")
Back to Journals » Journal of Pain Research » Volume 10
Identification of capsazepine as a novel inhibitor of system xc− and cancer-induced bone pain
Authors Fazzari J, Balenko MD , Zacal N, Singh G
Received 18 October 2016
Accepted for publication 13 January 2017
Published 18 April 2017 Volume 2017:10 Pages 915—925
DOI https://doi.org/10.2147/JPR.S125045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Jennifer Fazzari, Matthew D Balenko, Natalie Zacal, Gurmit Singh
Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
Abstract: The cystine/glutamate antiporter has been implicated in a variety of cancers as a major mediator of redox homeostasis. The excess glutamate secreted by this transporter in aggressive cancer cells has been associated with cancer-induced bone pain (CIBP) from distal breast cancer metastases. High-throughput screening of small molecule inhibitors of glutamate release from breast cancer cells identified several potential compounds. One such compound, capsazepine (CPZ), was confirmed to inhibit the functional unit of system xc− (xCT) through its ability to block uptake of its radiolabeled substrate, cystine. Blockade of this antiporter induced production of reactive oxygen species (ROS) within 4 hours and induced cell death within 48 hours at concentrations exceeding 25 μM. Furthermore, cell death and ROS production were significantly reduced by co-treatment with N-acetylcysteine, suggesting that CPZ toxicity is associated with ROS-induced cell death. These data suggest that CPZ can modulate system xc− activity in vitro and this translates into antinociception in an in vivo model of CIBP where systemic administration of CPZ successfully delayed the onset and reversed CIBP-induced nociceptive behaviors resulting from intrafemoral MDA-MB-231 tumors.
Keywords: glutamate, breast cancer, cancer-induced bone pain, system xc−
Introduction
Advanced cancers of the breast are the most common source of bone metastases in women.1 When in the bone, these cancers initiate a wide variety of pathological sequelae, with the primary symptom being debilitating pain.2 Once tumors metastasize to the skeleton, they are associated with a dramatic change in the bone microenvironment inducing a physiologically complex pain state, which can arise from a multitude of factors including altered bone remodeling, fracturing of the bone, damage to surrounding nerves, and release of nociceptive factors from the bone tissue itself or directly from the invading tumor.3–7 These skeletal-related effects correlate to a marked increase in patient morbidity and mortality2 with cancer-induced bone pain (CIBP) affecting up to 75% of cancer patients making it a key indicator of patient quality of life.8,9 The multiplicity of nociceptive pathways contributing to CIBP makes treatment difficult and often resistant to current analgesic therapies, with over 50% of patients having persistent, unresolved pain.10 New pharmacological targets are therefore crucial to advancing therapeutic strategies that can address this clinical problem and advance patients’ quality of life.
We hypothesize that cancer-specific factors are responsible for the complexity of CIBP and discovered that concentration of the nociceptive factor glutamate is greatly increased in the tumor microenvironment as a result of upregulated antioxidant machinery,6,7,11 namely, the cystine/glutamate antiporter, system xc− (xCT). System xc− is upregulated in many tumor types in response to the high concentrations of reactive oxygen species (ROS) produced as a consequence of their rampant cell growth and metabolism. Functional upregulation of this antiporter promotes cell survival under a high ROS burden by promoting cystine uptake and downstream production of glutathione (GSH). This is especially seen in cancers with aggressive phenotypes that readily metastasize to bone.11–13 The exchange of cystine for glutamate via system xc− across the plasma membrane occurs in a 1:1 stoichiometric ratio14,15 driven by the intracellular concentrations of glutamate. This secreted glutamate is believed to be a major stimulus for the initiation and propagation of CIBP following metastases to the bone. Glutamate is the most common neurotransmitter in the central nervous system, and is known to play a role in pain as well as modulating cellular homeostasis in the bone.16,17 With the nociceptive potential of glutamate implicated in a variety of painful disorders, targeting glutamate release via system xc− at the tumor site is a novel path to achieve analgesia in CIBP. Therefore, inhibition of system xc− in the cancer would limit the release of glutamate from the tumor, making it a pharmacologically relevant and pathology-specific target for reducing mechanical hyperalgesia associated with CIBP.7 Previously, our group has published the antinociceptive effects of a known xCT inhibitor, sulfasalazine (SSZ), in an animal model of CIBP where treatment with this drug delayed the onset of nociceptive behaviors. Slosky et al recently corroborated this in a syngeneic mouse model with complementary behavioral assessments.18 Despite the success of SSZ in animal models, it has poor translational capacity for treatment of CIBP due to the limited oral bioavailability of the parent drug relative to its colonic metabolites, which have no inhibitory action toward system xc−.18,19 In addition, common side effects of SSZ include nausea, vomiting, and anorexia, which need to be controlled not exacerbated in a cancer patient. Therefore, our aim is to identify novel inhibitors of system xc−. We previously ran a high throughput screen examining a chemical library of 30,000 compounds to identify lead molecules that effectively inhibit glutamate release from MDA-MB-231 cells relative to SSZ.20 Capsazepine (CPZ) was one of several lead compounds found to have a high inhibition of glutamate release. Originally, CPZ was the first reported competitive antagonist of the vanilloid receptor-1 (TRPV-1).21 Although commonly used as such in pharmacological studies, it lacks potency and selectivity toward TRPV-122,23 and has several known off-target effects.24–26 Therefore, it has since been replaced by newer, more selective compounds. Here, we present the first characterization of CPZ as an inhibitor of xCT in MDA-MB-231 cancer cells and corroborate this activity with antinociception in an in vivo murine model of CIBP induced by MDA-MB-231 bone colonization.
Methods
Cell culture
MDA-MB-231 human breast adenocarcinomas (American Type Culture Collection, Manassas, VA, USA) were maintained at sub-confluent densities with 5% CO2 at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1X antibiotic/antimycotic (Life Technologies, Burlington, ON, USA). Cultures tested negative for mycoplasma contamination following testing outlined by van Kuppeveld et al.27
Cell treatments
Capsazepine (CPZ; Cayman, Madison, WI, USA) and 5-iodoresiniferatoxin (IRTX; Alomone Labs, Jerusalem, Israel) were prepared in accordance with manufacturer’s recommendations using dimethyl sulfoxide (DMSO).
Uptake of [14C]-cystine in MDA-MB-231 cells
The uptake of radiolabeled [14C]-cystine was measured as previously described.28,29 MDA-MB-231 cells were plated in 6-well plates, 24 hours prior to testing, in complete culture medium. Prior to drug treatment, cells were washed with Hank’s balanced salt solution (HBSS) and incubated with drug diluted in HBSS for 20 minutes at 37°C. [14C]-cystine (0.03 μCi/mL) was then added and incubated for an additional 20 minutes at 37°C. Cells were then washed with ice-cold HBSS and lysed in 220 μL lysis buffer (0.1% Triton X-100, 0.1 N NaOH29) for 30 minutes. A 100 μL aliquot of lysate was added to 1 mL scintillation fluid (Ecoscint-H) and read in a Beckman LS6000 liquid scintillation counter. Total protein per sample was quantified using the BioRad reagent and used to normalize scintillation data.
Measurement of intracellular reactive oxygen species
Intracellular ROS was quantified using a choloromethyl 2′,7′–dichlorofluorescein diacetate derivative (DCFDA) after 4 and 24 hours of treatments with 12.5–50 μM CPZ. DCFDA is loaded into the cells at a concentration of 10 μM in HBSS for 30 minutes prior to drug treatment. Following incubation, cells were washed with HBSS and treated in phenol-red free DMEM supplemented with 10% FBS, sodium pyruvate (1 mM), and L-glutamine (4 mM). Fluorescence was then read at 529 nm following the indicated time points.
Quantification of xCT mRNA by quantitative real-time polymerase chain reaction
MDA-MB-231 cells treated with 25 μM CPZ for 48 hours were harvested by trypsinization followed by mRNA extraction and purification. mRNA was then subject to reverse transcription to generate cDNA, and quantitative real-time polymerase chain reaction (RT-PCR) was carried out using the following primers for system xc− [SLC7A11-forward (5′-CCTCTATTCGGACCCATTTAGT) and reverse (5′-CTGGGTTTCTTGTCCCATATAA)]. Results were quantified using the 2−[Δ][Δ]Ct method with the housekeeping gene β-actin and presented as fold changes relative to vehicle (DMSO)-treated control. RT-PCR was also used to confirm the presence of TRPV-1 mRNA in the MDA-MB-231 cells with the following primers: forward (5′-CAGGCTCTATGATCGCAGGAG-3′) and reverse (5′-TTTGAACTCGTTGTCTGTGAGG-3′).
Animals
Female athymic BALB/c nu/nu homozygous nude mice (Charles River, Montreal, QC, Canada) were used for developing MDA-MB-231 xenograft model. The mice ranged from 4–6 weeks of age and were sterile housed in groups of five. The mice were maintained at 24°C with a 12-hour light/dark cycle and were provided a sterile setting using autoclaved food and water ad libitum. All procedures were conducted according to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain and guidelines established by the Canadian Council on Animal Care with ethical approval from the McMaster University Animal Research Ethics Board. Humane endpoints dictate euthanization if the tumor interference with the normal function of the animals causes significant pain or distress or leads to infection/risk of infection. This is monitored by frequent examination, pain behavioral testing, and body weight recordings.
Tumor cell xenografts
Three days prior to cell implantation, all mice had a 0.25 mg, 21-day release 17β-estradiol pellet (Innovative Research of America, Sarasota, FL, USA) implanted subcutaneously. At experimental day 0, mice were randomized to tumor or sham-injected groups and subject to isoflurane anesthesia followed by subcutaneous administration of buprenorphine (0.05 mg/kg) prior to cell injection. Animals in the tumor group were inoculated with 2×106 cells in a 50 µL solution of phosphate-buffered saline (PBS) (tumor mice) and sham animals received an injection of only 50 µL of PBS into the right distal epiphysis of the femur, as previously reported.7
Experimental groups
Both tumor and sham-injected mice were randomized into treatment groups on experimental day 14 following tumor cell inoculation to allow for tumor establishment (Tumor injected: n=11, 5 mg/kg CPZ; n=10, 10 mg/kg CPZ; n=13 DMSO vehicle; Sham injected: n=3, 5 mg/kg CPZ; n=5 DMSO vehicle). Drugs were delivered via Alzet model 1004 mini-osmotic pumps (0.11 μL/hour for 28 days; Durect, Cupertino, CA, USA), which were implanted intraperitoneally to allow for stable drug delivery without constant surgical interference that may otherwise skew behavioral results. Final CPZ doses were equal to 5 and 10 mg/kg, which have previously been shown to be safe for animal use.19,30,31
Behavioral testing
The behavioral testing period started 8 days prior to cancer cell inoculation and was performed on alternate days to acquire a total of four baseline tests. The average of these four tests represents the baseline pain score prior to tumor development and treatment. After cell implantation, behavioral testing was performed 3 days/week until endpoint was reached and involved the use of two behavioral systems; the Dynamic Plantar Aesthesiometer (DPA) (Ugo Basile, Comerio, Italy) and the Dynamic Weight Bearing (DWB) (BioSeb, Vitrolles, France) systems. All animals were randomly assigned to treatment groups during baseline behavioral testing, which also accounted for their random cage assignment. Researchers conducting behavioral tests and assessment of radiography/histology remained blinded to treatment status through each experiment. All data collected following cancer cell inoculation were normalized to these baseline scores. All animals were given a 5- and 7-minute acclimatization period in the DPA and DWB chambers, respectively.
Dynamic Plantar Aesthesiometer
The DPA is an electronic Von Frey instrument measuring mechanical withdrawal thresholds as indicators of allodynia and hyperalgesia. The mice are placed individually in holding areas with grated floors and the device is manually moved under the cell-injected paw of the mouse and the actuator is triggered, raising the filament to the plantar surface of the paw. Once contact is made, the applied force increases steadily until the paw is withdrawn. An average of five withdrawal thresholds were collected on each testing day to represent the mechanical withdrawal threshold for that day. Only mice that had visible tumors based on radiographic and histological identification were used for final data analysis. In this study, tumor implantation was successful in all animals. The final animal numbers for each group, therefore, were: vehicle: n=13; 5 mg/kg CPZ: n=11; 10 mg/kg CPZ: n=10; sham vehicle: n=5; sham 5 mg/kg CPZ: n=3.
Dynamic Weight Bearing system
The DWB system records weight distribution and time spent on each limb over the course of 3 minutes using specialized weight sensors that are calibrated to the weight of the mouse and is equipped with a video camera mounted overhead to analyze the mouse’s movements as the experiment progresses. The mean weight applied by each limb was measured separately, as well as the mouse as a whole, but only the rear right (tumor-bearing) limb was used to analyze changes in applied mechanical force. The daily mean weight average of the rear right paw was then compared to the baseline mean weight to calculate the difference in weight distribution placed on the affected limb. The digital recordings from the overhead camera were also used to manually validate the mouse’s orientation on the sensor. The time spent on each paw was also analyzed as it provides a more specific measurement of nociceptive behavior as it highlights limb favoring and impaired ambulation of the tumor-bearing limb. Similarly to the DPA results, only mice showing confirmed radiographic tumor development in the injected limb were used, making the final group numbers as follows: vehicle: n=13; 5 mg/kg CPZ: n=11; 10 mg/kg CPZ: n=10; sham vehicle: n=5; sham 5 mg/kg CPZ: n=3. Only results of tumor-bearing mice are reported as no differences were seen among sham-injected groups.
Immunohistochemistry
Ipsilateral and contralateral tibiae, fibulae, femora, and surrounding tissues with tumor growth confirmed by radiographic analysis were dissected and fixed in formalin for 48 hours followed by decalcification in 10% EDTA and 4% formalin-buffered solution for 2 weeks. Fixed and decalcified samples were embedded in paraffin wax, and 4 µm sections were prepared for hematoxylin and eosin staining to assess the extent of tumor invasion. Sections were mounted on glass slides and heated at 65°C for 25 minutes. Sections were then de-paraffinized and rehydrated with multiple xylene and ethanol prior to staining and coverslipped with xylene miscible Permount (Fisher Scientific, Pittsburgh, PA, USA).
Statistical analysis
All in vitro data were measured using one-way analysis of variance (ANOVA) followed by a Tukey’s test to compare all groups or by unpaired t-test with P<0.05. All behavioral data were measured using a one-way repeated-measures ANOVA followed by a Tukey’s test comparing all data sets to one another. Behavioral data were calculated from day 25 until endpoint to limit the effects of the long lag phase from tumor implantation to onset of pain symptoms. All data presented are in terms of mean ± standard error of mean. GraphPad Prism software version 5 for Windows (GraphPad Software, Inc., La Jolla, CA, USA) was used for all graphing and statistical analyses.
Results
Radiolabeled cystine uptake identifies capsazepine as an inhibitor of xCT activity
CPZ decreases the uptake of [14C]-cystine in MDA-MB-231 cells within 20 minutes of compound addition showing a maximal inhibitory effect at 6 μM. This mimics the effect of the known xCT inhibitor, SSZ, indicating that CPZ does in fact show specificity for xCT (Figure 1). CPZ did not exhibit inhibition when drug was removed prior to substrate addition (data not shown).
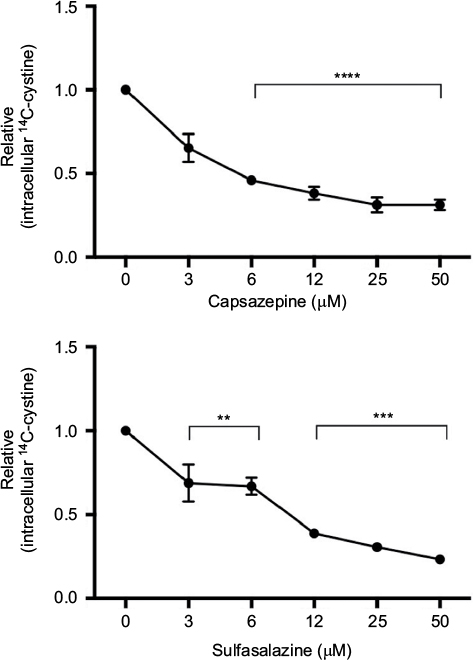 | Figure 1 Quantification of cystine acquisition in MDA-MB-231 cells in the presence of capsazepine (CPZ). Notes: CPZ inhibits the uptake of 14C-cystine when incubated with 0–50 μM of the compound. This effect is comparable to intracellular levels of 14C-cystine after incubation with sulfasalazine, a known system xc− inhibitor. Data are expressed as the fold change in counts per minute/mg protein (± standard error of the mean) relative to vehicle control (dimethyl sulfoxide). **P<0.01; ***P<0.001; ****P<0.0001. |
Intracellular ROS levels are modulated by capsazepine in MDA-MB-231 cells
ROS levels increased over the course of 48 hours following CPZ treatment, relative to vehicle-treated controls (Figure 2A). Within 24 hours, 25 μM CPZ increased intracellular ROS levels by approximately three-fold (P<0.005). At 48 hours, this same dose increased ROS levels dramatically relative to the 24-hour time point (P<0.0001) and by greater than 15-fold relative to the vehicle-treated control (P<0.05). By contrast, treatment with IRTX, a molecule of the same TRPV-1 antagonist class as CPZ but with approximately 10× higher affinity for TRPV-1, did not result in significant increases in ROS production over the course of 24 and 48 hours relative to the vehicle-treated control (Figure 2B). Furthermore, the observed CPZ-induced increase in ROS is abolished by the addition of 5 mM N-acetyl cysteine (NAC; Figure 2C), which is a cyst(e)ine pro-drug that supplies the cell with an exogenous source of cysteine in the absence of cystine uptake activity via system xc−. Furthermore, CPZ treatment induces cell death after 48 hours, and co-treatment with 5 mM NAC significantly reduces cell death at the highest concentration of 100 μM CPZ (P<0.001; Figure 2D).
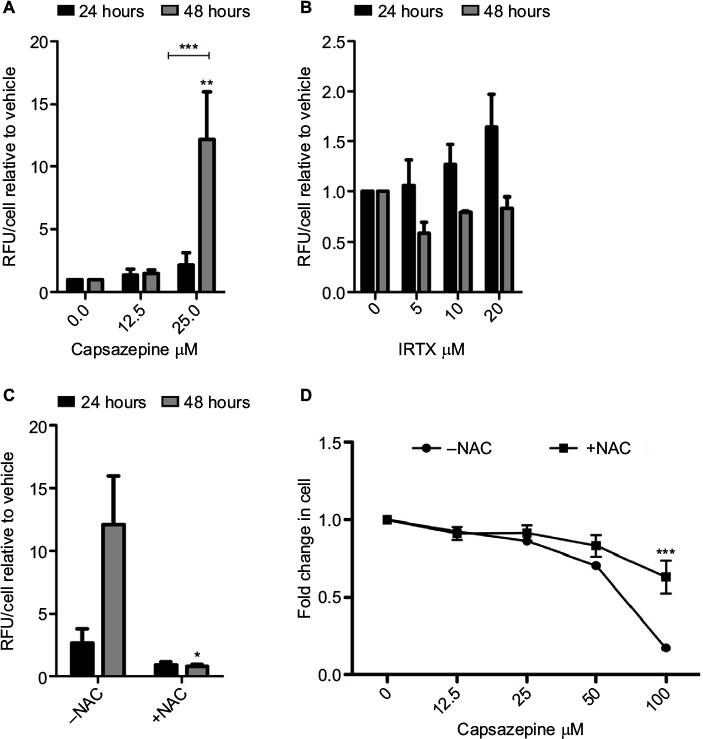 | Figure 2 Capsazepine treatment induces the production of reactive oxygen species. Notes: Quantification of intracellular levels of reactive oxygen species (ROS) as measured by DCFDA over 24 and 48 hours of treatment with capsazepine (CPZ) and 5′-iodoresiniferatoxin (IRTX; A). After 48 hours, 25 μM of CPZ induces a significant increase in ROS relative to dimethyl sulfoxide-treated cells (P<0.001). Treatment with IRTX does not result in significant changes in ROS over this time course. These data are represented as mean fold change ± standard error of mean relative to vehicle-treated cells. One-way analysis of variance (ANOVA) was used to measure significant increases in ROS relative to vehicle-treated control. CPZ-induced ROS production is abolished by co-treatment with 5 mM N-acetyl cysteine (NAC; B). An unpaired t-test was used to assess significance of NAC addition at each time point. Cell survival decreases in a dose-dependent manner after treatment for 48 hours with CPZ. Co-treatment with 5 mM NAC increases cell survival relative to treatment with CPZ alone (C). Cell survival at 100 μM CPZ is significantly higher in the presence of 5 mM NAC (P<0.001; one-way ANOVA comparing each concentration of CPZ without NAC to those with the addition of NAC). *P<0.05; **P<0.01; ***P<0.001. Abbreviations: DCFDA, dichlorofluorescein diacetate derivative; RFU, relative fluorescent units. |
xCT transcription increases in response to CPZ treatment
Treatment of MDA-MB-231 cells with 25 μM CPZ results in a significant increase in xCT mRNA over the course of 24 (P<0.001) and 48 hours (P<0.05; Figure 3). Similarly, this increase in expression is reversed to baseline by treatment with 5 mM NAC (data not shown). This increase in xCT mRNA does not result in a functional increase in system xc− activity nor does it resolve the increased ROS load induced by CPZ treatment. Interestingly, it also does not induce an antioxidant response through the induction of GSH production (data not shown).
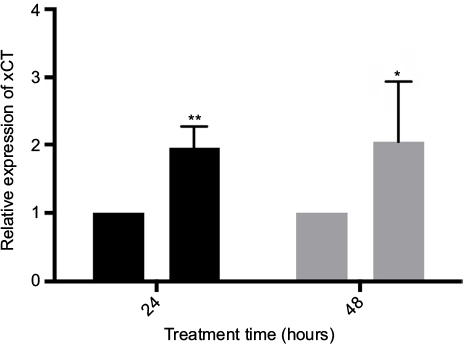 | Figure 3 Treatment with 25 μM capsazepine significantly induces xCT expression by 24 and 48 hours relative to dimethyl sulfoxide (DMSO) treatment. Notes: These data are expressed as the fold change in xCT mRNA levels relative to DMSO ± standard error of mean and analyzed using a one-way analysis of variance (24 hours P<0.001, 48 hours P<0.05). *P<0.05; **P<0.01. |
Behavioral analysis
BALB/c nude mice bearing successful MDA-MB-231 grafts in the right femur as determined by histological analyses were included in the study (Figure 4). The DPA and DWB tests were consistent, showing a steady increase in nociceptive behavior with tumor development. Treatment with CPZ at both a high dose (10 mg/kg) and a low dose (5 mg/kg) exhibited pain-modulating effects with the prevention/delay in the onset of nociceptive behaviors. To confirm that CPZ treatment alone did not affect the behavioral responses, a small group (n=3) of sham-injected animals were treated with vehicle or CPZ (5 mg/kg) with no significant differences in behavior between these two groups.
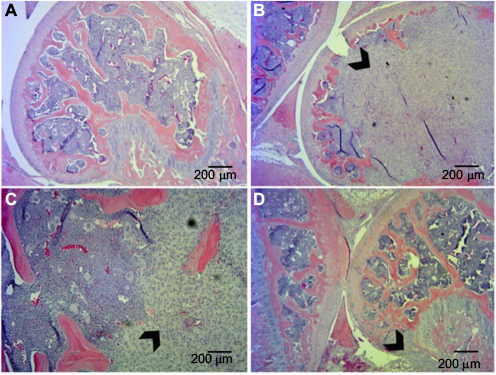 | Figure 4 Histological analysis of tumor xenografts in bone represented by hematoxylin and eosin staining. Notes: (A) Sham-injected mice. Tumor-injected mice show extensive invasion of xenograft into the femur with destruction of growth plate (B, D) and in some cases breaching of the periosteum (C). Arrowheads indicate tumor tissue. |
Dynamic Plantar Aesthesiometer
The DPA analysis showed that both the doses of CPZ prevented the onset of pain-related behaviors as paw withdrawal thresholds did not significantly deviate from baseline measurements over the course of the experiment (Figure 5), while vehicle-treated animals experienced a significant decrease in paw withdrawal thresholds relative to baseline at day 27 signifying the onset of pain behavior (day 27 P<0.05, day 29–36 P<0.001). Both the 5 and 10 mg/kg doses of CPZ significantly decreased the paw withdrawal threshold from day 25 (P<0.05) relative to the vehicle-treated group, and the withdrawal thresholds of the 10 mg/kg-treated group did not differ significantly from the sham-injected group (results not shown), indicating that this dose was trending toward the development of a nociceptive free state. However, both the 5 and 10 mg/kg dose did not differ significantly from one another.
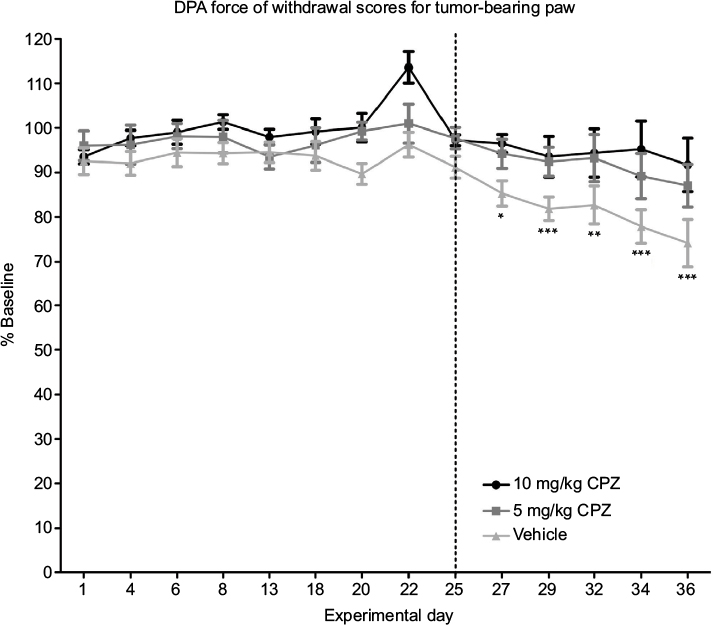 | Figure 5 Dynamic Plantar Aesthesiometer (DPA) measurements of force required for withdrawal of the injected limb compared to the baseline results in capsazepine (CPZ)-treated and non-treated mice (100% on the y-axis is therefore equivalent to the animal’s behavior pre-tumor implantation surgery). A significant decrease in the force required to induce paw withdrawal relative to baseline is only seen in the vehicle-treated group beginning on day 27 as indicated by the asterisks. Both the 5 and 10 mg/kg CPZ-treated groups do not show any significant deviation from baseline measurements. A one-way analysis of variance (ANOVA) with a Dunnett post-test was used to show significant differences between time points relative to the baseline control. Paw withdrawal thresholds are increased in CPZ-treated mice increase in force required for paw withdrawal in the CPZ-treated mice relative to vehicle-treated mice (10 mg/kg) n=10; (5 mg/kg) n=11; (vehicle) n=13. One-way repeated measures ANOVA was used on the measurements past day 25 (marked by dotted line) showing significant differences between groups (P<0.0001). While both doses were significantly different from vehicle mice (P<0.05), differences between doses were not significant. Data are expressed as the mean required force as a percentage of the baseline score ± standard error of mean. *P<0.05; **P<0.01; ***P<0.001. |
Dynamic Weight Bearing analysis
In this weight-based test, the mice experiencing nociception showed a dramatic decrease over time in the weight exerted on the tumor-bearing limb when compared to their baseline weight distribution obtained prior to cancer cell inoculation (Figure 6A). The onset of pain behavior in this test was marked on day 29 in which the percentage of body weight placed on the tumor-bearing limb significantly deviated from baseline (day 29 P<0.01, day 32–36 P<0.001). Treatment with 5 mg/kg CPZ delayed the onset of pain behavior assessed by this method until day 36 (P<0.01), while treatment with 10 mg/kg prevented the onset completely. When compared to the pain score of the vehicle-treated mice, both doses of CPZ were found to reverse this nociceptive behavior (P<0.05) in tumor-bearing mice. Furthermore, the ratio of weight applied to the contralateral (rear, left) limb and the ipsilateral (rear, right) limb was also diminished with CPZ treatment relative to vehicle-treated controls (data not shown). Furthermore, measurement of the time spent on each limb was indicative of limb preference, and it was shown that only mice experiencing severe nociception in the other behavioral tests would favor or lift their rear, right (tumor-bearing) limb (Figure 6B), consequently putting more weight on the contralateral leg. During this experiment, only the mice with the most severe pain were shown to raise their hind leg for time periods that significantly differed from that of their baseline preferences. Similarly to the other tests, both doses of CPZ were found to significantly increase the time spent on the tumor-bearing limb relative to the vehicle group (P<0.05), but failed to create a dose-specific response. Similarly to the DPA, the weight applied to the tumor-bearing limb of CPZ-treated mice (both 5 and 10 mg/kg treatments) did not differ significantly from that of the sham-injected mice (results not shown).
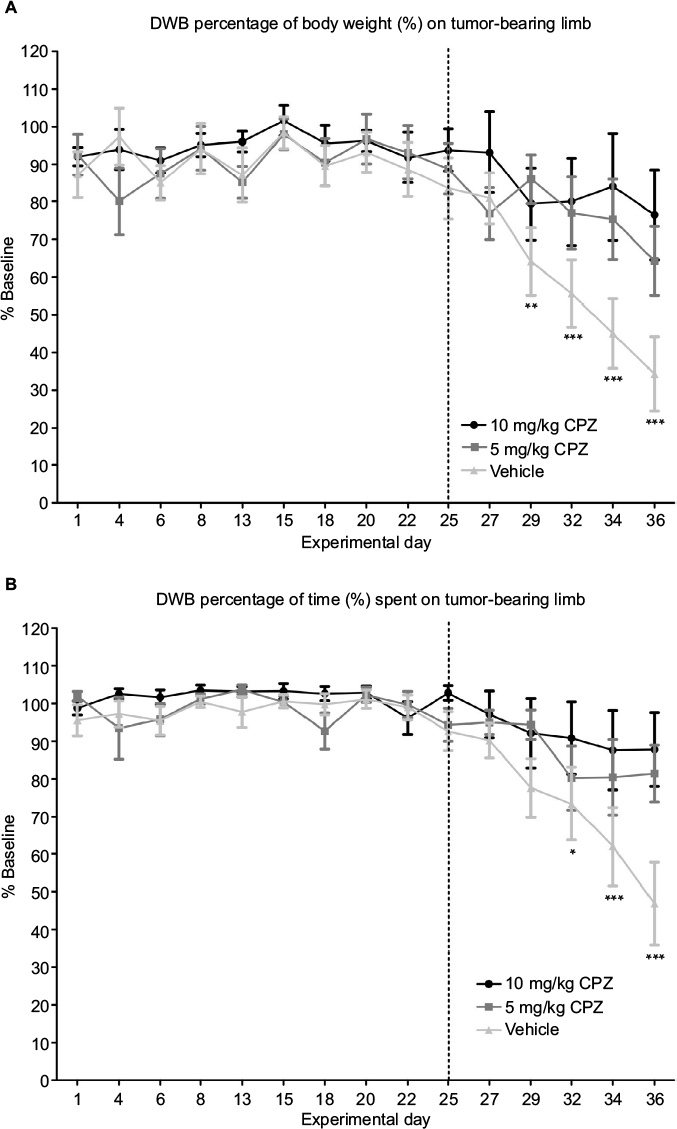 | Figure 6 (A) Capsazepine (CPZ)-treated mice and non-treated mice. A significant decrease in weight applied to the tumor-bearing paw relative to baseline marked the onset of pain behavior in vehicle-treated mice on day 29 as indicated by the asterisks. This is delayed until day 36 for the 5 mg/kg CPZ-treated mice, and no significant changes from baseline are seen in the 10 mg/kg CPZ-treated group. A one-way analysis of variance (ANOVA) with a Dunnett post-test was used to show significant differences between time points relative to the baseline control. This graph shows a significant increase in weight distribution in the CPZ-treated mice, relative to vehicle-treated mice (10 mg/kg) n=10; (5 mg/kg) n=11; (vehicle) n=13. A one-way repeated measures ANOVA was used on the measurements past day 25 (marked by dotted line) showing significant differences between groups (P=0.0008). While both doses were significantly different from vehicle mice (P<0.05), differences between doses were not significant. Data are expressed as the mean weight bearing in the injected limb as a percentage of the baseline score ± SEM. (B) DWB measurements of time spent on the injected limb compared to the baseline results in CPZ-treated mice and non-treated mice. A decrease in the time spent on the tumor-bearing limb significantly deviates from baseline at day 32 with the CPZ-treated group not showing any deviation from baseline at any time point. Relative to the vehicle-treated group, an increase in time spent on injected limb is seen in the CPZ-treated mice (10 mg/kg) n=10; (5 mg/kg) n=11; (vehicle) n=13. A one-way repeated measures ANOVA was used on the measurements past day 25 (marked by dotted line) showing significant differences between groups (P<0.005). While both doses were significantly different from vehicle mice (P<0.05), differences between doses were not significant. Data are expressed as the mean time spent on the injected limb as a percentage of the baseline time ± SEM. *P<0.05; **P<0.01; ***P<0.001. |
For both measurements of limb use, a one-way ANOVA with repeated measures was used past day 25 when pain-related behaviors manifest.
Discussion
From a clinical perspective, development of novel treatment strategies for CIBP is becoming more pressing as a significant proportion of oncology outpatients are reaching advanced disease stages and as a result experience greater degrees of pain coupled with inadequate pain control.32 Often, achieving analgesia is at the expense of a patient’s quality of life; therefore, having a peripheral and pathological target for blocking the source of a nociceptive stimuli rather than manipulating the physiological response to such stimuli offers a novel therapeutic approach that exploits pathological changes in the tumor while sparing physiological systems such as the central nervous system. In this study, a novel inhibitor of system xc−, CPZ, is reported and is shown to delay and, in some cases, prevent the onset of CIBP. The ability of CPZ to have similar analgesic effects as the known xCT inhibitor, SSZ, is important due to the limited bioavailability of SSZ making it an unattractive clinical therapeutic for CIBP.
To correlate the CPZ-induced decrease in glutamate release from MDA-MB-231 cells with system xc− inhibition, CPZ was tested for its ability to inhibit the uptake of radiolabeled cystine, a specific system xc− substrate. These data show that CPZ rapidly prevents cystine uptake indicating blockade of system xc−. Like SSZ, CPZ must inhibit system xc− sterically or by another fast acting signaling mechanism, as the inhibitory action of both drugs is lost after washout (data not shown). Because system xc− is one of the mechanisms responsible for ROS detoxification, downstream effects of its inhibition often manifest as rising levels of intracellular ROS and decreasing levels of GSH, the major intracellular antioxidant, as a consequence of impaired cystine acquisition.33,34 Cancer cells generate high titers of ROS as a byproduct of increased cellular metabolism and rapid growth, which requires upregulation of antioxidant machinery, such as system xc−, to survive. Therefore, blocking antioxidant synthesis through xCT inhibition results in a rise in intracellular ROS over time. This is observed following CPZ treatment in conjunction with a temporal increase in xCT mRNA likely in response to ROS production by way of antioxidant response elements in the xCT promoter. Similarly, recent data have proven that the reactive nitrogen species peroxynitrite, which is elevated in the tumor tissues, also drives system xc− expression and glutamate release from the tumor cell and that elimination of this species can attenuate CIBP.30
Furthermore, when cells are treated with another TRPV-1 antagonist (IRTX), a compound with greater than ten times more specificity for TRPV-1 than CPZ, this increase in ROS is not observed (Figure 2B) corroborating an off-target action of CPZ on xCT, not on TRPV-1. Considering that CPZ is known to lose its specificity for TRPV-1 and exhibit off-target effects at doses exceeding 700 nM as used here, as well as the fact that a highly selective TRPV-1 antagonist does not mirror the effect of CPZ, suggests that CPZ modulates intracellular redox levels not via TRPV-1, but through an off-target mechanism, likely system xc−.
Clinically, the treatment of bone pain is often met with little success as breakthrough pain interrupts periods of analgesia even in the presence of various stages of pharmacological intervention. In order to see if CPZ’s ability to modulate system xc− activity in vitro translates to effective analgesia in vivo, CPZ was tested in an experimental mouse model of CIBP for its ability to attenuate the nociceptive behaviors associated with an established intrafemoral tumor. There is evidence that bone cancer pain is often accompanied by skin hypersensitivity and as a result may be responsible for the manifestation of skeletal pain-related behaviors measured by common tests of nociceptive behaviors including the DWB.35 Here, we include tests of both cutaneous stimulus-evoked pain and postural equilibrium/limb weight bearing using the DPA and DWB tests, respectively, thus analyzing both features associated with skeletal pain. Treatment with CPZ has a significant effect in both tests confirming that analgesia obtained by administration of this molecule is not merely associated with a decrease in skin hypersensitivity.
In vivo, the non-selective nature of CPZ has proven to be more effective in reversing the nociceptive effects of neurogenic inflammation relative to a more selective TRPV-1 antagonist SB-366791. The use of TRPV-1 inhibitors, such as CPZ, is one of the fastest growing branches of analgesic study in the last decade, with a widely accepted rationale for the development of TRPV-1 antagonists for the treatment of various inflammatory pain conditions.31 However, the role of TRPV-1 antagonists for chronic pain states, where conditions of tactile, mechanical, and spontaneous pain predominate, is less clear. TRPV-1 was identified as the primary mediator of thermal hyperalgesia following tissue injury with its expression increasing both centrally and peripherally in accordance with specific pain states including those initiated by cancers of the bone. However, absence or attenuation of this receptor does not seem to influence mechanical allodynia/hyperalgesia,31,36,37 as is measured in this study, suggesting that attenuation of mechanical-based nociception is via a novel mechanism, which we propose to be a result of diminished glutamate secretion from xCT inhibition at the tumor. Furthermore, CPZ has successfully inhibited temperature-derived behaviors relating to pain, but failed to reverse hyperalgesia induced by Complete Freund’s Adjuvant, suggesting CPZ does not play a role in mitigating inflammatory-induced nociception.38 CPZ also does not reverse chronic inflammatory and neuropathic pain in rats and mice, where it only blocks capsaicin-induced TRPV-1 hyperalgesia.39
The need for system xc− in cancer cells and its association with pain states make it a novel, peripheral, and tumor-specific target for pharmacological development. Previously, we, and Slosky et al,18 have shown that system xc− inhibition, decreased the onset of nociceptive behaviors in an animal model of CIBP.7 This murine model of CIBP accurately mimics human metastatic breast cancer to the bone and has the advantage of being able to isolate the affected limb. Mechanical allodynia develops in conjunction with tumor development, as evidenced by a decrease in the force required to trigger paw withdrawal in the DPA test (Figure 5) and reduced weight bearing on the affected limb as measured by DWB testing (Figure 6A). We have shown that this behavior can be reversed toward baseline scores by systemic administration of CPZ at both 5 and 10 mg/kg. The DWB test offers advantages over the DPA testing as it reduces human interaction and produces a greater standardization of the testing procedure, limiting human error and subjectivity. This weight distribution measurement is a method of validating mechanical nociception in mice, indirectly measuring both weight bearing and spontaneous breakthrough pain, a clinically relevant assessment of CIBP. Measurements of the time spent on each limb were also found to be a useful parameter when measuring nociception in this model. The weight-based measurements of the DWB showed that CPZ-treated mice were able to apply more weight to the tumor-bearing limb relative to vehicle treated mice. The higher dose of CPZ (10 mg/kg) prevented the development of this nociceptive behavior unlike the lower dose, which only delayed the onset of this behavior until day 36 (5 mg/kg; Figure 6). The reduction in the ratio of the weight applied to the left limb relative to the right (tumor-bearing) limb suggests that CPZ-treated mice showed normal postural equilibrium relative to vehicle-treated animals indicating the CPZ-treated mice can bear more weight on their tumor-bearing limb. This was the most sensitive behavioral test used for indirectly measuring pain behavior as its high selectivity and low sensitivity ensure that a large deviation exists between mice experiencing a high degree of pain-related behavior and those that fall into the parameters of a pain-free state (baseline; Figure 6).
Similar to CPZ’s minimal selectivity for TRPV-1 in vitro at doses exceeding 700 nM, in vivo, CPZ is no longer considered selective for TRPV-1 at doses exceeding 2 mg/kg.40 Doses used in this study exceed this threshold and therefore this suggests that CPZ modulates nociceptive behavior through off-target interactions.
The ability of CPZ to modulate system xc− activity in vitro strongly suggests that its ability to limit progression of pain behaviors occurs at least in part through system xc− inhibition at the tumor. This is especially relevant considering CPZ’s limited affinity for TRPV-1. Overall, targeting glutamate release in the periphery as a means of treating cancer pain suggests a novel therapeutic strategy to relieving CIBP with fewer side effects and increased effectiveness. In the future, it would be interesting to expand this study using other cancers associated with metastatic bone pain and syngeneic animal models. Further investigation on the exact mode of action of CPZ in vivo is therefore warranted and may yield promising analgesic potential in the clinic.
Disclosure
The authors report no conflicts of interest in this work.
References
Abdelaziz DM, Stone LS, Komarova SV. Osteolysis and pain due to experimental bone metastases are improved by treatment with rapamycin. Breast Cancer Res Treat. 2014;143(2):227–237. | ||
Jimenez-Andrade JM, Mantyh WG, Bloom AP, Ferng AS, Geffre CP, Mantyh PW. Bone cancer pain. Ann N Y Acad Sci. 2010;1198:173–181. | ||
Guise TA, Kozlow WM, Heras-Herzig A, Padalecki SS, Yin JJ, Chirgwin JM. Molecular mechanisms of breast cancer metastases to bone. Clin Breast Cancer. 2005;5 Suppl(2):S46–S53. | ||
Lozano-Ondoua AN, Symons-Liguori AM, Vanderah TW. Cancer-induced bone pain: mechanisms and models. Neurosci Lett. 2013;557 Pt A:52–59. | ||
Schmidt BL. The neurobiology of cancer pain. Neurosci. 2014;20(5):546–562. | ||
Seidlitz EP, Sharma MK, Saikali Z, Ghert M, Singh G. Cancer cell lines release glutamate into the extracellular environment. Clin Exp Metastasis. 2009;26(7):781–787. | ||
Ungard RG, Seidlitz EP, Singh G. Inhibition of breast cancer-cell glutamate release with sulfasalazine limits cancer-induced bone pain. Pain. 2014;155(1):28–36. | ||
Portenoy RK. Treatment of cancer pain. Lancet. 2011;377(9784):2236–2247. | ||
Sabino MA, Mantyh PW. Pathophysiology of bone cancer pain. J Support Oncol. 2005;3(1):15–24. | ||
Wilson J, Stack C, Hester J. Recent advances in cancer pain management. F1000Prime Rep. 2014;6:10. | ||
Sharma MK, Seidlitz EP, Singh G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun. 2010;391(1):91–95. | ||
Banda M, Speyer CL, Semma SN, et al. Metabotropic glutamate receptor-1 contributes to progression in triple negative breast cancer. PLoS One. 2014;9(1):e81126. | ||
Timmerman LA, Holton T, Yuneva M, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–465. | ||
Chillarón J, Roca R, Valencia A, Zorzano A, Palacín M. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Physiol Renal Physiol. 2001;281(6):F995–F1018. | ||
Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274(17):11455–11458. | ||
Brakspear KS, Mason DJ. Glutamate signaling in bone. Front Endocrinol (Lausanne). 2012;3:97. | ||
Cowan RW, Seidlitz EP, Singh G. Glutamate signaling in healthy and diseased bone. Front Endocrinol (Lausanne). 2012;3:89. | ||
Slosky LM, BassiriRad NM, Symons AM, et al. The cystine/glutamate antiporter system xc− drives breast tumor cell glutamate release and cancer-induced bone pain. Pain. 2016;157(11):2605–2616. | ||
Doxsee DW, Gout PW, Kurita T, et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate. 2007;67(2):162–171. | ||
Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–1640. | ||
Fazzari J, Lin H, Murphy C, Ungard R, Singh G. Inhibitors of glutamate release from breast cancer cells; new targets for cancer-induced bone-pain. Sci Rep. 2015;5:8380. | ||
Bevan S, Hothi S, Hughes G, et al. Capsazepine: a competitive antagonist of the sensory neurone excitant capsaicin. Br J Pharmacol. 1992;107(2):544–552. | ||
Teng HP, Huang CJ, Yeh JH, et al. Capsazepine elevates intracellular Ca2+ in human osteosarcoma cells, questioning its selectivity as a vanilloid receptor antagonist. Life Sci. 2004;75(21):2515–2526. | ||
Docherty RJ, Yeats JC, Piper AS. Capsazepine block of voltage-activated calcium channels in adult rat dorsal root ganglion neurones in culture. Br J Pharmacol. 1997;121(7):1461–1467. | ||
Gill CH, Randall A, Bates SA, et al. Characterization of the human HCN1 channel and its inhibition by capsazepine. Br J Pharmacol. 2004;143(3):411–421. | ||
Liu L, Simon SA. Capsazepine, a vanilloid receptor antagonist, inhibits nicotinic acetylcholine receptors in rat trigeminal ganglia. Neurosci Lett. 1997;228(1):29–32. | ||
van Kuppeveld FJ, Johansson KE, Galama JM, Kissing J, Bölske G, van der Logt JT, Melchers WJ. Detection of mycoplasma contamination in cell cultures by a mycoplasma group-specific PCR. Appl Environ Microbiol. 1994;60(1):149–152. | ||
Bridges CC, Kekuda R, Wang H, et al. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42(1):47–54. | ||
Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59(17):4383–4391. | ||
Menéndez L, Juárez L, García E, García-Suárez O, Hidalgo A, Baamonde A. Analgesic effects of capsazepine and resiniferatoxin on bone cancer pain in mice. Neurosci Lett. 2006;393(1):70–73. | ||
Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20 Pt 2):6243s–6249s. | ||
Brandt MR, Beyer CE, Stahl SM. TRPV1 antagonists and chronic pain: beyond thermal perception. Pharmaceuticals (Basel). 2012;5(2):114–132. | ||
Fisch MJ, Lee JW, Weiss M, et al. Prospective, observational study of pain and analgesic prescribing in medical oncology outpatients with breast, colorectal, lung, or prostate cancer. J Clin Oncol. 2012;30(16):1980–1988. | ||
Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261(5):2256–2263. | ||
Ishii T, Sugita Y, Bannai S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J Cell Physiol. 1987;133(2):330–336. | ||
Guedon JM, Longo G, Majuta LA, Thomspon ML, Fealk MN, Mantyh PW. Dissociation between the relief of skeletal pain behaviors and skin hypersensitivity in a model of bone cancer pain. Pain. 2016;157(6):1239–1247. | ||
Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288(5464):306–313. | ||
Domercq M, Sánchez-Gómez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178(10):6549–6556. | ||
Christoph T, Bahrenberg G, De Vry J, et al. Investigation of TRPV1 loss-of-function phenotypes in transgenic shRNA expressing and knockout mice. Mol Cell Neurosci. 2008;37(3):579–589. | ||
Walker KM, Urban L, Medhurst SJ, et al. The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;304(1):56–62. | ||
Rami HK, Thompson M, Wyman P, et al. Discovery of small molecule antagonists of TRPV1. Bioorg Med Chem Lett. 2004;14(14):3631–3634. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.