Back to Journals » Journal of Inflammation Research » Volume 13
Identification of Key Genes Associated with Changes in the Host Response to Severe Burn Shock: A Bioinformatics Analysis with Data from the Gene Expression Omnibus (GEO) Database
Authors Fang X, Duan SF, Gong YZ, Wang F, Chen XL ![]()
Received 18 September 2020
Accepted for publication 13 November 2020
Published 1 December 2020 Volume 2020:13 Pages 1029—1041
DOI https://doi.org/10.2147/JIR.S282722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Xiao Fang,* Shu-Fang Duan,* Yu-Zhou Gong,* Fei Wang, Xu-Lin Chen
Department of Burns, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xu-Lin Chen
Department of Burns, The First Affiliated Hospital of Anhui Medical University, 120 Wanshui Road, Hefei, Anhui 230088, People’s Republic of China
Tel/Fax +86-551-65908495
Email [email protected]
Background: Patients with severe burns continue to display a high mortality rate during the initial shock period. The precise molecular mechanism underlying the change in host response during severe burn shock remains unknown. This study aimed to identify key genes leading to the change in host response during burn shock.
Methods: The GSE77791 dataset, which was utilized in a previous study that compared hydrocortisone administration to placebo (NaCl 0.9%) in the inflammatory reaction of severe burn shock, was downloaded from the Gene Expression Omnibus (GEO) database and analyzed to identify the differentially expressed genes (DEGs). Functional enrichment analyses of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were performed. The protein–protein interaction (PPI) network of DEGs was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING) database and then visualized in Cytoscape. In addition, important modules in this network were selected using the Molecular Complex Detection (MCODE) algorithm, and hub genes were identified in cytoHubba, a Cytoscape plugin.
Results: A total of 1059 DEGs (508 downregulated genes and 551 upregulated genes) were identified from the dataset. The DEGs enriched in GO terms and KEGG pathways were related to immune response. The PPI network contained 439 nodes and 2430 protein pairs. Finally, important modules and hub genes were identified using the different Cytoscape plugins. The key genes in burn shock were identified as arginase 1 (ARG1), cytoskeleton-associated protein (CKAP4), complement C3a receptor (C3AR1), neutrophil elastase (ELANE), gamma-glutamyl hydrolase (GGH), orosomucoid (ORM1), and quiescin sulfhydryl (QSOX1).
Conclusion: The DEGs, functional terms and pathways, and hub genes identified in the present study can help shed light on the molecular mechanism underlying the changes in host response during burn shock and provide potential targets for early detection and treatment of burn shock.
Keywords: burn shock, in silico study, host response, immune response
Introduction
With the development of the economy and improvements in public safety awareness, the incidence of burn in less-developed and developing areas is gradually decreasing.1 However, burns are still the fourth most common type of trauma worldwide, after traffic injuries, falls, and interpersonal violence, with up to 90% of the burns occurring in low- and middle-income areas.2 According to an estimate released by the World Health Organization in 2018, 18,000 burn-related deaths occur annually.3 Burn injuries are associated with a high mortality rate in patients with severe burn shock which is a combination of distributive and hypovolemic shock accompanied by immune and inflammatory responses, and metabolic changes.4 Hence, the state of shock experienced following burn injuries can be a challenge in patient management and may result in multiple organ failure.5 Notably, the number of deaths within the burn shock period has been greatly reduced through improvements in the available treatment and resuscitation procedures for burn injuries.6–8 However, the state of burn shock is associated with numerous complications, as patients experiencing an unstable period of shock will have a greatly weakened defense and repair system due to tissue ischemia, hypoxia, and reperfusion injury that accompany the state of shock. As a result, these patients are at a greater risk of experiencing concurrent severe systemic infections and multiple organ failure, which are currently the primary cause of death in severe burn cases.9,10 Therefore, exploring the precise mechanism that underlies the host response to severe burn shock is crucial.
Numerous studies have indicated that gene expression plays an important role in different burn stages. It has been reported that the expression of high-mobility group box protein 1 (HMGB1) is elevated in burn patients and has an important impact on the immune function of postburn patients.11 Carter found that the melanocortin 1 receptor (MC1R) gene polymorphism was associated with wound infection after burn injury.12 In the repair stage after burn injury, nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3 (NLRP3) promote wound healing through regulation of macrophage polarization and inflammation.13 However, there has been little information regarding the important genes expressed during burn shock. Given the popularity of gene detection technology, gene chips are widely used in research on many diseases.14 Bioinformatic analysis of the data generated from the use of gene chips has become a promising and effective tool for screening the significant genetic or epigenetic variations associated with the development and progression of diseases.15,16 The differentially expressed genes (DEGs), signaling pathways, and hub genes that are potentially related to the host response to burn shock have not been identified. Moreover, to our knowledge, no studies have been performed to analyze the DEGs, enrichment analysis of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and protein–protein interaction (PPI) networks associated with burn shock using datasets obtained from the Gene Expression Omnibus (GEO) database.
This study, to our knowledge, is the first to identify 1059 DEGs by screening the gene expression dataset GSE77791, which included 30 samples from burn shock patients and 13 samples from healthy volunteers. The analyzed biological functions, signal pathways, constructed PPI network, and selected hub genes were used to understand the molecular mechanism underlying the host response to burn shock, which may potentially be insightful for studying burn shock, including early detection and treatment.
Materials and Methods
Gene Expression Microarray Data
We acquired the microarray expression profiles of severe burn shock patients from the National Center for Biotechnology Information GEO database (http://www.ncbi.nlm.nih.gov/geo), which contains high-throughput gene expression data, chips, and microarrays, under the accession number GSE77791. The GSE77791 dataset, based on GPL570 (a platform for Affymetrix Human Genome U133 Plus 2.0 Array), contains 117 samples, including 30 samples collected from patients with burn shock prior to any treatment and 13 samples collected from healthy volunteers that served as controls. These 43 samples were selected for deep analysis in this study in accordance with our aims. The remaining samples collected from patients with different medical conditions were excluded.
Data Preprocessing: Identification of DEGs
First, the probe sets were correspondingly converted into gene symbols by the platform with annotation information. The mean value of multiple probe sets was calculated if they corresponded to the same gene. The data were normalized, and DEGs were subsequently identified using the linear models for microarray data (LIMMA) package in R software (version 3.6.1). Individual p-values were calculated and converted to adjusted p-values (adj. p. val) for comparisons by false discovery rate correction of the Benjamini and Hochberg test. DEGs were selected with the commonly used thresholds of∣log2fold change (FC)> 1 and adj. p. val < 0.05. The heatmap and volcano plot of the DEGs were constructed using the ggplot2 package in R.
Functional and Pathway Enrichment Analyses of DEGs
The functions of up- and down-regulated DEGs in severe burn shock were predicted based on GO function and KEGG pathway enrichment analyses and visualized using the ClusterProfiler package in R. The GO database (http://www.geneontology.org) primarily comprises the following three categories: biological processes (BP), cellular components (CC), and molecular functions (MF). The KEGG database (http://www.genome.ad.jp/kegg/) collects systematic functional, genomic, and chemical information. The significance criterion to screen the significantly enriched GO terms and KEGG pathways was set at p <0.05.
PPI Network Construction and Module Analysis
Functional interactions between proteins were analyzed to shed light on the exact mechanisms of generation and development of diseases. In this study, the PPI network of DEGs was predicted in an online database (Search Tool for the Retrieval of Interacting Genes database, STRING, version 11.0, http://string-db.org) and subsequently visualized in Cytoscape (version 3.7.2). DEGs were uploaded onto the database, and single protein nodes were removed. A combined score > 0.9 was set as the cutoff criterion. The PPI network was downloaded and further analyzed in Cytoscape. The important modules were identified and visualized by Molecular Complex Detection (MCODE), which is a plugin in Cytoscape for discovering densely connected nodes in a given network. The module selection was in accordance with the following criteria: k-score=2, MCODE score > 10, degree cut-off=2, max depth=100, and node score cut-off=0.2.17 The nodes in the key modules are presented as highly connected proteins that have important biological functions. Moreover, the pathway enrichment analysis of the DEGs in selected modules was performed using the ClusterProfiler package.
Hub Gene Selection
The hub genes in the PPI network were characterized by the cytoHubba plugin in Cytoscape. The top 10 genes were identified by three methods (Maximal Clique Centrality (MCC), Degree, and Maximum Neighborhood Component (MNC)) and overlapped to identify the hub genes.
Results
Patients in GSE77791
Thirty-two patients were enrolled in Plassais’s study,18 however, two of them were discarded due to a major batch effect related to a technical issue. The demographics of the patients are shown in Table 1. Briefly, these patients were severely burned with a total burn surface area (TBSA) range from 30 to 98 and registered for hospitalization between 24 and 72 h after injury. Few data about the severity of the burns in the original article were obtained: the mean abbreviated burn severity index (ABSI) score was 11, 12 patients had inhalation injuries, and 3 patients had infections during hospitalization. The exact time points of injury were not recorded. Samples were immediately collected from hospitalized patients with severe burn shock, which was defined by a norepinephrine dosage above 0.5 µg/kg/min.
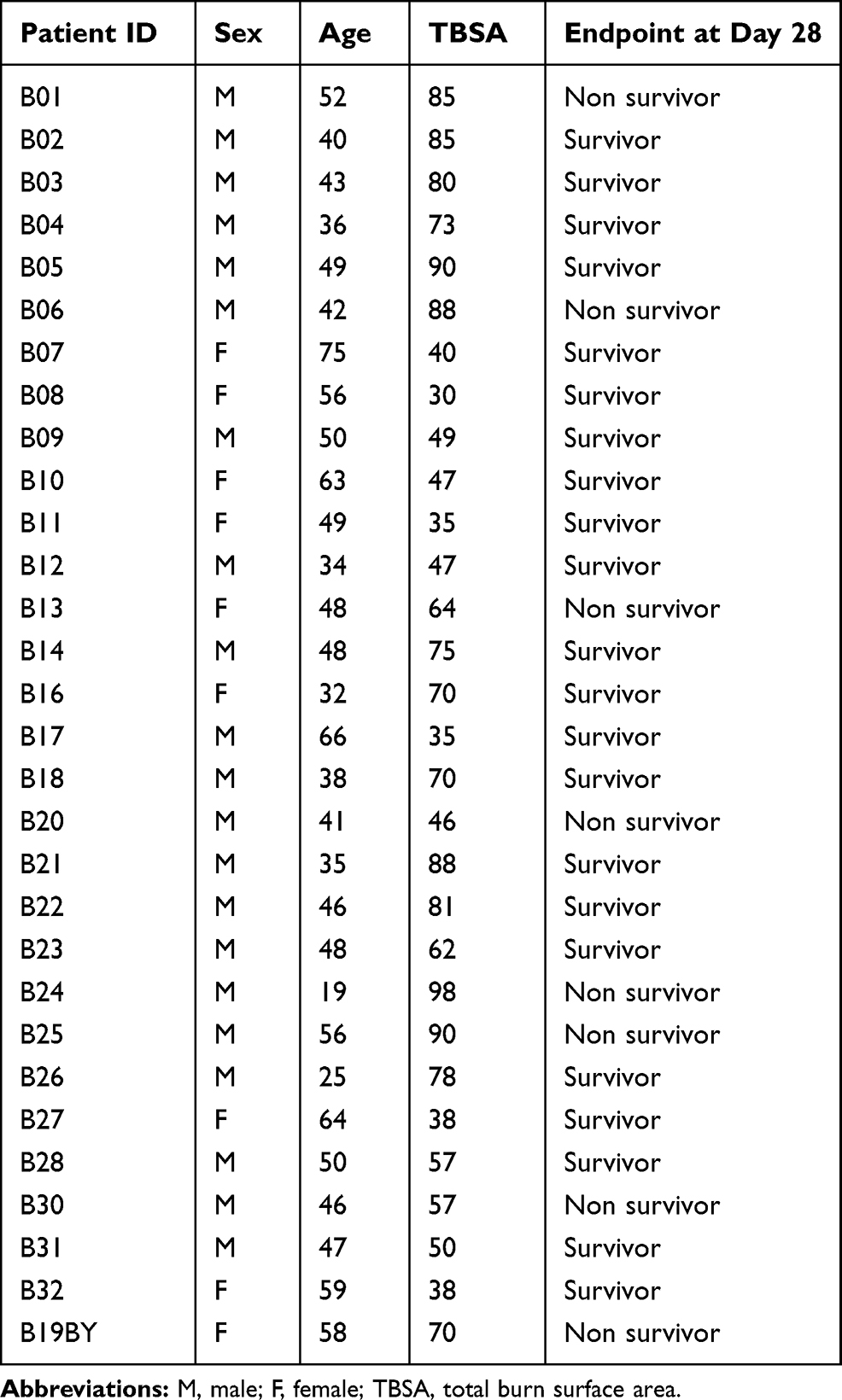 |
Table 1 Demographics of the Patients in Jonathan Plassais’s Study |
Identification of DEGs
The microarray data of 30 burn shock patients and 13 healthy volunteers from the GSE77791 dataset were analyzed to identify DEGs according to the following criteria:∣log2fold change (FC)∣> 1 and adj. p. val < 0.05. A total of 1059 DEGs from the samples of patients with burn shock were identified by comparing with the controls. This total included 508 downregulated genes and 551 upregulated genes, which are indicated in the volcano plot shown in Figure 1A. The top 100 DEGs of all samples were clustered and represented as a heatmap (Figure 1B).
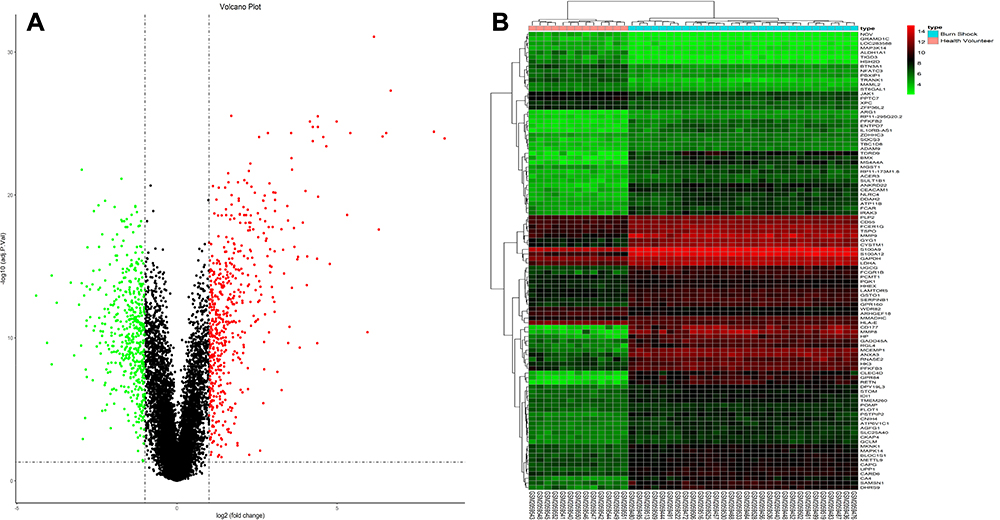 |
Figure 1 DEGs from GSE77791. (A) 1059 DEGs are showed in the volcano plot that containing 551 upregulated genes in red and 508 downregulated genes in green. (B) The heatmap shows the top 100 most significant DEGs. Red indicates a relatively high expression and blue indicates a relatively low expression. DEGs were identified by the criteria of∣log2fold change (FC)∣>1 and adj. p. val <0.05. DEGs, differentially expressed gene. |
Analyses of GO Function and KEGG Enrichment of DEGs
Analyses of GO function and KEGG enrichment were performed using the ClusterProfiler package to further understand the functions and mechanisms of the identified DEGs. The top eight enriched terms in BP, CC, and MF are shown in Figure 2A. For BP, the DEGs were enriched in neutrophil activation, neutrophil activation involved in immune response, neutrophil degranulation, neutrophil-mediated immunity, T cell receptor signaling pathway, T cell activation, T cell differentiation, and lymphocyte differentiation. In terms of CC, the DEGs were enriched in specific granules, tertiary granules, secretory granule lumen, tertiary granule lumen, specific granule lumen, secretory granule membranes, vesicle lumen, and cytoplasmic vesicle lumen. Regarding MF, the DEGs were enriched in carbohydrate binding, MHC protein complex binding, immunoglobulin binding, cytokine receptor activity, MHC class II protein complex binding, non-membrane spanning protein tyrosine kinase activity, cytokine binding, and chemokine binding.
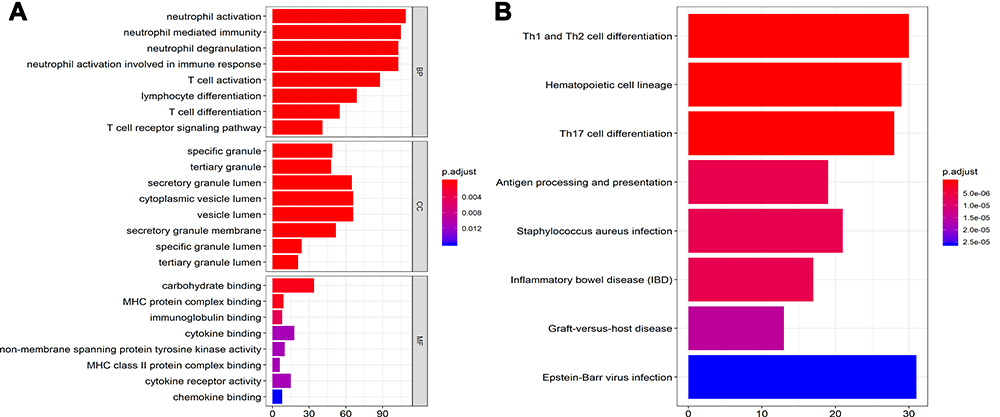 |
Figure 2 The GO terms and KEGG pathway enrichment analysis of all DEGs. (A) The top 8 terms enriched in BP, CC, and MF. (B) The top 8 enriched terms of KEGG pathway. Cutoff value is adj. p. val <0.05. GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, differentially expressed gene; BP, biological process; CC, cellular component; MF, molecular function. |
Based on KEGG pathway analysis, the DEGs were significantly enriched in Th1 and Th2 cell differentiation, hematopoietic cell lineage, antigen processing and presentation, Th17 cell differentiation, and Staphylococcus aureus infection (Figure 2B). The top six terms associated with the up- and down-regulated DEGs, enriched in BP, CC and MF as well as various KEGG pathways are shown in Tables 2 and 3, respectively. The details of the enriched DEGs in BP category of GO analysis and KEGG pathways are shown in Supplementary Tables 1 and 2, respectively.
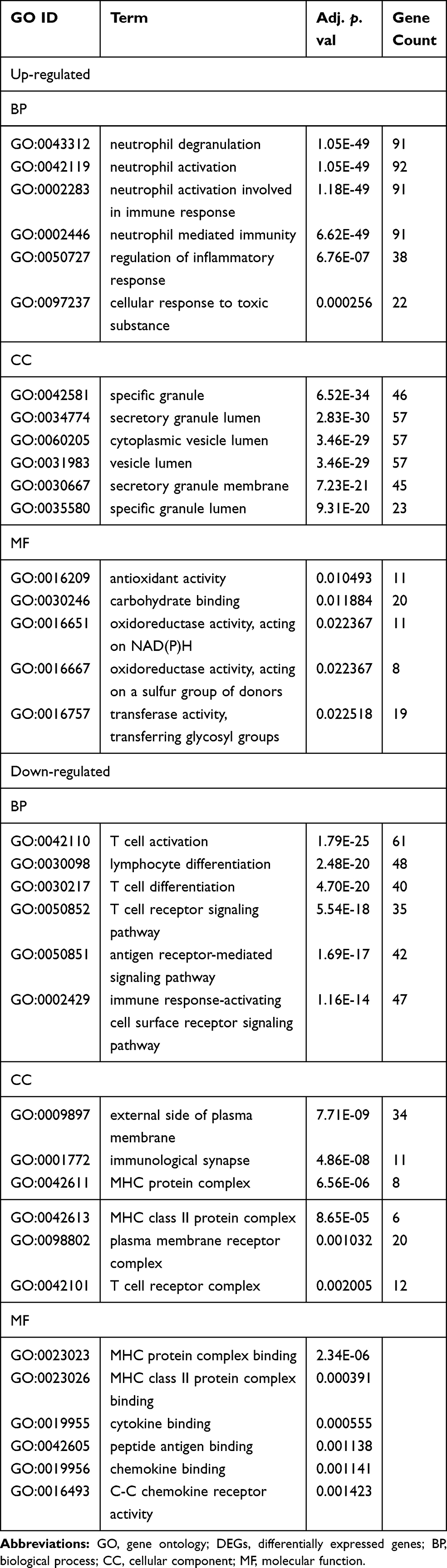 |
Table 2 GO Terms Enrichment Analysis of DEGs (Top 6 of Each) |
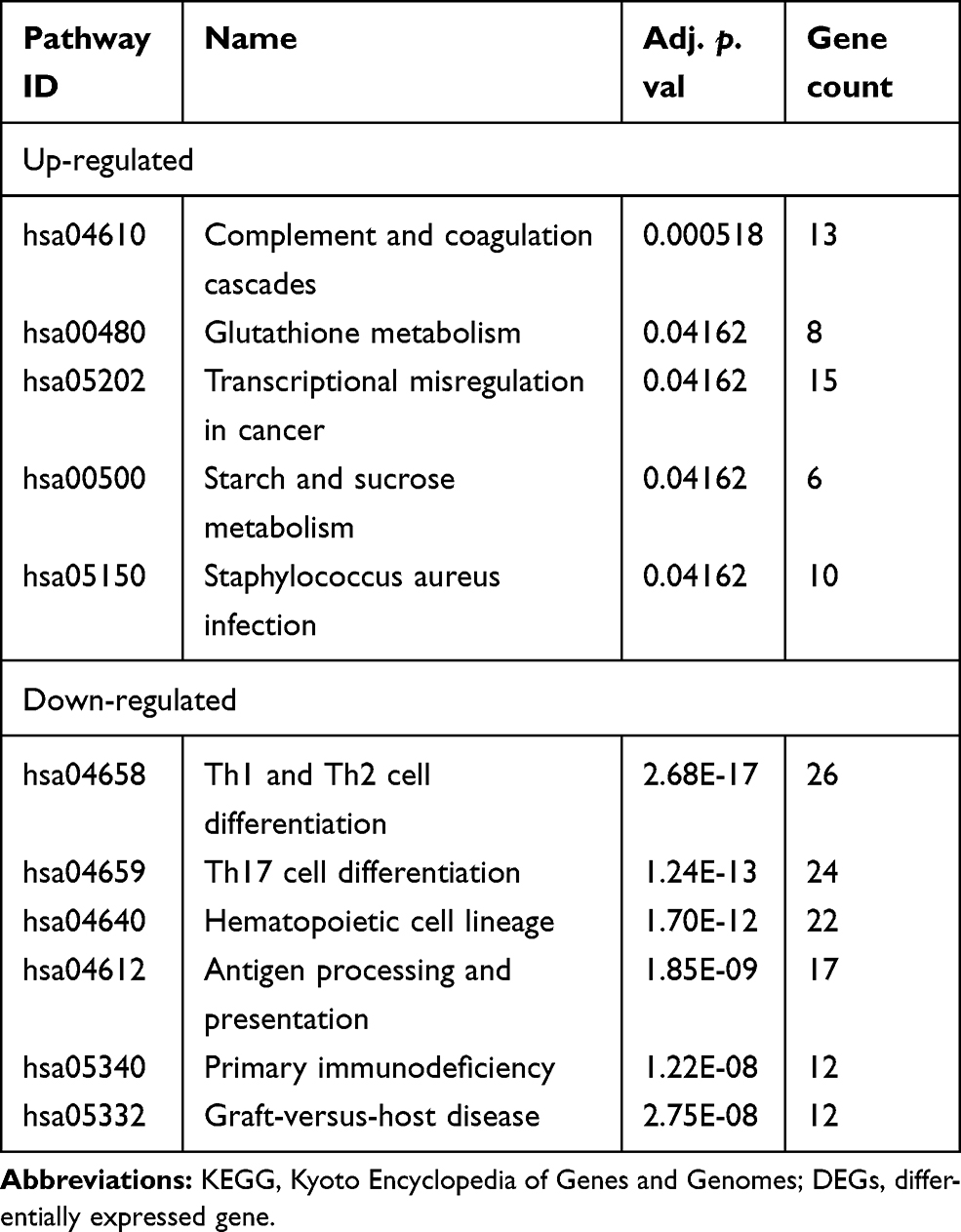 |
Table 3 KEGG Pathway Enrichment Analysis of DEGs (Top 6 of Each) |
PPI Network Construction and Module Analysis
The interactions between DEGs, which consisted of 439 nodes and 2430 edges (Figure 3A), were downloaded from the STRING database and visualized using Cytoscape. Four significant modules (Figure 3B) were identified by cluster analyzing the PPI network in MCODE based on the aforementioned criteria. KEGG pathway enrichment analysis was then performed for the DEGs in the four modules, and the results are listed in Table 4. The DEGs in module 1 were significantly enriched in S. aureus infections. The DEGs in module 2 were enriched in chemokine signaling pathways, viral protein interactions with cytokines, and cytokine receptors. The DEGs in module 3 were mainly enriched in ubiquitin-mediated proteolysis. Finally, the DEGs in module 4 were enriched in Th1 and Th2 cell differentiation, Th17 cell differentiation, primary immunodeficiencies, and T cell receptor signaling pathways.
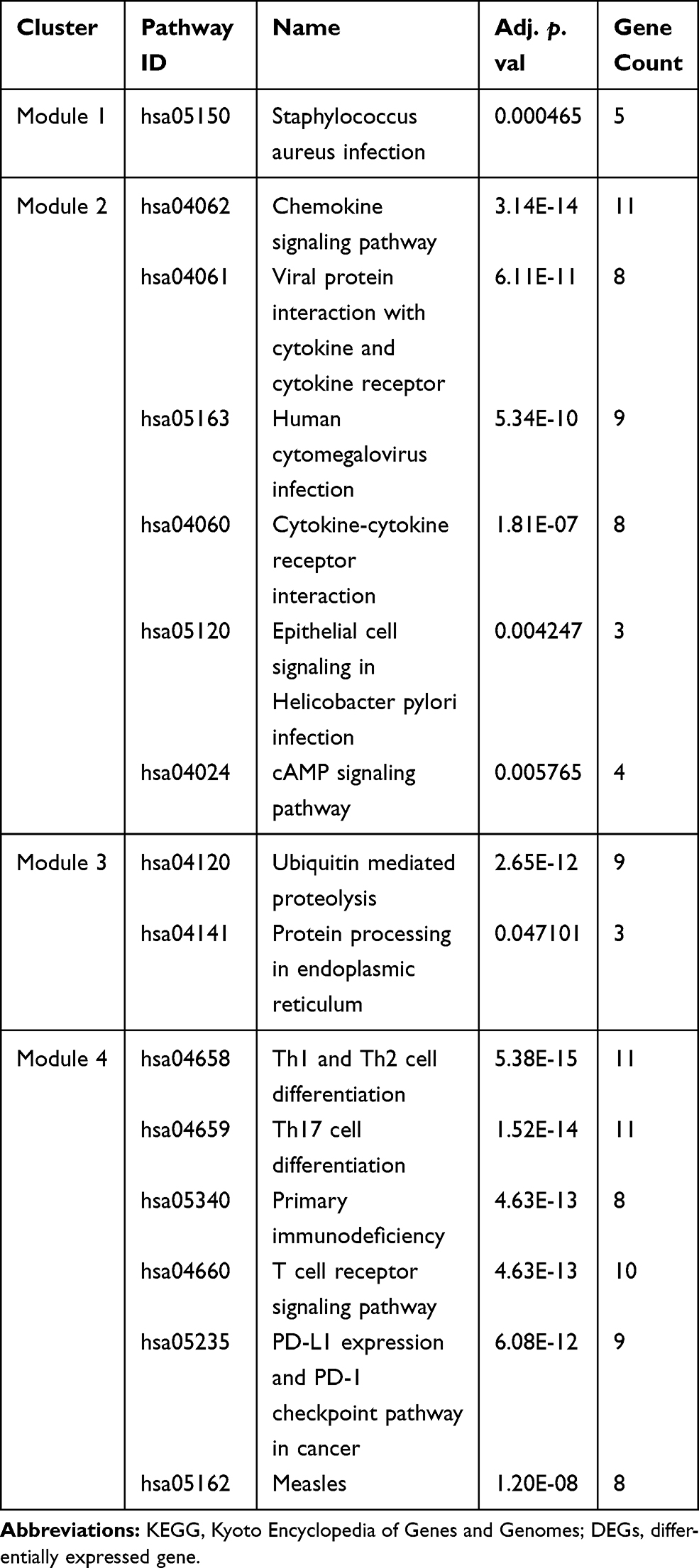 |
Table 4 KEGG Pathway Enrichment Analysis of the DEGs in the Four Modules (Top 6 of Each) |
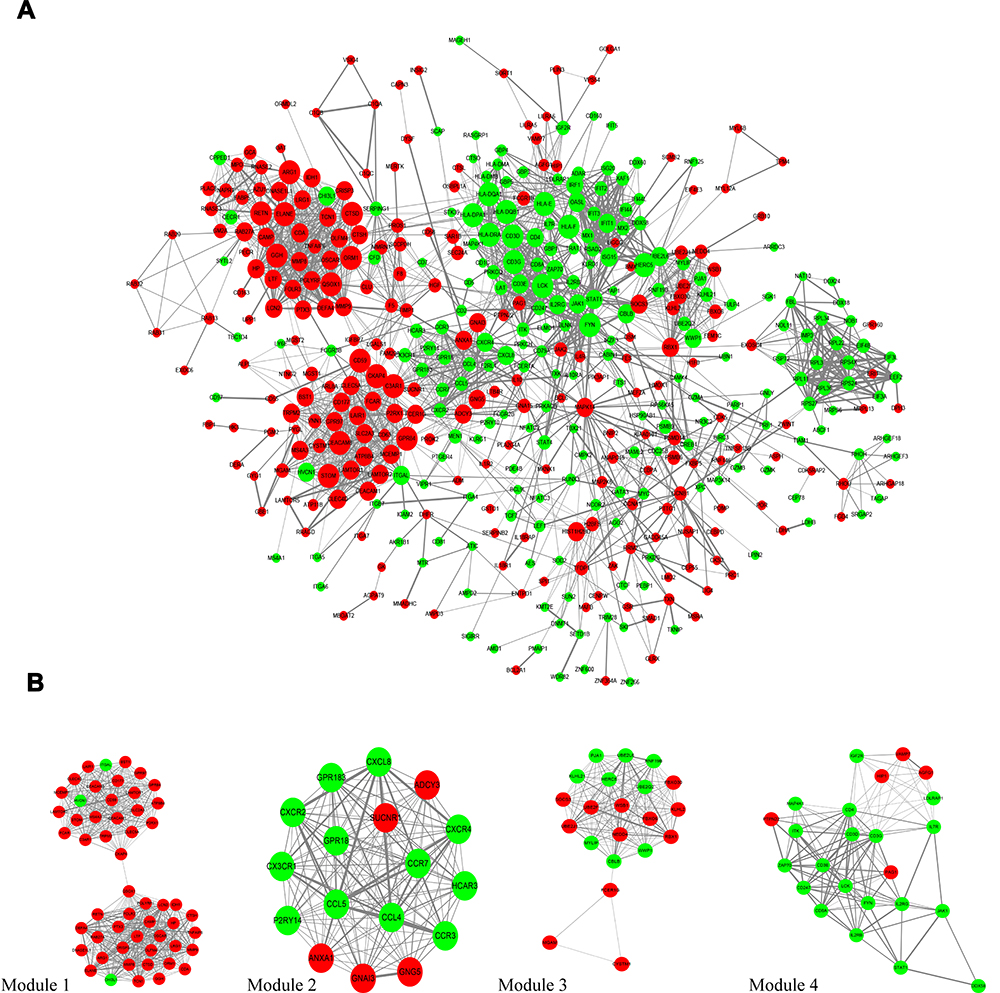 |
Figure 3 PPI network analysis. (A) The PPI network consists 439 nodes and 2430 edges were visualized in Cytoscape. Red notes the upregulated genes and green notes the downregulated genes. The size of each node is positive correlated with log2fold change (FC). The width of each edge is positively correlated with the combine score. (B) It shows the most significant modules in this PPI network. PPI, protein-protein interaction. |
Hub Gene Selection
Hub genes were identified by CytoHubba in the present study. The top 10 hub genes, which were selected based on the 3 most commonly used classification methods in cytoHubba, are displayed in Table 5. By overlapping the first 15 genes, 7 central genes (ARG1, CKAP4, C3AR1, ELANE, GGH, ORM1, and QSOX1) were consequently identified as shown in Figure 4A. After being marked on selected modules, we observed that the hub genes were upregulated and gathered in module 1 (Figure 4B).
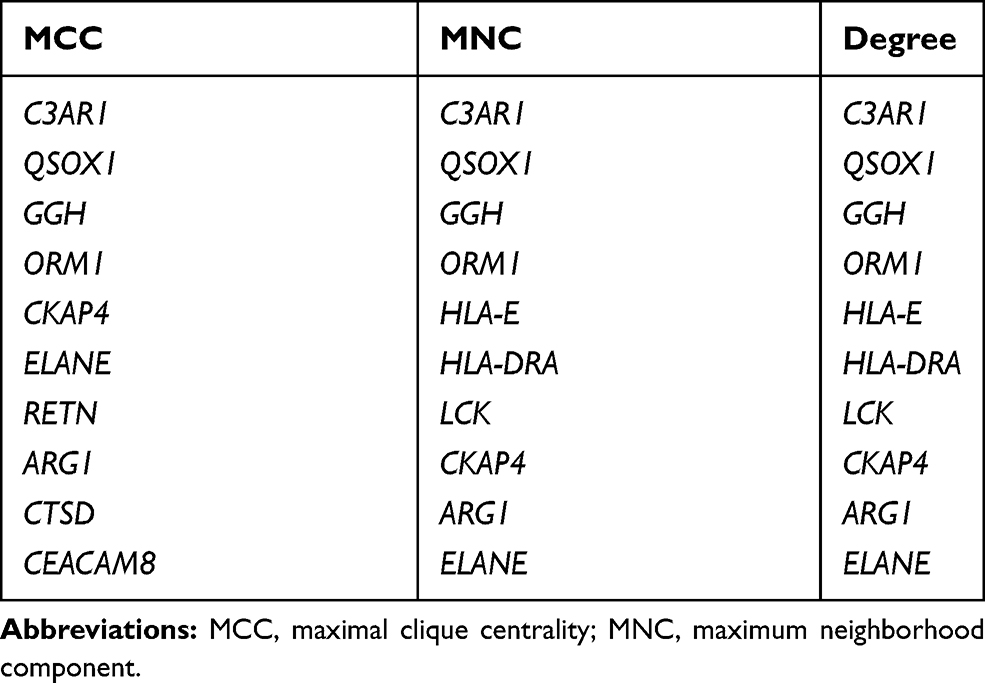 |
Table 5 List of the Top 10 Hub Genes Selected by MCC, MNC, and Degree Methods in cytoHubba |
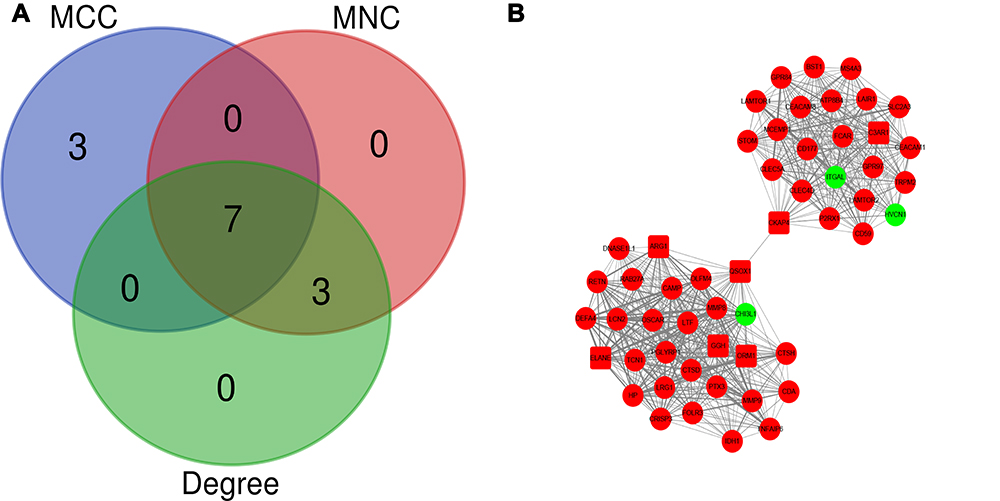 |
Figure 4 Identification of the hub genes. (A) Hub genes were identified by the overlapping of the three methods in cytoHubba. (B) From the module analysis we find that seven of the hub genes upregulated and gathered in module 1 in which showed by shape of square. |
Discussion
Severe burn injuries usually result in distributive shock,19 an abnormal physical state that is marked by capillary leakage of fluid from the intravascular to the interstitial space, and is commonly accompanied by multiple host responses.20 In the initial stages following a severe burn injury, burn shock presents complex pathophysiological changes under the influence of chemical transmitters, injury factors, and toxins due to the effects of injury, stress, and acute phase and immune-inflammatory reactions.21,22 Severe burn injury is often accompanied by smoke inhalation injury, and there were 12 of 30 patients have inhalation injury in this study. In most cases, inhalation injury aggravates burn shock by increasing vascular permeability, and edema formation.23 Burn shock is the earliest serious systemic complication of severe burns and could lead to multiple organ failure, especially in instances of delayed resuscitation for severe burns which not only has serious consequences in the early stages of the injury, but also has a significant impact on the entire course of the disease.24 However, sufficient fluid resuscitation alone does not ameliorate the effects of burn shock.25 Therefore, the exact molecular mechanism underlying the changes in host response during burn shock needs to be understood.
In the present study, the gene expression data of the GSE77791 dataset were obtained from the GEO database and bioinformatically analyzed to explore the key pathways and genes involved in the host response to burn shock. The GSE77791 microarray dataset contained 117 samples, including 13 samples from healthy volunteers, 30 samples from patients without any treatment administration, and the remaining 74 samples from 1 day, 5 days and 7 days after hydrocortisone or NaCl 0.9% administration. The dataset was utilized for assessing the hydrocortisone-induced transcriptional modulation of the immune response in severe burn shock without a deep microarray analysis in a previous study, which found that hydrocortisone administration may worsen the immunosuppression associated with severe burn shock.18 The present study, to our knowledge, is the first to extract data from the 13 healthy volunteer samples and the 30 burn shock patient samples before any treatment administration for further analysis and we subsequently identified the key genes associated with the host response to severe burn shock.
A total of 1059 DEGs, included 508 downregulated genes and 551 upregulated genes, were identified. The GO analysis annotates each DEG and enriches the DEGs with the same attribute into one term. As shown in Figure 2A, the DEGs enriched terms in the BP category were neutrophil activation, neutrophil activation involved in immune response, neutrophil degranulation, neutrophil-mediated immunity, T cell activation, T cell differentiation, lymphocyte differentiation, and T cell receptor signaling pathway. Interestingly, Table 2 shows that the upregulated genes were mostly enriched in the GO terms of neutrophil activation, neutrophil activation involved in immune response, neutrophil degranulation, and neutrophil-mediated immunity, which are associated with neutrophil activation, while the downregulated genes were mostly enriched in terms of T cell activation, T cell differentiation, lymphocyte differentiation, and T cell receptor signaling pathway, all of which are associated with T cell activation. There were many DEGs enriched in neutrophil activation and T cell activation and Supplementary Table 1 was added to show the specific DEGs. The key genes (ARG1, CKAP4, C3AR1, ELANE, GGH, ORM1, and QSOX1) identified by cytoHubba were all enriched in terms associated with neutrophil activation. Further in vivo and in vitro studies are warranted to determine the role of these key genes in burn shock. These GO analysis results are consistent with the postburn immune response, in which the neutrophil is over activated and T cell activation is suppressed. Neutrophils are immediately activated and migrate to the injury site to remove debris and pathogens as well as promote wound healing after burn injury.26,27 However, they also accumulate in other remote organs, such as the lung and small intestine and eventually lead to the injury of these organs.28 Overactivated neutrophils can secrete free radicals and inflammatory mediators such as interleukin (IL)-1, tumor necrosis factor (TNF), IL-6, and IL-8 after severe burn injury and result in systemic inflammatory response syndrome and multiple organ failure.29 Meanwhile, T cell proliferation is also suppressed in severe burn injuries.30 According to the p value in Figure 2B, the top three pathways with the highest reliability were Th1 and Th2 cell differentiation, Th17 cell differentiation, and hematopoietic cell lineage, in which the downregulated DEGs were enriched. There were many DEGs enriched in the various pathways and Supplementary Table 2 was added to show the specific DEGs. The signal transduction in T cell activation in burn shock was suppressed according to KEGG pathway analysis.
After severe burn injury, immune functions are severely compromised, including the decline of neutrophil killing of invading pathogens and macrophage antigen presentation as well as suppression of T cell activation.5 Together, these events lead to the development of immune suppression during the burn stage.5 According to the results of GO function analysis and KEGG pathway enrichment analysis, the most downregulated DEGs were enriched in T cell activation. Neutrophils are activated in burn shock, and the granules released by overactivated neutrophils can suppress T cell activation.31 Therefore, immunity may be impaired by the suppression of T cell activation.31 Our findings are consistent with the pathophysiology of burn shock in terms of neutrophil overactivation and lymphocyte suppression.32,33 This can explain the increased risk of severe systemic infection and multiple organ failure in the severe burn patients with an unstable shock period.
Four significant modules were selected from the PPI network using MCODE in Cytoscape. Next, KEGG pathway enrichment analysis was performed for the DEGs in these modules. As shown in Table 4, the DEGs in modules 1, 2, and 4 were mostly enriched in the pathways associated with immune response, and the DEGs in module 3 were mostly enriched in the pathways associated with protein metabolism. This is consistent with the hypermetabolic state in severe burn injury.34 Jeschke suggested that the continued release of TNF, IL-6, and other inflammatory mediators could further contribute to the hypermetabolic state of severe burn injury patients.35 Furthermore, immune dysregulation could also affect the metabolic state of the body.
Seven hub genes (ARG1, CKAP4, C3AR1, ELANE, GGH, ORM1, and QSOX1) were identified using cytoHubba and gathered in module 1 with a high degree. ARG1, which is contained in cytoplasmic azurophil granules and the gelatinase grains of neutrophils,36,37 hydrolyzes L-arginine to L-ornithine and urea in the cytosol. ARG1-induced depletion of arginine, suppression of T lymphocyte proliferation, and secretion of cytokines via down-regulated expression of the CD3 zeta chain in T cells31 have emerged as fundamental mechanisms underlying inflammation-associated immunosuppression.38 In addition, ARG1 competes with inducible nitric oxide synthase for substrate L-arginine to inhibit the production of nitric oxide, which can also suppress the inflammatory response.39 C3AR1 is a G protein-coupled receptor protein that is predominantly distributed on neutrophils, eosinophils, basophils, mast cells, and monocytes/macrophages.40 Previous studies have described C3a as a proinflammatory mediator in some chronic inflammatory responses.41,42 However, several studies have indicated that C3AR1 exerts a potent anti-inflammatory response with a high expression of C3AR1 on neutrophils by inhibiting the mobilization of neutrophils from the bone marrow to circulation following acute injury.43,44 Neutrophil elastase (NE) is coded by ELANE and secreted by activated neutrophils to kill bacteria and promotes the secretion of pro-inflammatory mediators, such as TNFα and IL-1β. Numerous studies have indicated that NE is a marker of neutrophil activation.45 ORM1, also known as alpha–1–acid glycoprotein (AGP1), encodes an acute-phase protein in activated neutrophil that mediates neutrophil migration failure and inhibition of neutrophil activation in the acute inflammatory response.46,47 The latest study indicates that ORM1 serves as a potential diagnostic marker for sepsis.48
CKAP4 is a transmembrane protein residing predominantly in the endoplasmic reticulum of eukaryotes that can promote cellular proliferation through the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway.49 Research has shown that PI3K plays a vital role in the regulation of neutrophil activation through AKT activation.50,51 Therefore, CKAP4 may activate neutrophils through the PI3K/AKT pathway in burn shock patients. GGH is a lysosomal enzyme involved in the metabolism of folates and the disposition of methotrexate that plays a role as a tumor marker in breast and lung cancer.52,53 GGH also catalyzes the hydrolysis of folylpoly-gamma-glutamates to generate glutamate, which is a substrate of glutamine.54 Glutamine, a substrate of enterocytes, plays a vital role in the normal immunological function and the structure of the gut.55 The intense stress and persistent inflammation caused by severe burn injury may result in intestinal mucosal barrier damage which is sensitive to infection.56 Wang and colleagues found that glutamine improved tissue perfusion and increased energy synthesis in enterocytes, thus alleviating the intestinal injury after burn injury.57 QSOX1 is mainly expressed in several tumor cells and can promote tumor growth and metastasis.58,59 A recently study reported that the peripheral blood level of QSOX1 related to neutrophil infiltration is a novel independent predictor of left ventricular dysfunction.60
To date, the clinical value of ARG1, CKAP4, C3AR1, ELANE, GGH, ORM1, and QSOX1 in burn shock has not been documented. From the above description, we know that ARG1, C3AR1, ELANE, and ORM1 are released from activated neutrophils and have different effects on immune response. Neutrophil activation is considered the most dominant host response to burn shock; thus, it is crucial to conduct further studies to explore the diagnostic or therapeutic value of these four genes in burn shock. Although CKAP4, GGH, and QSOX1 play crucial roles in tumor proliferation and metastasis, these genes were also significantly upregulated in neutrophil in burn shock and had close interactions with ARG1, C3AR1, ELANE, and ORM1 in the PPI network constructed using the DEGs. Furthermore, an increasing amount of research indicates that CKAP4, GGH, and, QSOX1 are associated with the immune response of neutrophils. Therefore, we speculate these three genes play crucial effects on the immune response to severe burn injury, but more studies are needed to confirm this hypothesis. Our study revealed the host response to burn shock and identified the key genes that provide a new direction for further in vivo and in vitro studies to explore the mechanism of occurrence and development of burn shock.
There are no precise methods to identify hub genes. Some researchers select the higher-degree nodes in the PPI network as hub genes,61 while others identify the hub genes via cytoHubba in Cytoscape using a single method.62 In addition, some researchers believe that the overlapping results from several methods in cytoHubba is a more reasonable method for detecting hub genes.63 In this study, we overlapped the top 10 genes selected by the MCC, MNC, and degree methods in cytoHubba to identify the hub genes.
Several previous studies have used the bioinformatic analysis of microarray datasets to explore gene expression after burn injury. Xu64 and Wu65 reanalyzed the GSE19743 dataset and determined the time course change in gene expression after burn injury. The time points of sample collection in the GSE19743 dataset were separated into two groups: early-stage for < 11 days postburn and middle-stage for 11–49 days after thermal injury. Bioinformatic analysis of the GSE19743 dataset was preformed to reveal the gene changes over the whole burn stage which ranged from 0 to 49 days after thermal injury. However, the 30 samples in GSE77791 were collected within 72 h after burn injury, and the analysis of these samples was utilized to identify gene changes in the shock stage after burn injury. The GSE77791 dataset, to our knowledge, contains unique burn shock data in the GEO database and this study is the first to use GSE77791 to explore the different gene expression levels during the burn shock stage after burn injury.
Compared to previous studies, the present study has several advantages. First, our study successfully identified novel hub genes associated with severe burn shock. Second, we further analyzed and presented the functions and pathway enrichment of the main DEGs. Our findings can help to elucidate the molecular mechanism underlying the changes in host response during burn shock as well as provide potential targets for early detection and treatment of burn shock. However, some limitations are also present in our study. We only made use of a single dataset from the GEO database, which is the only one we could find that was associated with burn shock. We will repeat the experiment in the future should other datasets, associated with burn shock, become available. Moreover, we did not verify the expression of these hub genes in patients with severe burn injury because samples from patients with severe burn injury are limited. However, we will verify the expression levels of the hub genes in severe burn injuries and perform further studies on the mechanisms of these genes in the occurrence of severe burn injury shock once the samples are available.
Conclusions
In conclusion, following the bioinformatic analyses of DEGs, GO terms, KEGG pathway enrichment, and the PPI network, our study found that the immune response of the host is altered during burn shock, which may be mediated by specific hub genes. The present study provides a new direction for future research to investigate the underlying mechanism of burn shock occurrence and development.
Disclosure
Xiao Fang, Shu-Fang Duan, and Yu-Zhou Gong contributed equally to this work and are joint first authors.
The authors report no conflicts of interest in this work.
References
1. Smolle C, Cambiaso-Daniel J, Forbes AA, et al. Recent trends in burn epidemiology worldwide: A systematic review. Burns. 2017;43(2):249–257. doi:10.1016/j.burns.2016.08.013
2. Greenhalgh DG, Longo DL. Management of Burns. N Engl J Med. 2019;380(24):2349–2359. doi:10.1056/NEJMra1807442
3. WHO Burns. https://www.who.int/news-room/fact-sheets/detail/burns. 2020
4. Brusselaers N, Monstrey S, Snoeij T, et al. Morbidity and mortality of bloodstream infections in patients with severe burn injury. Am J Crit Care. 2010;19(6):e81.
5. Jeschke MG, van Baar ME, Choudhry MA, Chung KK, Gibran NS, Logsetty S. Burn injury. Nat Rev Dis Primers. 2020;6(1):11.
6. Barrow RE, Jeschke MG, Herndon DN. Early fluid resuscitation improves outcomes in severely burned children. Resuscitation. 2000;45(2):91–96.
7. Latenser BA. Critical care of the burn patient: the first 48 hours. Crit Care Med. 2009;37(10):2819–2826.
8. Salinas J, Chung KK, Mann EA, et al. Computerized decision support system improves fluid resuscitation following severe burns: an original study. Crit Care Med. 2011;39(9):2031–2038.
9. Wanis M, Walker SAN, Daneman N, et al. Impact of hospital length of stay on the distribution of Gram negative bacteria and likelihood of isolating a resistant organism in a Canadian burn center. Burns. 2016;42(1):104–111.
10. Hasan DJA, Eriby DQH, Hammoodi S. Causes of mortality in burns, prospective study. International Journal of Surgery Science. 2019;3(3):261–264. doi:10.33545/surgery.2019.v3.i3e.178
11. Lantos J, Foldi V, Roth E, Weber G, Bogar L, Csontos C. Burn trauma induces early HMGB1 release in patients: its correlation with cytokines. Shock. 2010;33(6):562–567. doi:10.1097/SHK.0b013e3181cd8c88
12. Carter DW, Sood RF, Seaton ME, et al. MC1R gene polymorphisms are associated with dysfunctional immune responses and wound infection after burn injury. J Surg Res. 2018;231:448–452.
13. Vinaik R, Abdullahi A, Barayan D, Jeschke MG. NLRP3 inflammasome activity is required for wound healing after burns. Transl Res. 2020;217:47–60. doi:10.1016/j.trsl.2019.11.002
14. Adomas A, Heller G, Olson Å, et al. Comparative analysis of transcript abundance in Pinus sylvestris after challenge with a saprotrophic, pathogenic or mutualistic fungus. Tree Physiology. 2008;28(6):885–897. doi:10.1093/treephys/28.6.885
15. Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5(10):588–599. doi:10.1038/ncponc1187
16. Chen Y, Jiang P, Wen J, et al. Integrated bioinformatics analysis of the crucial candidate genes and pathways associated with glucocorticoid resistance in acute lymphoblastic leukemia. Cancer Med. 2020.
17. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4(1):2. doi:10.1186/1471-2105-4-2
18. Plassais J, Venet F, Cazalis M-A, et al. Transcriptome modulation by hydrocortisone in severe burn shock: ancillary analysis of a prospective randomized trial. Crit Care. 2017;21(1):158. doi:10.1186/s13054-017-1743-9
19. Demling RH. Fluid replacement in burned patients. Surg Clin North Am. 1987;67(1):15–30. doi:10.1016/S0039-6109(16)44130-7
20. Guillory AN, Clayton RP, Herndon DN, Finnerty CC. Cardiovascular Dysfunction Following Burn Injury: what We Have Learned from Rat and Mouse Models. Int J Mol Sci. 2016;17(1):1. doi:10.3390/ijms17010053
21. Kramer GC, Lund T, Herndon D. Pathophysiology of Burn Shock and Burn Edema. Total Burn Care.
22. Vaughn L, Beckel N. Severe burn injury, burn shock, and smoke inhalation injury in small animals. Part 1: burn classification and pathophysiology. Journal of Veterinary Emergency and Critical Care. 2012;22(2):179–186. doi:10.1111/j.1476-4431.2012.00727.x
23. Navar PD, Saffle JR, Warden GD. Effect of inhalation injury on fluid resuscitation requirements after thermal injury. Am J Surg. 1985;150(6):716–720. doi:10.1016/0002-9610(85)90415-5
24. Wolf SE, Rose JK, Desai MH, Mileski JP, Barrow RE, Herndon DN. Mortality determinants in massive pediatric burns. An analysis of 103 children with > or = 80% TBSA burns (> or = 70% full-thickness). Ann Surg. 1997;225(5):554–565. doi:10.1097/00000658-199705000-00012
25. Rae L, Fidler P, Gibran N. The Physiologic Basis of Burn Shock and the Need for Aggressive Fluid Resuscitation. Critical Care Clinics. 2016;32(4):491–505. doi:10.1016/j.ccc.2016.06.001
26. Parihar A, Parihar MS, Milner S, Bhat S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns. 2008;34(1):6–17. doi:10.1016/j.burns.2007.04.009
27. Sood RF, Gibran NS, Arnoldo BD, et al. Early leukocyte gene expression associated with age, burn size, and inhalation injury in severely burned adults. J Trauma Acute Care Surg. 2016;80(2):250–257. doi:10.1097/TA.0000000000000905
28. Toth B, Alexander M, Daniel T, Chaudry IH, Hubbard WJ, Schwacha MG. The role of γδ T cells in the regulation of neutrophil-mediated tissue damage after thermal injury. J Leukoc Biol. 2004;76(3):545–552. doi:10.1189/jlb.0404219
29. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
30. Schluter B, Konig W, Koller M, Erbs G, Muller FE. Differential regulation of T- and B-lymphocyte activation in severely burned patients. J Trauma. 1991;31(2):239–246. doi:10.1097/00005373-199102000-00015
31. Munder M, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006;108(5):1627–1634. doi:10.1182/blood-2006-11-010389
32. Deveci M, Sengezer M, Bozkurt M, Eski M, Inal A. Comparison of lymphocyte populations in cutaneous and electrical burn patients: a clinical study. Burns. 2000;26(3):229–232. doi:10.1016/S0305-4179(99)00124-2
33. Hampson P, Dinsdale RJ, Wearn CM, et al. Neutrophil Dysfunction, Immature Granulocytes, and Cell-free DNA are Early Biomarkers of Sepsis in Burn-injured Patients: A Prospective Observational Cohort Study. Ann Surg. 2017;265(6):1241–1249.
34. Jeschke MG. Postburn Hypermetabolism: past, Present, and Future. J Burn Care Res. 2016;37(2):86–96. doi:10.1097/BCR.0000000000000265
35. Jeschke MG, Gauglitz GG, Kulp GA, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One. 2011;6(7):e21245. doi:10.1371/journal.pone.0021245
36. Munder M. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105(6):2549–2556. doi:10.1182/blood-2004-07-2521
37. Jacobsen LC, Theilgaard-Mönch K, Christensen EI, Borregaard BN. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood. 2007;109(7):3084–3087. doi:10.1182/blood-2006-06-032599
38. Thewissen M, Damoiseaux J, van de Gaar J, Tervaert JWC. Neutrophils and T cells: bidirectional effects and functional interferences. Mol Immunol. 2011;48(15–16):2094–2101. doi:10.1016/j.molimm.2011.07.006
39. Santiago-Olivares C, Rivera-Toledo E, Gomez B. Nitric oxide production is downregulated during respiratory syncytial virus persistence by constitutive expression of arginase 1. Arch Virol. 2019;164(9):2231–2241. doi:10.1007/s00705-019-04259-0
40. Martin U, Bock D, Arseniev L. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. 1997;186(2):199–207. doi:10.1084/jem.186.2.199
41. Fischer WH, Jagels MA, Hugli TE. Regulation of IL-6 Synthesis in Human Peripheral Blood Mononuclear Cells by C3a and C3adesArg. J Immunol. 1999;162(1):453–459.
42. Asgari E, Friec GL, Yamamoto H, et al. C3a modulates IL-1 b secretion in human monocytes by regulating ATP ef fl ux and subsequent NLRP3 in fl ammasome activation. J Histochem Cytochem. 2015;122(20):3473–3482.
43. Wu MC, Brennan FH, Lynch JP, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci U S A. 2013;110(23):9439–9444.
44. Brennan FH, Jogia T, Gillespie ER, Blomster LV, Ruitenberg MJ. Complement receptor C3aR1 controls neutrophil mobilization following spinal cord injury through physiological antagonism of CXCR2. JCI Insight. 2019;4:9.
45. Kramps JA, van der Valk P, van der Sandt MM, Lindeman J, Meijer CJ. Elastase as a marker for neutrophilic myeloid cells. J Histochem Cytochem. 1984;32(4):389–394.
46. Costello MJ, Gewurz H, Siegel JN. Inhibition of neutrophil activation by alpha1-acid glycoprotein. Clin Exp Immunol. 1984;55(2):465–472.
47. Mestriner FL, Spiller F, Laure HJ, et al. Acute-phase protein alpha-1-acid glycoprotein mediates neutrophil migration failure in sepsis by a nitric oxide-dependent mechanism. Proc Natl Acad Sci U S A. 2007;104(49):19595–19600.
48. Hull C, Dekeryte R, Buchanan H, et al. NLRP3 inflammasome inhibition with MCC950 improves insulin sensitivity and inflammation in a mouse model of frontotemporal dementia. Neuropharmacology. 2020;180:108305.
49. Kimura H, Fumoto K, Shojima K, et al. CKAP4 is a Dickkopf1 receptor and is involved in tumor progression. J Clin Invest. 2016.
50. Rane MJ, Coxon PY, Powell DW, et al. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem. 2001;276(5):3517–3523.
51. Liao CH, Chen JJ. 5-hydroxy-2-(4-hydroxy-3-methoxyphenyl)-3,7-dimethoxy-4H-chromen-4-one (MSF-2) suppresses fMLP-mediated respiratory burst in human neutrophils by inhibiting phosphatidylinositol 3-kinase activity. J Cell Physiol. 2011;226(6):1519–1530.
52. Schneider E, Ryan TJ. Gamma-glutamyl hydrolase and drug resistance. Clin Chim Acta. 2006;374(1–2):25–32.
53. Wang SM, Kong XY, Li M, Sun LL, Yan D. Association of GGH Promoter Methylation Levels with Methotrexate Concentrations in Chinese Children with Acute Lymphoblastic Leukemia. Pharmacotherapy. 2020;40(7):614–622.
54. GGH gamma-glutamyl hydrolase. https://www.ncbi.nlm.nih.gov/gene/8836. 2020
55. Ziegler TR, Bazargan N, Leader LM, Martindale RG. Glutamine and the gastrointestinal tract. Curr Opin Clin Nutr Metab Care. 2000;3(5):355–362.
56. Parry-Billings M, Calder PC, Newsholme EA, Evans J. Does glutamine contribute to immunosuppression after major burns? Lancet. 1990;336(8714):523–525.
57. Wang ZE, Wu D, Zheng LW, et al. Effects of glutamine on intestinal mucus barrier after burn injury. Am J Transl Res. 2018;10(11):3833–3846.
58. Sung HJ, Ahn JM, Yoon YH, et al. Quiescin Sulfhydryl Oxidase 1 (QSOX1) Secreted by Lung Cancer Cells Promotes Cancer Metastasis. Int J Mol Sci. 2018;19:10.
59. Geng Y, Xu C, Wang Y, Zhang L. Quiescin Sulfhydryl Oxidase 1 Regulates the Proliferation, Migration and Invasion of Human Glioblastoma Cells via PI3K/Akt Pathway. Onco Targets Ther. 2020;13:5721–5729.
60. Vanhaverbeke M, Vausort M, Veltman D, et al. Peripheral Blood RNA Levels of QSOX1 and PLBD1 Are New Independent Predictors of Left Ventricular Dysfunction After Acute Myocardial Infarction. Circ Genom Precis Med. 2019;12(12):e002656.
61. Wang S, Wang H, Liu W, Wei B. Identification of Key Genes and Pathways Associated with Sex Differences in Osteoarthritis Based on Bioinformatics Analysis. Biomed Res Int. 2019;2019:3482751.
62. Liu L, He C, Zhou Q, Wang G, Lv Z, Liu J. Identification of key genes and pathways of thyroid cancer by integrated bioinformatics analysis. J Cell Physiol. 2019;234(12):23647–23657.
63. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
64. Xu HT, Guo JC, Liu HZ, Jin WW, Time-Series A. Analysis of Severe Burned Injury of Skin Gene Expression Profiles. Cell Physiol Biochem. 2018;49(4):1492–1498.
65. Wu D, Zhou M, Li L, et al. The Time Course Pathological Changes After Burn Injury. Inflammation. 2018;41(5):1864–1872.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.