Back to Journals » OncoTargets and Therapy » Volume 11
Identification of differentially expressed genes and signaling pathways in ovarian cancer by integrated bioinformatics analysis
Authors Yang X, Zhu S, Li L, Zhang L, Xian S, Wang Y, Cheng Y
Received 21 September 2017
Accepted for publication 28 November 2017
Published 15 March 2018 Volume 2018:11 Pages 1457—1474
DOI https://doi.org/10.2147/OTT.S152238
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Samir Farghaly
Xiao Yang,1 Shaoming Zhu,2 Li Li,3 Li Zhang,1 Shu Xian,1 Yanqing Wang,1 Yanxiang Cheng1
1Department of Obstetrics and Gynecology, 2Department of Urology, Renmin Hospital of Wuhan University, 3Department of Pharmacology, Wuhan University Health Science Center, Wuhan, Hubei, People’s Republic of China
Background: The mortality rate associated with ovarian cancer ranks the highest among gynecological malignancies. However, the cause and underlying molecular events of ovarian cancer are not clear. Here, we applied integrated bioinformatics to identify key pathogenic genes involved in ovarian cancer and reveal potential molecular mechanisms.
Results: The expression profiles of GDS3592, GSE54388, and GSE66957 were downloaded from the Gene Expression Omnibus (GEO) database, which contained 115 samples, including 85 cases of ovarian cancer samples and 30 cases of normal ovarian samples. The three microarray datasets were integrated to obtain differentially expressed genes (DEGs) and were deeply analyzed by bioinformatics methods. The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichments of DEGs were performed by DAVID and KOBAS online analyses, respectively. The protein–protein interaction (PPI) networks of the DEGs were constructed from the STRING database. A total of 190 DEGs were identified in the three GEO datasets, of which 99 genes were upregulated and 91 genes were downregulated. GO analysis showed that the biological functions of DEGs focused primarily on regulating cell proliferation, adhesion, and differentiation and intracellular signal cascades. The main cellular components include cell membranes, exosomes, the cytoskeleton, and the extracellular matrix. The molecular functions include growth factor activity, protein kinase regulation, DNA binding, and oxygen transport activity. KEGG pathway analysis showed that these DEGs were mainly involved in the Wnt signaling pathway, amino acid metabolism, and the tumor signaling pathway. The 17 most closely related genes among DEGs were identified from the PPI network.
Conclusion: This study indicates that screening for DEGs and pathways in ovarian cancer using integrated bioinformatics analyses could help us understand the molecular mechanism underlying the development of ovarian cancer, be of clinical significance for the early diagnosis and prevention of ovarian cancer, and provide effective targets for the treatment of ovarian cancer.
Keywords: ovarian cancer, GEO data, integrated bioinformatics, differentially expressed genes
Introduction
The mortality rate of ovarian cancer ranks the first among the malignant tumors that occur in female reproductive organs, and the incidence of this cancer is increasing each year. More than 200,000 new cases of ovarian cancer occur each year in the world, resulting in >140,000 deaths.1 The incidence of ovarian cancer is occult, early diagnosis is difficult, and invasion and metastasis occur easily. When ovarian cancer is detected, the patient is usually at an advanced stage of the disease. The 5-year survival rate of patients with advanced ovarian cancer is only ~20%, but for patients in the early stage, it can reach 85%–90%.2 At present, the commonly used methods for the early diagnosis and monitoring of ovarian cancer are ultrasonography combined with serum tumor marker assays, but there are some limitations, and the specificity is not high. Computed tomography (CT) and positron emission tomography (PET) can only detect lesions with a volume of ≥1 cm, and they cannot detect early tumor metastasis. At present, the treatment of ovarian cancer includes surgical staging, surgery, reoperation, staging surgery, cytoreductive surgery, postoperative combined chemotherapy, radiotherapy, and biological treatment. In recent years, the clinical diagnosis and treatment of ovarian cancer have improved, but the 5-year survival rate of patients is still 30%.3 The reasons that lead to a failure of treatment, tumor recurrence, and the low survival rate include its insidious onset, the fact that ovarian cancer is not usually detected at an early stage and cannot be removed effectively by surgery, and the fact that tumor cells have a primary or secondary tolerance to radiotherapy and chemotherapy.4 Therefore, it is important to study the underlying molecular mechanisms of the malignant biological behavior of ovarian cancer cells and therefore identify more effective early diagnostic techniques and more reliable molecular markers for monitoring recurrence and evaluating prognosis, as well as to explore a more effective way to block and control tumor cell proliferation, metastasis, and the reversal of drug resistance in cancer cells. As an efficient and large-scale technique for acquiring genetic data, gene expression microarrays have been widely used to collect gene chip expression profiling data and to study gene expression profiles in many human cancers. These microarrays provide a new method for studying tumor-related genes and offer promising prospects for molecular prediction, drug-based molecular targeting, and molecular therapy.5,6 With the widespread application of gene expression microarray technology, a large amount of data have been published on public database platforms, and integrating these databases can allow a deeper study of molecular mechanisms.
At present, a large number of studies have been performed on ovarian gene expression profiles, and these studies have screened thousands of differentially expressed genes (DEGs) that may be involved in the development and progression of ovarian cancer.7 However, the results for the identification of significantly expressed mRNAs are inconsistent or discrepant among different studies due to tissue or sample heterogeneity among each independent experiment, different technological detection platforms, different data processing methods, and the fact that the samples come from different backgrounds. Thus, there are still some limitations in a single-cohort study, and we should integrate their results using an unbiased approach. The integration and analysis of microarray data from several gene expression profiles may resolve these problems and enable the discovery of effective and reliable molecular markers. The RobustRankAggreg (RRA) approach has been specifically designed for the comparison of several ranked gene lists.8 The RRA method uses a probabilistic model for aggregation that is robust to noise and facilitates the calculation of significance probabilities for all the elements in the final ranking. The RRA method is able to view the ranking of each item in each list, and it compares this ranking with the baseline case where all the preference lists are randomly ordered. The P-value can represent the rank location and significance of a gene. The higher the gene ranks, the smaller the P-value is. RRA is a suitable and effective integrative analysis solution for the identification of statistically significant genes. In addition, it is useful when different kinds of genes are obtained by different technology platforms and full rankings of mRNAs are not available.
In this study, we have downloaded three original microarray datasets, GDS3592,9 GSE54388,10 and GSE66957 (Cheng et al, unpublished data, 2015), from the NCBI-Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), which contained a total of 115 samples, with 85 cases of ovarian cancer samples and 30 cases of normal ovarian samples. DEGs in ovarian cancer and normal ovarian samples were screened using the R software, and the gene ontology (GO) pathway enrichment analysis of DEGs was performed on DAVID (https://david.ncifcrf.gov/) and KOBAS-Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (http://kobas.cbi.pku.edu.cn/). Then, the STRING online database protein–protein interaction (PPI) network was used to analyze the association of DEGs and discover the molecular interactions involved in tumorigenesis. In conclusion, the DEGs associated with the carcinogenesis and development of ovarian cancer were screened through the GEO database of ovarian cancer and an integrated analysis was conducted. The biological functions and key signaling pathways of these DEGs are discussed, and the encoding protein interaction network was analyzed. Our study provides reliable molecular markers for early detection and prognosis, as well as effective drug targets for treating ovarian cancer.
Materials and methods
Microarray data
DNA microarray is a new technique that can analyze genome and characteristic map of gene expression. A variety of DNA microarray and DNA chip devices and systems have now been developed and commercialized. DNA microarray analysis includes an oligonucleotide chip, cDNA chip, and genomic chip, and is divided into the following two modes: one is to fix the target DNA on the support, which is suitable for the analysis of a large number of different target DNAs, and another involves fixing a large number of probes on the support material, which is suitable for the analysis of different probe sequences of the same target DNA.11 There are various platforms available (http://arrayconsortium.tgen.org/np2/home.do; http://www1.amershambiosciences.com/APTR1X/upp01077.nsf/Contentand/codelink_bioarray_system; http://www.affymetrix.com; http://www.illumina.com/prod_expression.htm; and http://www.nimblegen.com; http://www.xeotron.com).12
Using the keywords “ovarian cancer geo accession” to search on the GEO DataSets database (https://www.ncbi.nlm.nih.gov/geo/), the gene expression profiles of GDS3592,9 GSE54388,10 and GSE66957 (Cheng et al, unpublished data, 2015) were downloaded. The platform for GDS3592 is GPL570, [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2 Array, which includes 12 normal ovarian surface epithelial samples and 12 serous papillary ovarian adenocarcinomas specimens. The platform for GSE54388 is GPL570, [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2 Array, which consists of six ovarian surface epithelium samples and 16 high-grade serous ovarian cancer samples. The platform for GSE66957 is GPL15048 Rosetta/Merck Human RSTA Custom Affymetrix 2 microarray [HuRSTA_2a520709.CDF], which includes 12 normal ovarian surface epithelium samples and 57 high-grade serous ovarian cancer samples. Platform and series matrix file(s) were downloaded as TXT files. The dataset information is shown in Table 1. The R software package was used to process the downloaded files and to convert and reject the unqualified data. The data were calibrated, standardized, and log2 transformed.
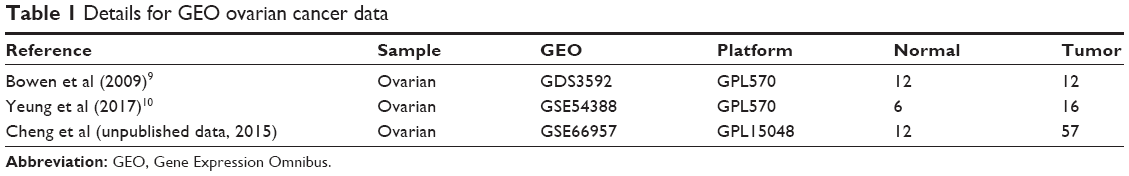 | Table 1 Details for GEO ovarian cancer data |
Screening for DEGs
The downloaded platform and series of matrix file(s) were converted using the R language software and annotation package. The ID corresponding to the probe name was converted into an international standard name for genes (gene symbol) and saved in a TXT file. Gene differential expression analysis was performed using the limma package in the Bioconductor package (available online: http://www.bioconductor.org/). The related operating instruction codes were put into R, and the DEGs in ovarian cancer and normal ovarian samples of the three microarray datasets were analyzed by the limma software package. Samples with a corrected P-value of <0.05 and log fold change (FC) >2 were considered DEGs. The TXT results were preserved for subsequent analysis.
Integration of microarray data
The list of DEGs from the three microarray datasets obtained by limma packet analysis was saved as a TXT file. The RRA software package was downloaded, and R was used to run the instruction code. A list of genes that were up- or downregulated in the three chips were used for subsequent analysis. The RRA approach is openly available in the Comprehensive R Network (http://cran.r-project.org/).
GO and KEGG pathway enrichment analyses of DEGs
The DAVID database (https://david.ncifcrf.gov/) is an essential foundation for the success of any high-throughput gene function analysis. The functional and pathway enrichment of the proteins encoded by candidate genes were analyzed, and these genes were annotated using the DAVID database.13 GO annotations were performed using a DAVID online tool on the screened DEGs. KEGG pathway analysis of DEGs was performed using the KOBAS online analysis database (available online: http://kobas.cbi.pku.edu.cn/). In this study, we analyzed the DEGs that were significantly up- and downregulated as determined from integrated microarray ovarian cancer data, and a P-value of <0.05 was considered statistically significant.
PPI network integration
The STRING database (http://string-db.org/) is a software system that is commonly used to identify the interactions between known proteins and predicted proteins. The results in this database are obtained from experimental data, databases, text mining, and predictive bioinformatics data.14 In addition, the core of the Cytoscape software is a network. Each node is a gene, protein, or molecule, and the connections between nodes represent the interaction of these biological molecules, which can be used to identify interactions and pathway relationships between the proteins encoded by DEGs in ovarian cancer. The corresponding proteins in the central node may be core proteins or key candidate genes with important physiological regulatory functions.
Results
Microarray data information and identification of DEGs in ovarian cancer
The ovarian cancer expression microarray datasets GDS3592, GSE54388, and GSE66957 were standardized, and the results are shown in Figure 1. When the GDS3592 dataset was screened by the limma package (corrected P-value <0.05, logFC >2), 839 DEGs were obtained. Among them, 289 downregulated genes and 550 upregulated genes were identified. Overall, 255 DEGs were screened from the GSE54388 dataset, including 96 upregulated genes and 159 downregulated genes. Additionally, 1,498 DEGs were screened from the GSE66957 dataset, including 709 upregulated genes and 789 downregulated genes. The differential expression of multiple genes from the two sets of sample data included in each of the three microarrays is shown in Figure 2. The cluster heatmaps of the top 200 DEGs are shown in Figure 3.
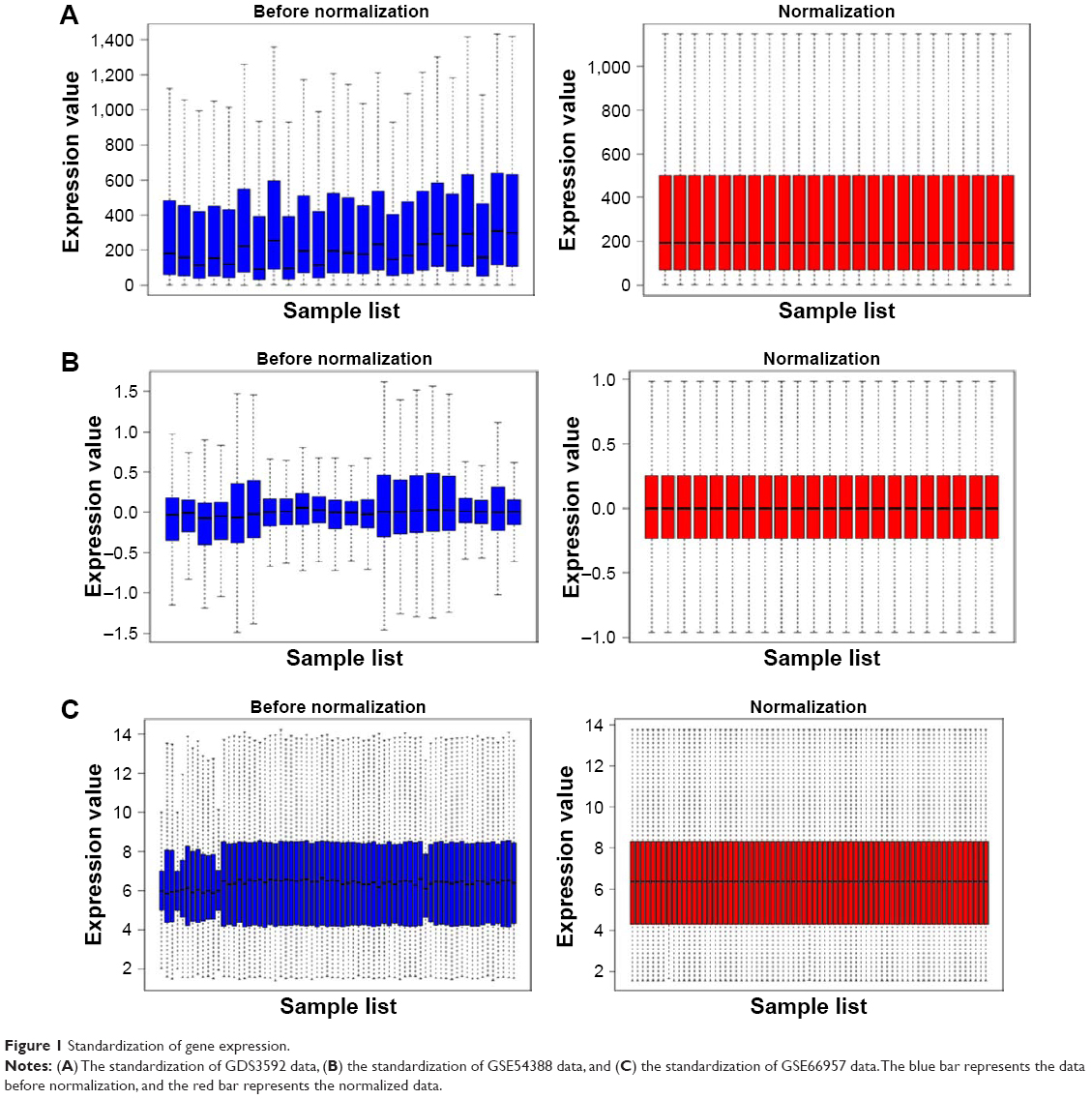 | Figure 1 Standardization of gene expression. |
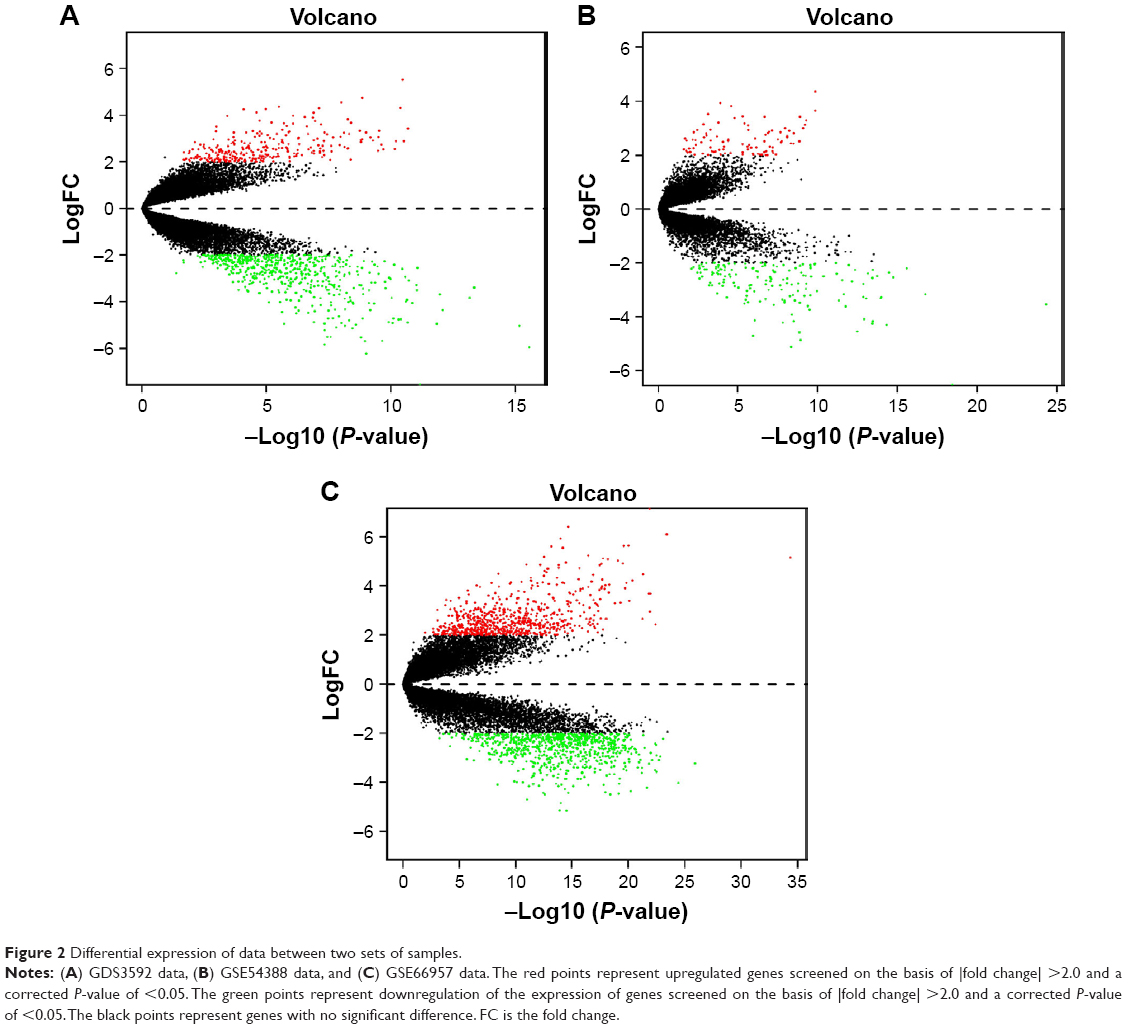 | Figure 2 Differential expression of data between two sets of samples. |
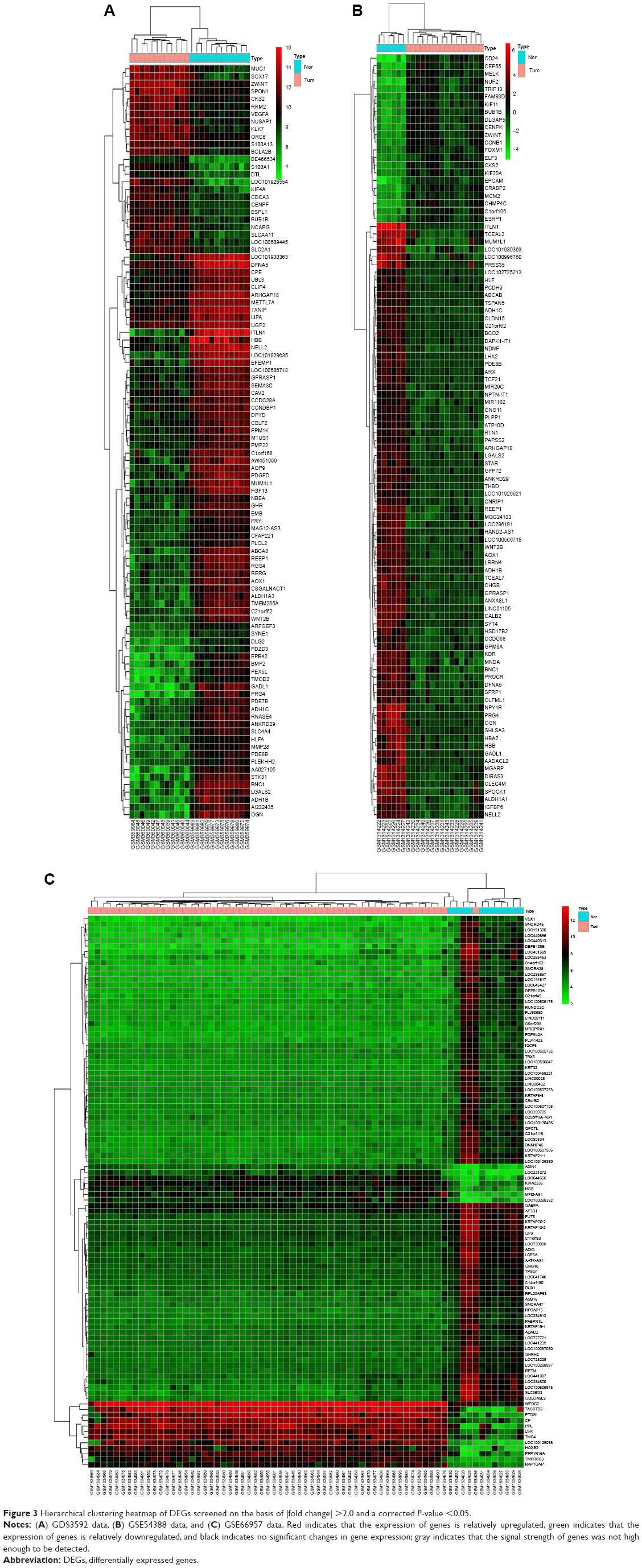 | Figure 3 Hierarchical clustering heatmap of DEGs screened on the basis of |fold change| >2.0 and a corrected P-value <0.05. |
Identification of DEGs in ovarian cancer using integrated bioinformatics
The three ovarian cancer gene expression microarray datasets were analyzed by the limma package and sorted according to logfold-change value and then analyzed by RRA (corrected P-value <0.05). The RRA method is based on the hypothesis that each gene is randomly ordered in each experiment. If a gene ranked high in all experiments, then the smaller its P-value is, the greater the likelihood of differential gene expression. Through rank analysis, we identified 190 DEGs, with 99 upregulated genes and 91 downregulated genes; the DEGs are shown in Table 2. R-heatmap software was used to draw a heatmap of the top 20 up- and downregulated genes, as shown in Figure 4.
 | Table 2 Screening DEGs in ovarian cancer by integrated microarray |
 | Figure 4 LogFC heatmap of the image data of each expression microarray. |
GO term enrichment analysis of DEGs
Biological annotation of the DEGs in ovarian cancer identified from an integrated analysis of microarray data was performed using the DAVID online analysis tool, and GO functional enrichments of up- and downregulated genes with a P-value of <0.05 were obtained. GO analysis of DEGs was divided into three functional groups, including molecular function, biological processes, and cell composition. The results are shown in Figures 5 and 6. Significant results of the GO enrichment analysis of DEGs in ovarian cancer are shown in Tables 3 and 4. In the biological process group, the upregulated genes were mainly enriched in cell proliferation, regulation of transcription, proteolysis, and epithelial cell differentiation. The downregulated genes were mainly concentrated in ethanol oxidation, mesenchymal–epithelial cell signaling, and the Wnt signaling pathway. In the molecular function group, the upregulated genes were mainly enriched in DNA binding, transcriptional activator activity, and endopeptidase activity. The downregulated genes were mainly enriched in binding, including oxygen binding, iron ion binding, and calcium ion binding, as well as peptidase activator activity and growth factor activity. In the cell composition group, the upregulated genes were mainly enriched in plasma membranes, extracellular space, extracellular exosomes, and vesicles. The downregulated genes were mainly enriched in the extracellular matrix, extracellular exosomes, and the hemoglobin complex. These results indicate that most DEGs are significantly enriched in cell proliferation, binding, cell cycle regulation, and transcriptional activity.
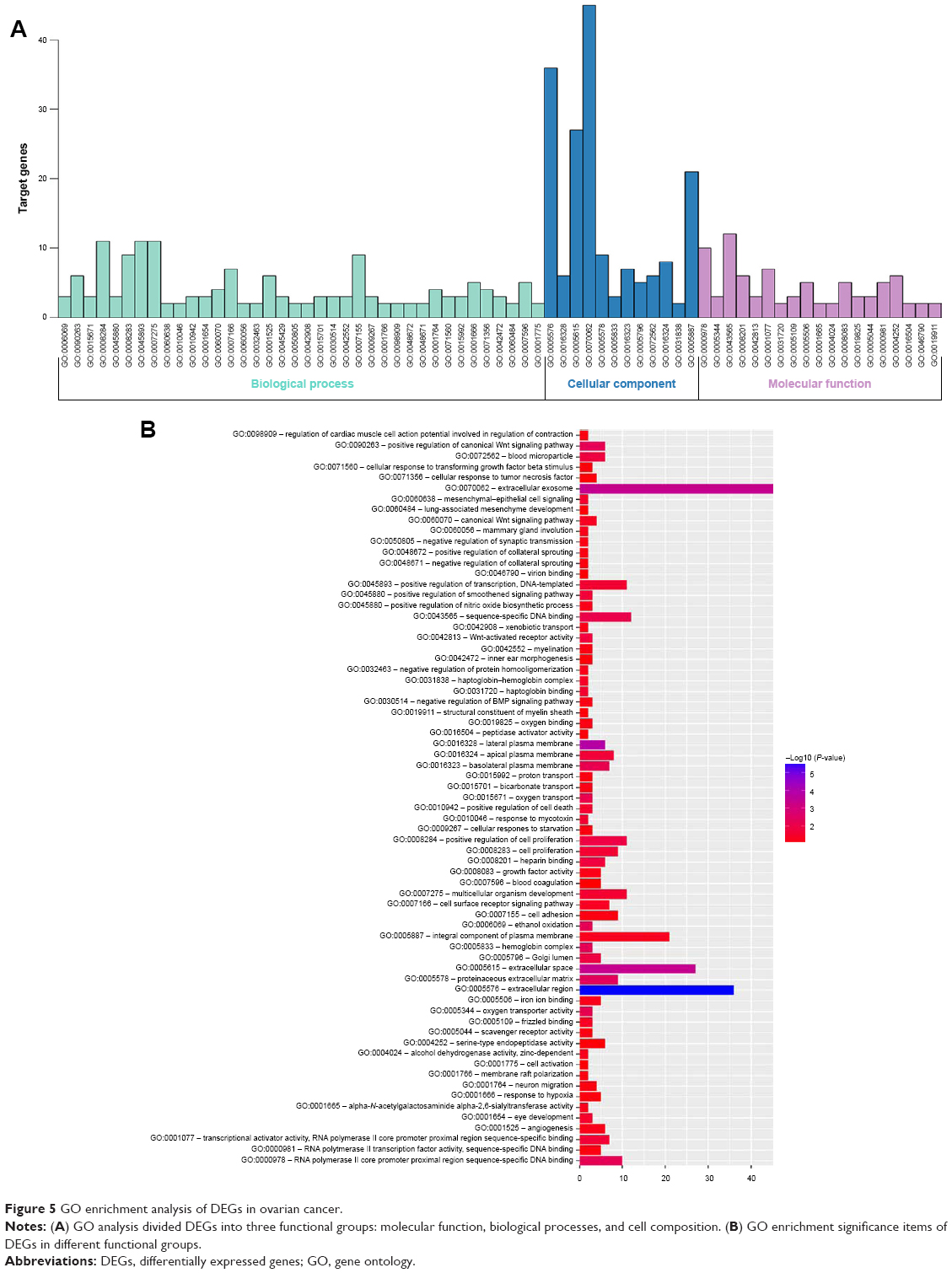 | Figure 5 GO enrichment analysis of DEGs in ovarian cancer. |
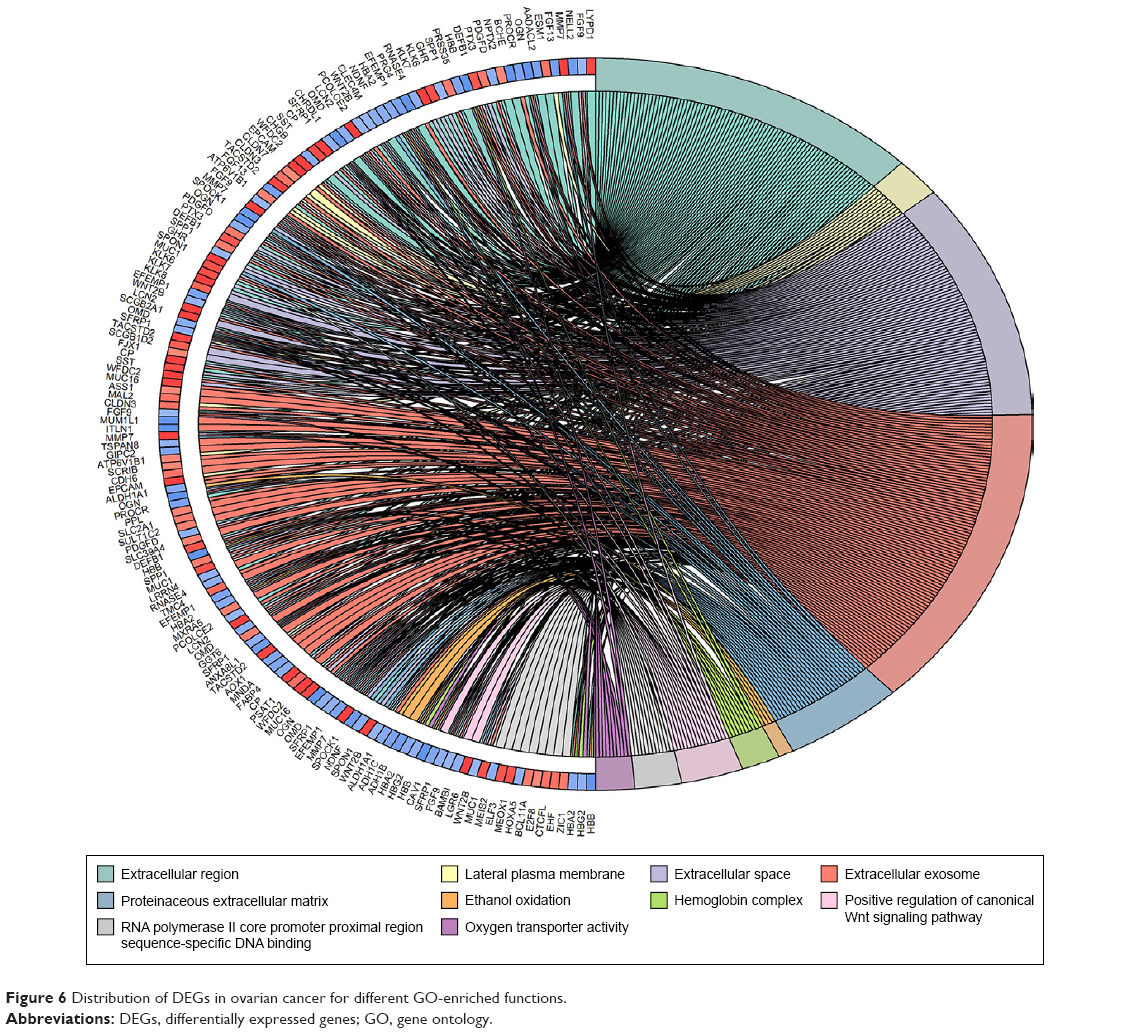 | Figure 6 Distribution of DEGs in ovarian cancer for different GO-enriched functions. |
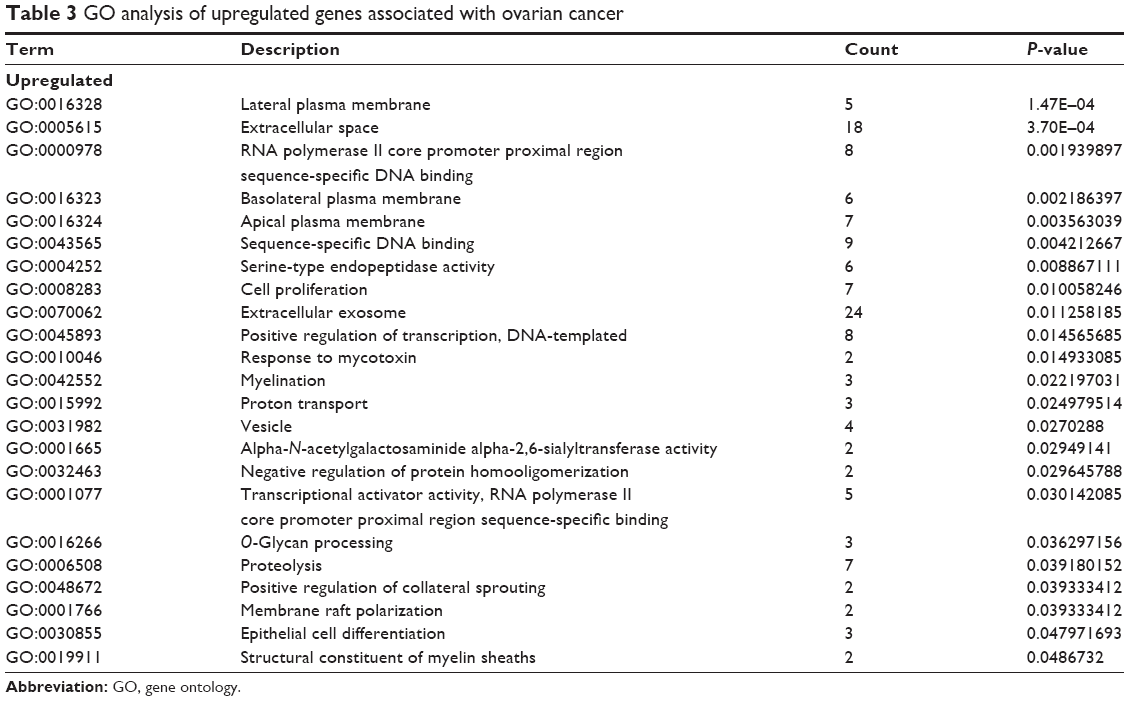 | Table 3 GO analysis of upregulated genes associated with ovarian cancer |
 | Table 4 GO analysis of downregulated genes associated with ovarian cancer |
KEGG pathway analysis of DEGs
Using the KOBAS online analysis database (http://kobas.cbi.pku.edu.cn/) to analyze the DEGs identified from ovarian cancer-integrated gene microarrays, the most significantly enriched pathways of the DEGs were submitted to KEGG analysis. The results are shown in Table 5. The signaling pathways of DEGs were mainly enriched in the Wnt signaling pathways, metabolic pathways, and pathways in cancer. The data were imported into Cytoscape to calculate the topological characteristics of the network and determine each node. The genes and pathway nodes are represented by semiellipses. The results are shown in Figure 7.
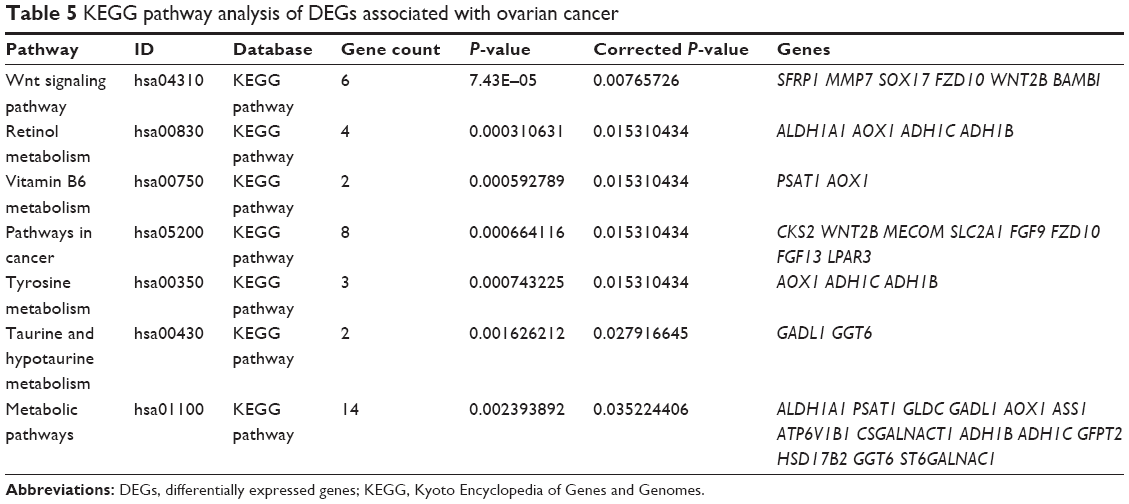 | Table 5 KEGG pathway analysis of DEGs associated with ovarian cancer |
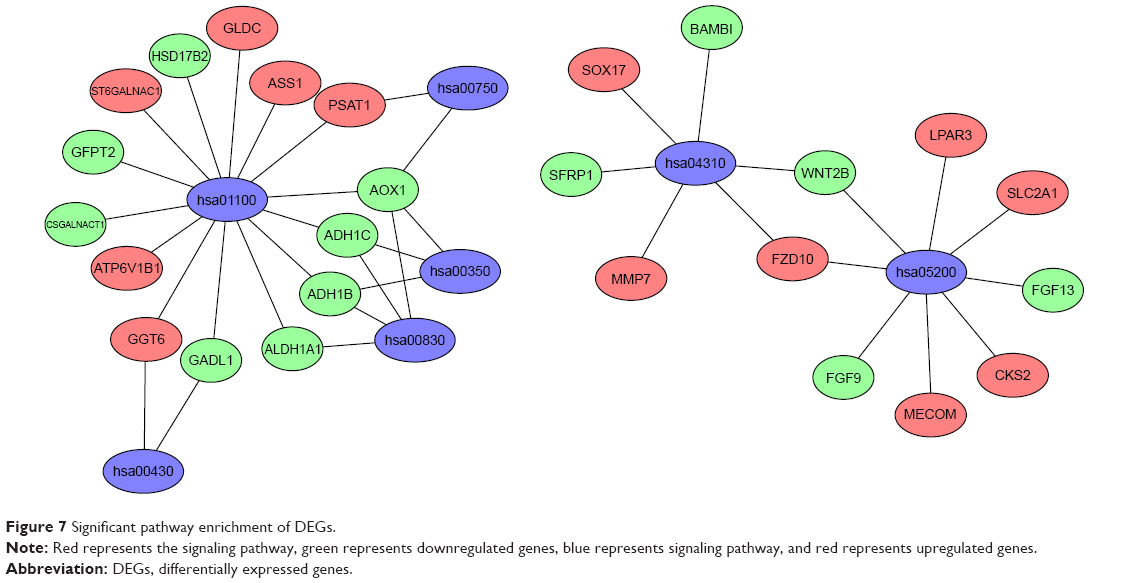 | Figure 7 Significant pathway enrichment of DEGs. |
Analyzing DEGs in ovarian cancer using a PPI network
The DEG expression products in ovarian cancer were constructed using the STRING database (http://string-db.org) to construct PPI networks, with a total of 190 DEGs, including 99 upregulated genes and 91 downregulated genes. After removing the isolated and partially connected nodes, a complex network of DEGs was constructed, as shown in Figure 8. The 17 most significant genes showing significant interaction were HBB, ZWINT, WNT2B, SPP1, HBA2, NUF2, ALDH1A1, FZD10, MMP7, MUC16, MUC1, OMD, OGN, AOX1, ADH1B, HBG2, and TTK.
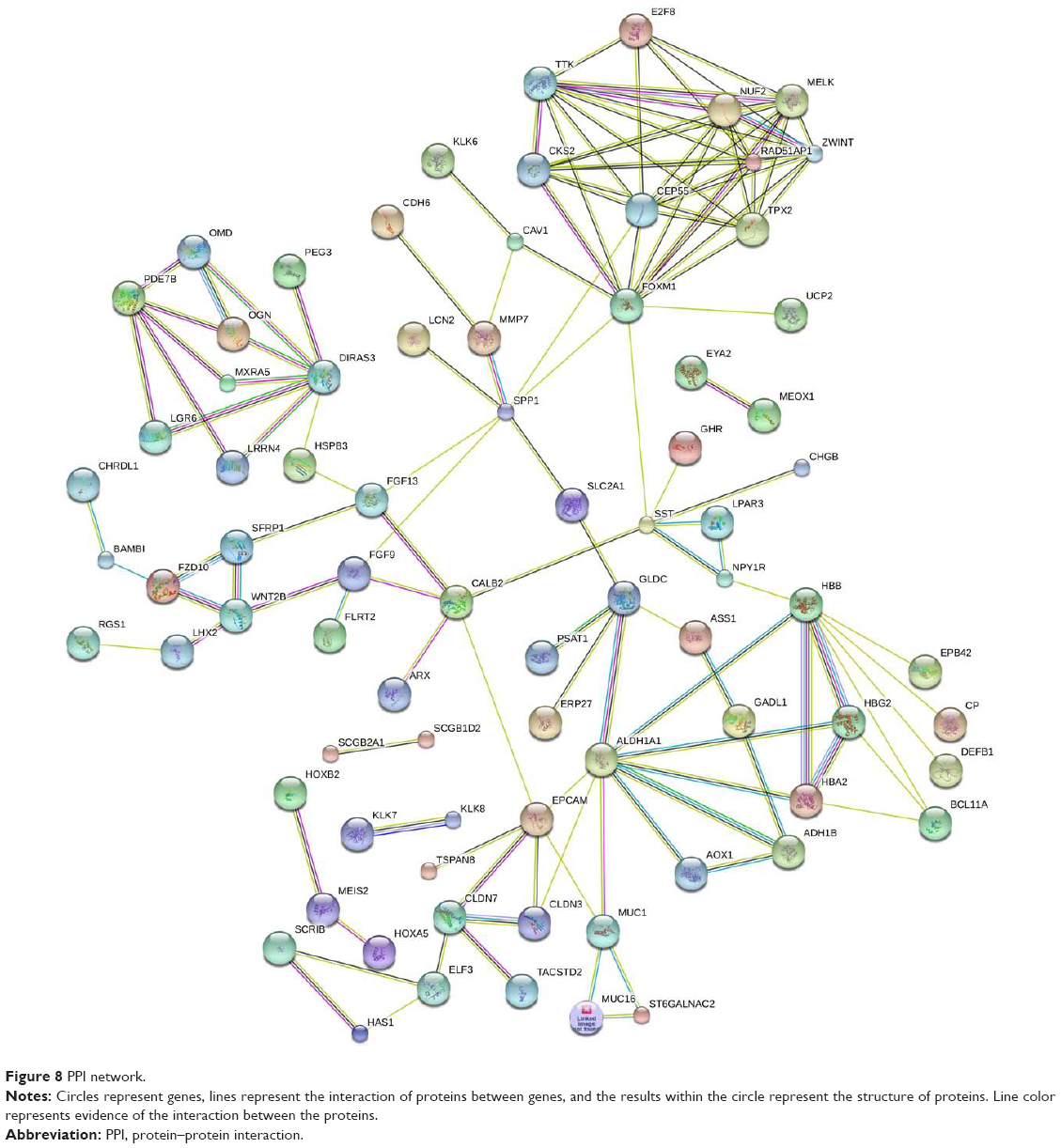 | Figure 8 PPI network. |
Discussion
Ovarian cancer is one of the most common tumors in the female reproductive system. The incidence of ovarian cancer is the second highest among gynecologic malignancies, but the mortality rate is the highest among gynecologic tumors.15 The early onset of ovarian cancer is difficult to identify, and tumors are often found in the late stages of the disease. Its occurrence and development are complex biological processes that can occur at any age, and the disease has a poor prognosis. Therefore, it is important to study the molecular mechanisms of the carcinogenesis and development of ovarian cancer.
Microarray and high-throughput sequencing technologies that detect the expression levels of tens of millions of genes in humans have been widely used to predict potential targets for ovarian cancer treatment. In recent decades, there have been many basic research reports on the mechanisms underlying the occurrence of ovarian cancer, but the 5-year survival rate of ovarian cancer is still relatively low, and there are no clear and effective treatment measures because most studies focus on a single genetic event or the results are generated from a single-cohort study.16 This study integrated three gene expression profile datasets from different groups and used R software and bioinformatics to deeply analyze these datasets. The results identified 255 DEGs using the RRA analysis method, including 96 upregulated genes and 159 downregulated genes. The top 20 most significantly upregulated genes were CP, CD24, KLK7, ST6GALNAC1, MMP7, EPCAM, SCGB2A1, PTH2R, PRAME, SPON1, MEOX1, ESRP1, MPZL2, TMPRSS4, SOX17, TNNT1, ELF3, SCGB1D2, SST, and LCN2. The top 20 most significantly downregulated genes were ITLN1, GADL1, PRG4, BCHE, SYT4, OGN, REEP1, GIPC2, MGARP, HOXA5, HBB, CHRDL1, SPOCK1, FABP4, MUM1L1, BNC1, VGLL3, SERTM1, TCEAL2, and ANXABL1. In addition, 255 DEGs were divided into groups by GO functional annotation, including molecular function, biological process, and cellular component groups. DEG enrichment was determined by KEGG signal pathway analysis to construct a PPI of DEG-encoding proteins and to screen the 17 most closely related genes. The DEGs in ovarian cancer were analyzed by GO functional annotation and showed that the upregulated DEGs were mainly involved in the regulation of cell proliferation and gene differentiation in ovarian cancer, transcription, different membranes, membrane raft polarization, proteolysis, and extracellular exosomes, and that the downregulated genes were mainly involved in the positive regulation of the canonical Wnt signaling pathway, oxygen transport, and mesenchymal–epithelial cell signaling, and in the negative regulation of the bone morphogenetic protein signaling pathway, and the cellular response to starvation and transforming growth factor β (TGF-β) stimulus. This finding is consistent with the knowledge that cell proliferation and differentiation, mesenchymal–epithelial cell signaling, and the cellular response to starvation and TGF-β play important roles in the tumor development and progression and that ion transport can contribute to cancer-related processes that differ substantially from processed in normal cells.17
The TGF-β signaling pathway is an important intracellular signal transduction pathway involved in the embryonic development, tumorigenesis, wound healing, inflammatory response, and physiological and pathophysiological processes.18 TGF-β can improve the adhesion and motility of tumor cells by promoting the epithelial-to-mesenchymal transition (EMT) and can inhibit the expression of E-cadherin, damaging the connection between the epithelium and weakening the adhesion of cancer cells. Through the expression of N-cadherin, interstitial characteristics can develop, and the invasion and metastasis of tumor cells from the primary site is facilitated.19 Therefore, the abnormal expression of TGF-β is closely related to tumorigenesis and cancer progression.
Furthermore, the enriched KEGG pathways of DEGs included the Wnt signaling pathway, metabolic pathways, and pathways in cancer. Recent study has shown that the Wnt/β-catenin signaling pathway can promote ovarian cancer resistance by promoting the EMT in ovarian cancer.20 Bodnar et al21 found that the activation of the Wnt/β-catenin signaling pathway could promote the proliferation and differentiation of ovarian cancer cells, inhibit apoptosis, and promote the growth of ovarian cancer. Multiple levels of negative modulators are involved in the Wnt/β-catenin signaling pathway. The DEGs that are closely associated with the Wnt/β-catenin signaling pathway in ovarian cancer were determined using the Cytoscape method, with SFRP1, MMP7, SOX17, FZD10, WNT2B, BAMBI, SOX17, FZD10, and MMP7 expressions upregulated and SFRP1, BAMBI, and WNT2B downregulated. Matrix metalloproteinases (MMPs) are closely related to tumor invasion and metastasis. The overexpression of MMPs can significantly promote the invasion and metastasis of tumor cells.22 MMP7 is an important member of the MMP family that can degrade the extracellular matrix, including the basement membrane, and inhibit the defense against tumor invasion and metastasis, thus increasing the invasive capacity of the tumor.23 The FZD10 gene is a member of the Frizzled gene family, which encodes the receptor protein of the Wnt pathway and plays an important role in the pathway. A study has found that BRMS1L mediates FZD10 silencing by promoting the recruitment of HDAC1 in the FZD10 promoter region and the acetylation of histone H3K9, thereby inhibiting the migration, invasion, and adhesion of breast cancer cell lines.24 Another study found that silencing the expression of FZD10 in ovarian cancer cells could significantly increase the sensitivity to chemotherapy drugs, but the specific mechanism was not clear.25 Our study suggests that FZD10 is closely related to Wnt and tumor-related pathways, which can provide a theoretical basis for further studies. SFRP1 is a Wnt antagonist that competes with the Frizzled protein receptor for Wnt ligands and can block Wnt signal transduction.26 Therefore, when SFRP1 is downregulated, it can promote the activation of the Wnt signaling pathway and the proliferation of tumor cells and accelerate the occurrence and development of tumors. The downregulation of BAMBI can promote tumor invasion and metastasis. Overexpression of BAMBI can reduce the TGF-β-induced EMT, and the invasion and migration of tumor cells and can slow tumor growth.27 A large number of studies have reported that WNT2B is upregulated in a variety of tumor tissues and is capable of activating the β-catenin-dependent Wnt signaling pathway.28,29 However, in our study, we found that the expression of WNT2B was downregulated in ovarian cancer, suggesting that it requires further detection in tissue samples to identify its expression patterns and explore the molecular mechanism underlying its role in ovarian cancer. Detecting these pathways and the expression of related molecules can help predict the occurrence and development of tumors.
We constructed a PPI network of protein encoded by DEGs and identified the following 17 closely related genes: HBB, ZWINT, WNT2B, SPP1, HBA2, NUF2, ALDH1A1, FZD10, MMP7, MUC16, MUC1, OMD, OGN, AOX1, ADH1B, HBG2, and TTK. The proteins encoded by these genes are key nodes in the PPI network. Pathway enrichment analysis revealed that the genes were mainly involved in the Wnt signaling pathway, retinol metabolism, pathways in cancer, and metabolic pathways. Endo et al found that ZWINT was overexpressed in breast cancer cell lines and could promote tumor cell growth, and that the degradation of ZWINT negatively regulated cell proliferation. Increasing evidence shows that SPP1 is closely related to the tumorigenesis and metastasis of tumors.30 A high expression of SPP1 was detected in tumor tissues, such as colon cancer, gastric cancer, prostate cancer, and breast cancer.31–33 NUF2 is expressed in a variety of malignancies and plays an important role in tumorigenesis and progression.34 NUF2 is highly expressed in epithelial ovarian cancer and is involved in many characteristics, such as the metastasis, invasion, division, and proliferation of cancer cells.35 However, the specific molecular mechanisms underlying the abnormal expression of NUF2 in tumorigenesis and progression remain to be studied.
In recent years, acetaldehyde dehydrogenases (ALDHs) have been found to play an important role in tumor cell metabolism and tumorigenesis. Among them, ALDH1 is closely related to tumor cell stemness. Among the numerous ALDH1 families, aldehyde dehydrogenase 1A1 (ALDH1A1) is one of the important members of the superfamily. The physiological function of ALDH1A1 is to participate in the metabolism of retinoic acid and promote the activation of the metabolic pathway of retinoic acid. In recent years, the role and function of this molecule in malignant tumors have attracted research attention. Studies have reported that ALDH1A1 is highly expressed in ovarian cancer cells and is involved in the resistance to chemoradiotherapy in ovarian cancer, resulting in recurrence and metastasis.36 Another study reported that ALDH1A1 is the target molecule of β-catenin and β-catenin knockdown can disrupt ovarian cancer spheroid formation, cell viability, and tumor growth and metastasis.37 Therefore, developing more specific ALDH1A1 inhibitors could increase chemotherapy effectiveness in ovarian cancer.
MUC16 is an important tumor marker for the early diagnosis of epithelial ovarian cancer. It is widely used in clinical practice. A study found that 80% of patients with ovarian cancer had elevated serum MUC16 levels.38 In addition, The Cancer Genome Atlas (TCGA) effort aimed at ovarian cancer found that both the amplification of the gene encoding MUC16 and the expression of MUC16 mRNA are closely related to the poor prognosis of patients with ovarian cancer,39 suggesting that the abnormal increase of tumor marker MUC16 in ovarian cancer plays an important role in the development and progression of ovarian cancer.
The Wnt signaling pathway is an important signaling pathway in biological development and tumorigenesis. β-Catenin is the major effector of the Wnt signaling pathway. After silencing MUC1, the levels of E-cadherin and E-cadherin/β-catenin complexes were elevated, and the expression levels of nuclear β-catenin, cyclin D1 (cyclinD1), and c-myc were decreased.40 In renal cell carcinoma, MUC1-C and β-catenin interact with the promoter region of SNAIL and increase SNAIL transcription, thereby facilitating the EMT of tumor cells.41 In recent years, MUC1-N monoclonal antibodies, MUC1-C peptides, and MUC1 vaccines have been extensively studied in preclinical, experimental, and clinical trials.
Osteomodulin (OMD) plays an important role in the extracellular matrix in tooth and cartilage tissue and is reportedly involved in bone mineralization.42 Osteoglycin (OGN) is a member of proteoglycans (PGs) called small leucine-rich PGs (SLRPs), which are a group of extracellular matrix molecules related in structure and function that are involved in matrix assembly, the regulation of cellular growth, and migration.43 OGN, also known as osteoinductive factor, plays an important role in maintaining normal bone tissue.44 As an extracellular matrix molecule, OGN is involved in the formation and regulation of extracellular matrix and has a potential impact on tumor cell metastasis. In vivo experiments have shown that OGN is involved in the regulation of collagen fiber formation associated with tumor metastasis, suggesting that OGN may inhibit tumor metastasis by a number of mechanisms.45 MMPs are important regulators of extracellular matrix metabolism and are closely related to tumor metastasis.46 Future studies should focus on the relationship between OGN expression and MMPs secreted by tumor cells.
Many studies have shown that the protein expression of the ADH1B gene is downregulated in many tumors.47,48 Compared with levels in normal tissues, the mRNA levels of ADH1B in colorectal cancer tissues were low.49 The risk of residual lesions in patients with high-grade ovarian cancer after cytoreductive surgery is closely related to the high expression of ADH1B.50 HBB is one of the globin strands of hemoglobin, whose basic function is to transport oxygen.51 A study found that the expression of HBB proteins in a rat model of ovarian cancer was significantly lower, which suggests that it can be used for early diagnosis and disease detection, and as a treatment target.52 Thus, ADH1B and HBB are negatively correlated with patient outcome. This conclusion is also consistent with previous studies reporting that HBB and ADH1B are biomarkers for early detection and found in other microarrays retrieved from oncomine.53
The important role of protein kinases in cellular activity has received increased attention. Protein kinases can phosphorylate target proteins deliver and amplify signals, and then regulate the activities of normal epithelial cells, inflammatory cells, tumor cells, and other cells, such as proliferation, migration, apoptosis, and metastasis.54 TTK kinase is a dual-specificity kinase that can phosphorylate tyrosine and serine/threonine residues and is critical for the recruitment of SAC proteins to unattached kinetochores, mitotic checkpoint complex (MCC) formation, and mitotic arrest.55 Studies have reported that TTK is overexpressed in many cancers, such as breast cancer, lung cancer, prostate cancer, and liver cancer.56–59 Another report showed that TTK is a favorable prognostic biomarker associated with triple negative breast cancer (TNBC) survival, and a high level of TTK expression predicts good survival and may safely spare the patient from adjuvant chemotherapy. A low level of TTK activates B-Raf/ERK signaling, which contributes to the invasiveness of cancer cells and poor survival of patients with TNBC.60
Conclusion
We downloaded multiple microarray datasets from the NCBI GEO database and integrated three microarray datasets. Then, we used R software and bioinformatics analysis to further investigate these datasets. We have identified 190 candidate DEGs, which may be involved in the progression of ovarian cancer. Among them, 99 genes were upregulated and 91 genes were downregulated. Using R software, we identified the top 20 most significantly up- and downregulated genes that could be the most related to the occurrence and development of ovarian cancer. By analyzing the GO and KEGG pathways, we found that DEGs were mainly enriched in the Wnt signaling pathway, metabolic pathways, and pathways in cancer, which provide a theoretical basis for studying the biological processes of ovarian cancer. We successfully constructed a PPI network of DEGs in epithelial ovarian cancer and screened several key genes encoding proteins in the network that are involved in the process of ovarian cancer in the form of molecular populations. Further study of this network would be beneficial for understanding the interaction between DEGs. These findings improve our understanding of the pathogenesis of ovarian cancer and the occurrence and development of the underlying molecular mechanisms. Our study has important clinical significance for the early diagnosis and treatment, as well as the prevention, of ovarian cancer and provides effective targets for the treatment of ovarian cancer. However, further molecular biological experiments are required to confirm the function of the identified genes associated with ovarian cancer.
Acknowledgments
This work was supported by the Independent Research Project of Wuhan University (grant no 413000117) and the National Natural Science Foundation of China (grant no 81302273).
Disclosure
The authors report no conflicts of interest in this work.
References
Alipour S, Zoghi S, Khalili N, Hirbod-Mobarakeh A, Emens LA, Rezaei N. Specific immunotherapy in ovarian cancer: a systematic review. Immunotherapy. 2016;8(10):1193–1204. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Bookman MA. Optimal primary therapy of ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i58–i62. | ||
Petryszak R, Burdett T, Fiorelli B, et al. Expression Atlas update – a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42(Database issue):D926–D932. | ||
Nannini M, Pantaleo MA, Maleddu A, Astolfi A, Formica S, Biasco G. Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives. Cancer Treat Rev. 2009;35(3):201–209. | ||
Bitler BG, Aird KM, Garipov A, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21(3):231–238. | ||
Vosa U, Kolde R, Vilo J, Metspalu A, Annilo T. Comprehensive meta-analysis of microRNA expression using a robust rank aggregation approach. Methods Mol Biol. 2014;1182:361–373. | ||
Bowen NJ, Walker LD, Matyunina LV, et al. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med Genomics. 2009;2:71. | ||
Yeung TL, Leung CS, Wong KK, et al. ELF3 is a negative regulator of epithelial–mesenchymal transition in ovarian cancer cells. Oncotarget. 2017;8(10):16951–16963. | ||
Marzancola MG, Sedighi A, Li PC. DNA microarray-based diagnostics. Methods Mol Biol. 2016;1368:161–178. | ||
Hollingshead D, Lewis DA, Mirnics K. Platform influence on DNA microarray data in postmortem brain research. Neurobiol Dis. 2005;18(3):649–655. | ||
Sherman BT, Huang da W, Tan Q, et al. DAVID knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007;8:426. | ||
von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31(1):258–261. | ||
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. | ||
Duffy MJ. Use of biomarkers in screening for cancer. Adv Exp Med Biol. 2015;867:27–39. | ||
Djamgoz MB, Coombes RC, Schwab A. Ion transport and cancer: from initiation to metastasis. Philos Trans R Soc Lond B Biol Sci. 2014;369(1638):20130092. | ||
Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. | ||
Kim AN, Jeon WK, Lim KH, Lee HY, Kim WJ, Kim BC. Fyn mediates transforming growth factor-beta1-induced down-regulation of E-cadherin in human A549 lung cancer cells. Biochem Biophys Res Commun. 2011;407(1):181–184. | ||
Zhang C, Zhang Z, Zhang S, Wang W, Hu P. Targeting of Wnt/beta-catenin by anthelmintic drug pyrvinium enhances sensitivity of ovarian cancer cells to chemotherapy. Med Sci Monit. 2017;23:266–275. | ||
Bodnar L, Stanczak A, Cierniak S, et al. Wnt/beta-catenin pathway as a potential prognostic and predictive marker in patients with advanced ovarian cancer. J Ovarian Res. 2014;7:16. | ||
Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696–1701. | ||
Li K, Ying M, Feng D, et al. Fructose-1,6-bisphosphatase is a novel regulator of Wnt/beta-catenin pathway in breast cancer. Biomed Pharmacother. 2016;84:1144–1149. | ||
Gong C, Qu S, Lv XB, et al. BRMS1L suppresses breast cancer metastasis by inducing epigenetic silence of FZD10. Nat Commun. 2014;5:5406. | ||
Tomar T, Alkema NG, Schreuder L, et al. Methylome analysis of extreme chemoresponsive patients identifies novel markers of platinum sensitivity in high-grade serous ovarian cancer. BMC Med. 2017;15(1):116. | ||
Malinauskas T, Jones EY. Extracellular modulators of Wnt signalling. Curr Opin Struct Biol. 2014;29:77–84. | ||
Marwitz S, Depner S, Dvornikov D, et al. Downregulation of the TGFbeta pseudoreceptor BAMBI in non-small cell lung cancer enhances TGFbeta signaling and invasion. Cancer Res. 2016;76(13):3785–3801. | ||
Jiang H, Li F, He C, Wang X, Li Q, Gao H. Expression of Gli1 and Wnt2B correlates with progression and clinical outcome of pancreatic cancer. Int J Clin Exp Pathol. 2014;7(7):4531–4538. | ||
Schwab RHM, Amin N, Flanagan DJ, Johanson TM, Phesse TJ, Vincan E. Wnt is necessary for mesenchymal to epithelial transition in colorectal cancer cells. Dev Dyn. Epub 2017 May 30:doi:10.1002/dvdy.24527. | ||
Endo H, Ikeda K, Urano T, Horie-Inoue K, Inoue S. Terf/TRIM17 stimulates degradation of kinetochore protein ZWINT and regulates cell proliferation. J Biochem. 2012;151(2):139–144. | ||
Xu C, Sun L, Jiang C, et al. SPP1, analyzed by bioinformatics methods, promotes the metastasis in colorectal cancer by activating EMT pathway. Biomed Pharmacother. 2017;91:1167–1177. | ||
Zhuo C, Li X, Zhuang H, et al. Elevated THBS2, COL1A2, and SPP1 expression levels as predictors of gastric cancer prognosis. Cell Physiol Biochem. 2016;40(6):1316–1324. | ||
Fedarko NS, Jain A, Karadag A, Van Eman MR, Fisher LW. Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin Cancer Res. 2001;7(12):4060–4066. | ||
Hu P, Chen X, Sun J, Bie P, Zhang LD. siRNA-mediated knockdown against NUF2 suppresses pancreatic cancer proliferation in vitro and in vivo. Biosci Rep. 2015;35(1):e00170. | ||
Sethi G, Pathak HB, Zhang H, et al. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS One. 2012;7(10):e47086. | ||
Januchowski R, Wojtowicz K, Sterzynska K, et al. Inhibition of ALDH1A1 activity decreases expression of drug transporters and reduces chemotherapy resistance in ovarian cancer cell lines. Int J Biochem Cell Biol. 2016;78:248–259. | ||
Condello S, Morgan CA, Nagdas S, et al. Beta-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene. 2015;34(18):2297–2308. | ||
Burki TK. CA-125 blood test in early detection of ovarian cancer. Lancet Oncol. 2015;16(6):e269. | ||
Rao TD, Tian H, Ma X, et al. Expression of the carboxy-terminal portion of MUC16/CA125 induces transformation and tumor invasion. PLoS One. 2015;10(5):e0126633. | ||
Xu H, Inagaki Y, Seyama Y, et al. Expression of KL-6/MUC1 in pancreatic ductal carcinoma and its potential relationship with beta-catenin in tumor progression. Life Sci. 2011;88(23–24):1063–1069. | ||
Gnemmi V, Bouillez A, Gaudelot K, et al. MUC1 drives epithelial–mesenchymal transition in renal carcinoma through Wnt/beta-catenin pathway and interaction with SNAIL promoter. Cancer Lett. 2014;346(2):225–236. | ||
Rehn AP, Cerny R, Sugars RV, Kaukua N, Wendel M. Osteoadherin is upregulated by mature osteoblasts and enhances their in vitro differentiation and mineralization. Calcif Tissue Int. 2008;82(6):454–464. | ||
Williamson RE, Darrow KN, Giersch AB, et al. Expression studies of osteoglycin/mimecan (OGN) in the cochlea and auditory phenotype of Ogn-deficient mice. Hear Res. 2008;237(1–2):57–65. | ||
Tanaka K, Matsumoto E, Higashimaki Y, et al. Role of osteoglycin in the linkage between muscle and bone. J Biol Chem. 2012;287(15):11616–11628. | ||
Pollard JW. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol. 2008;84(3):623–630. | ||
Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. | ||
Hansel DE, Zhang Z, Petillo D, Teh BT. Gene profiling suggests a common evolution of bladder cancer subtypes. BMC Med Genomics. 2013;6:42. | ||
Han SS, Kim WJ, Hong Y, et al. RNA sequencing identifies novel markers of non-small cell lung cancer. Lung Cancer. 2014;84(3):229–235. | ||
Chiang CP, Jao SW, Lee SP, et al. Expression pattern, ethanol-metabolizing activities, and cellular localization of alcohol and aldehyde dehydrogenases in human large bowel: association of the functional polymorphisms of ADH and ALDH genes with hemorrhoids and colorectal cancer. Alcohol. 2012;46(1):37–49. | ||
Tucker SL, Gharpure K, Herbrich SM, et al. Molecular biomarkers of residual disease after surgical debulking of high-grade serous ovarian cancer. Clin Cancer Res. 2014;20(12):3280–3288. | ||
Giardina B, Messana I, Scatena R, Castagnola M. The multiple functions of hemoglobin. Crit Rev Biochem Mol Biol. 1995;30(3):165–196. | ||
Huang Y, Zhang X, Jiang W, et al. Discovery of serum biomarkers implicated in the onset and progression of serous ovarian cancer in a rat model using iTRAQ technique. Eur J Obstet Gynecol Reprod Biol. 2012;165(1):96–103. | ||
Liu X, Gao Y, Zhao B, et al. Discovery of microarray-identified genes associated with ovarian cancer progression. Int J Oncol. 2015;46(6):2467–2478. | ||
Lavoie H, Li JJ, Thevakumaran N, Therrien M, Sicheri F. Dimerization-induced allostery in protein kinase regulation. Trends Biochem Sci. 2014;39(10):475–486. | ||
Xu Q, Zhu S, Wang W, et al. Regulation of kinetochore recruitment of two essential mitotic spindle checkpoint proteins by Mps1 phosphorylation. Mol Biol Cell. 2009;20(1):10–20. | ||
Daniel J, Coulter J, Woo JH, Wilsbach K, Gabrielson E. High levels of the Mps1 checkpoint protein are protective of aneuploidy in breast cancer cells. Proc Natl Acad Sci U S A. 2011;108(13):5384–5389. | ||
Landi MT, Dracheva T, Rotunno M, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3(2):e1651. | ||
Shiraishi T, Terada N, Zeng Y, et al. Cancer/testis antigens as potential predictors of biochemical recurrence of prostate cancer following radical prostatectomy. J Transl Med. 2011;9:153. | ||
Liang XD, Dai YC, Li ZY, et al. Expression and function analysis of mitotic checkpoint genes identifies TTK as a potential therapeutic target for human hepatocellular carcinoma. PLoS One. 2014;9(6):e97739. | ||
Xu Q, Xu Y, Pan B, et al. TTK is a favorable prognostic biomarker for triple-negative breast cancer survival. Oncotarget. 2016;7(49):81815–81829. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.