in treatment selection.3
Current studies have shown that the blood supply, function, and environmental carcinogens of colon and rectum are different. Therefore, clinical characteristics, pathological stage, and the prognosis of colorectal cancer are always different among primary sites.4–6 The analysis of the differences in the molecular biology of colorectal cancer among different primary sites is necessary in the selection of individualized treatment for patients with colorectal cancer. At present, with the deepening understanding of tumor biology and the complexity of tumor metabolism, metabolic reprogramming has been found to be a marker of malignancy.7 Compared with normal tissues, cancer cells have a high degree of metabolic heterogeneity.8 Metabolic phenotypes evolve at the different stages of tumorigenesis, early tumor growth requires nutrient uptake and biosynthesis, and other subtypes of selective metabolic requirements occur during local invasion and distant metastasis.9 Two studies proposed prognostic models of prognosis-related metabolic genes for colorectal cancer prognosis.10,11 In the present study, we modeled the prognosis genes of colorectal adenocarcinoma (COAD) and rectum adenocarcinoma (READ) separately for the individualized detection of patients with colorectal cancer.
We constructed the prognostic models of prognosis-related metabolic genes for COAD and READ, utilizing the expression profiles of TCGA databases. The prognostic models were further optimized through Lasso regression and Cox regression analyses, and the specificity and sensitivity of the models were evaluated through ROC curve analysis. Meanwhile, metabolism-related genes (MRGs) related to OS were screened out. Our data established two models specific to COAD and READ, which are useful in guiding individualized prognosis and treatment.
Materials and Methods
Data Collection
The transcriptome data and the relevant clinical data of COAD (including 398 cancer and 39 normal tissues) and READ (including 84 cancer and 2 normal tissues) were downloaded from TCGA database (https://portal.gdc.cancer.gov/) to construct risk prediction models, and GSE17538 of GEO database (https://www.ncbi.nlm.nih.gov/geo/) including 238 COAD patients was used as the validation dataset. Meanwhile, the gene mutation data were obtained from GSCALite (http://bioinfo.life.hust.edu.cn/web/GSCALite/). The clinical and pathological characteristics of patients in TCGA are summarized in Table 1. We obtained MRGs by searching “metabolism” from the Molecular Signatures Database v7.0 (http://software.broadinstitute.org/gsea/msigdb) and selected the C2 sub-collection (c2.cp.kegg.v7.0.symbols.gmt) as the needed gene set, then 944 MRGs were obtained. Then, MRGs expression was extracted from GSE17538 dataset through R language merge package. Finally, a gene expression matrix consisting of 772 gene expression values was obtained.
 |
Table 1 Clinicopathological Parameters of COAD Patients in the TCGA Database |
Differential Expression Analysis of MRGs and Functional Enrichment Analyses
For COAD, the differences of MRGs between cancer tissues and non-tumor samples were analyzed by Wilcoxon signed-rank test. An adjusted P < 0.01 and a |log2 (FC) |> 2 were considered the cutoffs for identifying differential expression MRGs (DEMRGs). KEGG analysis was then performed to discover the primary biological characteristics of these genes.
Construction and Validation of Risk Prediction Models Using MRGs
We first used univariate Cox analysis to initially identify potential MRGs, and least absolute shrinkage and selection operator (Lasso) regression analysis was subsequently applied by using “glmnet” and “survival” packages to eliminate false parameters caused by overfitting. Multivariable regression analysis was used to screen to MRGs with independent prognostic significance, and these MRGs were used to construct prognostic models with the following formula: Risk score = (CoefficientmRNA1×mRNA1 expression) + (CoefficientmRNA2×mRNA2 expression) + ⋯ + (CoefficientmRNAn× mRNAn expression). Finally, Cox regression was used to establish OS prognostic risk model. In addition, univariate and multivariate regressions were performed to determine whether the model was an independent prognostic factor.
Comprehensively Analyze MRGs of the Risk-Specific Model
The independent prognostic value of MRGs was verified by Kaplan–Meier analysis using GEPIA (http://gepia.cancer-pku.cn/). Meanwhile, the gene mutation data were obtained from GSCALite (http://bioinfo.life.hust.edu.cn/web/GSCALite/).
Gene Set Enrichment Analysis (GSEA)
GSEA was performed to explore the characteristics and pathways that were enriched in the high-risk group and low-risk group. Using normalized enrichment score (NES) and normalized P-value calculation predefined tags and KEGG pathway enrichment. Terms with |NES|>1 and P<0.05 were considered significantly enriched.
Patient Samples
A total of 117 COAD and 114 READ were obtained from Tianjin cancer hospital. All tissue sections were confirmed by specialists to make a final diagnosis. Histopathological diagnoses were made using the World Health Organization criteria. Table 2 summarizes all patients’ characteristics. This study has been approved by the Tianjin cancer hospital’s Human Ethics and Research Ethics Committee and obtained written informed consent from all patients.
 |
Table 2 Clinicopathological Parameters of COAD and READ Patients |
Immunohistochemistry Staining and Evaluation
Paraffin-embedded tissue sections (4µm) were dewaxed and rehydrated with xylene and fractional alcohol solution, the endogenous peroxidase activity was hardened with 3% hydrogen peroxide, and the sections were exposed to antigen by boiling in 10 mM citric acid buffer (pH 6.0). The sections were incubated with primary antibody at 4°C for 18 h, incubated with PV6001at 37°C for 30 min, and stained with DAB (Zhongshan Goldbridge Biotechnology Company) for 1 ~ 2 min. The control group was incubated with PBS instead of primary antibody.
To score staining of tissue sections, we selected 5 consecutive high-power fields on each section to estimate the average percentage of stained cells. For the expression of GSR in the nucleus, the mean value of expression in all cancer tissues was truncated, expression level ≤50% in tissues was classified as low expression group, and 50% of > was classified as high expression group (Figure 1A). For the expression of GSR in cytoplasm, ENPP2 and GAMT, both percentage and intensity were considered in the semi-quantitative evaluation. The percentage of positive cells was divided into 0(0% positive cells), 1(1–25% positive cells), 2(26–50% positive cells), 3(50–75% positive cells), 4(>75% positive cells), and the strength was divided into 0(negative), 1(weak), 2(medium), and 3(strong). The intensity score (0–3) is multiplied by the percentage score (0–4), 0–4 being low expression and 5–12 being high expression.
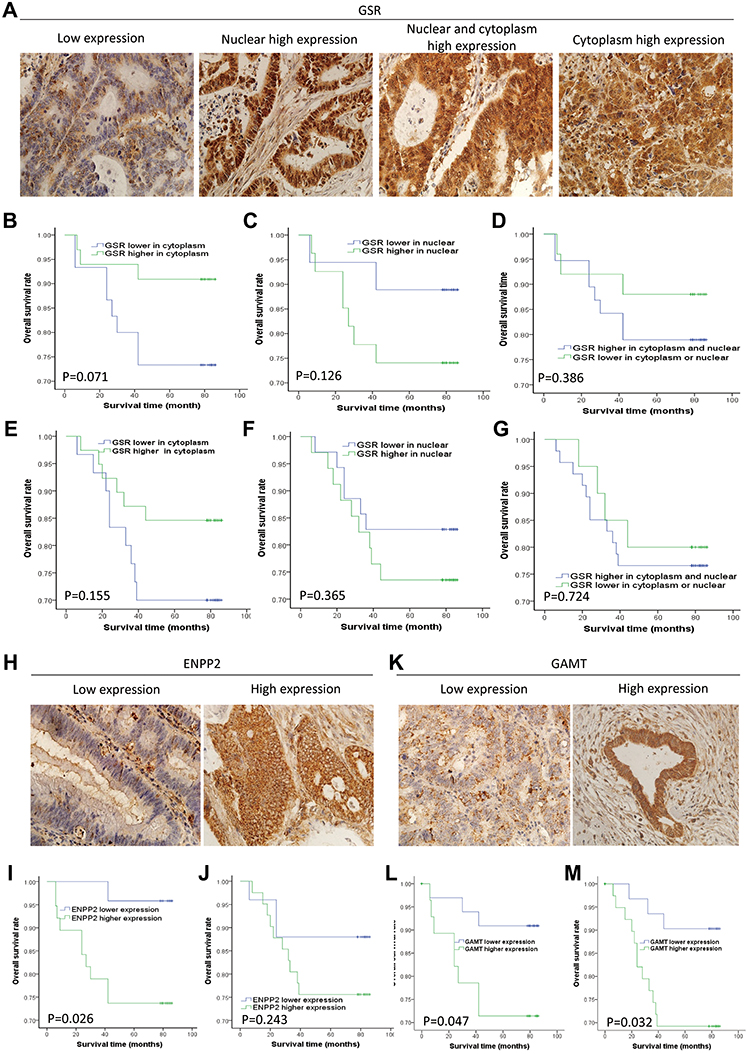 |
Figure 1 The expression of GSR, ENPP2 and GAMT in colon cancer and rectal cancer detected by immunohistochemistry stain. (A) the present pictures of GSR detected by immunohistochemistry stain (low expression, nuclear high expression, nuclear and cytoplasm high expression, cytoplasm high expression, respectively). (B) the relationship between GSR expression level in cytoplasm and survival of patients with colon cancer. (C) the relationship between GSR expression level in nuclear and survival of patients with colon cancer. (D) the relationship between GSR expression level in both nuclear and cytoplasm and survival of patients with colon cancer. (E–G) the relationship between GSR expression level and survival of patients with rectal cancer. (H) the present pictures of ENPP2 detected by immunohistochemistry stain. (I) the relationship between ENPP2 expression level and survival of patients with colon cancer. (J) the relationship between ENPP2 expression level and survival of patients with rectal cancer. (K) the present pictures of GAMT detected by immunohistochemistry stain. (L) the relationship between GAMT expression level and survival of patients with colon cancer. (M) the relationship between GAMT expression level and survival of patients with rectal cancer. |
Statistical Analysis
All statistical analyses were performed using R software (version 3.6.0), and the related source codes were provided in Supplementary File 3. Wilcox test or Kruskal–Wallis test was used to evaluate the distribution differences among variables. Kaplan–Meier survival curve analysis and Log Rank test were used to analyze OS. Cox regression model was used to analyze the factors affecting patient survival. Cox proportional hazard regression model was used for univariate and multivariate analysis. Prognostic accuracy of the model was assessed by time-dependent ROC analysis. P<0.05 was considered statistically significant.
Results
Flow Chart of This Study
The detailed workflow for constructing the risk models and downstream analysis was shown in Figure 2. We first used the TCGA database to extract MRGs from the COAD and READ data sets and then observed the differential expression of MRGs at the gene expression level. The specific prognostic risk models of COAD and READ were constructed using the MRG set data, and the predictive ability of each risk model was tested through ROC analysis. The risk model of COAD was validated in the GEO database. Finally, Kaplan–Meier analysis and expression level detection of the genes in the risk models were performed, and the mutation information of each gene in COAD and READ in the models was examined.
 |
Figure 2 Flow chart of this study. |
Differential Expression and Functional Annotation of MRGs in COAD
From the TCGA database, we first downloaded mRNA expression data and the clinical information of 398 COAD tissue samples and 39 non-tumor tissues (Table 1). A total of 772 MRGs were detected in this database, and 196 differentially expressed MRGs were obtained. The expression pattern of the differentially expressed MRGs in COAD and non-tumor tissues was shown in the volcanic and heat maps (Figure 3A and B). Among the 196 differentially expressed genes, 98 genes were down-regulated, and 98 genes were up-regulated in the tumor tissues. In order to observe metabolic differences between colon cancer and normal tissues, we used the KEGG pathway to conduct a functional enrichment analysis of the differentially expressed MRGs. We found that the up-regulated genes are mainly involved in the following metabolic pathways: purine metabolism, amino acid biosynthesis, carbon metabolism, and cysteine and methionine metabolism (Figure 3C). Meanwhile, the down-regulated genes are mainly involved in fatty acid degradation, starch and sucrose metabolism, retinol metabolism, and galactose metabolism (Figure 3D). We also downloaded the mRNA expression data and corresponding clinical information of 84 READ tissue samples and 2 non-tumor tissues (Table 1). Owing to the small number of normal rectal tissue samples, we cannot obtain valid differential gene data.
 |
Figure 3 Expression and functional annotation of MRGs in COAD. (A) Clustered heatmap of differentially expressed MRGs expression level between colon cancer and normal colon tissues of TCGA database. (B) Volcano plot for the MRGs. (C) KEGG analysis of upregulated MRGs. (D) KEGG analysis of downregulated MRGs. |
Construction and Verification of the COAD Risk Model
To investigate the relationship between MRGs and COAD prognosis, we constructed a prognostic risk model. Initially, univariate regression analysis was performed on 13 genes that were significantly associated with prognosis, including 2 low-risk MRGs (GSR and SUCLG2P2) and 11 high-risk MRGs (CPT1C, PLCB2, PLCG2, PLA2G2D, PTGDS, GAMT, ENPP2, SU LG2P2, PIK3CD, PIP4K2B, GPX3, and MAT1A; Figure 4A). Then, Lasso regression and multivariate regression were used to generate a final prognosis model with eight MRGs, including CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR (Figure 4B and C). The specific information of these genes, including full name, coefficient, and relevant pathway, was shown in Table 3. Then, the patients with COAD in the TCGA database were divided into low- and high-risk groups, and Kaplan–Meier survival analysis was performed. The results showed that patients with high-risk scores had significantly worse OS than patients with low-risk scores (Figure 4D). Figure 4E shows the association between survival time and risk score, and survival time decreased significantly with increasing risk score. Time-dependent ROC analysis indicated that this prognosis model can accurately predict the OS for patients with COAD (Figure 4F). The heatmap developed to show the gene expression profiles indicated that CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, and GPX3 were identified as positive risk-correlated genes, whereas GSR was identified as negative risk-correlated gene (Figure 4G).
 |
Figure 4 Construction of COAD risk model using TCGA database. (A) The univariate Cox regression was performed to calculate HR and 95% CI of MRGs. (B and C) The Lasso regression was performed using prognosis-significant MRGs. (D) Kaplan–Meier analysis was used to represent that the patients in the high-risk group had a significantly shorter overall survival time, while the patients in the low-risk group had longer overall survival time. (E) The association between survival time and risk score was detected. (F) 5-year ROC curve analysis of the sensitivity and specificity of the OS for the combination of risk score and clinical characteristics. (G) The heatmap of the key MRGs expression profiles was showed. |
 |
Table 3 Lasso Cox Analysis for colon cancer |
To verify the validity of the prediction model, we used the COAD data of GSE17538 from the Gene Expression Omnibus (GEO) as validation data. Consistent with the results obtained from TCGA database, our finding showed that the prediction model could provide reliable prognoses for the patients in GSE17538, and the patients with high-risk scores had considerably poor OS (Figure 5A). ROC curve analysis showed the sensitivity and specificity of the risk score for the OS when combined with clinical characteristics (Figure 5B). Furthermore, the association between survival time and risk score indicated that the survival time decreased with the significant increase in risk score (Figure 5C). Meanwhile, the results of the heat map were consistent with those in the TCGA database (Figure 5D).
 |
Figure 5 Verification of COAD risk model using GSE17538 data from GEO. (A) Kaplan–Meier analysis was used to certificate the risk model for prognosis. (B) ROC curve analysis of the sensitivity and specificity of the OS for the combination of risk score and clinical characteristics. (C) The association between survival time and risk score was detected. (D) The heatmap of the key MRGs expression profiles was showed. |
Construction of the READ Risk Model
By the approach used in COAD analysis, we performed univariate regression analysis to construct a prognostic model for READ, and seven MRGs were used (Figure 6A). Then, a final prognosis model including six MRGs (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) was constructed using Lasso regression and multivariate Cox regression (Figure 6B and C). Specific information on the genes is shown in Table 4, including full name, coefficient, and relevant pathways. Meanwhile, the results of Kaplan–Meier survival analysis showed that patients with low-risk scores had significantly better OS than those with high-risk scores, suggesting that the established prognostic model was available (Figure 6D). Survival time increased when the risk score decreased significantly (Figure 6E), and ROC curve analysis showed the sensitivity and specificity of the risk score for OS when combined with clinical characteristics (Figure 6F). Furthermore, the heatmap showed that TDO2, PKLR, and GAMT were identified as positive risk-correlated genes, while EARS2, ACO1, and WAS were identified as negative risk-correlated genes (Figure 6G). Given the small number of READ cases in the GEO database, we were unable to use these data to validate the risk model.
 |
Figure 6 Construction of READ risk model using TCGA database. (A) The univariate Cox regression was performed to calculate HR and 95% CI of MRGs. (B and C) The Lasso regression was performed using prognosis-significant MRGs. (D) Kaplan–Meier analysis was used to represent the prognosis of patients. (E) The association between survival time and risk score was detected. (F) 5-year ROC curve analysis of the sensitivity and specificity of the OS for the combination of risk score and clinical characteristics. (G) The heatmap of the key MRGs expression profiles was showed. |
 |
Table 4 Lasso Cox Analysis for rectal cancer |
Prognostic Risk Models of COAD and READ Were Independently Related to OS
To further validate this prediction model, univariate and multivariate regression analyses were performed, and TCGA and GSE17538 were used to investigate the correlations of clinical parameters and risk score with OS in patients with COAD. The TCGA results of univariate Cox regression showed that pathological stage; T, N, and M stages; and risk score were all significantly correlated with OS (all P<0.001; Figure 7A), and multivariate Cox regression showed that T stage and risk score were correlated with OS in patients with COAD (P<0.05; Figure 7B). The GSE17538 results showed that not only univariate Cox regression analysis but also multivariate regression analysis indicated that the prognostic risk model of COAD was independently related to OS (P<0.05; Figure 7C and D). As to READ, the results of univariate regression and multivariate Cox regression analysis indicated that the obtained risk model was independently related to OS (P<0.05; Figure 7E and F).
 |
Figure 7 Univariable and multivariable Cox regression analyses of OS in COAD and READ. (A) univariable cox regression analyses of OS in COAD using TCGA data. (B) multivariable cox regression analyses of OS in COAD using TCGA data. (C) univariable cox regression analyses of OS in COAD using GEO data. (D) multivariable cox regression analyses of OS in COAD using GEO data. (E) univariable cox regression analyses of OS in READ using TCGA data. (F) multivariable cox regression analyses of OS in READ using TCGA data. |
Comprehensive Analysis of All the Genes in the Prognostic Models of COAD and READ
According to the previous results, the COAD risk model has eight genes, and the READ risk model has six genes. To further evaluate the prognostic value of the above genes, we used the GEPIA database in performing Kaplan–Meier analysis. We found that high expression of CPT1C, PLCB2, or GPX3 indicated significantly poor prognosis, whereas the high expression of GSR indicated excellent prognosis in patients with COAD in the GEPIA database (Figure 8A). Although PLA2G2D, GAMT, ENPP2, and PIP4K2B cannot significantly predict prognosis, patients with high expression level of PLA2G2D, GAMT, ENPP2, or PIP4K2B always had poor prognosis (Figure 8A). In READ, high TDO2 and GAMT expression levels were correlated with poor prognosis, whereas high EARS2 and ACO1 expression levels predicted good prognosis (Figure 8B). Overall, the results of Kaplan–Meier analysis were generally consistent with the above univariable Cox analysis results.
 |
Figure 8 Kaplan–Meier analyses of MRGs in prognosis models of COAD and READ. (A) Kaplan–Meier analyses of CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3 and GSR in COAD. (B) Kaplan–Meier analyses of TDO2, PKLR, GAMT, EARS2, ACO1 and WAS in READ. |
Meanwhile, we analyzed the mRNA expression levels of the genes in relative tumor and normal tissues with the GEPIA database. As shown in Figure 9A, in the COAD risk model, the gene expression levels of PLA2G2D, ENPP2, and GPX3 in COAD tissues were significantly lower than corresponding normal tissues, but changes in the other genes were not obvious. As to the READ risk model, only GAMT had significantly lower expression in READ tissues than in corresponding normal tissues (Figure 9B).
 |
Figure 9 Expression levels of MRGs mRNA in prognosis models of COAD and READ. (A) the expression level of genes in COAD risk model in colon cancer tissues and corresponding normal tissues. (B) the expression level of genes in READ risk model in rectal cancer tissues and corresponding normal tissues. (*P<0.05). |
In addition, we investigated genetic alterations of genes in the prognosis models with the data of Gene Set Cancer Analysis Lite (http://bioinfo.life.hust.edu.cn/web/GSCALite/). The results showed that the genes in the COAD model were changed in all the 38 queried samples (100%; Figure 10A), and the genes in the READ model were changed in all the 5 queried samples (100%; Figure 10B). In COAD, genetic alterations included single nucleotide polymorphism (SNP), delete mutation, and insert mutation, whereas the genetic alterations in READ just included SNP (Figure 10). Meanwhile, on Copy Number Variation (CNV) module, the statistics of heterozygous and homozygous CNV of each cancer type are displayed in Figure 10C.
 |
Figure 10 The genetic alterations of the genes in the prognosis models of COAD and READ. (A) The genetic alterations of the genes in the COAD model. (B) The genetic alterations of the genes in the READ model. (C) the statistics of heterozygous and homozygous CNV of each cancer type. |
Involved Metabolic Pathways of the Genes in the Risk Models Analyzed by GSEA
To understand the metabolic characteristics of COAD and READ tissues, we used the GSEA approach to analyze the major differences in metabolic pathways between high- and low-risk patients. In COAD, inositol phosphate metabolism, tyrosine metabolism, arachidonic acid metabolism, glycerophospholipid metabolism, and ether lipid metabolism were activated in high-risk tumors, whereas fatty acid metabolism, butanoate metabolism, amino sugar and nucleotide sugar metabolism, propanoate metabolism, and glycolysis gluconeogenesis were inhibited in low-risk tumors (Figure 11A). In READ, glycerophospholipid metabolism, arachidonic acid metabolism, alpha linoleic acid metabolism, linoleic acid metabolism, and glutathione metabolism were activated in high-risk tumors, whereas the TCA cycle, propanoate metabolism, one carbon pool by folate, lysine, valine leucine and isoleucine degradation were inhibited in low-risk tumors (Figure 11B). All the number of genes and full details for the GSEA analysis of COAD and READ were provided in Supplementary Files 1 and 2.
 |
Figure 11 KEGG enrichment pathway analysis. (A) Five representative KEGG pathways in high-risk patients and five representative KEGG pathways in low-risk patients were showed in colon cancer. (B) Five representative KEGG pathways in high-risk patients and five representative KEGG pathways in low-risk patients were showed in rectal cancer. |
Expression of GSR, ENPP2 and GAMT in COAD and READ Detected Through Immunohistochemistry Staining
To test the validity of the prognostic models, we detected the protein expression levels of GSR, ENPP2 and GAMT through immunohistochemistry staining in clinical colorectal cancer specimens and analyzed the relationship between the expression levels and patient prognosis. Immunohistochemical studies had demonstrated that GSR was expressed in the cytoplasm and nuclei of cancer cells (Figure 1A), ENPP2 and GAMT were mainly expressed in the cytoplasm of cancer cells (Figure 1H and K). Then, all the patients were divided into two groups (low expression group and high expression group) based on these protein expression levels in the tumor tissue. Kaplan–Meier survival analysis showed that the high expression levels of GSR in the cytoplasm was significantly correlated with the poor prognosis of patients with COAD (Figure 1B) but not with the prognosis of patients with READ (Figure 1E). No significant correlation was found between the expression levels of GSR in nucleus and prognoses of patients with COAD (Figure 1C) and READ (Figure 1F). Meanwhile, the high expression of GSR in both cytoplasm and nucleus could not indicate the prognoses of patients of COAD (Figure 1D) or READ (Figure 1G). In addition, high ENPP2 expression levels in cancer tissues was significantly correlated with the poor prognosis for COAD (Figure 1I), and no significant correlation between ENPP2 expression and patient prognosis was found in READ (Figure 1J). Furthermore, the significant correlations were found between the expression levels of GAMT and the prognoses of patients with COAD (Figure 1L) and READ (Figure 1M).
Discussion
In this study, the prognosis model of COAD with eight MRGs (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR), and the prognosis mode of READ with six MRGs (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) were identified successfully, and multivariable Cox regression analysis showed that the risk score calculated by the model could independently predicted the prognosis of patient with COAD or READ. Furthermore, some protein expression levels of these MRGs in prognosis models were also significantly correlated with the prognosis of patients with COAD or READ.
The Warburg effect is one of the important characteristics of tumor cells, which shows hyperactive glycolysis even when oxygen is sufficient. However, this effect is only a part of metabolic reprogramming, and metabolic abnormalities in tumor cells are far more complex than previously thought. Tumor cells reprogram many metabolic pathways to the meet nutrient, energy, and REDOX requirements of rapid proliferation.12 Metabolic reprogramming causes changes in the levels and types of specific metabolites inside and outside tumor cells. Such changes can promote tumor growth and metastasis by influencing gene expression, tumor microenvironment, and cell status.13 Currently, drugs targeting these metabolic reprogramming can inhibit the malignant progression of tumors and promote the death of tumor cells.14–16 Therefore, using metabolic gene changes in constructing a prognosis model for cancer is feasible. More and more studies have focused on the relationship between metabolic reprogramming and the progression of colorectal cancer. Compared with normal colorectal tissues, glucose, lipid, amino acid and nucleotide metabolism have been found significantly perturbed in cancer tissues.17,18 Currently, metabolomics’ tests have identified a variety of dysregulated metabolites associated with metabolic pathways in colorectal cancer samples, and these metabolites may serve as useful biomarkers for indicating the prognosis of patients with colorectal cancer, but lots of researches are still needed before clinical application.19,20
Recently, two study teams analyzed DEMRGs between colorectal cancer and normal tissues using TCGA database, then used DEMRGs in constructing a prognosis MRG model. One study constructed a model comprising six genes (NAT2, XDH, GPX3, AKR1C4, SPHK1, and ADCY5) for colorectal cancer10 and another model comprising AOC2, ENPP2, ADA, ACADL, GPD1L, and CPT2.11 Although they used the same data, they obtained completely different prognostic models. We considered that the difference might be due to differences in methods used in data processing or different P values defined. In these studies, the authors modeled COAD and READ together for prognosis. As the blood supply, function, and environmental carcinogens vary among different sites, the prognosis of colorectal cancer varies among different primary sites.21–23 Thus, in our study, we constructed a prognostic model of prognosis-related metabolic genes for COAD and another for READs, using the expression profiles of TCGA databases. Differentially expressed genes in the tumor and relative normal tissues have been used in constructing prognostic models in many studies. We think that by doing so, numerous genes that actually have prognostic significance would be overlooked. The reason is that genes that predict prognosis in cancer patients are in fact not necessarily expressed differently in tumors unlike in normal tissues. We did not directly use differential genes in constructing the prognostic models. Instead, we analyzed all the genes involved in metabolism. Finally, we constructed models using Cox regression and Lasso regression in COAD and READ, respectively. The final prognosis model of COAD with eight MRGs (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR), and the mode READ with six MRGs (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) were identified, and multivariable Cox regression analysis showed that the risk score calculated by the model independently predicted the prognosis of patient with COAD or READ. Thus, prognostic models for COAD and READ are quite different. Only one gene, GAMT, appeared in the risk models for COAD and READ. This finding further showed that modeling COAD and READ separately for the prediction of the prognoses of patients is necessary and reliable.
Among the eight genes in the prognostic model of COAD, three genes (ENPP2, CPT1C, and PLA2G2D) are involved in lipid metabolism, two genes (PIP4K2B and PLCB2) are involved in phosphoinositol metabolism, and two genes (GPX3 and GSR) are involved in glutathione metabolism, indicating that the metabolism of lipid, phosphoinositol, and glutathione is significantly important to COAD. GPX3, as a risk gene, is responsible for the peroxide metabolism of glutathione, and GSR, as a protect gene, is responsible for the reductive metabolism of glutathione. Thus, glutathione peroxidation may be associated with the malignancy of COAD. In our study, we used immunohistochemistry to verify the relationship between GSR and the prognosis of colorectal cancer. We found that high GSR expression level of the cytoplasm was significantly correlated with the poor COAD prognosis but not with READ prognosis. Among the six genes in the model of READ, two genes (TDO2 and GAMT) are involved in amino acid metabolism, suggesting that amino acid metabolism is active, and the prognosis of READ patient is poor. Meanwhile, two genes (EARS2 and WARS) are involved in tRNA aminoacylation of tryptophany and glutamine, which is associated with good READ prognosis. In addition, PKLR is involved in pyruvate metabolism, and ACO1 is involved in the TCA cycle. GAMT, which is mainly involved in creatine synthesis, was present in the two tumor prognostic models, but the creatine system in relation to malignancy requires extensive investigation.24 In addition to supporting the biosynthesis of creatine, GAMT supports the metabolism of polyamine and methionine synthesis in tumor cells, both of which are in high demand in proliferating sarcoma cells.25 The relationship between GAMT protein expression level and the prognosis of colorectal cancer was also detected in this study, and we found that high GAMT expression was significantly correlated with the poor COAD prognosis and READ prognosis.
Notably, two genes, GPX3 and ENPP2, were also identified in the other two studies for MRGs in colorectal cancer.10,11 GPX3, the only extracellular GPX of the family of oxidoreductases, can catalyze the detoxification of lipid hydroperoxides by reduced glutathione. GPX3 has dual properties in tumors, acting as a tumor suppressor in some tumors and as a tumor promoter in others.26 In colorectal cancer, tumor cells with low GPX3 expression were more sensitive to chemotherapy drugs, such as oxaliplatin and cisplatin.27 ENPP2 can encode a secreted glycoprotein autotaxin, which converts lysophosphatidylcholine into lysophosphatidic acid, and the autotaxin–LPA axis is essential for cell survival, migration, proliferation, and differentiation in many cancers.28–31 Moreover, ATX expression is positively correlated with microvascular density and tumor angiogenesis in colorectal cancer.32 In our study, patients with COAD and high ENPP2 expression in cancer tissues always had poor survival, but no significant correlation between ENPP2 expression and patient prognosis was found in READ.
Gene mutations were significantly varied between the two cancer types. In COAD, genetic alterations included SNP, delete mutation, and insert mutation, whereas the genetic alterations in READ included SNP only. Moreover, the major differences in metabolic pathways between the low- and high-risk patients with COAD or READ also differed significantly. In COAD, arachidonic acid metabolism, tyrosine metabolism, glycerophospholipid metabolism, inositol phosphate metabolism, and ether lipid metabolism were activated in the high-risk tumors, whereas in READ, glycerophospholipid metabolism, arachidonic acid metabolism, linoleic acid metabolism, alpha linoleic acid metabolism, and glutathione metabolism were activated in the low-risk tumors. Basing on this result, we found that arachidonic acid metabolism and glycerophospholipid metabolism were all activated in high-risk groups for COAD and READ. However, no changes in the same metabolic pathway were found in low-risk patients with COAD or READ. In addition, we screened DEMRGs between COAD and non-tumor tissues and performed KEGG analysis on the DEMRGs to understand metabolism in COAD. We found that the genes involved in amino acids biosynthesis and carbon metabolism were up-regulated, whereas the genes involved in fatty acid degradation were down-regulated. These results suggested that COAD cells are more prone to anabolism than normal tissues and inhibit catabolism.
Our study has some limitations. Although our prognostic models were conducted based on massive data mining, additional functional and mechanism experiments are still needed for exploring the effect of each gene in the prognostic model on the metabolism of colorectal cancer. Moreover, some important clinical factors and lifestyle habits, such as dietary habits, obesity, smoking, and diabetes, were not included in our analysis. However, these factors influenced the prognoses of patients with colorectal cancer.33–36
Conclusions
In conclusion, applying TCGA and GEO data sets, we successfully identified and validated the risk prediction models with metabolism-related genes for colon cancer and rectal cancer, respectively. Meanwhile, through immunohistochemistry, we verified the relationship between the expression levels of some proteins in the models and colorectal cancer prognosis. The results of our study may provide novel insights for further research on the metabolic characteristics and provide potential metabolic targets for treatment of colorectal cancer. However, there are still many survival analyses and functional experiments that need to be validated.
Abbreviations
MRG, metabolism-related gene; COAD, colon adenocarcinoma; READ, rectum adenocarcinoma; OS, overall survival; GSEA, gene set enrichment analysis.
Data Sharing Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The present study was approved by the Ethics Committee of the Tianjin Medical University Cancer Institute and Hospital, and in accordance with the Declaration of Helsinki.
Consent for Publication
All the authors agree to publish the research.
Acknowledgments
The authors thank the patients for their willingness to cooperate with our study.
Funding
This research was supported by grants from the National Natural Science Foundation of China (Numbers 81602446, 82073085, 82073252 and 81772804), Tianjin Municipal Science and Technology Commission (Number 20JCZDJC00030).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Weiser MR. AJCC 8th edition: colorectal cancer. Ann Surg Oncol. 2018;25(6):1454–1455. doi:10.1245/s10434-018-6462-1
2. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14(2):89–103. doi:10.5114/pg.2018.81072
3. Usher-Smith J, Miller R, Griffin S. Predicting survival in patients with colorectal cancer. BMJ. 2017;357:j2772. doi:10.1136/bmj.j2772
4. Feng Z, Shi X, Zhang Q, et al. Analysis of clinicopathological features and prognosis of 1315 cases in colorectal cancer located at different anatomical subsites. Pathol Res Pract. 2019;215(10):152560. doi:10.1016/j.prp.2019.152560
5. Murphy N, Ward HA, Jenab M, et al. Heterogeneity of colorectal cancer risk factors by anatomical subsite in 10 European countries: a multinational cohort study. Clin Gastroenterol Hepatol. 2019;17(7):1323–1331. doi:10.1016/j.cgh.2018.07.030
6. Phipps AI, Chan AT, Ogino S. Anatomic subsite of primary colorectal cancer and subsequent risk and distribution of second cancers. Cancer. 2013;119(17):3140–3147. doi:10.1002/cncr.28076
7. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368(6487):eaaw5473. doi:10.1126/science.aaw5473
8. Allison KE, Coomber BL, Bridle BW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology. 2017;152(2):175–184. doi:10.1111/imm.12777
9. Sciacovelli M, Frezza C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017;284(19):3132–3144. doi:10.1111/febs.14090
10. Miao Y, Li Q, Wang J, et al. Prognostic implications of metabolism-associated gene signatures in colorectal cancer. PeerJ. 2020;8:e9847. doi:10.7717/peerj.9847
11. Sun YL, Zhang Y, Guo YC, Yang ZH, Xu YC. A prognostic model based on six metabolism-related genes in colorectal cancer. Biomed Res Int. 2020;2020:5974350. doi:10.1155/2020/5974350
12. Yang HC, Stern A, Chiu DT. G6PD: a hub for metabolic reprogramming and redox signaling in cancer. Biomed J. 2020. doi:10.1016/j.bj.2020.08.001
13. Yoo HC, Park SJ, Nam M, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020;31(2):267–283. doi:10.1016/j.cmet.2019.11.020
14. Wen SS, Zhang TT, Xue DX, et al. Metabolic reprogramming and its clinical application in thyroid cancer. Oncol Lett. 2019;18(2):1579–1584. doi:10.3892/ol.2019.10485
15. Cha JY, Lee HJ. Targeting lipid metabolic reprogramming as anticancer therapeutics. J Cancer Prev. 2016;21(4):209–215. doi:10.15430/JCP.2016.21.4.209
16. Morandi A, Taddei ML, Chiarugi P, Giannoni E. Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front Oncol. 2017;7:40. doi:10.3389/fonc.2017.00040
17. Jeong KY. Cancer-specific metabolism: promising approaches for colorectal cancer treatment. World J Gastrointest Oncol. 2019;11(10):768–772. doi:10.4251/wjgo.v11.i10.768
18. Brown RE, Short SP, Williams CS. Colorectal cancer and metabolism. Curr Colorectal Cancer Rep. 2018;14(6):226–241. doi:10.1007/s11888-018-0420-y
19. Brown DG, Rao S, Weir TL, et al. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016;4:11. doi:10.1186/s40170-016-0151-y
20. Li J, Li J, Wang H, Qi LW, Zhu Y, Lai M. Tyrosine and glutamine-leucine are metabolic markers of early-stage colorectal cancers. Gastroenterology. 2019;157(1):257–259. doi:10.1053/j.gastro.2019.03.020
21. Tokunaga R, Xiu J, Johnston C, et al. Molecular profiling of appendiceal adenocarcinoma and comparison with right-sided and left-sided colorectal cancer. Clin Cancer Res. 2019;25(10):3096–3103. doi:10.1158/1078-0432.CCR-18-3388
22. Zhao Y, Ge X, He J, et al. The prognostic value of tumor-infiltrating lymphocytes in colorectal cancer differs by anatomical subsite: a systematic review and meta-analysis. World J Surg Oncol. 2019;17(1):85. doi:10.1186/s12957-019-1621-9
23. Xi Y, Yuefen P, Wei W, et al. Analysis of prognosis, genome, microbiome, and microbial metabolome in different sites of colorectal cancer. J Transl Med. 2019;17(1):353. doi:10.1186/s12967-019-2102-1
24. Patra S, Ghosh A, Roy SS, et al. A short review on creatine-creatine kinase system in relation to cancer and some experimental results on creatine as adjuvant in cancer therapy. Amino Acids. 2012;42(6):2319–2330. doi:10.1007/s00726-011-0974-3
25. Bera S, Wallimann T, Ray S, Ray M. Enzymes of creatine biosynthesis, arginine and methionine metabolism in normal and malignant cells. FEBS J. 2008;275(23):5899–5909. doi:10.1111/j.1742-4658.2008.06718.x
26. Chang C, Worley BL, Phaeton R, Hempel N. Extracellular glutathione peroxidase GPx3 and its role in cancer. Cancers. 2020;12(8):2197. doi:10.3390/cancers12082197
27. Pelosof L, Yerram S, Armstrong T, et al. GPX3 promoter methylation predicts platinum sensitivity in colorectal cancer. Epigenetics. 2017;12(7):540–550. doi:10.1080/15592294.2016.1265711
28. Kaffe E, Katsifa A, Xylourgidis N, et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology. 2017;65(4):1369–1383. doi:10.1002/hep.28973
29. Lee D, Suh DS, Lee SC, Tigyi GJ, Kim JH. Role of autotaxin in cancer stem cells. Cancer Metastasis Rev. 2018;37(2–3):509–518. doi:10.1007/s10555-018-9745-x
30. Brindley DN, Tang X, Meng G, Benesch MGK. Role of adipose tissue-derived autotaxin, lysophosphatidate signaling, and inflammation in the progression and treatment of breast cancer. Int J Mol Sci. 2020;21(16):5938. doi:10.3390/ijms21165938
31. Jinno N, Yoshida M, Hayashi K, et al. Autotaxin in ascites promotes peritoneal dissemination in pancreatic cancer. Cancer Sci. 2021;112(2):668–678. doi:10.1111/cas.14689
32. Kazama S, Kitayama J, Aoki J, Mori K, Nagawa H. Immunohistochemical detection of autotaxin (ATX)/lysophospholipase D (lysoPLD) in submucosal invasive colorectal cancer. J Gastrointest Cancer. 2011;42(4):204–211. doi:10.1007/s12029-010-9186-4
33. Ye P, Xi Y, Huang Z, Xu P. Linking obesity with colorectal cancer: epidemiology and mechanistic insights. Cancers. 2020;12(6):1408. doi:10.3390/cancers12061408
34. Soltani G, Poursheikhani A, Yassi M, Hayatbakhsh A, Kerachian M, Kerachian MA. Obesity, diabetes and the risk of colorectal adenoma and cancer. BMC Endo Dis. 2019;19(1):113. doi:10.1186/s12902-019-0444-6
35. Lee S, Woo H, Lee J, Oh JH, Kim J, Shin A. Cigarette smoking, alcohol consumption, and risk of colorectal cancer in South Korea: a case-control study. Alcohol. 2019;76:15–21. doi:10.1016/j.alcohol.2018.06.004
36. Makino A, Tsuruta M, Okabayashi K, et al. The impact of smoking on pulmonary metastasis in colorectal cancer. Onco Targets Ther. 2020;13:9623–9629. doi:10.2147/OTT.S263250
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.