")
Back to Journals » Cancer Management and Research » Volume 15
Identification of Immune-Related Seven-Long Non-Coding RNA Signature for Overall Survival and Validation of the Effect of LINC01270 in Malignant Phenotypes of Clear Cell Renal Carcinoma
Authors Liu C, Xiong W, Song J , Ouyang X, Fu Y
Received 28 October 2022
Accepted for publication 4 February 2023
Published 14 February 2023 Volume 2023:15 Pages 131—145
DOI https://doi.org/10.2147/CMAR.S394100
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Chengxuan Liu,1 Weijian Xiong,1 Jing Song,2 Xiaoqin Ouyang,1 Yang Fu3
1Department of Nephrology, Chongqing Traditional Chinese Medicine Hospital, Chongqing, People’s Republic of China; 2Molecular Medicine and Cancer Research Centre, Chongqing Medical University, Chongqing, People’s Republic of China; 3Department of Rehabilitation, Chongqing Traditional Chinese medicine Hospital, Chongqing, People’s Republic of China
Correspondence: Yang Fu, Department of Rehabilitation, Chongqing Traditional Chinese Medicine Hospital, No. 6 Panxi Branch Road, Jiangbei District, Chongqing, 400021, People’s Republic of China, Email [email protected]
Introduction: The overall survival of patients with high-stage clear cell renal carcinoma (ccRCC) is poor. However, promising molecular-level prognostic marker is still lacking to date.
Methods: We systematically evaluated the prognostic potential of immune-related long non-coding RNAs (lncRNAs) in ccRCC. The expressions of lncRNAs were validated in clinical tissues of ccRCC. Functional experiments were performed to investigate the role of lncRNAs in ccRCC.
Results: Eight hundred and ninety-three lncRNAs were differentially expressed in ccRCC and compared with normal controls and were screened out using three independent cohorts. Among them, 290 immune-related lncRNAs were identified. We identified a seven-lncRNA signature (LINC01270, FIRRE, RP11-37B2.1, RP11-253I19.3, RP11-438L19.1, RP11-504P24.9, and CTB-41I6.1) associated with the overall survival of late-stage ccRCC patients. Further multivariate Cox analysis using clinical factors as covariates showed that our lncRNA signature was an independent biomarker in training (P < 0.001, Log rank test) and validation cohorts (P = 0.003). The seven lncRNAs were closely related to the major targets (PD-1, PD-L1, and CTLA4) of immune checkpoint blockade drugs, implying that they may have potential value in predicting immunotherapy response. The seven lncRNAs may play an important role in tumor-infiltrating immune cells (eg, T/B cells) and tumor progression through regulating the binding of protein receptors/complexes, as revealed by functional analysis. qRT-PCR showed that LINC01270 was upregulated in ccRCC tissues (n=20) compared with paired normal samples. Functional experiments showed that LINC01270 silencing inhibited the proliferation, invasion, and migration of ccRCC cells.
Discussion: In summary, the seven-lncRNA signature has great potential in prognosis for patients with late-stage ccRCC, which could be a novel clinical biomarker. LINC01270 could be a novel therapeutic target of ccRCC.
Keywords: long non-coding RNA FIRRE, prognosis, renal cell carcinoma, tumor immune
Introduction
Kidney cancer is a serious threat to human health. Approximately 73,750 new kidney cancer cases were diagnosed in the United States in 2020.1 The 5-year survival rates of patients with localized kidney cancer (ie, stage I/II in clinical) have increased to 90%.2 However, the overall prognosis of patients who present with high-stage disease (ie, stage III/IV) is still poor.3 Clear cell renal cell carcinoma (ccRCC) is the most common subtype of kidney cancer.4 Patients with localized RCC usually have a better prognosis after surgical removal of part or all of the kidney.5 However, late-stage ccRCC (stage III/IV) generally cannot be cured because they usually have metastatic lesions.5 Patients with late-stage ccRCC may be treated with surgery (cytoreductive nephrectomy and/or metastasectomy), sometimes combined with anti-angiogenic therapy and immunotherapy or with exclusive systemic treatment.4,6 To date, several prognostic scores have been proposed for the prognosis of ccRCC patients, such as the stage, size, grade, and necrosis (SSIGN) score7 and The University of California Los Angeles Integrated Staging System (UISS) score.8 However, they were less useful for individualized risk prediction.3 For the prognosis of advanced RCC, an IMDC prognostic score was also proposed.9 Moreover, these prognostic systems depend on evaluating clinical parameters without consideration of molecular markers.10 Therefore, the identification of molecular-level prognostic biomarkers is warranted for the clinical management of late-stage ccRCC.
In recent years, medical treatment for ccRCC has transitioned to a novel immunotherapy era.3 Identification of immune-related genes contributes to understanding the molecular basis of the effects of immunotherapy in ccRCC. Protein-coding genes that can be immunotherapy targets are under investigation, such as immune checkpoint molecules.11 Long non-coding RNA (lncRNA) is an important type of RNA. LncRNAs were closely associated with cell cycle, proliferation, metastasis, and drug resistance of cancer cells.12 Functionally, they may act as signal, decoy, scaffold, guide, enhancer RNAs, and short peptides. For example, the classic lncRNAs HOTAIR, upregulated in diverse cancer types, was found to promote invasion and metastasis by regulating the H3K27 methylation of tumor suppressor genes.13 Recently, lncRNAs have been linked to PD-L1/PD-1 checkpoint pathways, implying the clinical potential of lncRNAs.14 Thus, lncRNAs may serve as novel biomarkers in the future.12 However, the prognostic value of immune-related lncRNAs has not been fully investigated. Thus, we systematically assessed the prognostic potential of immune-related lncRNAs in ccRCC by leveraging the knowledge of MSigDB C7 immunologic signatures.15
Materials and Methods
Transcriptome Data
The raw CEL files (GSE5375716 and GSE66270)17 from the Affymetrix Human Genome U133 Plus 2.0 Array platform were retrieved from the Gene Expression Omnibus (GEO) database. For the GSE53757 dataset, a total of 144 ccRCC samples (72 tumor samples and 72 normal samples) were downloaded. For the GSE66270 dataset, a total of 28 ccRCC samples (14 tumor samples and 14 normal samples) were downloaded. Moreover, the level 3 gene expression data (HT-Seq read counts) of TCGA-KIRC (n = 603) were retrieved from the Genomic Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/repository). The RNA-Seq data (read count) of late-stage ccRCC (stage III/IV, n = 186) of the ICGC project were downloaded from the Xena data hub (https://xena.ucsc.edu/public). Follow-up clinical data were also retrieved for analysis. The.gtf annotation file from GENCODE (v22) database was used to obtain the lncRNA records. The genes with the identifier “lincRNA” in the annotation file were extracted and analyzed in this study.
Differential Expression Analysis
For microarray expression profiles, the background correction was performed for the signal intensities. For comparison between sample groups, quantile normalization was performed for samples in the same dataset. The expression matrix of lncRNA genes was retained for differential expression analysis. The limma package18 was used for differential expression analysis of microarray data. For the read count of RNA-Seq data, the DESeq2 package19 was used. Genes with Benjamini–Hochberg (BH) adjusted P-value <0.05 were statistically significant.
Identification of Immune-Related lncRNAs and the Function Analysis
Differentially expressed protein-coding genes in significant enriched Molecular Signatures Database (MSigDB) C7 immunologic signatures15 were considered immune-related genes. The lncRNAs with Pearson correlation |r| ≥0.4 and FDR-value <0.05 with BH adjustment were determined as immune-related lncRNAs. A functional analysis of the lncRNA signature was performed based on the lncRNA-associated protein-coding genes. Protein-coding genes correlated with lncRNAs were determined using Pearson correlation analysis. The mRNA-lncRNA pairs with FDR <0.05 were considered. The mRNAs in the “pathway in cancer” category from the PathCards database were included in this correlation analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were included in the enrichment analysis.
Statistical Analysis
Univariate and Multivariate Cox regressions were used to identify prognostic lncRNAs in ccRCC. LncRNAs with BH-adjusted P < 0.05 in univariate analysis were subjected to multivariate analysis to obtain the lncRNA signature. Similar to Wang et al,20 The prognostic index (Pindex) was calculated using the lncRNA expression levels and Cox regression coefficients of the seven lncRNAs:
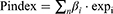
where n is the number of lncRNAs,  is the expression of lncRNA i, and
is the expression of lncRNA i, and  is the estimated regression coefficient of lncRNA i in the multivariate Cox model. The ccRCC patients were divided into two groups based on Pindex value (median as cut-off). Clinical parameters were added into the multivariate Cox hazard model to assess whether the Pindex was an independent factor associated with the prognosis of ccRCC, such as TNM stage, grade, age, and gender.
is the estimated regression coefficient of lncRNA i in the multivariate Cox model. The ccRCC patients were divided into two groups based on Pindex value (median as cut-off). Clinical parameters were added into the multivariate Cox hazard model to assess whether the Pindex was an independent factor associated with the prognosis of ccRCC, such as TNM stage, grade, age, and gender.
Clinical Specimens
The ccRCC tissues (n = 20) and the paired normal tissues from 20 patients were obtained in the First Affiliated Hospital with Chongqing Medical University (Chongqing, China). The use of tissues has been abided by the ethics committee.
Cell Lines and Cell Culture
The ccRCC cell lines ACHN, Caki-1, 769-P, Caki-2, and 786-O (RPMI 1640) and human renal tubular epithelial cells HK-2 were purchased from the Cell Bank of the Chinese Academy of Sciences. The HK-2 cells were cultured in Keratinocyte Serum Free Medium (K-SFM), the ACHN cells were cultured in DMEM, the Caki-1 and Caki-2 cells were cultured in McCoy’s 5A, and 786-P and 786-O cells were cultured in RPMI 1640 with 10% fetal bovine serum FBS (PAN-Biotech, Adenbach, Germany) and 100 U/mL penicillin-streptomycin. All cells were cultured at 37°C with 5% CO2.
Transfection
Si-LINC01270 and NC were purchased from Gene Pharma (Shanghai, China). Transfections were performed using GP-transfect-mate (Gene Pharma, China) according to the manufacturer’s instructions. During the transfection process, many negatively charged proteins in the serum may interfere with the adsorption of cationic liposomes on nucleic acids, affecting the transfection efficiency. We use a serum-free medium for transfection. After 6h of transfection, cells were cultured in complete media for 48h.
RNA Isolation and qRT-PCR
According to the manufacturer’s instructions, total RNA was extracted by Trizol (Invitrogen Corporation, Carlsbad, CA, USA), and 0.5 ug RNA was reverse transcribed using a Reverse Transcription Kit (Takara, China). PCR used SYBR Green Master Mix (Takara, China). GAPDH was used as endogenous control, and all primers used for qRT-PCR are listed as follows: LINC01270 Forward: 5’-CTCACGAAAGCGCAGGAATG-3’; Reverse: 5’-GCTCCAAAAGCAGACAAGCC-3’. FIRRE Forward: 5’-CACTGGAGCAATCCTGGCTG −3’; Reverse: 5’-TGCCTAACTACCTCTATTGGCT-3’. RP11-37B2.1 Forward: 5’-GCGATTTGGGGAGTGTTGTG-3’; Reverse: 5’-GTTGGGGCAGTAGGCAACTA-3’. RP11-253I19.3 Forward: 5’-TGTCGGATTGGTTAGCGACC-3’; Reverse: 5’-CTTAAGTGTGGAGCCCTCGG-3’. RP11-438L19.1 Forward: 5’-TGGTTCTGCTCCTGGTAACG-3’; Reverse: 5’- AGGATAACAGGTCTGCCTGC-3’. RP11-504P24.9 Forward: 5’-TGTGTCCCCAAGGAAGGATG-3’; Reverse: 5’-TCCACACAGTGTAGTCAAGCC-3’. CTB-41I6.1 Forward: 5’-GTGTCCCCAAGGAAGGATGAG-3’; Reverse: 5’- GTAGTCAAGCCGACTCTCCA −3’. GAPDH Forward: 5’-CCATGGGGAAGGTGAAGGTC-3’; Reverse: 5’- AGTGATGGCATGGACTGTGG −3’.
Cell Proliferation and Colony Formation Assays
Cell proliferation was measured by Cell Counting Kit-8 reagent (CCK-8) (Bimake, China). Briefly, the cells were cultured in 96-wells plates, and 10 ul CCK-8 solution was added to each well every day. Then, the cells were cultured at 37°C for 1h and were detected at 450 nm on a Microplate Reader (BIO-TEK, USA). For colony formation assays, 1.5×103 cells were plated in 6-wells plates and cultured for 2 weeks. Then, the cell colonies were fixed, stained, and counted.
Cell Migration and Invasion Assays
The cells were suspended in 200 ul serum-free medium and inoculated into the upper chamber (BD Company, USA) uncoated (for migration analysis) or coated with Matrigel. Then, the lower chambers were fixed with 600 ul 20% FBS serum. Migrating and invading cells were fixed and stained after 36h. Chambers were randomly selected from three fields of view to count, and the average count was considered the number of permeable cells.
Results
Identification of ccRCC-Related lncRNAs and Immune-Associated Coding Genes
In the present study, we analyzed the differentially expressed immune-related lncRNAs with prognostic potential by leveraging the knowledge of immune-related gene signatures from the MSigDB database (Figure 1). First, we identified 2342 protein-coding genes (FDR < 0.05 with BH adjustment, Figure 2A) and 893 lncRNAs (FDR < 0.05 with BH adjustment, Figure 2B) differentially expressed between tumor samples and normal controls based on one TCGA dataset (TCGA-KIRC, Table 1) and two GEO datasets (GSE53757 and GSE66270, Table 1). To determine immune-related protein-coding genes, gene set enrichment analysis based on MSigDB C7 immunologic signatures showed that 1105 differentially expressed protein-coding genes were significantly enriched in 51 signatures (FDR < 0.01 with BH adjustment, Figure 2C), including CD8 T cell, CD4 T cell, and NK cell-associated signatures. Here, 1105 genes were considered immune-related genes associated with ccRCC.
 |
Table 1 Patient Clinical Information of ccRCC Datasets |
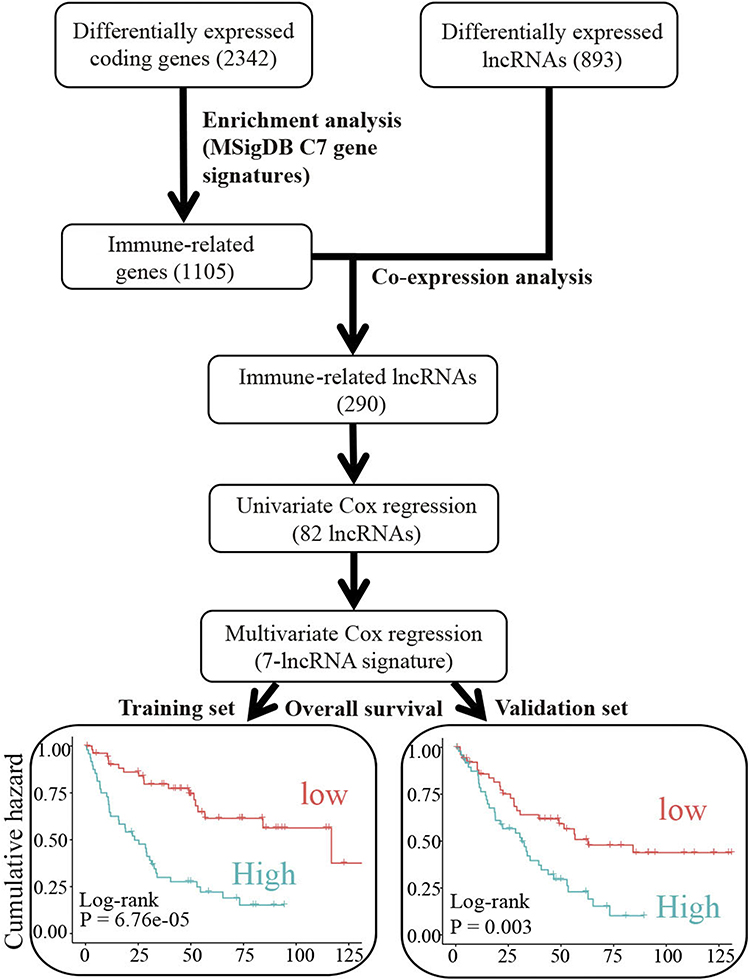 |
Figure 1 Flowchart of the strategy to identify immune-related prognostic lincRNAs in ccRCC. Abbreviation: MSigDB, Molecular Signature Database. |
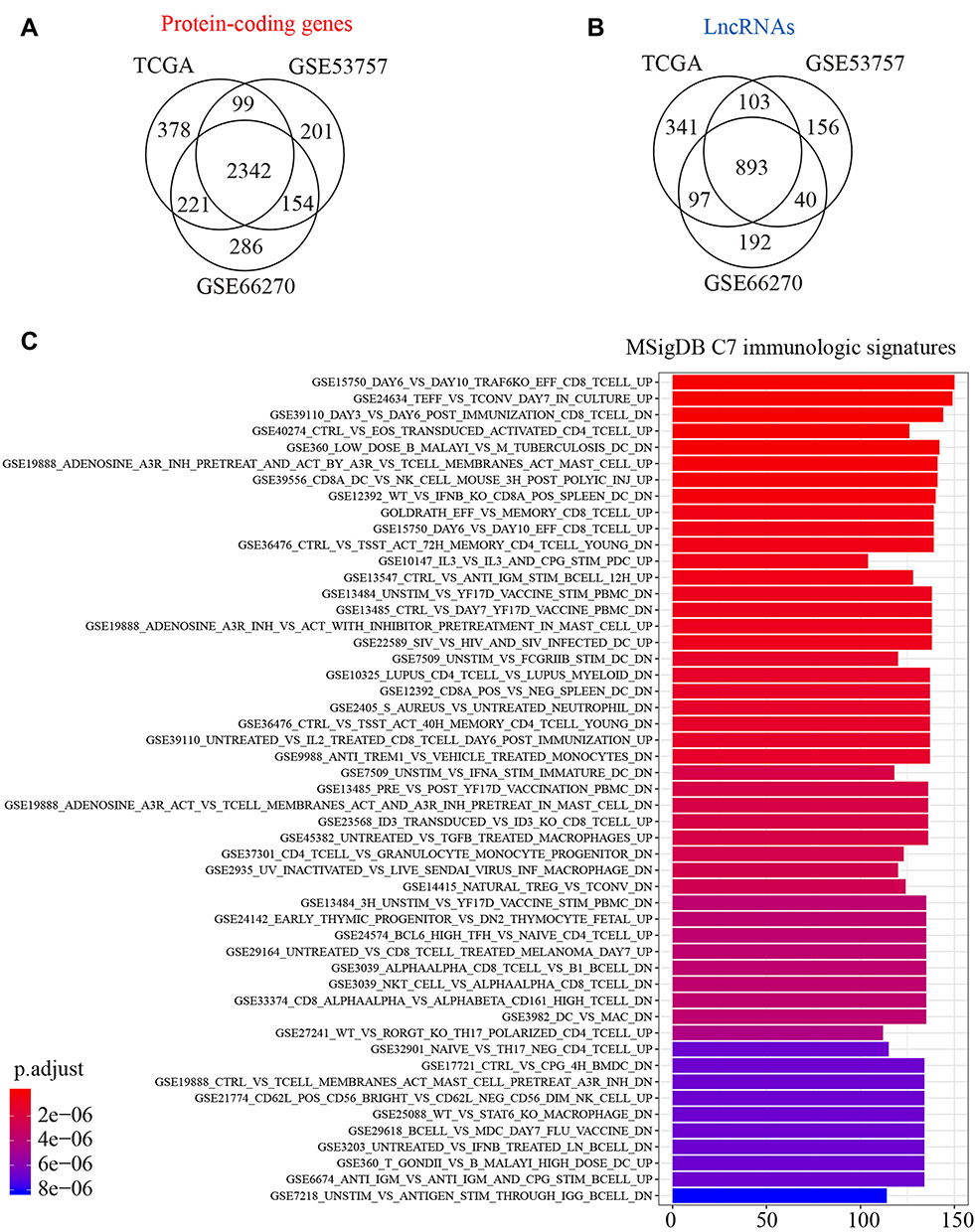 |
Figure 2 Identification of ccRCC-related lncRNAs and immune-associated coding genes. (A) Identification of differentially expressed (DE) protein-coding genes between ccRCC tumor sample and normal controls based on TCGA and GEO databases. (B) Identification of DE lncRNA between tumor and normal. (C) Gene set enrichment analysis of DE protein-coding genes based on MSigDB C7 immunologic signatures. Genes and signatures with Benjamini-Hochberg (BH) adjusted P-value < 0.05 were significant. |
Identification of Immune-Related lncRNAs
We performed a Pearson correlation analysis to identify differentially expressed lncRNAs with potentially functional relevance to immune-related genes. The hypothesis is that if a lncRNA is functionally associated with a protein-coding gene, it should be co-expressed. The lncRNA-gene pairs with Pearson correlation coefficients |r| ≥0.4 and BH-adjusted P <0.05 were significant co-expression. As a result, a total of 290 differentially expressed lncRNAs were co-expressed with immune-related genes (Figure 3). Hierarchical clustering of the Pearson correlation matrix showed that 290 lncRNA was associated with immune-related genes involved in activities of CD4/CD8 T cells, B cells, and natural killer (NK) cells (Figure 3).
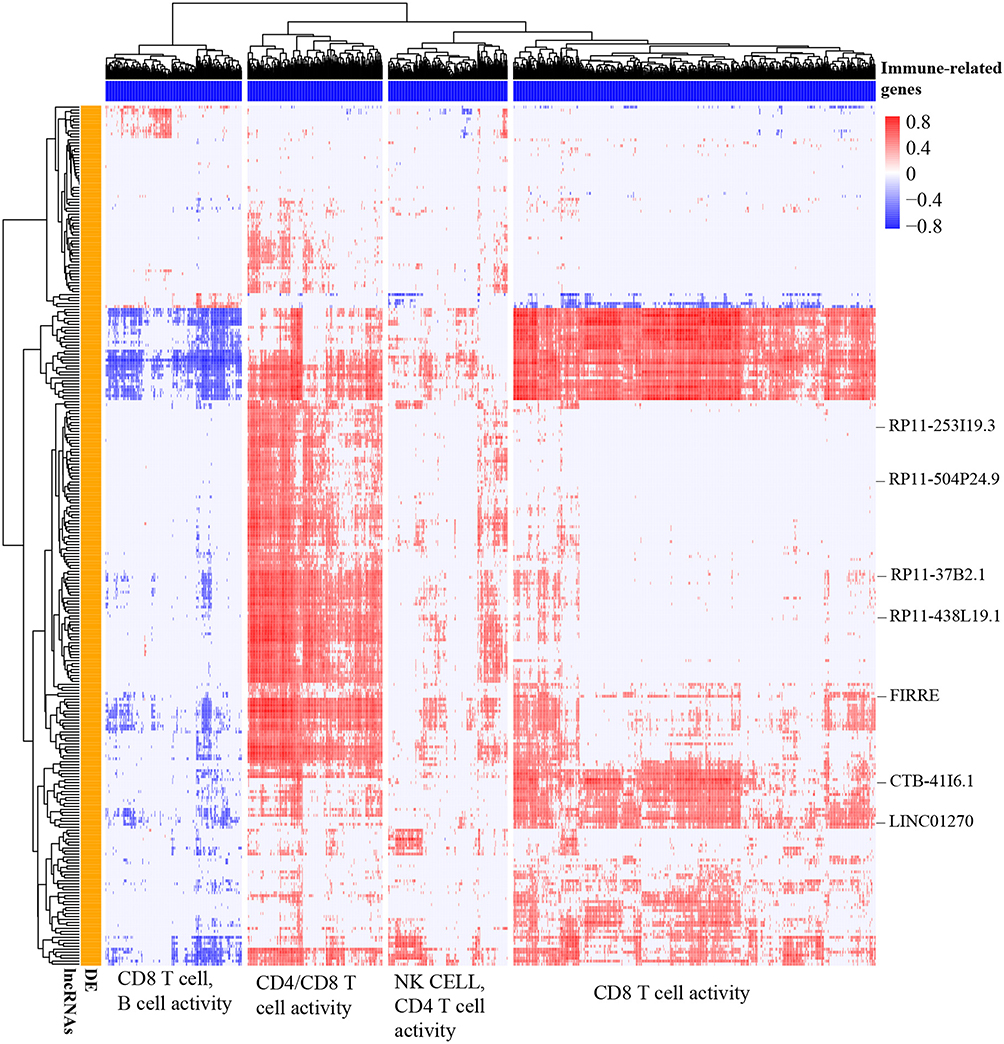 |
Figure 3 Identification of immune-related lncRNAs in ccRCC. Pearson correlation coefficient matrix of 290 differentially expressed lncRNAs (y-axis) and 1105 immune-related protein-coding genes (x-axis). For better visualization, the cells in the heatmap (correlation of specific gene-lncRNA pair) with BH-adjusted P < 0.05 and Pearson correlation coefficients |r| < 0.4 were considered no significance and were replaced with zero. The Euclidean distance metric with Ward linkage was used for hierarchical clustering of the correlation matrix. The functional classification of the four clusters of immune-related genes was performed based on MSigDB C7 signatures (BH-adjusted P < 0.05 was used as a cut-off value). |
7-lncRNA Signature Could Be an Independent and Unfavorable Prognostic Biomarker for Late-Stage ccRCC
Univariate Cox proportional hazard regression based on 225 immune-related lncRNAs was performed to identify lncRNAs associated with overall survival of patients with late-stage ccRCC. Here, to make full use of available data and make the results more robust, the TCGA cohort and the ICGC cohort were used for training and validation, respectively. Results showed that 82 lncRNAs were associated with overall survival of patients with late-stage ccRCC (P < 0.05, Log rank test). Further multivariate analysis using the 82 lncRNAs showed a 7-lncRNA signature (LINC01270, FIRRE, RP11-37B2.1, RP11-253I19.3, RP11-438L19.1, RP11-504P24.9 and CTB-41I6.1, log-rank P = 6.04E-06, Figure 4A). The scoring method (Pindex) was calculated based on the 7-lncRNA signature. To investigate whether the 7-lncRNA model was affected by clinical factors, multivariate regression was performed using the Pindex and clinical parameters as candidate variables. The Kaplan–Meier curves were used to evaluate the survival rates of the 7-lncRNA signature. In this multivariate model, the Pindex values of patients were divided into two groups (median as cut-off value). Results showed that our 7-lncRNA signature could be an independent factor for the overall survival prognosis of late-stage ccRCC in both training (P = 2.14E-04, Figure 4B, Table 2) and validation datasets (P = 0.003, Figure 4C, Table 2). The Pindex-high group patients have significantly poor survival compared with low-Pindex patients (Figure 4B and C).
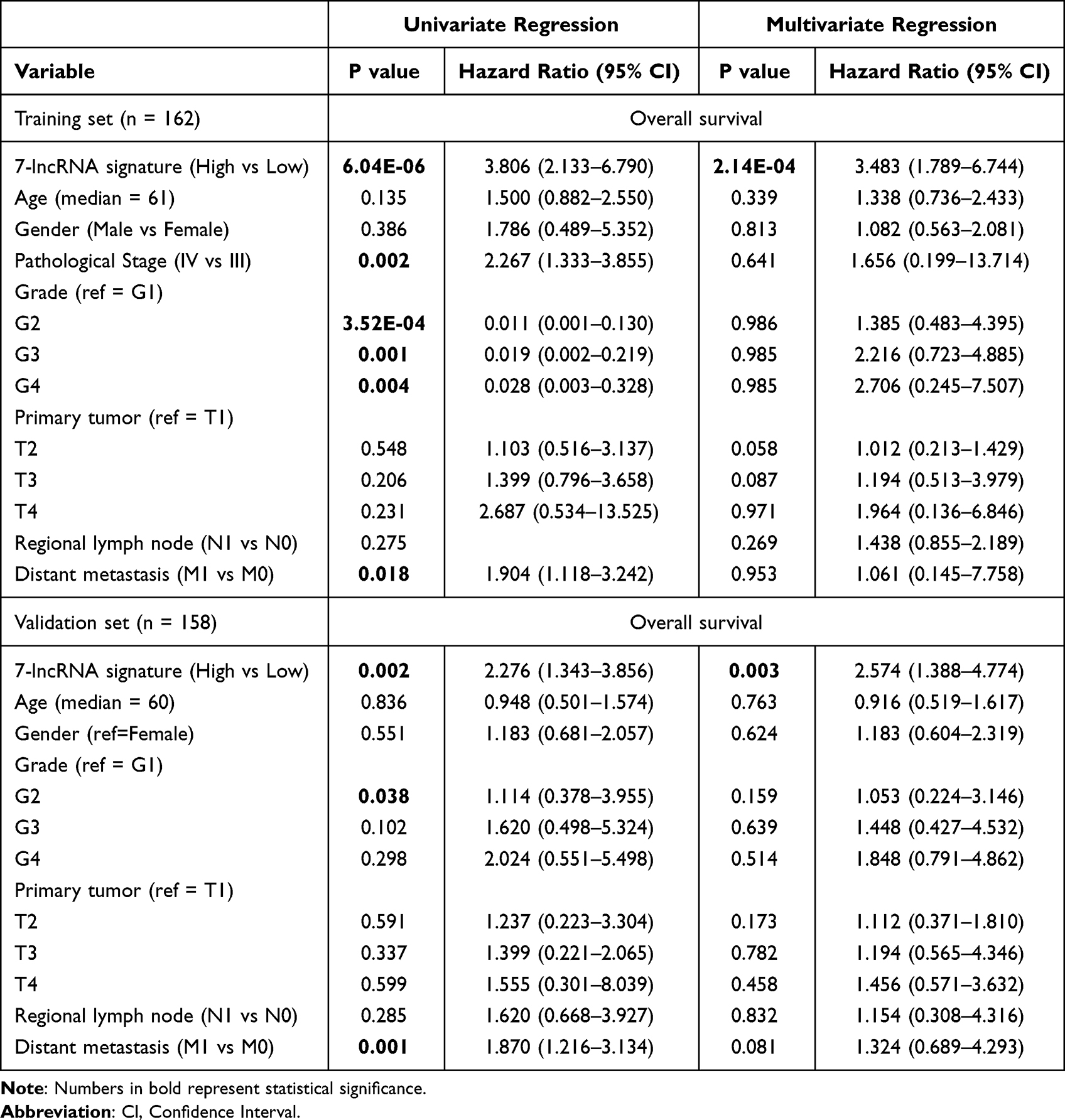 |
Table 2 The Univariate and Multivariate Cox Proportional Hazard Model for Stage III/IV ccRCC Patients |
 |
Figure 4 The prognostic potential of our 7-lncRNA signature. (A) The expression of the seven lncRNAs in the TCGA cohort. (B and C) Overall survival of patients with late-stage ccRCC in training (TCGA, n = 162) and validation datasets (ICGC, n = 158). The prognostic index (Pindex) was calculated based on the expression and Cox regression coefficients of the seven lncRNAs. The median value of Pindex was used as a cut-off to divide patients into high- and low-Pindex groups. |
The Functions of the 7 lncRNAs and the Correlations with Immunotherapy Targets
Immune checkpoint blockade therapies were effective for diverse cancer types, such as ccRCC. The major targets of immune checkpoint blockade therapies are PD-1 (PDCD1 or CD279), PD-L1 (CD274), and CTLA4. We performed correlation analysis between immunotherapeutic targets and the seven lncRNAs. It showed that they have universal associations with immunotherapeutic targets (Figure 5). For example, both RP11−504P24.9 and CTB-41I6.1 were associated with the expression of PD-1 and CTLA4 (P values <0.05, r >0.2). RP11−37B2.1 was associated with the expression of PD-L1 and CTLA4 (P values <0.05, r >0.2, Figure 5). We further explored the potential functions and pathways to investigate how the 7-lncRNA signature is involved in tumor immunity. We showed that 260 genes of “pathway in cancer” were related to the seven lncRNAs (FDR <0.05 and Pearson |r| >0.4). They were involved in many cancer-related signaling pathways, such as MAPK, cell adhesion-related pathways, ERBB, Wnt, apoptosis, T/B cell, and chemokine signaling (Figure 6A). Based on GO enrichment analysis, the cellular components are protein receptors/complexes in the cell surface and extracellular matrix, such as immune-related CD40 receptor complex (Figure 6B). Moreover, based on the enrichment results of molecular functions, the protein receptors/complexes were growth factors, transcriptional factors, kinase, and cytokine/cytokine receptors (Figure 6C). Biological process enrichment results indicated that these protein receptors/complexes were involved in proliferation and apoptosis (Figure 6D). In summary, the seven lncRNAs may play an important role in tumor immune cells and tumor progression through regulating the binding of protein receptors/complexes.
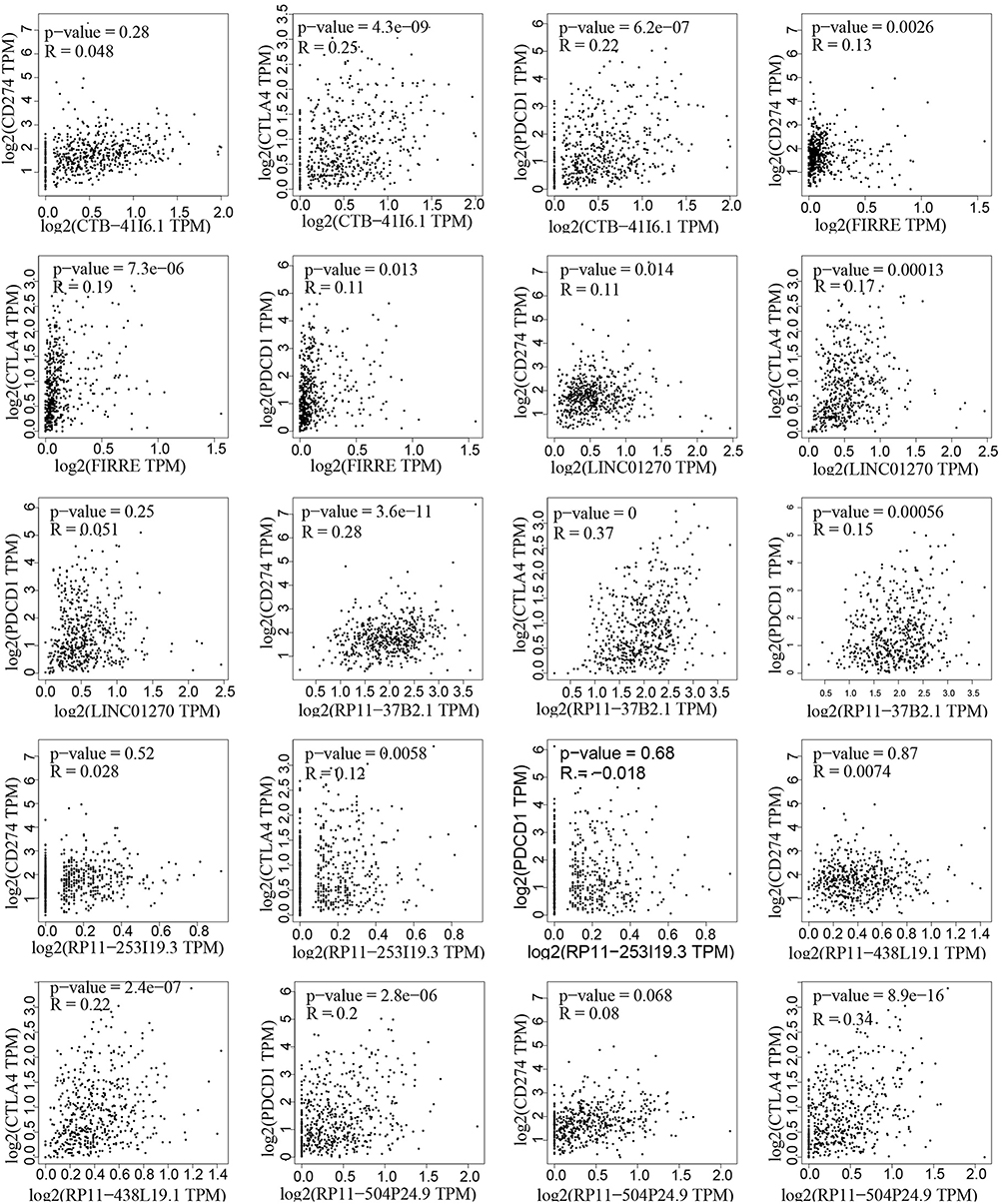 |
Figure 5 The correlations between PD-1/PD-L1/CTLA4 and the 7 lncRNAs in ccRCC. The three genes PD-1/PD-L1/CTLA4 are the most effective drug targets of immune checkpoint blockade therapy. The Pearson correlation was used in this analysis. |
 |
Figure 6 The functions and pathways of the 7 lncRNAs. (A) The KEGG pathways analysis enriched based on the protein-coding genes associated with the 7 lncRNAs. (B–D) The GO cellular components, molecular function, and biological process enrichment results, respectively. |
Experimental Validation of the Expression of the Seven lncRNAs in ccRCC Tissues and Cell Lines
qRT-PCR was performed to examine the expression level of the seven lncRNAs in ccRCC tissues (n = 20). We found that the seven lncRNAs had a higher expression level in ccRCC tissues than in adjacent normal tissues (Figure 7A). Based on the lncRNA expression level in ccRCC tissues, we finally selected LINC01270 as the object of subsequent research. qRT-PCR showed that the expression level of LINC01270 was increased in ccRCC cell lines than in human renal tubular epithelial cells HK-2 cells (Figure 7B).
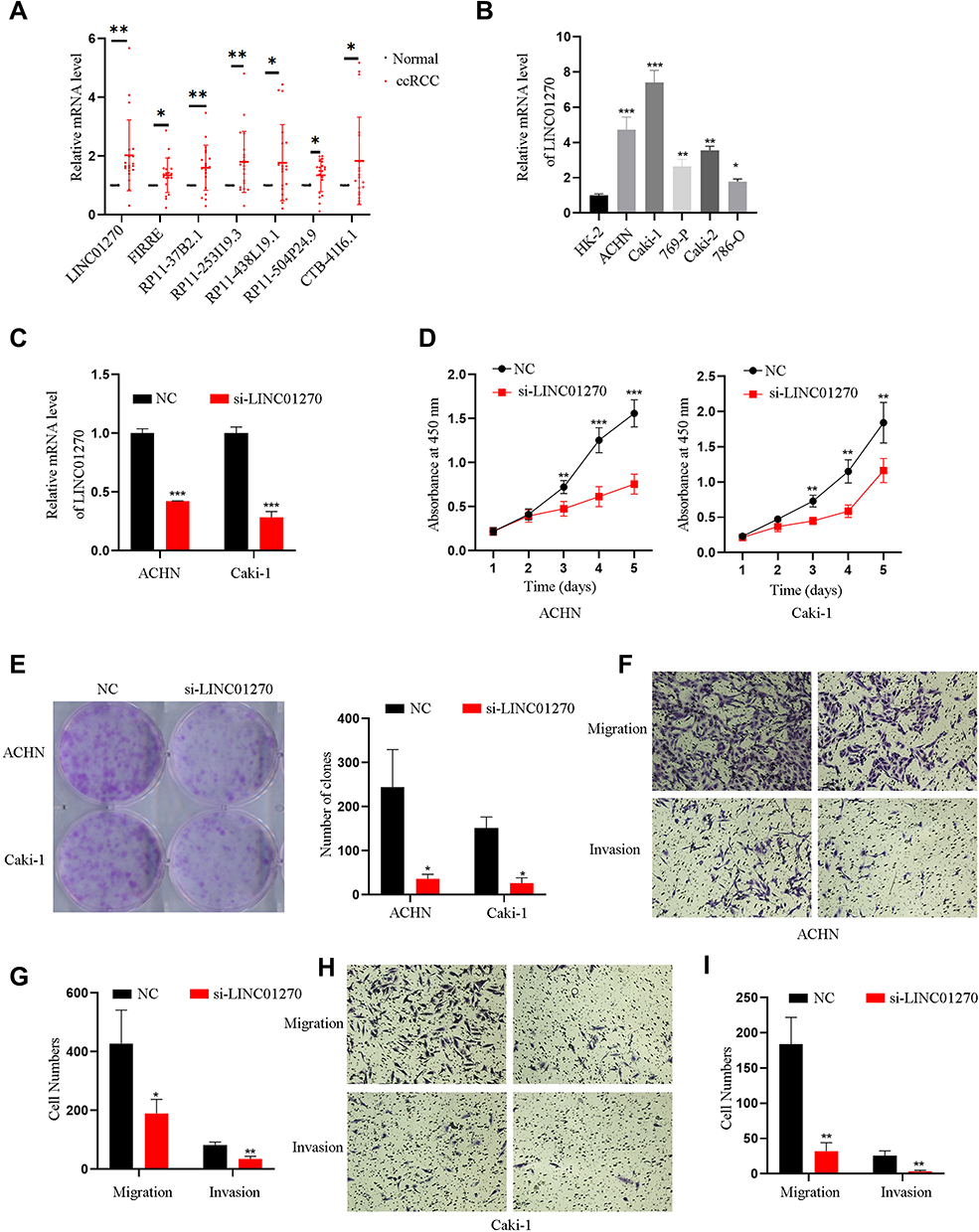 |
Figure 7 Experimental validation for the expression and function of LINC01270. (A) The differential expression level of seven lncRNAs between renal clear cell carcinoma tissues (n=20) and their paired normal tissues were detected by qRT-PCR. (B) The differential expression levels of LINC01270 between renal clear cell carcinoma cells and human renal tubular epithelial cells HK-2 cells were measured by qRT-PCR. (C) The transfected efficiency of LINC01270 was measured using qRT-PCR. (D and E) The CCK-8 and colony formation assays were used to detect cell proliferation ability. (F–I) The migration and invasion were used to measure the cell migration and invasion ability. *p<0.05; **p<0.01; ***p<0.001. |
The Effect of LNC01270 in Proliferation, Invasion, and Migration of ccRCC Cell Lines
We further investigate the function of LINC01270 in ccRCC cell lines. The transfected efficiency of LINC01270 was verified. The expression of LINC01270 was significantly decreased in both ACHN and Caki-1 cells by siRNA transfection (Figure 7C). To explore whether LINC01270 plays an important role in ccRCC progression, CCK-8, colony formation assays, and Migration and Invasion assays were used. CCK-8 assays and colony formation assays showed that the cell proliferation of ccRCC cells was inhibited in si-LINC01270 group compared with the control group (Figure 7D and E). Moreover, results of the transwell suggested that LINC01270 knockdown inhibited migration and invasion of ccRCC cells (Figure 7F–I). Taken together, LINC01270 may act as an oncogene to promote ccRCC progression.
Discussion
This study comprehensively evaluated the prognostic potential of immune-related lncRNAs in ccRCC by leveraging the knowledge of MSigDB C7 immunologic signatures. First, we identified ccRCC-related lncRNAs using differential expression analysis. Next, the immune-related genes were identified by gene set enrichment analysis of immunologic signatures. The lncRNAs co-expressed with immune-related genes were considered immune-related lncRNAs and were used to perform an overall survival analysis. Finally, a 7-lncRNA signature was identified, which could be an unfavorable biomarker for the overall survival of late-stage ccRCC patients. Patients in the high-Pindex group showed poorer survival than those in the low-Pindex group. This signature provided a novel clinical tool for the prognosis of late-stage ccRCC at the molecular level. Importantly, we found that the LINC01270 was upregulated in ccRCC tissues and cell lines. Silence of LINC01270 resulted in inhibition of cell proliferation, invasion, and migration of ccRCC. Although current prediction model has significant limitations in clinical application, prognostic models provide information on which risk factors are associated with disease outcomes and the relative magnitude of their influence on disease outcomes. Moreover, prospective validation of this signature is necessary for future clinical applications.
The polygenic molecular signature is a composite signature that has shown strong ability in tumor diagnosis, prognosis, and classification of pathological subtypes, probably because it organically integrates the predictive roles of multiple key genes. An expression analysis using three independent cohorts showed that the seven lncRNAs were upregulated in ccRCC compared with normal controls. Based on the functional analysis, the 290 immune-related lncRNAs showed strong relevance to immune cell activity, including B cells, NK cells, and CD4/CD8 T cells. The seven prognostic lncRNAs were mainly associated with CD4/CD8 T cells activity. It was reported that RP11-37B2.1 was associated with thrombocytopenia development during anti-TB therapy and suggested that its genetic variants could be potential biomarkers for thrombocytopenia during anti-TB treatment.21 FIRRE can serve as a platform for trans-chromosomal co-localization in that the loci located on different chromosomes are brought together around the transcription site of FIRRE to modulate nuclear architecture across chromosomes.22,23 Another study reported that FIRRE helped position the inactive X chromosome near the nucleolus and was related to the stabilization of H3K27me3 histone modifications.24 Notably, it was revealed that FIRRE was involved in NF-kappaB signaling to positively regulate the expression of inflammatory genes in macrophages following LPS stimulation, suggesting that FIRRE was associated with the innate immune system.25 There is no functional study for other lncRNAs to date. Thus, the 7-lncRNA signature could be a novel prognostic biomarker for the overall survival of ccRCC.
Cancer immune checkpoint blockade therapy is the most valuable cancer immunotherapy by blocking the activation of co-suppressive information molecules by T cells and releasing the anti-cancer immune response in a suppressed state to attack cancer cells. At present, the most effective immune checkpoint blockade drugs mainly target three genes: PD-1, PD-L1, and CTLA4. Their expression levels in tumors are closely related to the efficacy of this immunotherapy. We found that the seven lncRNAs were universally correlated with the three genes. Notably, RP11−504P24.9 and RP11−37B2.1 have a high correlation with CTLA4, implying that they may be of value in predicting the anti-CTLA4 immunotherapy.
Conclusions
In summary, we identified a 7-lncRNA signature (LINC01270, FIRRE, RP11-37B2.1, RP11-253I19.3, RP11-438L19.1, RP11-504P24.9, and CTB-41I6.1) with great potential in overall survival prognosis for patients with late-stage ccRCC, which could be a novel clinical tool at the molecular level.
Abbreviations
ccRCC, clear cell renal cell carcinoma; lncRNA, long non-coding RNA; GEO, Gene Expression Omnibus; GDC, Genomic Data Commons; BH, Benjamini-Hochberg; MSigDB, Molecular Signatures Database; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; Pindex, prognostic index; CCK-8, Cell Counting Kit-8 reagent; FDR, false discovery rate; NK, cells natural killer cells.
Data Sharing Statement
All data supporting the results of this study were included in the manuscript.
Ethics Approval
This study has been reviewed by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University. The informed consent was obtained from the study participants. The guidelines outlined in the Declaration of Helsinki were followed.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the Chongqing Science and Technology Commission of the joint project of traditional Chinese medicine in China (Grant no.2019ZY3130 to Chengxuan Liu).
Disclosure
The authors declare that there is no conflict of interest for this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
3. Barata PC, Rini BI. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin. 2017;67(6):507–524. doi:10.3322/caac.21411
4. Choueiri TK, Motzer RJ. Systemic therapy for metastatic renal-cell carcinoma. N Engl J Med. 2017;376(4):354–366. doi:10.1056/NEJMra1601333
5. Bex A, Albiges L, Ljungberg B, et al. Updated European Association of Urology guidelines regarding adjuvant therapy for renal cell carcinoma. Eur Urol. 2017;71(5):719–722. doi:10.1016/j.eururo.2016.11.034
6. Sánchez-Gastaldo A, Kempf E, González Del Alba A, Duran I. Systemic treatment of renal cell cancer: a comprehensive review. Cancer Treat Rev. 2017;60:77–89. doi:10.1016/j.ctrv.2017.08.010
7. Parker WP, Cheville JC, Frank I, et al. Application of the Stage, Size, Grade, and Necrosis (SSIGN) score for clear cell renal cell carcinoma in contemporary patients. Eur Urol. 2017;71(4):665–673. doi:10.1016/j.eururo.2016.05.034
8. Zisman A, Pantuck AJ, Dorey F, et al. Improved prognostication of renal cell carcinoma using an integrated staging system. J Clin Oncol. 2001;19(6):1649–1657. doi:10.1200/JCO.2001.19.6.1649
9. Motzer RJ, Powles T, Burotto M, et al. Nivolumab plus cabozantinib versus sunitinib in first-line treatment f or advanced renal cell carcinoma (CheckMate 9ER): long-term follow-up results from an open-label, randomised, Phase 3 trial. Lancet Oncol. 2022;23(7):888–898. doi:10.1016/S1470-2045(22)00290-X
10. Ciccarese C, Brunelli M, Montironi R, et al. The prospect of precision therapy for renal cell carcinoma. Cancer Treat Rev. 2016;49:37–44. doi:10.1016/j.ctrv.2016.07.003
11. Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A systematic review of immunotherapy in urologic cancer: evolving roles for targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol. 2015;68(2):267–279. doi:10.1016/j.eururo.2015.02.032
12. Najafi S, Khatami SH, Khorsand M, et al. Long non-coding RNAs (lncRNAs); roles in tumorigenesis and potentials as biomarkers in cancer diagnosis. Exp Cell Res. 2022;418(2):113294. doi:10.1016/j.yexcr.2022.113294
13. Fang Y, Fullwood MJ. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genomics Proteomics Bioinformatics. 2016;14(1):42–54. doi:10.1016/j.gpb.2015.09.006
14. Nallasamy P, Chava S, Verma SS, et al. PD-L1, inflammation, non-coding RNAs, and neuroblastoma: immuno-oncology perspective.
15. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. doi:10.1016/j.cels.2015.12.004
16. von Roemeling CA, Radisky DC, Marlow LA, et al. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014;74(17):4796–4810. doi:10.1158/0008-5472.CAN-14-0210
17. Wotschofsky Z, Gummlich L, Liep J, et al. Integrated microRNA and mRNA signature associated with the transition from the locally confined to the metastasized clear cell renal cell carcinoma exemplified by miR-146-5p. PLoS One. 2016;11(2):e0148746–e0148746. doi:10.1371/journal.pone.0148746
18. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
19. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
20. Wang W, Song J, Zhang W, et al. Identification of long non-coding RNA signatures for specific disease-free prognosis in clear cell renal carcinoma. IEEE Access. 2019;7:99290–99298. doi:10.1109/ACCESS.2019.2929588
21. Song J, Liu T, Zhao Z, et al. Genetic polymorphisms of long non-coding RNA RP11-37B2.1 associate with susceptibility of tuberculosis and adverse events of antituberculosis drugs in west China. J Clin Lab Anal. 2019;33:e22880. doi:10.1002/jcla.22880
22. Hacisuleyman E, Goff LA, Trapnell C, et al. Topological organization of multichromosomal regions by the long intergenic non-coding RNA Firre. Nat Struct Mol Biol. 2014;21(2):198–206. doi:10.1038/nsmb.2764
23. Nakagawa S, Hirano T. Gathering around Firre. Nat Struct Mol Biol. 2014;21:207. doi:10.1038/nsmb.2782
24. Yang F, Deng X, Ma W, et al. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015;16(1):52. doi:10.1186/s13059-015-0618-0
25. Lu Y, Liu X, Xie M, et al. The NF-κB-responsive long noncoding RNA FIRRE regulates posttranscriptional regulation of inflammatory gene expression through interacting with hnRNPU. J Immunol. 2017;199(10):3571–3582. doi:10.4049/jimmunol.1700091
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.