")
Back to Journals » Journal of Inflammation Research » Volume 17
Identification of Key Immune Infiltration Related Genes Involved in Aortic Dissection Using Bioinformatic Analyses and Experimental Verification
Authors Zheng L , Yang Y , Liu J , Zhao T , Zhang X , Chen L
Received 11 August 2023
Accepted for publication 29 March 2024
Published 5 April 2024 Volume 2024:17 Pages 2119—2135
DOI https://doi.org/10.2147/JIR.S434993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Lin Zheng,1,* Yusi Yang,2,* Jie Liu,3 Tianliang Zhao,3 Xin Zhang,3 Lihua Chen3
1Department of Vascular Surgery, the Second Hospital, Shanxi Medical University, Taiyuan, 030001, People’s Republic of China; 2Department of Cardiology, Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences Tongji Shanxi Hospital, Taiyuan, 030032, People’s Republic of China; 3Department of Cardiac Surgery, the Second Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lihua Chen, The Second Hospital of Hebei Medical University, 215 Hepingxi Street, Shijiazhuang, 050000, People’s Republic of China, Tel +86 188 4679 1714, Fax +86 0311-87045297, Email [email protected]
Purpose: Immune microenvironment plays an important role in aortic dissection (AD). Therefore, novel immune biomarkers may facilitate AD prevention, diagnosis, and treatment. This study aimed at mining key immune-related genes and relevant mechanisms involved in AD pathogenesis.
Patients and Methods: Key immune cells in AD were identified by ssGESA algorithm. Next, genes associated with key immune cells were screened by weighted gene coexpression network analysis (WGCNA). Then hub immune genes were picked from protein-protein interaction network of overlapped genes from differential expression and WGCNA analyses by cytohubba plug-in. Their diagnostic potential was evaluated in two independent cohorts from GEO database. In addition, the expressions of hub immune genes were determined by quantitative RT-PCR, immunohistochemistry, and Western blotting in dissected and normal aortic tissues.
Results: Activated B cells, CD56dim natural killer cells, eosinophils, gamma delta T cells, immature B cells, natural killer cells and type 17 T helper cells were identified as key immune cells in AD. Thereafter, a gene module significantly correlated with key immune cells were found by WGCNA method. Subsequently, KDR, IGF1, NOS3, PECAM1, GAPDH, FLT1, DLL4, CDH5, VWF, and TEK were identified as hub immune cell related genes by PPI network analysis, which may be potential diagnostic markers for AD, as evidenced by ROC curves. Moreover, the decreased expression of VWF in AD was validated at both mRNA and protein levels, and its expression was significantly positive correlated with the marker of smooth muscle cells, ACTA2, in AD. Further immunofluorescent results showed that VWF was colocalized with ACTA2 in aortic tissues.
Conclusion: We identified key immune cells and hub immune cell-related genes involved in AD. Moreover, we found that VWF was co-expressed with the smooth muscle cell marker ACTA2, indicating the important role of VWF in smooth muscle cell loss in AD pathogenesis.
Keywords: aortic dissection, infiltrating immune cells, diagnosis, smooth muscle cells
Introduction
Aortic dissection (AD) is a fatal emergency triggered by aortic wall rupture. Briefly, an intimal tear permits blood to enter the media, thus forming a false channel that might enlarge proximally or widely, possibly leading to rupture as the outer layer worsens. AD is categorized as Stanford type A or Stanford type B depending on the location of the initial rupture and the area of dissection involved. Stanford type A AD, defined as ascending AD, is associated with a high risk of death and morbidity. AD-specific causes are currently unclear, and it has been reported that inflammatory responses serve as disease mediator.1 AD is often associated with hypertension, hereditary connective tissue disease, aortic inflammatory diseases, and other diseases that contribute to the destruction of the aortic media. Although AD is uncommon, it is associated with high mortality rates.2 The full aorta computed tomographic angiography (CTA) is the preferred tool to support a diagnosis and follow-up evaluation after treatment. Through imaging examinations, the type of dissection, extent of involvement, tear location, and involvement of the aortic valve can be understood, and treatment measures can be decided afterwards. The survival rate after acute AD has significantly improved through timely emergency surgery, but operative mortality remains high.3 Thus, it is necessary to establish efficient strategies to predict the risk and prevent or mitigate AD injury.
Previous studies have shown that autoimmune diseases trigger vascular inflammatory responses, which in turn cause further aortic injury and ECM destruction, which is a main risk factor for dissection.4 Furthermore, immune cell infiltration is associated with AD development.5 Immune cell infiltration in the vessel wall of dissection has been observed, including macrophages, dendritic cells, natural cells, B cells, and killer T cells. Furthermore, macrophage recall and activation have been identified as critical events in the early stages of Stanford type A AD.6 According on reports, the monocyte-macrophage system is primarily responsible for the immunological inflammatory response that leads to the development of AD.1 In addition, Liu et al discovered that necroptosis causes cell swelling and rupture, releasing potentially inflammatory cellular contents that influence immune infiltration and the onset and development of dissection.7 The inflammatory and immune response in AD may have important implications.
Therefore, to determine the pathophysiology of AD from an immune and inflammatory perspective, this study was designed to determine the key immune infiltration-related target genes implicated in AD and investigate their functions based on bioinformatics and in vivo and in vitro experiments. An overview of the study design and main findings is presented in Figure 1.
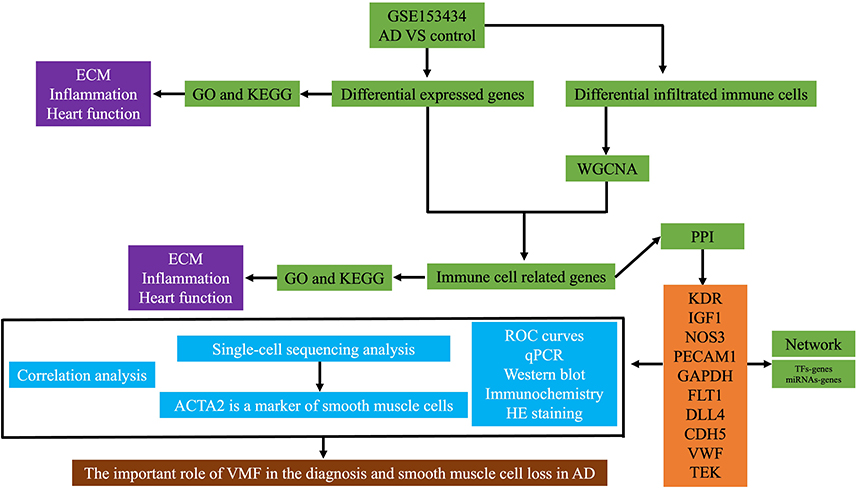 |
Figure 1 The schematic workflow of the current study. Abbreviations: ECM, extracellular matrix; GO, gene ontology; KEGG; Kyoto Encyclopedia of genes and genomes; AD, aortic dissection; WGCNA, weighted gene coexpression network analysis; PPI, protein-protein interaction; TF, transcription factor; KDR, kinase insert domain receptor; IGF1, insulin-like growth factor 1; NOS3, nitric oxide synthase 3; PECAM1, platelet and endothelial cell adhesion molecule 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; FLT1, Fms-related receptor tyrosine kinase 1; DLL4, delta-like canonical notch ligand 4; CDH5, cadherin 5; VWF, von Willebrand factor; TEK, TEK receptor tyrosine kinase; ROC, receiver operating characteristic; HE, haematoxylin and eosin; ACTA2, actin alpha 2, smooth muscle; CA9, carbonic anhydrase 9; DIO3, iodothyronine deiodinase 3; CXCL5, C-X-C motif chemokine ligand 5; GCKR, glucokinase regulator; CCL20, C-C motif chemokine ligand 20; NPY2R, neuropeptide Y receptor Y2; C2CD4A, C2 calcium dependent domain containing 4A; MMP1, matrix metallopeptidase 1; SCN7A, sodium voltage-gated channel alpha subunit 7; RELN, reelin; PI16, peptidase inhibitor 16; ADIPOQ, adiponectin, C1Q and collagen domain containing; LSAMP, limbic system associated membrane protein; PMP2, peripheral myelin protein 2; RAMP3, receptor activity modifying protein 3; STAB2, stabilin. |
Materials and Methods
Data Source
Gene expression data of ascending aortic tissues collected from patients with Stanford type A AD (N = 10) and normal ascending aortic tissues collected from patients without any aortic disease (N = 10) samples were downloaded from GSE153434 to identify potential AD biomarkers. The GSE52093 dataset, including seven ascending aortic tissues obtained from patients with Stanford type A AD and five normal control ascending aortic tissue samples, was also sourced from the GEO database to validate the diagnostic value of the markers identified in GSE153434. GSE222328 was used to perform single-cell analyses from human AD. Using the limma R package, DEGs between the AD and control samples in GSE153434 were identified using |log2FC| > 1 and a p-value < 0.05.
Identification of Immune Cell Types Involved in AD
The ssGSEA algorithm was applied to count the enrichment fractions of 28 immune cells based on relevant immune gene sets. Compared with other methods, ssGSEA has less noise and more biological interpretability.8 The levels of 28 immune cells evaluated in the current study were type 17 T helper cells, activated B cells, eosinophils, gamma delta T cells, immature B cells, natural killer cells, CD56dim natural killer cells, MDSCs, activated dendritic cells, plasmacytoid dendritic cells, central memory CD4 T cells, immature dendritic cells, macrophages, mast cells, memory B cells, neutrophils, type 2 T helper cells, activated CD4 T cells, activated CD8 T cells, effector memory CD8 T cells, T follicular helper cells, type 1 T helper cells, monocytes, CD56bright natural killer cells, and regulatory T cells. After calculating the above mentioned immune cells, we compared their levels between AD patients and controls using the Wilcoxon test, and significantly differentially infiltrated immune cells were identified using p-value <0.05.
Identifying and Analyzing the Functions of AD Hub Genes
Next, genes associated with differentially enriched immune cells were filtered using weighted gene coexpression network analysis (WGCNA). WGCNA is a method used for finding modules of highly correlated genes, relating modules to sample traits and calculating membership modules. One obvious advantage of WGCNA is that it transforms gene expression data into co-expression modules, which is also suitable for datasets having low abundance or small fold change genes without losing much information. In addition, WGCNA is performed in R, which is an open source programming language and user-friendly.9 Thus, we applied WGCNA to identify gene modules correlated with key immune cells. Briefly, to eliminate outliers, a clustering tree map of the samples was generated. Considering the sample size, we chose 8 as the soft threshold according to the instructions of the WGCNA. The adjacency matrix was transformed into a topological overlap matrix based on the soft threshold used to build the network, and the genetic dendrogram and modular colors were determined based on the degree of difference. Dynamic tree-cutting was used to isolate the initial modules. The coefficient of Pearson’s correlation involving the eigengenes of the module and the sample traits was calculated to identify the module that was most closely connected to the differentially infiltrated immune cells. The most positive module was selected, and module membership (MM) > 0.8 and gene significance (GS) > 0.2 were used to further screen genes.
Identification and Validation of Potential Diagnostic Biomarkers for AD
To further identify the important immune cell-related genes involved in AD, DEGs and WGCNA modular genes were overlapped, and the GO terms and KEGG pathways in which overlapping genes were enriched were analyzed. Meanwhile, the overlapped genes were uploaded to STRING database, which includes known and predicted protein-protein interactions from experimental validations and computational prediction. Those interactions in STRING database are critically assessed and scored.10 Next, Cytoscape plug-in “cytohubba”11 was applied to identify hub genes in the network by measuring and ranking the network features of overlapped genes. The diagnostic values of hub genes were evaluated using GSE153434 and validated using GSE52093. To determine the potential molecular mechanisms that regulate the expression of hub genes, we used the miRNet and NetworkAnalyst databases to search for miRNAs and transcription factors (TFs), respectively. Accordingly, regulatory networks of miRNA-hub genes and TFs-hub genes were established.
Sample Collection
Five ascending aortic tissues from Stanford A AD patients who underwent thoracic aortic replacement surgery were obtained from the Second Affiliated Hospital of the Shanxi Medical University. Three normal thoracic aorta tissues were obtained from patients undergoing heart coronary bypass surgery without any aortic disease. All arterial samples were collected following procedures authorized by the Office for Human Subjects Protection at Shanxi Medical University’s Second Affiliated Hospital.
Quantitative Real-Time PCR
AD and normal tissues were ground, and total RNA was extracted using an RNA extraction reagent according to the manufacturer’s instructions (Servicebio, China). A NanoDrop 2000 spectrophotometer was used to determine the concentration and purity of the total RNA (Thermo Fisher Scientific, Wilmington, DE, United States). A Servicebio RT First-Strand cDNA Synthesis Kit was used to create complementary DNA (cDNA). Quantitative real-time PCR was performed using the MonAmpTM ChemoHS qPCR Master Mix at 95 °C for 10 min, followed by 40 cycles of 95 °C for 10s and 55 °C for 30s. The primer sequences used for detection are listed in Table 1. Beta-actin expression was used as an internal standard. The 2−ΔΔCt method was used to calculate gene expression levels.
 |
Table 1 Primers for qPCR in the Current Study |
Western Blotting
AD and normal tissues were ground and lysed using a protein lysis buffer (Merck, Germany). Protein concentrations were measured by the BCA assay (Beyotime, China). Briefly, 25 μg of lysate samples was separated on NuPage 4–12% Bis-Tris Gels (Novex, Life Technologies, Carlsbad, CA, United States). After transfer, the blots were incubated in blocking buffer for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4 °C, PBST washing (three times, each for 10 min), secondary antibody for 1 h at room temperature, and PBST washing (three times, each for 10 min). Blots were imaged using the LiCor Odyssey imaging system. Primary antibodies against VWF (1:5000; 11778-1-ap) and PECAM1 (1:500; 28083-1-ap) were purchased from Sanying (Wuhan, China). GAPDH (1:2000, GB15002) and ACTIN (1:2000, GB113225) were purchased from ServiceBio (Wuhan, China). The blot image was then analyzed using ImageJ software, according to the user guidelines.12
Immunohistochemical and Histopathological and Analyses
Formalin-fixed, paraffin-embedded human AD and normal tissue specimens were used for immunohistochemical analyses. A 1:5000 dilution of anti-VWF (Sanying, Wuhan, China) was used as the primary antibody. Immunohistochemical results were evaluated using the mean density. At least three 200× fields were randomly selected for each slide in each group. Efforts were made to ensure that the field of vision was largely filled with tissue, and that each image had the same background light. Image-pro Plus 6.0, which was used to select the same brown-yellow color as the unified standard to judge positivity. The cumulative optical density (IOD) and pixel AREA (AREA) were obtained and the mean density (IOD/AREA) was calculated. For histopathological evaluations, AD and normal samples were fixed in 4% paraformaldehyde, sectioned at 2.5-µm thickness after fixation and paraffin embedding, stained with hematoxylin and eosin (H&E) following standard procedures, and examined under light microscopy. Masson’s trichrome staining kit (DC0032-100, Leagene, Beijing, China) and Picrosirius Red Staining Kit (G1018, Servicebio, Wuhan, China) were used to measure collagen deposition according to the manufacturer’s instructions.
Immunofluorescent Staining
Paraffin-embedded aortic tissues from AD and controls were cut into 4 µm sections, followed by deparaffinized, rehydrated and antigen retrieval. Then tissue sections were incubated with primary antibodies (anti-ACTA2, 1:200 dilution, GB12045; anti-VWF, 1:1000 dilution, GB11020; anti-SM22α, 1:2000 dilution, GB11366) overnight at 4°C and secondary antibodies (Cy3-labeled Goat Anti-Mouse IgG, 1:300 dilution, GB21201; Alexa Fluor 488 Goat Anti-Rabbit IgG, 1:300 dilution, GB25303;) for 1 hour at room temperature. Subsequently, the nuclei were counterstained with DAPI for visualization. All the antibodies were purchased from Servicebio company (Wuhan, China). Staining quantification was performed by Image-Pro Plus 6.0 software by counting cells at x400 magnification from randomly selected 4 fields in each slide.
Analysis of Single-Cell Sequencing Data
The Seurat packet (https://satijalab.org/seurat/ (accessed on June 11, 2023)) in R was used for cell filtration and analysis. We set the conditions for cell filtration as follows: the number of genes identified in single cells was greater than 500 and less than 30,000; the total number of UMI detected in a single cell was greater than 1000, the proportion of mitochondrial gene expression in single cells was less than 5%, and the DoubletFinder package was used to remove multicellular cells. After screening, 96,001 cells were obtained. Follow-up for further filtering, standardization, cell subgroup classification, subgroup analysis, and marker gene screening of differentially expressed genes was performed using Seurat.
Statistical Analysis
All analyses were performed using the R software. The Student’s t-test or Wilcoxon test was used to compare the results of qPCR, Western blotting, and immunohistochemistry between the AD and normal groups. The correlation between VWF and ACTA2 expression was analyzed using Spearman correlation coefficient. The P value less than 0.05 were considered statistically significant.
Results
DEGs Associated with ECM and Inflammation Were Identified Between AD and Controls
The GSE153434 dataset was first analyzed, and 1571 DEGs were identified between patients with AD and controls (Figure 2A). Among them, 566 genes were remarkably upregulated, whereas 1005 genes were downregulated in AD (Figure 2A). We then extracted the expression patterns of the top 10 upregulated [carbonic anhydrase 9 (CA9), iodothyronine deiodinase 3 (DIO3), C-X-C motif chemokine ligand 5 (CXCL5), glucokinase regulator (GCKR), C-C motif chemokine ligand 20 (CCL20), CCL7, neuropeptide Y receptor Y2 (NPY2R), C2 calcium dependent domain containing 4A (C2CD4A), matrix metallopeptidase 1 (MMP1), and MMP10] and downregulated [sodium voltage-gated channel alpha subunit 7 (SCN7A), reelin (RELN), peptidase inhibitor 16 (PI16), CXCL14, CCL15-CCL14, adiponectin, C1Q and collagen domain containing (ADIPOQ), limbic system associated membrane protein (LSAMP), peripheral myelin protein 2 (PMP2), receptor activity modifying protein 3 (RAMP3), and stabilin 2 (STAB2)] genes in AD, as shown in the heatmap (Figure 2B). Interestingly, both GO and KEGG pathway enrichment results indicated that these DEGs may play important roles in ECM modeling, ECM-mediated cellular functions, and inflammation, such as BPs of ECM organization, cell migration, CCs of collagen-containing ECM, cell-cell junction, signaling receptor activator activity, cytokine activity, and KEGG pathways of cytokine-receptor interaction, TNF signaling pathway, ECM-receptor interaction, and focal adhesion (Figure 2C and 2D), indicating that aberrant expression of these genes may regulate AD development via ECM and inflammation. In addition, some DEGs were enriched in heart-related functions and diseases, such as heart contraction, vascular processes in the circulatory system, heart morphogenesis, and hypertrophic cardiomyopathy (Figure 2C and 2D), suggesting that these DEGs may be common pathogenic factors in both AD and heart disease, which may guide further in vivo and in vitro studies.
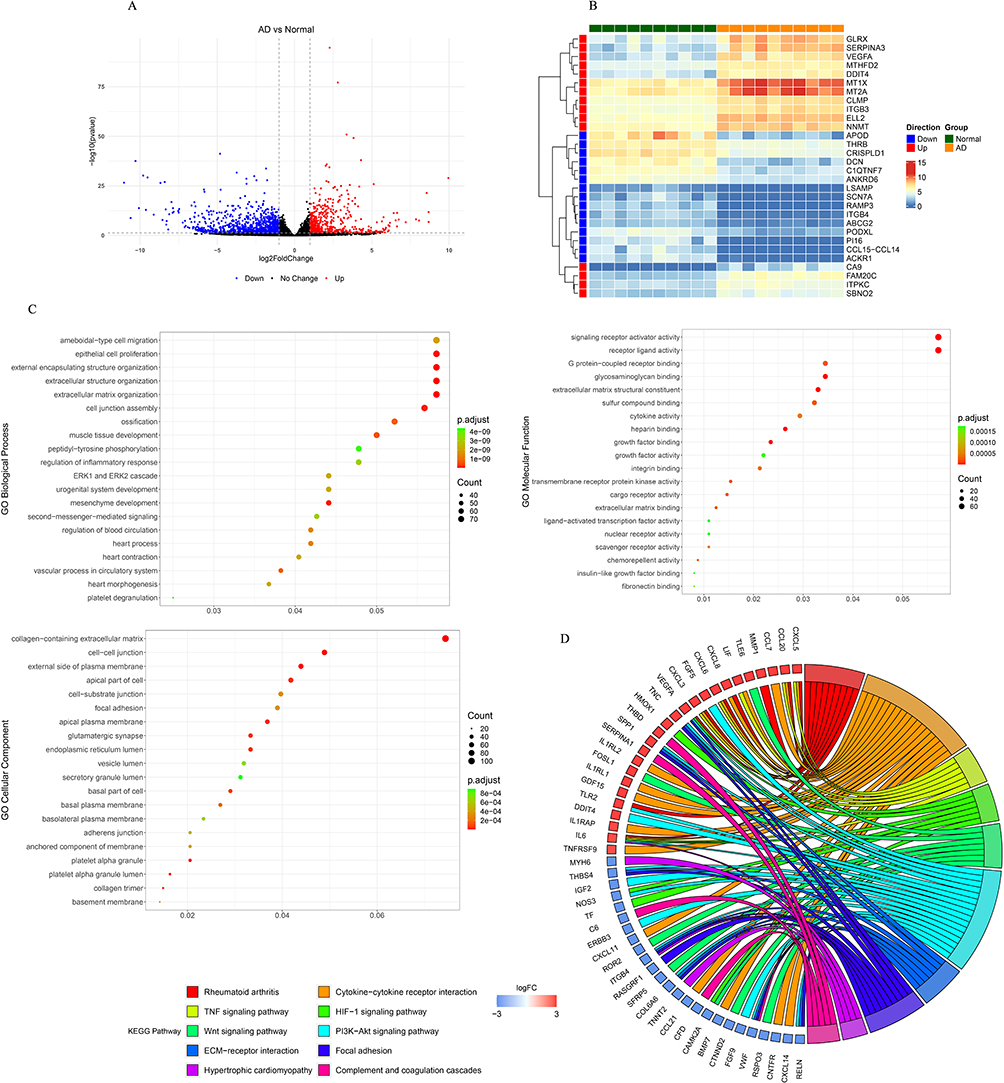 |
Figure 2 Identification and functional analysis of differentially expressed genes (DEGs) between aortic dissection (AD) and control samples. (A) Volcano plot for the differential expression analysis, in which red dots indicate upregulated DEGs and blue dots indicate downregulated DEGs in AD. (B) Heatmap showing the increased and decreased expressions of the most significant 15 up-regulated genes and 15 down-regulated genes (ranked according to the adjusted p-value). (C) Dot plot displaying GO terms sorted by adjusted p value in the category of biological process (BP), cellular component (CC) and molecular function (MF) that DEGs between AD and control are enriched in. (D) Chord diagram showing the corresponding relationship between the top 10 pathways with the highest significance and relevant DEGs involved in those pathways. The left are KEGG pathways, and the right is DEGs, and log2FC was displayed from top to bottom. When log 2 FC > 0, the larger the log 2 FC is, the greater the differential expression of up-regulated protein is; when log 2 FC < 0, the smaller the log 2 FC is, the greater the differential expression of down-regulated protein is. The closer the log 2 FC is to 0, the smaller the differential expression fold is. |
217 Genes Associated with Activated B Cells Were Mined by WGCNA
ssGSEA was used to calculate the enrichment score of 28 distinct cell types to compare cellular heterogeneity between the AD and control samples. We found a significant difference between AD and control samples in the infiltration of type 17 T helper cells, activated B cells, gamma delta T cells, immature B cells, eosinophils, and natural killer cells (Figure 3A and 3B). To identify genes associated with type 17 T helper cells, activated B cells, eosinophils, gamma delta T cells, immature B cells, and natural killer cells, we performed WGCNA. First, one outlier sample was removed after clustering (Figure 4A). To achieve a scale-free topology, 8 was selected as the optimal soft threshold power, according to the instructions of the WGCNA package (Figure 4B). Twenty modules from the coexpression network were identified after merging the similar modules (Figure 4C). According to the module-trait relationships in Figure 4D, the blue module and activated B cells had the strongest positive correlation. Thus, 217 activated B cell-related genes in the blue module were further obtained under module membership (MM)> 0.8 and gene significance (GS)> 0.2.
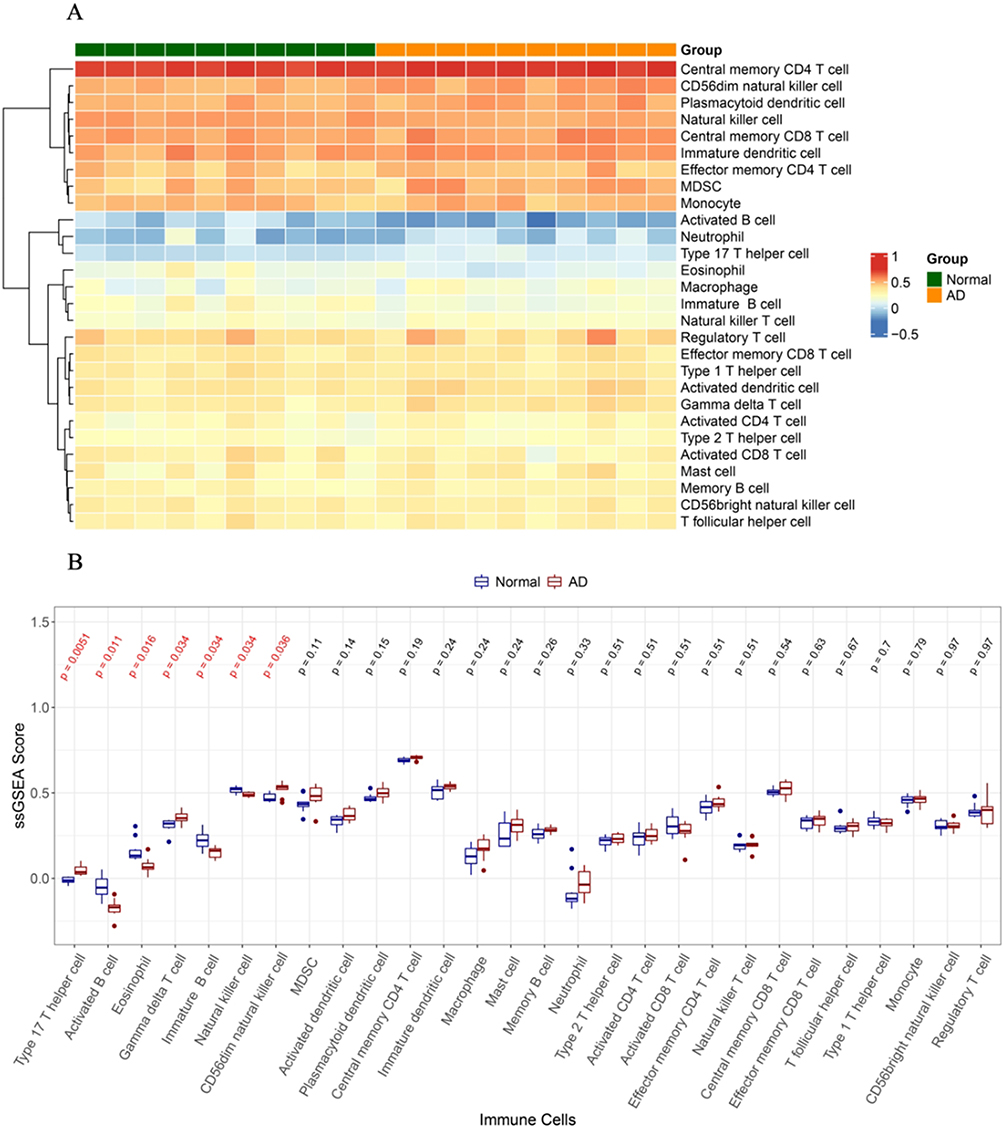 |
Figure 3 Analysis of immune cell infiltration in aortic dissection (AD) and control samples. (A) Heatmap showing the distribution of 28 immune cell in AD and control samples. The row means the type of immune cells, and the column corresponds to each sample. Red and blue represent high and low expression, respectively. (B) Box plot showing the difference of immune cell levels between AD and control group. Wilcoxon test was used, and P was corrected by false discovery rate (FDR). The red colored p values highlight the immune cells (type 17 T helper cell, activated B cell, eosinophil, gamma delta T cell, immature B cell, natural killer cell, CD56dim natural killer cell) that were significantly infiltrated between AD and control samples. |
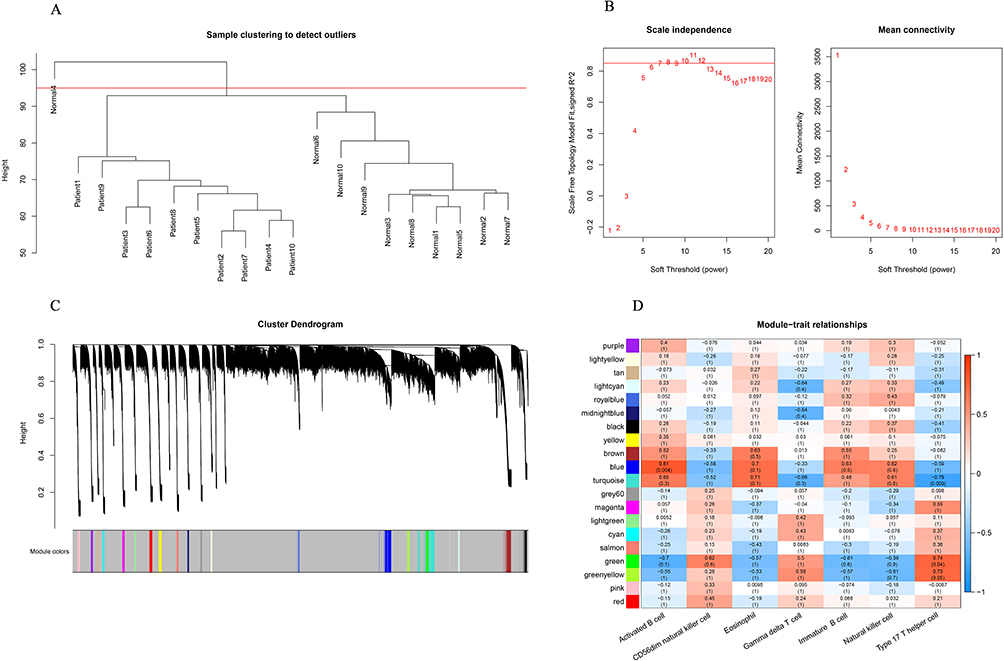 |
Figure 4 Identification of immune cell-related modules by weighted gene coexpression network analysis (WGCNA). (A) The clustering dendrogram of samples showing that one sample (Normal 4) was an outlier, and the others were well divided into two groups with no outlier. (B) Determination of the soft-threshold to achieve the scale-free network. The left panel shows the influence of soft-threshold power on the scale-free fit index, and the right panel shows the impact of soft-threshold power on the mean connectivity. (C) Gene dendrogram obtained by hierarchical clustering and modules with different colors assigned by dynamic tree cutting. (D) Heatmap displaying the correlation between modules and immune cells. Each row correlates to a module eigengene, and each cell includes the corresponding correlation and p value. |
Hub Activated B Cell Related Genes May Serve as Potential Diagnostic Biomarkers in AD
By overlapping 1571 DEGs with 217 genes related to activated B cells, 192 activated B cell-related DEGs were obtained (Figure 5A). GO and KEGG enrichment analyses again showed that these genes were mainly involved in ECM- and inflammation-related biological functions, such as ameboidal-type cell migration, cell junction assembly, the PI3K-Akt signaling pathway, focal adhesion, and complement and coagulation cascades (Figure 5B and 5C). Pathways associated with heart function and related pathways, such as the calcium signaling pathway, dilated cardiomyopathy, and vascular smooth muscle contraction, were detected (Figure 5C). To better understand their interactions, we constructed a PPI network (Figure 5D) in which kinase insert domain receptor (KDR), insulin-like growth factor 1 (IGF1), nitric oxide synthase 3 (NOS3), platelet and endothelial cell adhesion molecule 1 (PECAM1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Fms-related receptor tyrosine kinase 1 (FLT1), delta-like canonical notch ligand 4 (DLL4), cadherin 5 (CDH5), von Willebrand factor (VWF), and receptor tyrosine kinase (TEK) had the highest connection degrees calculated by cytohubba (Figure 5E). Thus, those genes were identified as hub activated B cell-related genes in AD. Next, we evaluated the potential of hub genes for AD diagnosis using ROC analysis. The AUCs of KDR, IGF1, NOS3, PECAM1, GAPDH, FLT1, DLL4, CDH5, VWF, and TEK were 1.00, 0.96, 1.00, 0.99, 0.99, 0.94, 0.95, 1.00, 1.00, and 1.00, respectively (Figure 6A), indicating that they all performed well in distinguishing AD and control samples and thus may serve as potential biomarkers for the diagnosis of AD. Furthermore, similar expression patterns were found in both the training and validation datasets (Figure 6B and 6C), further demonstrating their potential as diagnostic biomarkers. Due to their aberrant expression in AD, it is important to explore potential regulatory factors to guide future upstream molecular mechanisms of AD pathogenesis. Thus, we constructed networks of TFs-hub genes and miRNA-hub genes (Figure 6D and 6E) and found that some hub genes were regulated by common TFs and miRNAs. These shared TFs and miRNAs may play important roles in the initiation and development of AD; however, solid in vitro and in vivo experiments need to be performed to provide more information.
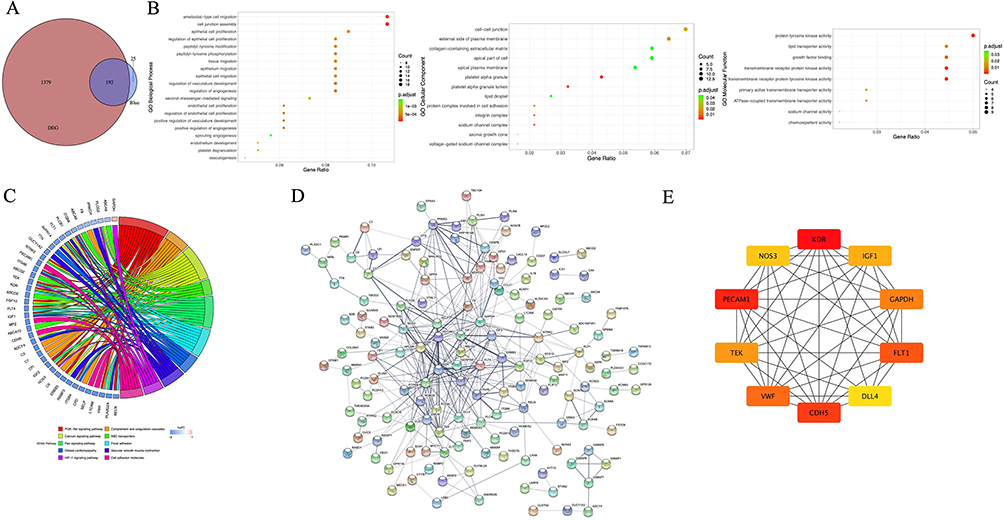 |
Figure 5 Identification of activated B cell-related hub genes in aortic dissection (AD). (A) Venn plot showing 192 overlapped genes between WGCNA modular genes and differentially expressed genes (DEGs). (B) Gene ontology (GO) enrichment of 192 activated B cell related DEGs. GO-biological processes (BPs) for the 20 pathways with the highest significance (according to the p value order after correction), all GO-cellular components (CCs) and GO-molecular functions (MFs) are displayed. (C) Kyoto Encyclopedia of genes and genomes (KEGG) pathway enrichment of 192 activated B cell related DEGs. The 10 pathways with the highest significance type were selected. (D) Visualization for the predicted results of protein-protein interaction (PPI) network among 192 activated B cell-related DEGs via STRING and Cytoscape. Each node represents a protein, and each line refers an interaction. Line thickness indicates the strength of interaction. (E) PPI subnetwork of hub genes identified by cytohubba plug-in. Hub genes were colored from yellow to red with red being the most important. |
 |
Figure 6 Analysis of hub genes in aortic dissection (AD). (A) Receiver operating characteristic ROC curves of hub genes on determining AD patients from normal control. (B, C) Box plots showing the expressions of hub genes in training and validation cohorts. (D) Transcription factors-hub genes regulation network, in which rectangles in cyan indicate hub genes and diamonds in violet indicate transcription factors. The lines between rectangles and diamonds mean that they have regulatory relationship. (E) MiRNAs-hub genes regulation network, in which rectangles in cyan indicate hub genes and circles in Orange indicate miRNAs. The lines between rectangles and circles mean that they have regulatory relationship. Abbreviations: KDR, kinase insert domain receptor; IGF1, insulin-like growth factor 1; NOS3, nitric oxide synthase 3; PECAM1, platelet and endothelial cell adhesion molecule 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; FLT1, Fms-related receptor tyrosine kinase 1; DLL4, delta-like canonical notch ligand 4; CDH5, cadherin 5; VWF, von Willebrand factor; TEK, TEK receptor tyrosine kinase. |
VWF Plays a Key Role in AD, Maybe via Regulating Loss of Smooth Muscle Cells
We first observed the histopathological characteristics of our clinical AD samples, including elevated deposition of total collagens (Figure 7A) and loss of smooth muscle cells (Figure 7B and 7C) during dissection. Next, we performed qPCR to validate the aberrant expression of hub genes in clinical samples. The results showed that VWF mRNA expression was significantly reduced in AD samples (Figure 7D), which was consistent with the RNA sequencing results in GSE153434. Moreover, the protein level of VWF was also remarkably decreased in the AD samples (Figure 7E). In addition, immunohistochemistry revealed that the VWF-positive area in the dissection was much smaller than that in normal artery walls (Figure 7F). These results indicate that aberrant expression of VWF is associated with AD development. To further investigate the role of VWF in AD, we analyzed human single-cell sequencing data of AD sourced from GSE222328. After removing the unqualified cells, the remaining 96,001 cells were grouped into 29 subpopulations using tSNE (Figure 8A). Using standard marker genes of established cell lineages, these 29 subpopulations were annotated into 8 cell types, including B cells, neutrophils, macrophages, NK cells, monocytes, endothelial cells, smooth muscle cells, and T cells (Figure 8A). We found that ACTA2 expression was significantly decreased in AD samples, and ACTA2 was mainly expressed in smooth muscle cells (Figure 8B), indicating that ACTA2 is a marker for muscle cells in AD, which was verified by co-staining with another smooth muscle cell marker, SM22α, in Figure 7C. Thus, to determine the relationship between muscle cells and VWF, we analyzed the correlation between ACTA2 and VWF expression. Interestingly, we found a significant positive correlation between VWF and ACTA2 (p = 0.001, cor = 0.67) (Figure 8C). Moreover, immunofluorescent staining showed that VWF was co-localized with ACTA2 in aortic tissues, and again we found that VWF expression was significantly reduced in aortic tissues from AD patients (Figure 8D). Those findings suggested that VWF may be involved in the pathology of AD by affecting the loss of smooth muscle cells.
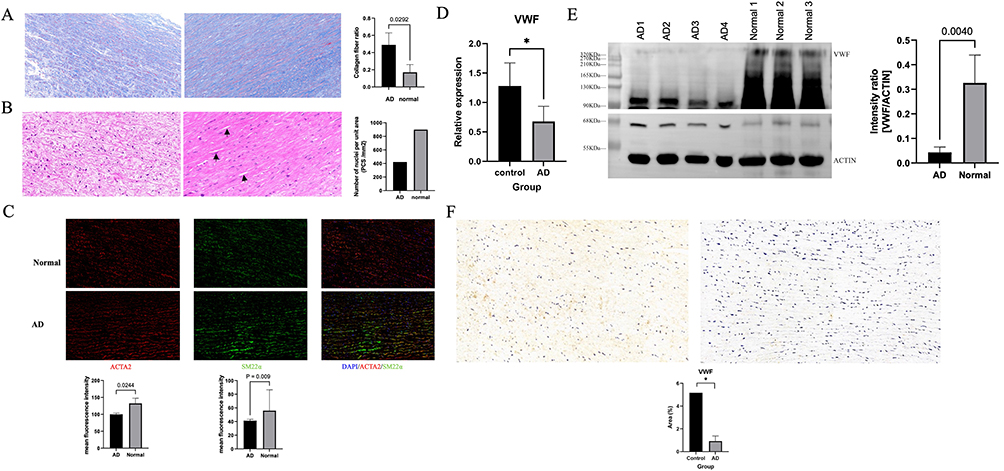 |
Figure 7 Validation of the expression of von Willebrand factor (VWF) in aortic dissection (AD). (A) Representative images of Masson staining in the aortic tissue from control (left) and AD patient (right). The collagens are stained blue and smooth muscle cells are stained red. In the AD (right), there was significantly elevated deposition of total collagens compared with controls (left). (B) Representative images of haematoxylin and eosin (HE) staining in the aortic tissue from control (left) and AD patient (right). In the control (left), the smooth muscle cells are evenly distributed and neatly arranged. In the AD (right), the disarrangement and the loss of smooth muscle cells are detected as indicated by black arrows. (C) Representative images of immunofluorescent staining of smooth muscle cells in aortic tissue from control and AD patients using specific marker proteins ACTA2 and SM22α. Nuclei (blue) were visualized with DAPI. ACTA2 (red) – actin alpha 2, smooth muscle, and SM22α (green) – smooth muscle protein 22 alpha were labelled with the respective antibodies. (D, E) Decreased VWF expressions in aortic tissues from AD patients were detected by PCR and Western blotting compared to controls. (F) Representative images of VWF immunohistochemistry in control (left) and AD (right) tissue. The staining for VWF was significantly decreased in AD. *p <0.05. |
 |
Figure 8 Analysis of the relationship between von Willebrand factor (VWF) and smooth muscle cells. (A) 29 cell clusters were identified from single-cell sequencing data of human aortic dissection (AD). Subsequently, 29 cell clusters were annotated into 8 cell types, including B cells, neutrophils, macrophages, NK cells, monocytes, endothelial cells, smooth muscle cells, and T cells. (B) Actin alpha 2, smooth muscle (ACTA2) was mainly expressed in smooth muscle cells, and its expression was significantly decreased in AD at single-cellular level. (C) The expression of VWF was significantly correlated with ACTA2 in human aortic tissues. (D) Representative images of immunofluorescent staining of VWF in aortic tissues from control and AD patients. Nuclei (blue) were visualized with DAPI. ACTA2 (red) – actin alpha 2, smooth muscle, and VWF (green) – von Willebrand factor were labelled with the respective antibodies. The images showed that VWF was co-localized with ACTA2, the marker of smooth muscle cells, in the aortic tissues. Again, the expressions of both VWF and ACTA2 were significantly reduced in aortic tissues from AD patients compared to controls. ****p <0.0001. |
Discussion
To date, few medical treatments are available for the prevention or treatment of AD. Therefore, clarifying the pathological mechanism of AD, determining the risk factors, early detection and diagnosis, and active prevention are important for improving the survival rate and prognosis of AD. Several studies have identified hub genes in AD using bioinformatic strategies.13–16 For example, Li et al calculated the immune scores of AD patients and identified key immune related genes by WGCNA and PPI analyses.14 Zhong et al16 and Luo et al17 first identified AD associated genes via WGCNA and then calculated their correlations with immune cells. In addition, Zeng et al18 and Ren et al19 performed mass spectrometry to identify biomarkers in the serum of AD patients via differential expression and correlation analyses. However, to the best of our knowledge, information directly on the role of infiltrated immune cells in AD pathogenesis and diagnosis using aortic tissues is still lacking. Thus, we first identified key immune cells involved in AD and performed a bioinformatic study directly focusing on identify core differentially expressed genes associated with key immune cells in AD.
In the current study, bulk sequencing results showed that infiltration of B cells (activated and immature) was significantly altered in the AD immune microenvironment. In addition, by analyzing single-cell sequencing data, we found that B cells were one of the main cell types in AD. Thus, both bulk and single-cell sequencing results indicate an important role of B cells in AD. Thereafter, using WGCNA, we identified 192 DEGs associated with activated B cells, which secrete antibodies and cytokines to regulate vascular immune and inflammation response.20,21 To further investigate the function of these activated B cell-related DEGs. We performed GO and KEGG pathway enrichment analyses and found that they were mainly enriched in cell migration and the PI3K-Akt signaling pathway. It has been reported that migration, proliferation, and phenotypic switching of smooth muscle cells contribute to the alteration of the structure and function of smooth muscle cells, thus leading to AD formation.22–24 PI3K-Akt signaling plays an important role in phenotypic switching and apoptosis of human aortic vascular smooth muscle cells.25–27 Those studies together with our results suggest that activated B cell-related DEGs may regulate the migration, apoptosis, and phenotype switch of smooth muscle cells to further affect the development of AD.
Next, using the PPI network and ROC curves, we screened 10 hub activated B-cell-related DEGs, including KDR, IGF1, NOS3, PEAM1, GAPDH, FLT1, DLL4, CDH5, TEK, and VWF. KDR, also known as vascular endothelial growth factor receptor 2 (VEGFR2), is mainly expressed on vascular endothelial cells. It binds to VEGFs to regulate the survival, proliferation and migration of endothelial cells.28 Many studies have shown its importance in cardiomyocyte growth,25 vasculogenesis, normal and pathological angiogenesis.29,30 In addition, KDR could form complexes with adhesion molecules, which destabilize endothelial junctions and increase vascular permeability when PI3K and p38 MAPK signaling are activated.31 However, the exact function of KDR in AD remains unclear. Wang et al have reported that knockdown of osteoglycin promotes the proliferation and migration of vascular smooth muscle cells in AD via VEGF/VEGFR2.32 IGF1 plays a critical role in regulating behaviors of endothelial cells, such as migration and tube formation, by binding to its receptors. In cancers and eye diseases, IGF1 has shown to be involved in angiogenesis.33,34 In addition, a recent study on atherosclerosis showed that IGF1 pathway could regulate the inflammation and proliferation of vascular smooth muscle cells.35 In AD-related disease, IGF1 has been reported to be positively correlated with an increased aortic diameters36 in Turner syndrome, which is a common cause of AD. NOS3 is a key player in governing angiogenesis, and its aberrant expression will result in the dysfunction of vascular endothelium.37 In addition, NO generated by endothelial NOS3 spread into vascular smooth muscle cells, thus leading to relaxation of smooth muscle cells and vasodilation.38 Özmen et al found higher expression of NOS3 intron 4b/4b in AD than in the control group.39 About 13% of NOS3 knockout mice had spontaneous AD in the sinotubular junction,40 indicating the important role of NOS3 in AD development. FLT1, also known as VEGFR1 is expressed in endothelial cells, vascular smooth muscle cells and some other non-endothelial cells. Like KDR, FLT1 plays an important role in the vasculogenesis and angiogenesis process via regulating the differentiation, migration and proliferation of endothelial cells and migration of immune cells.29,30 Additionally, it can bind with VEGF-A to activate cardiomyocytes.41 However, its role in AD has not been studied yet. PECAM1 is a key endothelial adhesion molecule, which is involved in endothelial force sensing.42 In a recent study from myocardial ischemia mice model, Chu et al found that the survival of cardiomyocytes may be associated with decreased PECAM1 in endothelial cells, which induced the permeability of endothelial cells and increased blood infiltration in subendocardial area.43 CDH5, also known as VE-cadherin, participates in maintaining the integrity of endothelial and vascular tissues via cell adhesion.44 A single-cell study on AD identified CDH5 as a core gene in smooth muscle cells,45 but its role in AD is unclear. DLL4 is an important regulator in angiogenesis and vascular morphogenesis by binding to Notch receptors.46 Multiple studies in cancers showed that DLL4/Notch signaling may interact with VEGF signaling to modulate tumor angiogenesis.46,47 Zou et al observed a significant reduction in the expression of DLL4 in vascular smooth muscle cells from thoracic AD samples.48 As for TEK, studies have shown that its mutation is associated with venous malformations,49,50 but its role in AD has not been identified. VWF, a large glycoprotein, is mainly secreted by endothelial cells and stored in Weibel-Palade bodies.51 The interactions of VWF with other cellular and extracellular proteins make it able to regulate blood clotting, would healing, arteriogenesis, leukocyte infiltration and angiogenesis.52 In addition, another factor affecting the function of VWF is its multimeric structure, which ranges from single dimer to 20 dimers.51 In preoperative acute type A AD patients, the activity of VWF was remarkably higher than control group.53 Taken together, hub genes identified in our study is involved in the cellular behaviors of epithelial cells and/ or vascular smooth muscle cells, which play important roles in angiogenesis and vasculogenesis. However, the precise molecular mechanisms of how they regulate AD development have not been studied or clarified.
To validate the bioinformatic results, we performed qPCR, Western blotting, and immunohistochemistry. Only VWF was successfully validated at both mRNA and protein levels, which were all decreased in AD. As mentioned above, VWF is mainly secreted by endothelial cells, we are just wondering if the decreased VWF expression is caused by endothelial cell dysfunction in AD. In addtion, we found that its expression was significantly correlated with the marker of smooth muscle cells, ACTA2. Qin et al reported that VWF treatment promotes smooth muscle cell proliferation in a dose-dependent manner.54 Further investigations by another group revealed that VWF may regulate smooth muscle cell proliferation via VWF/LRP4/αVβ3 axis.55 In addition, VWF was found to inhibit Notch signaling, which could further lead to the loss of smooth muscle cells.56 However, the correlation between VWF and ACTA2 found in the current study did not imply causation. Further work is required to determine what cause the decrease of VWF expression and whether VWF regulates smooth muscle cell loss in AD in the same manner as previously reported or depend other molecules and signaling. In consideration of our findings and other reports, it can be anticipated that VWF is involved in the pathogenesis of AD, at least via affecting the loss of smooth muscle cells.
Our study has several limitations. First, our study is mainly based on public transcriptome datasets, which are relatively small sample sizes, and may affect the reliability of the model and miss some biomarkers. Larger clinical samples from our own institution and external cohorts from another institution should be collected to determine and validate the diagnostic performance and clinical utility of hub genes, especially VWF. Second, in vitro and in vivo experiments are needed to investigate the exact molecular mechanism by which VWF regulates loss of smooth muscle cells obtained from patients with AD and also the relationship between infiltrated activated B cells and altered VWF expression in AD requires further clarification. In addition, further studies are required to determine whether those hub genes are specific for AD or shared by aortic aneurysm to better evaluate their clinical utility. Thus, based on above limitations, we plan to (1) collect fresh samples at different stages (acute, subacute and chronic) of AD to examine the expressions and locations of VWF by using qPCR and immunofluorescence and to detect whether there is endothelial cell dysfunction by immunofluorescence; (2) seek collaboration from other institutions to obtain a large external cohort to validate the diagnostic value of VWF in AD; (3) overexpress and knock down of VWF in aortic smooth muscle cells to evaluate its effect on the proliferation and survival of smooth muscle cells in vitro; (4) construct VWF-deficient mouse model to examine the role of VWF in immune cell infiltration and smooth muscle cell loss in vivo.
Conclusion
This study identified ten hub immune cell-related genes involved in AD by differential expression, WGCNA and PPI network analysis. Among them, by experimental validation, VWF was found to be colocalized with the marker of smooth muscle cells, ACTA2, and was significantly correlated with ACTA2 expression in AD. Those findings provide a basis to further investigate the role of VWF in AD’s smooth muscle cell loss, thus enriching our understanding of AD pathogenesis and also diagnosis, prevention and VWF targeted treatment for AD patients.
Abbreviations
DIIC, differentially infiltrated immune cells; AD, aortic dissection; GEO, gene expression omnibus; DEGs, differentially expressed genes; WGCNA, weighted gene coexpression network analysis; PPI, protein-protein interaction; CTA, computed tomographic angiography; MM, module membership; GS, gene significance; TF, transcription factor; H&E, hematoxylin and eosin; KDR, kinase insert domain receptor; IGF1, insulin-like growth factor 1; NOS3, nitric oxide synthase 3; PECAM1, platelet and endothelial cell adhesion molecule 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; FLT1, Fms-related receptor tyrosine kinase 1; DLL4, delta-like canonical notch ligand 4; CDH5, cadherin 5; VWF, von Willebrand factor; TEK, TEK receptor tyrosine kinase.
Data Sharing Statement
The datasets used in this study were downloaded from the GEO database. Further inquiries can be directed to the corresponding authors.
Ethics Approval and Informed Consent
This study was conducted in accordance with the principles of the Declaration of Helsinki. This study was approved by the Ethics Committee of the Second Hospital of Shanxi Medical University. Informed consent was obtained from all subjects involved in the study.
Acknowledgments
The authors thank BIOINFOR-MEDICAL CENTRE at the Second Hospital of Shanxi Medical University for their academic instruction and manuscript revision.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rylski B, Schilling O, Czerny M. Acute aortic dissection: evidence, uncertainties, and future therapies. Eur Heart J. 2023;44(10):813–821. doi:10.1093/eurheartj/ehac757
2. Pape LA, Awais M, Woznicki EM, et al. Presentation, diagnosis, and outcomes of acute aortic dissection: 17-year trends from the International Registry of Acute Aortic Dissection. Journal of the American College of Cardiology. 2015;66:350–358. doi:10.1016/j.jacc.2015.05.029
3. Juraszek A, Czerny M, Rylski B. Update in aortic dissection. Trends Cardiovasc Med. 2021;32:456–461. doi:10.1016/j.tcm.2021.08.008
4. Shen YH, LeMaire SA, Webb NR, et al. Aortic aneurysms and dissections series. Arteriosclerosis Thrombosis Vasc Biol. 2020;40:e37–e46. doi:10.1161/ATVBAHA.120.313991
5. Li S, Li J, Cheng W, et al. Independent and Interactive Roles of Immunity and Metabolism in Aortic Dissection. Int J Mol Sci. 2023;24:15908. doi:10.3390/ijms242115908
6. Liu J, Yang Y, Liu X, et al. Macrophage-biomimetic anti-inflammatory liposomes for homing and treating of aortic dissection. J Control Release. 2021;337:224–235. doi:10.1016/j.jconrel.2021.07.032
7. Liu F, Wei T, Liu L, et al. Role of Necroptosis and Immune Infiltration in Human Stanford Type A Aortic Dissection: novel Insights from Bioinformatics Analyses. Oxid Med Cell Longev. 2022;2022:6184802. doi:10.1155/2022/6184802
8. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:1–15. doi:10.1186/1471-2105-14-7
9. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):1–13. doi:10.1186/1471-2105-9-559
10. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–D646. doi:10.1093/nar/gkac1000
11. Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014;8:1–7. doi:10.1186/1752-0509-8-S4-S11
12. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
13. Ainiwan M, Wang Q, Yesitayi G, et al. Identification of FERMT1 and SGCD as key marker in acute aortic dissection from the perspective of predictive, preventive, and personalized medicine. EPMA J. 2022;13:597–614. doi:10.1007/s13167-022-00302-4
14. Li Z, Wang J, Yu Q, et al. Identification of Immune-Related Gene Signature in Stanford Type A Aortic Dissection. Front Genet. 2022;13:911750. doi:10.3389/fgene.2022.911750
15. Pan H, Lu W, Liu Z, et al. Identification of ferroptosis-associated biomarkers in Stanford type A aortic dissection based on machine learning. Am J Transl Res. 2023;15:3092–3114.
16. Zhong A, Cai Y, Zhou Y, et al. Identification and Analysis of Hub Genes and Immune Cells Associated with the Formation of Acute Aortic Dissection. Comput Math Methods Med. 2023;2023:8072369. doi:10.1155/2023/8072369
17. Luo J, Shi H, Ran H, et al. Identification of key biomarkers and immune infiltration in the thoracic acute aortic dissection by bioinformatics analysis. BMC Cardiovascular Disorders. 2023;23:75. doi:10.1186/s12872-023-03110-4
18. Zeng Q, Rong Y, Li D, et al. Identification of serum biomarker in acute aortic dissection by global and targeted metabolomics. Ann Vasc Surg. 2020;68:497–504. doi:10.1016/j.avsg.2020.06.026
19. Ren Y, Tang Q, Liu W, et al. Serum biomarker identification by mass spectrometry in acute aortic dissection. Cell. Physiol. Biochem. 2018;44:2147–2157. doi:10.1159/000485954
20. Treanor B. B-cell receptor: from resting state to activate. Immunology. 2012;136(1):21–27. doi:10.1111/j.1365-2567.2012.03564.x
21. Raza IGA, Clarke AJ. B Cell Metabolism and Autophagy in Autoimmunity. Front Immunol. 2021;12:681105. doi:10.3389/fimmu.2021.681105
22. An Z, Liu Y, Song ZG, et al. Mechanisms of aortic dissection smooth muscle cell phenotype switch. J Thorac Cardiovasc Surg. 2017;154:1511–1521. doi:10.1016/j.jtcvs.2017.05.066
23. Wang Z, Zhuang X, Chen B, et al. Osteoglycin knockdown promotes vascular smooth muscle cell proliferation and migration in aortic dissection via the VEGF/VEGFR2 axis. Mol Med Rep. 2021;23:65. doi:10.3892/mmr.2020.11703
24. Zhang K, Qi Y, Wang M, et al. Long non-coding RNA HIF1A-AS2 modulates the proliferation, migration, and phenotypic switch of aortic smooth muscle cells in aortic dissection via sponging microRNA-33b. Bioengineered. 2022;13:6383–6395. doi:10.1080/21655979.2022.2041868
25. Zhu SB, Zhu J, Zhou ZZ, et al. TGF-β1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am J Transl Res. 2015;7:2764–2774.
26. Wang L, Wang Z, Zhang R, et al. MiR-4787-5p Regulates Vascular Smooth Muscle Cell Apoptosis by Targeting PKD1 and Inhibiting the PI3K/Akt/FKHR Pathway. J Cardiovasc Pharmacol. 2021;78(2):288–296. doi:10.1097/FJC.0000000000001051
27. Xie X, Hong X, Hong S, et al. Progression of Thoracic Aortic Dissection Is Aggravated by the hsa_circ_0007386/miR-1271-5P/IGF1R/AKT Axis via Induction of Arterial Smooth Muscle Cell Apoptosis. Biomedicines. 2023;11(2):571. doi:10.3390/biomedicines11020571
28. Smith RO, Ninchoji T, Gordon E, et al. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. Elife. 2020;9:e54056. doi:10.7554/eLife.54056
29. Bowler E, Oltean S. Alternative Splicing in Angiogenesis. Int J Mol Sci. 2019;20(9):2067. doi:10.3390/ijms20092067
30. Melincovici CS, Boşca AB, Şuşman S, et al. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 2018;59:455–467.
31. Cebe-Suarez S, Zehnder-Fjällman A, Ballmer-Hofer K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 2006;63:601–615. doi:10.1007/s00018-005-5426-3
32. Wang Z, Zhuang X, Chen B, et al. Osteoglycin knockdown promotes vascular smooth muscle cell proliferation and migration in aortic dissection via the VEGF/VEGFR2 axis. Mol Medicine Rep. 2021;23:1.
33. Seo SH, Hwang SY, Hwang S, et al. Hypoxia‐induced ELF3 promotes tumor angiogenesis through IGF1/IGF1R. EMBO Rep. 2022;23:e52977. doi:10.15252/embr.202152977
34. Truong T, Silkiss RZ. The Role of Insulin-like Growth Factor-1 and Its Receptor in the Eye: a Review and Implications for IGF-1R Inhibition. Ophthalmic Plastic Reconstructive Surg. 2023;39(1):4–12. doi:10.1097/IOP.0000000000002146
35. Li J, Li Y, Yuan X, et al. The effective constituent puerarin, from Pueraria lobata, inhibits the proliferation and inflammation of vascular smooth muscle in atherosclerosis through the miR-29b-3p/IGF1 pathway. Pharm Biol. 2023;61:1–11. doi:10.1080/13880209.2022.2099430
36. Uçar A, Tuğrul M, Erol BO, et al. Determinants of Increased Aortic Diameters in Young Normotensive Patients With Turner Syndrome Without Structural Heart Disease. Pediatr Cardiol. 2018;39(4):786–793. doi:10.1007/s00246-018-1821-z
37. Liu K, Chen B, Zeng F, et al. ApoE/NOS3 Knockout Mice as a Novel Cardiovascular Disease Model of Hypertension and Atherosclerosis. Genes. 2022;13(11):1998. doi:10.3390/genes13111998
38. Pautz A, Li H, Kleinert H. Regulation of NOS expression in vascular diseases. Front Bioscience-Landmark. 2021;26:85–101.
39. Özmen R, Tunçay A, Şener EF, et al. Relationship of genetic factors with development of aortic dissection and aneurysm. Turk Gogus Kalp Damar Cerrahisi Derg. 2018;26:557–564. doi:10.5606/tgkdc.dergisi.2018.16424
40. Peterson JC, Wisse LJ, Wirokromo V, et al. Disturbed nitric oxide signalling gives rise to congenital bicuspid aortic valve and aortopathy. Dis Model Mech. 2020;13:dmm044990. doi:10.1242/dmm.044990
41. Braile M, Marcella S, Cristinziano L, et al. VEGF-A in Cardiomyocytes and Heart Diseases. Int J Mol Sci. 2020;21(15):5294. doi:10.3390/ijms21155294
42. Chuntharpursat-Bon E, Povstyan OV, Ludlow MJ, et al. PIEZO1 and PECAM1 interact at cell-cell junctions and partner in endothelial force sensing. Commun. Biol. 2023;6(1):358. doi:10.1038/s42003-023-04706-4
43. Chu Q, Song X, Xiao Y, et al. Alteration of endothelial permeability ensures cardiomyocyte survival from ischemic insult in the subendocardium of the heart. Exp Biol Med. 2023;248:1364–1372. doi:10.1177/15353702231194344
44. Li Y, Lv J. A comprehensive pan-cancer analysis of CDH5 in immunological response. Front Immunol. 2023;14:1239875. doi:10.3389/fimmu.2023.1239875
45. Xu C, Liu X, Fang X, et al. Single-cell RNA sequencing reveals smooth muscle cells heterogeneity in experimental aortic dissection. Front Genetics. 2022;13:836593. doi:10.3389/fgene.2022.836593
46. Park S, Kim J, Jang W, et al. Clinicopathologic significance of the delta-like ligand 4, vascular endothelial growth factor, and hypoxia-inducible factor-2α in gallbladder cancer. J Pathol Translational Med. 2023;57:113–122. doi:10.4132/jptm.2023.02.01
47. Xia S, Menden HL, Mabry SM, et al. HDAC6 and ERK/ADAM17 Regulate VEGF-Induced NOTCH Signaling in Lung Endothelial Cells. Cells. 2023;12(18):2231. doi:10.3390/cells12182231
48. Zou S, Ren P, Nguyen M, et al. Notch signaling in descending thoracic aortic aneurysm and dissection. PLoS One. 2012;7:e52833. doi:10.1371/journal.pone.0052833
49. Ye C, Pan L, Huang Y, et al. Somatic mutations in exon 17 of the TEK gene in vascular tumors and vascular malformations. J Vascular Surg. 2011;54:1760–1768. doi:10.1016/j.jvs.2011.06.098
50. Mahajan P, Bergstrom KL, Phung TL, et al. The genetics of vascular birthmarks. Clin Dermatol. 2022;40:313–321. doi:10.1016/j.clindermatol.2022.02.006
51. Randi AM, Smith KE, Castaman G. von Willebrand factor regulation of blood vessel formation. Blood J Am Soc Hematol. 2018;132:132–140.
52. Laboyrie SL, de Vries MR, de Jong A, et al. von Willebrand factor: a central regulator of arteriovenous fistula maturation through smooth muscle cell proliferation and outward remodeling. J Am Heart Assoc. 2022;11:e024581. doi:10.1161/JAHA.121.024581
53. Zindovic I, Sjögren J, Bjursten H, et al. The role of von Willebrand factor in acute type A aortic dissection and aortic surgery. Thromb Res. 2019;178:139–144. doi:10.1016/j.thromres.2019.04.018
54. Qin F, Impeduglia T, Schaffer P, et al. Overexpression of von Willebrand factor is an independent risk factor for pathogenesis of intimal hyperplasia: preliminary studies. J Vascular Surg. 2003;37:433–439. doi:10.1067/mva.2003.63
55. Lagrange J, Worou ME, Michel JB, et al. The VWF/LRP4/αVβ3-axis represents a novel pathway regulating proliferation of human vascular smooth muscle cells. Cardiovascular Res. 2022;118:622–637. doi:10.1093/cvr/cvab042
56. Meng H, Zhang X, Lee SJ, et al. Von Willebrand factor inhibits mature smooth muscle gene expression through impairment of Notch signaling. PLoS One. 2013;8:e75808. doi:10.1371/journal.pone.0075808
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.