Back to Journals » OncoTargets and Therapy » Volume 12
Identification of an Activating Mutation in the Extracellular Domain of HER2 Conferring Resistance to Pertuzumab
Authors Zhang Y, Wu S ![]() , Zhuang X, Weng G
, Zhuang X, Weng G ![]() , Fan J, Yang X, Xu Y, Pan L, Hou T, Zhou Z
, Fan J, Yang X, Xu Y, Pan L, Hou T, Zhou Z ![]() , Chen S
, Chen S ![]()
Received 30 September 2019
Accepted for publication 12 December 2019
Published 30 December 2019 Volume 2019:12 Pages 11597—11608
DOI https://doi.org/10.2147/OTT.S232912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Ying Zhang,1 Shanshan Wu,2 Xinlei Zhuang,1 Gaoqi Weng,1 Jiansheng Fan,1 Xiaoyue Yang,1 Yingchun Xu,1 Liqiang Pan,1 Tingjun Hou,1,3 Zhan Zhou,1 Shuqing Chen1
1Institute of Drug Metabolism and Pharmaceutical Analysis and Zhejiang Provincial Key Laboratory of Anti-Cancer Drug Research, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, People’s Republic of China; 2Precision Medicine Center, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325015, People’s Republic of China; 3State Key Laboratory of Computer Aided Design and Computer Graphics (CAD&CG), Zhejiang University, Hangzhou 310058, People’s Republic of China
Correspondence: Zhan Zhou; Shuqing Chen
Institute of Drug Metabolism and Pharmaceutical Analysis and Zhejiang Provincial Key Laboratory of Anti-Cancer Drug Research, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, People’s Republic of China
Tel +86-0571-8820-8410
Email [email protected]; [email protected]
Background: The aberrant expression of HER2 is highly associated with tumour occurrence and metastasis, therefore HER2 is extensively targeted for tumour immunotherapy. For example, trastuzumab and pertuzumab are FDA-approved monoclonal antibodies that target HER2-positive tumour cells. Despite their advances in clinical applications, emerging resistance to these two HER2-targeting antibodies has hindered their further application. Somatic mutations in HER2 receptor have been identified as one of the major reasons for resistance to anti-HER2 antibodies.
Methods: We analysed the frequency of somatic mutations in various tumour types based on TCGA and COSMIC databases. Then, the effect of the most frequent mutation (S310F) on the interaction between pertuzumab and HER2 was analysed by molecular modelling analysis. The effect of the S310F mutation was further evaluated through multiple in vitro binding experiments and antitumour activity assays.
Results: We found through bioinformatics analysis that S310F, an activating mutation in the HER2 extracellular domain, was the most frequent mutation in HER2. The S310F mutation was shown to confer resistance of HER2-positive tumour cells to pertuzumab treatment. With molecular modelling analysis, we confirmed the possibility that the S310F mutation might disrupt the interaction between pertuzumab and HER2 as a result of a significant change in the critical residue S310. Further functional analyses revealed that the S310F mutation completely abolished pertuzumab binding to HER2 receptor and inhibited pertuzumab antitumour efficacy.
Conclusion: We demonstrated the loss-of-function mechanism underlying pertuzumab resistance in HER2-positive tumour cells bearing the S310F mutation.
Keywords: HER2, mutation, pertuzumab, drug resistance, tumour cells
Introduction
Human epidermal growth factor receptor 2 (HER2) belongs to the ErbB/HER receptor tyrosine kinase family. As a transmembrane glycoprotein, it is divided into three domains: an extracellular domain (ECD) which includes four subdomains (I-IV), a transmembrane domain and a tyrosine kinase domain.1 HER2 amplification/overexpression is implicated in carcinogenesis and increased risk for progression,2 promoting its use as a promising target for immunotherapy across a variety of tumour types.3–5 For example, there are already two FDA-approved monoclonal antibodies targeting HER2, trastuzumab and pertuzumab. Trastuzumab, a humanized antibody targeting subdomain IV of the HER2 extracellular domain,6 combined with chemotherapy serves as a first-line treatment in HER2-positive breast/gastric cancer.7,8 Pertuzumab is a HER2 dimerization inhibitor that specifically binds to extracellular subdomain II,6 and its combination with trastuzumab and chemotherapy has been approved for treating HER2-positive breast cancer in the neoadjuvant, adjuvant and metastatic settings (Figure 1).9–11 Despite their advances in clinical applications, the emergence of primary and acquired drug resistance to HER2-targeted antibodies has hindered their further application.12–14 Previous studies have reported that the drug resistance mechanisms of trastuzumab and pertuzumab include dysregulation of ErbB family receptors,15,16 loss of PTEN,17 and mutations of PI3KCA that result in the activation of the PI3K/Akt signal pathway.18
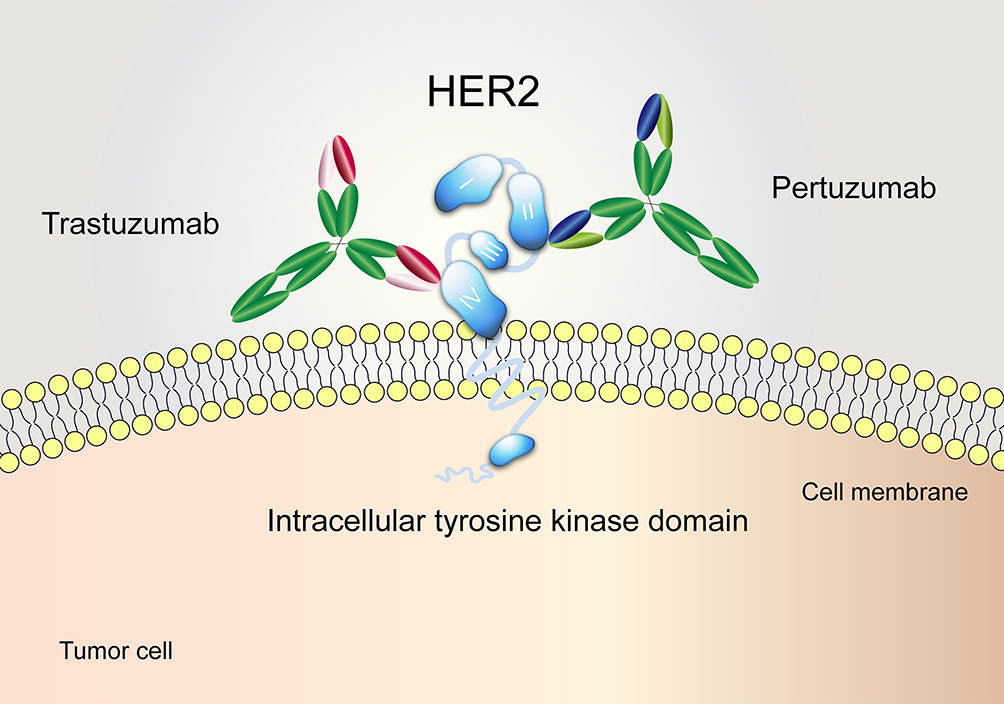 |
Figure 1 The distinct binding epitopes of HER2-targeted monoclonal antibodies approved by the FDA. Trastuzumab binds to subdomain IV of the HER2 extracellular domain. Pertuzumab binds to an epitope in subdomain II, the dimerization domain of HER2. |
In addition to the well-studied intrinsic/acquired resistance mechanisms, anti-HER2 antibody resistance can also be caused by somatic mutations of the HER2 receptor. As reported by Ou and colleagues,19 mutations at the amino acid residues V659 and G660 (located in the HER2 transmembrane domain) have been shown to reduce HER2 protein degradation and stabilize HER2 dimerization, thus these mutations are associated with resistance to trastuzumab. Drug resistance driven by somatic mutations also exists with other therapies targeting ErbB family members. The S492R mutation in the EGFR extracellular domain was found to be the key factor in cetuximab treatment resistance.20 Tumours with a HER2 tyrosine kinase mutation (L755S, L755P, T798M or T798I) showed primary or acquired resistance to lapatinib.21–23 Therefore, somatic mutations are emerging as important factors in the development of resistance to targeted therapies.
In this study, we analysed the frequency of somatic mutations across various tumour types based on TCGA and COSMIC databases and discovered that the S310F mutation, located in subdomain II of HER2 ECD, was the most frequent substitution among all tumour types and HER2 mutations. We analysed the effect of the S310F mutation on the interaction between pertuzumab and HER2 by molecular modelling analysis. Then, we further evaluated the effect of the S310F mutation through multiple functional assays.
Materials and Methods
Cancer Somatic Mutation Analysis
The somatic mutations of 33 cancer types were downloaded from TCGA (https://gdc.cancer.gov/, MC3 project) and COSMIC (https://cancer.sanger.ac.uk/cosmic, V89) databases. The list of cancer-associated genes was extracted from the Cancer Gene Census of COSMIC (https://cancer.sanger.ac.uk/census). The missense mutations of tumour-associated genes that lead to altered amino acid property within the extracellular domain of membrane proteins were extracted by TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), and further screening was carried out via UniProt (https://www.uniprot.org/) database. Then, the selected mutations were analysed by R software (https://www.r-project.org/) to depict the distribution across 33 cancer types.
Cell Culture and Reagents
NIH3T3 and MCF7 cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences (China), and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (FBS, Gibco) and antibiotic (1% penicillin/streptomycin (P/S)). HEK293F cell line was a generous gift from Qilin Zhang’s laboratory of Tsinghua University and was cultured in SMM 293-TI (Sino Biological, China) with 0.5% FBS and 1% P/S. All cells were maintained in a 5% CO2 humidified atmosphere at 37°C. Pertuzumab and trastuzumab monoclonal antibodies were kindly provided by Zhejiang Hisun Pharmaceutical Co., Ltd. (China).
Molecular Modelling Analysis
Using the X-ray structure of the HER2-pertuzumab complex (Protein Data Bank (PDB) ID: 1S78) as a template, the Ser310 of HER2 was mutated to Phe310 by PyMOL software (The PyMOL Molecular Graphics System, Schrödinger, LLC, NY, USA). In addition, the HER2-pertuzumab complex was also analysed by using the HawkDock server.24–27
Plasmid Construction and Transfection
The pCMV3-HER2 cDNA and pCMV3-HER3 cDNA plasmids were purchased from Sino Biological. The HER2 929C_T point mutation (from the first ATG of HER2 cDNA) was constructed by polymerase chain reaction (PCR) with primers 1 and 2 and confirmed by DNA sequencing. HER3 cDNA gene was amplified from the pCMV3-HER3 plasmid, and EcoRI and NotI restriction enzyme sites were added at both ends with primers 3 and 4. Then, the full-length sequence was inserted into the pMH3 expression vector. All primers are listed in Table S1.
NIH3T3 cells were co-transfected with the wild-type (WT)/S310F-mutant HER2 and HER3 constructs using Lipofectamine 3000 according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). MCF7 cells were transfected with the WT/S310F-mutant HER2 construct using the above method. For stable ectopic expression, stable WT/S310F-mutant HER2-expressing NIH3T3 cells were selected with Hygromycin B (Solarbio, China) and G418 (Sangon Biotech, China). Stable WT/S310F-mutant HER2-expressing MCF7 cells were selected with Hygromycin B.
Flow Cytometry Analysis
NIH3T3 cells expressing WT/S310F-mutant HER2 were used to evaluate the anti-HER2 antibody affinity. WT and mutant HER2 cells (6×105) were harvested using 0.25% trypsin-EDTA solution and washed twice with ice-cold phosphate-buffered saline (PBS, pH 7.4). Then, cells were incubated with pertuzumab or trastuzumab in staining medium (PBS with 1% bovine serum albumin (BSA)) for 30 min on ice. After incubation, cells were washed twice and then incubated with FITC-conjugated goat anti-human IgG (H+L) (Beyotime, China) for 30 min. After washing, cells were suspended in 500 µL PBS and examined on a Cytomics FC500 MCL flow cytometer (Beckman Coulter, Brea, CA, USA).
Expression and Purification of Recombinant Fusion Proteins
Recombinant fusion proteins for enzyme-linked immunosorbent assay (ELISA) and surface plasmon resonance (SPR) analysis were produced in HEK293F cells. Briefly, DNA encoding the human WT (HER2-ECD-WT) or S310F-mutant (HER2-ECD-S310F) HER2 extracellular domain (ECD, amino acids 23–652) was fused with the human IgG1 Fc gene and cloned into the pMH3 expression vector. Then, the obtained HER2-ECD-WT/S310F Fc fusion plasmid was transiently transfected into HEK293F cells using polyethylenimine (PEI). Cell culture supernatants containing the above fusion protein were collected at three days post transfection, and HER2-ECD-WT/S310F Fc fusion protein was purified by using a HiTrap Protein A column (GE Healthcare, PA, USA). Purified fusion proteins were analysed by SDS-PAGE.
Enzyme-Linked Immunosorbent Assay
The relative affinities of pertuzumab and trastuzumab to WT/S310F-mutant HER2 were measured by ELISA. HER2-ECD-WT/S310F Fc-tagged protein (1 µg/mL) was coated on a 96-well plate at 4°C overnight, and the plate was blocked with PBS containing 2% BSA for 2 h at 37°C to avoid non-specific binding. Serial dilutions of pertuzumab and trastuzumab were added into the plate and incubated for 2 h at 37°C. Then, the plate was washed with PBST (PBS containing 0.05% Tween 20) three times, and polyclonal goat anti-human kappa horseradish peroxidase (HRP)-conjugated antibody was added into the plate and incubated for 2 h. After washing, tetramethyl benzidine (TMB) was added into the plate and incubated for 20 min in the dark. The absorbance at 450 nm was measured by a 680 Microplate Reader (Bio-Rad, CA, USA).
Surface Plasmon Resonance Assay
SPR assay was performed on a Biacore 3000 instrument (GE Healthcare) at 25°C. WT and S310F-mutant HER2-ECD Fc-tagged proteins were immobilized on a CM5 sensor chip using the amine coupling reaction following the manufacturer’s instructions. The coupling density of HER2-ECD-WT/S310F Fc antigen was typically restricted to 300 response units (RU). All analytes (trastuzumab and pertuzumab) were prepared by serial dilution from 0.96 nM to 600 nM in HBS-EP buffer (pH 7.4, GE Healthcare) and injected over the CM5 sensor chip at a flow rate of 30 µL/min for 3 min. The dissociation step lasted for 450 s. The sensor chip was regenerated with 10 mM citrate sodium and 1 M NaCl (pH 3.0) at each cycle. Kinetic parameters were analysed using Biacore’s evaluation software.
Western Blot Analysis
NIH3T3 cells co-expressing HER2 (WT/S310F) and HER3 receptors were seeded in 6-well plates in DMEM culture medium containing 1% FBS. Cell medium was replaced with serum-free medium, and cells were pretreated with or without 600 nM pertuzumab or trastuzumab for 2 h. Then, cells were treated with 5 nM HRG-β1177-244 (HRG) or culture medium (negative control). All cells were washed with PBS and lysed using cell lysis buffer (P0013, Beyotime) supplemented with PMSF (1 mM) and protease inhibitor (1 mM) on ice for 1 h. Supernatants were collected after centrifugation at 14,000 ×g for 10 min at 4°C and the protein concentration was measured with the Enhanced BCA Protein Assay Kit (Beyotime). Cell lysates were separated on a 8% SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with TBST (Tris-buffered saline with 0.1% Tween 20) containing 5% BSA for 2 h at 37°C and washed three times with TBST. Then, the membranes were incubated with the following primary antibody: anti-phospho-HER2/ErbB2 (Y1221/1222) monoclonal antibody, anti-HER2/ErbB2 or anti-β-actin polyclonal antibody (Beyotime) at 4°C overnight. After washing, the membranes were incubated with goat anti-rabbit/mouse IgG (H+L) HRP-conjugated antibody for 2 h at 37°C. Protein bands were visualized using ECL reagent and exposed on a gel imaging system (Bio-Rad).
Cell Viability Test
MCF7 cells and MCF7 cells expressing WT (MCF7-HER2 WT) or S310F-mutant (MCF7-HER2 S310F) HER2 were collected for the cellular activity test. MCF7, MCF7-HER2 WT and MCF7-HER2 S310F cells were seeded at a density of 3×103 cells per well into 96-well microtiter plates in DMEM culture medium with 1% FBS. After overnight incubation, 2 nM HRG with or without 300 nM pertuzumab/trastuzumab or culture medium was added into the wells, and cells were treated with various conditions for 5 days. Cell viability was measured by using the Cell Counting Kit-8 (CCK-8, Japan) and the absorbance values at 450 nm were analysed by a 680 Microplate Reader.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 software. One-way analysis of variance (ANOVA) was applied for multiple sets of data to determine statistical significance (***P < 0.001).
Results
The Frequency of Somatic Mutations Across Various Tumours
According to the TCGA MC3 project, there were more than 1.5 million missense mutations in 10,033 cancer samples. Among these mutations, we focused on the mutations with altered amino acid property, occurring in the extracellular domains of membrane proteins which are encoded by tumour-associated genes. Recurrent mutations with more than three occurrences among all cancer samples were further analysed. HER2 S310F was the most frequent (n=43) among these mutations, followed by FGFR2 S252R (n=27) and EGFR G598V (n=15) (Figure 2A, Figure S1). Among all cancer types, bladder urothelial carcinoma (BLCA) and stomach adenocarcinoma (STAD) tended to have a higher probability of acquiring the S310F mutation (Figure S2A). The mutation landscape of HER2 was also analysed in BLCA, STAD and breast invasive carcinoma (BRCA). The S310F mutation turned out to be the most frequent mutation in STAD and BLCA, with 6 occurrences among the 440 cancer samples and 21 occurrences among the 412 cancer samples, respectively (Figure S2B–C). The S310F mutation did not show up in the mutation landscape of BRCA, with only 1 occurrence among the 986 cancer samples (Figure S2D). Furthermore, the distribution of HER2 mutations extracted from COSMIC database also indicated that the frequency of the S310F mutation was the highest among all HER2 mutations (Figure 2B). Similarly, the S310F mutation was also the most frequent mutation within all HER2 mutations in BLCA, STAD and BRCA (Figure S3A–C). Overall, we found that the S310F mutation of HER2 ECD was the most frequent mutation among all tumour types and HER2 mutations.
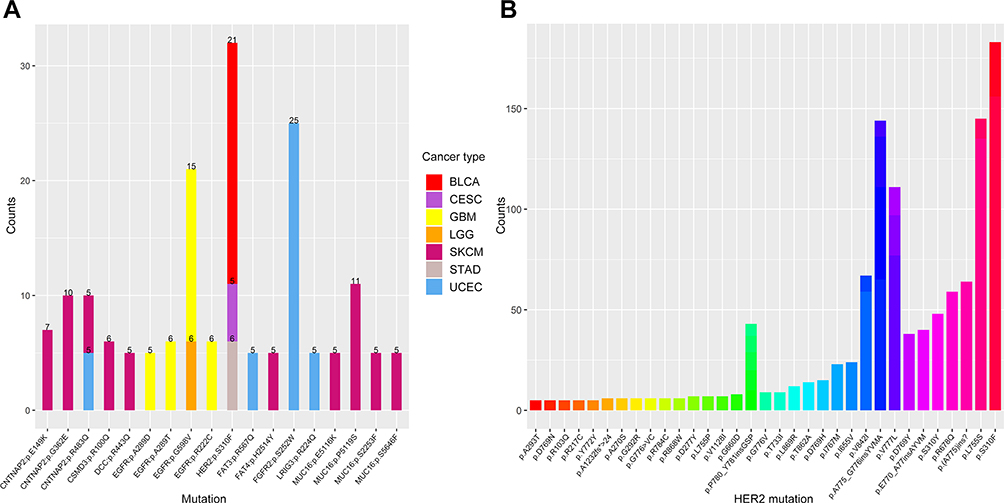 |
Figure 2 The distribution and frequency of HER2 S310F mutation in tumours. (A) The distribution of recurrent mutations (more than five occurrences in an individual cancer type) in the extracellular domain of membrane proteins. The numbers mean the occurrences of each mutation in the same tumour. Different cancer types are represented by different colours. (B) The distribution of HER2 mutations based on COSMIC database.Abbreviations: BLCA, bladder urothelial carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; GBM, glioblastoma multiforme; LGG, brain lower grade glioma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UCEC, uterine corpus endometrial carcinoma. |
Molecular Modelling Analysis of the Interaction Between Pertuzumab and S310F-Mutant HER2
The S310F mutation causes a substitution of serine to phenylalanine at amino acid 310, which maps to the pertuzumab epitope on HER2 (Figure 3A). We performed molecular modelling to predict the effect of the S310F mutation on pertuzumab binding to HER2. Compared to WT HER2, the S310F-mutant HER2 introduced a nonpolar side chain, which was predicted to decrease the interaction between pertuzumab and HER2 (Figure 3B).
 |
Figure 3 Molecular modelling of the interaction between pertuzumab and HER2. (A) Structure visualization of the HER2-pertuzumab complex (PDB: 1S78), including the HER2 extracellular domain (bluish-green), pertuzumab’s light chain (purple) and heavy chain (green). The position of HER2 Ser310 was confirmed at the interface of HER2 and pertuzumab. Selected side chains from HER2 and pertuzumab are shown in stick representation, with carbons coloured by domain, nitrogens blue, oxygens red and hydrogens grey. The backbones are shown in ribbon representation. (B) Molecular modelling of pertuzumab bound to WT/S310F-mutant HER2. Hydrogen bonds are shown as red dashed lines. (C) The top 10 critical residues of the HER2-pertuzumab interaction predicted by the HawkDock server. |
In addition, to further explore the importance of HER2 residue S310, the HawkDock server was used to analyse the key residues of the binding interface of the HER2-pertuzumab complex. As shown in Figure 3C, the energy contribution of Ser310 ranked third and reached −5.26 kcal mol−1, mainly coming from the electrostatic potentials (−7.53 kcal mol−1), suggesting that the residue S310 was quite important for the formation of the HER2-pertuzumab complex.
S310F Mutation Prevented Pertuzumab Binding to HER2
NIH3T3 cells are a mouse fibroblast cell line that lacks endogenous EGFR, HER2, and HER3.23 We expressed WT or S310F-mutant HER2 in NIH3T3 cells to investigate the exact effect of the S310F mutation on pertuzumab binding affinity. We preliminarily measured the pertuzumab affinity to these two cell lines by flow cytometry analysis. The binding epitope of trastuzumab on HER2 is completely inconsistent with that of pertuzumab. Thus, trastuzumab was used as the positive control, and its binding ability to these two cell lines was also measured. As depicted in Figure 4A, pertuzumab could efficiently bind to cells expressing WT HER2 but not to cells that expressed S310F-mutant HER2. Unlike pertuzumab, trastuzumab retained the ability to bind to both WT and S310F-mutant HER2 cells. We found that the affinity of pertuzumab for NIH3T3-HER2 WT cells was dose-dependent, but no detectable increase in pertuzumab affinity for NIH3T3-HER2 S310F cells was observed (Figure 4B). Consistent with the abovementioned molecular modelling, the S310F mutation led to a significant reduction in pertuzumab binding relative to WT HER2.
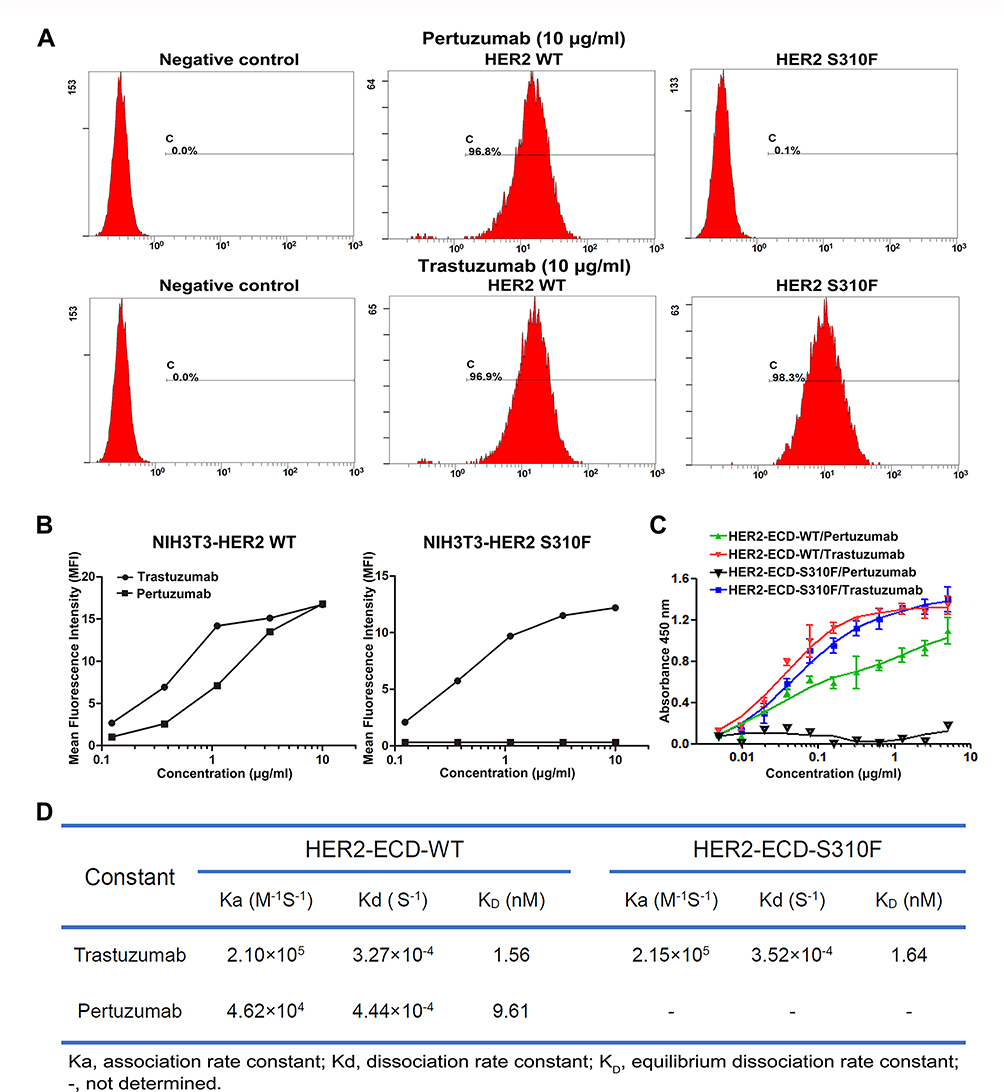 |
Figure 4 The binding affinities of anti-HER2 antibodies for WT/S310F-mutant HER2. (A) The binding affinities of pertuzumab and trastuzumab for NIH3T3 cells expressing WT (HER2 WT) or S310F-mutant (HER2 S310F) HER2. NIH3T3 cells were used as a negative control. (B) The affinities of serial concentrations of pertuzumab and trastuzumab for WT (NIH3T3-HER2 WT) or S310F-mutant (NIH3T3-HER2 S310F) HER2 cells. (C) Comparisons of pertuzumab and trastuzumab interacting with WT (HER2-ECD-WT) or S310F-mutant (HER2-ECD-S310F) HER2 ECD protein by ELISA as described in the materials and methods. (D) The kinetic parameters for trastuzumab and pertuzumab derived from the fitted data models according to SPR analyses. |
Pertuzumab Affinity for WT/S310F-Mutant HER2 ECD Protein
We further compared the affinities of pertuzumab and trastuzumab for WT/S310F-mutant HER2 ECD protein through ELISA. WT and S310F-mutant HER2 ECD Fc-tagged proteins were successfully expressed by HEK293F cells and analysed by SDS-PAGE under reductive condition (Figure S4). As shown in Figure 4C, the S310F-mutant HER2 ECD was selectively defective in binding pertuzumab but not trastuzumab. The binding affinity of pertuzumab for S310F-mutant HER2 ECD was substantially unchanged with increasing antibody concentration. As expected, WT HER2 ECD could bind to both two antibodies. An in vitro biochemical binding study proved once again that the S310F mutation interfered with the binding of pertuzumab to HER2.
Kinetic Characteristics of Pertuzumab Interaction with WT/S310F-Mutant HER2 Antigen
The kinetics of anti-HER2 antibodies binding to WT/S310F-mutant HER2-ECD-Fc antigen were determined by SPR assay. The binding curves were fitted using a bivalent analyte model (Figure S5). The apparent association, dissociation and equilibrium dissociation rate constants obtained from the fitted data models are shown in Figure 4D. Trastuzumab and pertuzumab exhibited high affinities for the WT HER2 antigen, and the KD values were determined to be 1.56 nmol/L and 9.61 nmol/L, respectively. Both antibodies binding towards WT HER2 antigen showed similar association and dissociation rate constants. These values were very close to the previously reported apparent bivalent affinities for trastuzumab and pertuzumab.28 In addition, trastuzumab was still able to bind to the S310F-mutant HER2 antigen, with a high affinity (KD=1.64 nmol/L). Pertuzumab did not have any detectable binding to the S310F-mutant HER2 antigen, and the KD value could not be determined. The SPR results also showed that the S310F mutation disrupted the interaction between pertuzumab and HER2 receptor.
Effect of the S310F Mutation on Pertuzumab-Mediated Inhibition of HER2 Signalling
We then tested whether the efficacy of pertuzumab could be completely abolished by the S310F mutation. WT/S310F-mutant HER2 and HER3 constructs were cotransfected into NIH3T3 cells (Figure 5A). We assessed the effect of the S310F mutation on pertuzumab-mediated inhibition of HER2 signalling via these two cell lines. Previous studies have shown that pertuzumab inhibits HER2 signalling that is initiated by ligand-mediated dimerization, whose downstream signals include HER2 intracellular phosphorylation, MAPK and PI3K/Akt.29 In contrast to pertuzumab, trastuzumab is only effective in blocking the signalling induced by increased HER2 dimerization in the absence of ligand binding. In this assay, the intracellular phosphorylation signalling of WT or S310F-mutant HER2 was activated by HER3-ligand heregulin (HRG)-induced dimerization (Figure 5B). Pertuzumab significantly reduced ligand-stimulated WT HER2 signalling. However, there was no reduction in S310F-mutant HER2 signalling in response to pertuzumab. Trastuzumab had little effect on ligand-activated HER2 signalling, which is consistent with previous studies.30
 |
Figure 5 Effect of S310F mutation on the antitumour activity of pertuzumab. (A) NIH3T3 cells were used as the parental cell line and negative control, pCMV3-HER2 WT/S310F-mutant and pMH3-HER3 constructs were cotransfected into NIH3T3 cells. (B) Different ability of pertuzumab to inhibit HER2 phosphorylation signalling in WT/S310F-mutant HER2 cell lines after stimulation with or without HRG. NIH3T3 cells co-expressing WT/S310F HER2 and HER3 were incubated with culture medium or pertuzumab/trastuzumab for 2 h and then stimulated with or without HRG. Western blotting was performed using antibodies for the indicated proteins. (C) HER2 expression levels of MCF7 cells and MCF7 cells transfected with WT (MCF7-HER2 WT) or S310F-mutant (MCF7-HER2 S310F) HER2 gene, respectively. (D) Cell viability of MCF7, MCF7-HER2 WT and MCF7-HER2 S310F cell lines treated with culture medium (basal), HRG, HRG+pertuzumab or HRG+trastuzumab. Cell viability (%) is represented as the mean ± SD. One-way analysis of variance (ANOVA) was applied for statistical comparisons on multiple groups, ***p < 0.001. Abbreviation: NS, no significance. |
Antitumour Activity of Pertuzumab Against WT/S310F-Mutant HER2-Positive Tumour Cells
We further evaluated the in vitro efficacy of pertuzumab in the HER2-overexpressing breast cancer cell line bearing the S310F mutation. MCF7 cells which have low level of endogenous HER2 and high endogenous HER3, were chosen as the parental breast cancer cell line.23 WT or S310F-mutant HER2 construct was transfected into MCF7 cells to establish a model of high expression, the WT/S310F-mutant HER2 breast cancer cell line (Figure 5C). Cell viability of MCF7, MCF7-HER2 WT and MCF7-HER2 S310F cell lines was measured under different treatment conditions (Figure 5D). The addition of HRG significantly increased the growth of MCF7, MCF7-HER2 WT and MCF7-HER2 S310F cells compared with that of the basal controls, which were incubated in the absence of HRG. In the presence of pertuzumab and HRG, the growth of MCF7-HER2 WT cells was significantly reduced to the basal level, while the cell viability of MCF7-HER2 S310F cell line was unaffected. Trastuzumab barely decreased HRG-induced cell viability in all cell lines.
Discussion
In this study, we used bioinformatics to discover that S310F, an extracellular domain mutation of HER2, was the most frequent mutation within HER2 and among various tumours. We also unexpectedly found that the S310F mutation alone completely abolished the binding ability of pertuzumab to HER2 ECD, which provides different insights into the important role of somatic mutations in the resistance mechanism of anti-HER2 antibodies.
Taken together, the molecular modelling results revealed that residue S310 significantly contributed to the tight binding of pertuzumab with HER2 ECD. The S310F mutation disrupted the essential hydrogen bonds between residue S310 of HER2 and residue D31 of pertuzumab by introducing a hydrophobic benzene ring. A previous study conducted by Franklin et al.30 also showed that these two hydrogen bonds play a major role in the interaction of pertuzumab and HER2, indicating that disruption of hydrogen bonds in key contacting residues could dramatically attenuate pertuzumab binding affinity. Therefore, in addition to residue S310, we hypothesize that there might be similar effects when somatic mutation happens in other key residues such as V308 (based on our analysis by using the HawkDock server).
Previous studies reported that the S310F is an activating mutation that has been considered an oncogenic driver, or a promoter of cancer cell growth.23,31 Greulich et al.31 showed that the S310F mutation exhibited a significant increase in HER2 phosphorylation signalling and was implicated in colony formation and oncogenic potency when studied in NIH3T3, AALE and Ba/F3 cell lines. Activating mutations of the HER2 gene identified in various cancer types were shown to play an important role in tumorigenesis, and serve as promising targets in preclinical models.32,33 Recently, the focus on new antineoplastic strategies has turned to targeting activating mutations.34,35 However, we discovered that an activating mutation S310F within the HER2 ECD conferred resistance to pertuzumab by abolishment of the binding. In addition to the S310F mutation, G309A/E and S310Y mutations that were also verified as activating mutations mapping to the pertuzumab epitopes on HER2,23,31 suggesting that tumours bearing these mutations may be refractory to pertuzumab.
Trastuzumab is known to bind to extracellular subdomain IV, which results in the downregulation of ligand-independent HER2 signalling. However, trastuzumab is not effective in impairing ligand-activated HER2 heterodimerization,29,36 which is in agreement with our observation that trastuzumab did not significantly inhibit ligand-activated HER2 phosphorylation signalling. Therefore, it seems reasonable to argue that trastuzumab does not block important sites of HER2 receptor dimerization. The action-mechanism of pertuzumab is complementary to that of trastuzumab.37 Pertuzumab binds to the extracellular dimerization subdomain, prevents ligand-induced HER2 heterodimer formation with other HER receptors and inhibits subsequent HER2 downstream signalling. The combination of pertuzumab and trastuzumab has a synergistic anticancer effect, as it provides a more complete block of HER2 downstream signalling.37 In addition, a previous study showed that either pertuzumab or trastuzumab did not affect the response to the other.38 Despite the effectiveness of trastuzumab, clinical observations have shown that not all patients with HER2-positive tumours benefit from trastuzumab-based therapies. Moreover, a proportion of patients relapsed after a period of therapeutic response.36 These observations are the basis for combination with additional therapy, especially pertuzumab. Currently, pertuzumab is always used in combination with trastuzumab as a treatment for patients with HER2-positive breast cancer who have progressed on trastuzumab therapy, and the combination therapy has significantly prolonged the survival time of these patients.37 This means that pertuzumab plays an important role in cancer treatment. However, our discovery confirmed that HER2-positive tumour cells bearing the S310F mutation were resistant to pertuzumab therapy, which provides a theoretical basis and a guide for clinically avoiding the irrational use of pertuzumab in patients with HER2-positive breast cancer bearing the S310F mutation.
Recently, the JACOB study, a Phase III trial that evaluated the efficacy of adding pertuzumab to trastuzumab-based therapy for HER2-positive gastric cancer, did not meet its primary endpoint.39 As a consequence, more studies are needed to further screen out patients with gastric cancer who would benefit from dual targeting therapy. Here, we found that the S310F mutation occurred more frequently in gastric cancer than other HER2 mutations, and its frequency in gastric cancer was almost 10 times higher than that in breast cancer. Based on these data, thus we think that the S310F mutation is worthy to be considered in the clinical trial to classify gastric cancer patients more accurately, and it might be conducive to increasing the success rate of the clinical trial. However, we have no specific experimental data of the JACOB trial, and this assumption needs to be verified by extensive studies.
Many cancer-related genetic alterations, such as EGFR tyrosine kinase mutations L858R and T790M, have been included in clinical screening for treatment selection. Considering its destructive effect on pertuzumab binding in this study, it is rational to perform S310F mutation screening in patients with HER2-positive tumours prior to antibody therapy. Our study also further suggests the necessity of analysing somatic mutations in the extracellular domain of target receptors during antibody-based therapy.
Conclusions
Our study demonstrated the loss-of-function mechanism underlying pertuzumab resistance in HER2-positive tumour cells bearing the S310F mutation. Our findings provide a molecular explanation for the potential clinical implications of the HER2 S310F mutation, and assist in identifying patients who would benefit from pertuzumab treatment.
Abbreviations
ECD, extracellular domain; ELISA, enzyme-linked immunosorbent assay; HRG, heregulin; PDB, protein data bank; SPR, surface plasmon resonance; WT, wild-type.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No. 2017YFC0908600), the State Key Program of National Natural Science of China (Grant No. 81430081), and the National Natural Science Foundation of China (Grant No. 81502971).
Disclosure
The authors declare no conflicts of interest.
References
1. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26(45):6469–6487. doi:10.1038/sj.onc.1210477
2. Neve RM, Lane HA, Hynes NE. The role of overexpressed HER2 in transformation. Ann Oncol. 2001;12(suppl 1):S9–S13. doi:10.1093/annonc/12.suppl_1.S9
3. Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19(9):1523–1529. doi:10.1093/annonc/mdn169
4. Yan M, Schwaederle M, Arguello D, et al. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34(1):157–164. doi:10.1007/s10555-015-9552-6
5. Tagliabue E, Balsari A, Campiglio M, Pupa SM. HER2 as a target for breast cancer therapy. Expert Opin Biol Ther. 2010;10(5):711–724. doi:10.1517/14712591003689972
6. Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–275. doi:10.1016/j.jconrel.2010.04.009
7. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi:10.1056/NEJM200103153441101
8. Bang YJ, Van CE, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a Phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. doi:10.1016/S0140-6736(10)61121-X
9. Swain SM, Kim S-B, Cortés J, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14(6):461–471. doi:10.1016/S1470-2045(13)70130-X
10. Amiri-Kordestani L, Wedam S, Zhang L, et al. First FDA approval of neoadjuvant therapy for breast cancer: pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin Cancer Res. 2014;20(21):5359–5364. doi:10.1158/1078-0432.CCR-14-1268
11. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med. 2017;377(2):122–131. doi:10.1056/NEJMoa1703643
12. Gagliato DM, Jardim DL, Marchesi MS, Hortobagyi GN. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget. 2016;7(39):64431–64446. doi:10.18632/oncotarget.7043
13. Luque-Cabal M, Garcia-Teijido P, Fernandez-Perez Y, Sanchez-Lorenzo L, Palacio-Vazquez I. Mechanisms behind the resistance to trastuzumab in HER2-amplified breast cancer and strategies to overcome it. Clin Med Insights Oncol. 2016;10(Suppl 1):21–30. doi:10.4137/CMO.S34537
14. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17(1):1–16. doi:10.1615/CritRevOncog.v17.i1.20
15. Gallardo A, Lerma E, Escuin D, et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br J Cancer. 2012;106(8):1367–1373. doi:10.1038/bjc.2012.85
16. Hellyer NJ, Kim MS, Koland JG. Heregulin-dependent activation of phosphoinositide 3-kinase and Akt via the ErbB2/ErbB3 co-receptor. J Biol Chem. 2001;276(45):42153–42161. doi:10.1074/jbc.M102079200
17. Esteva FJ, Guo H, Zhang S, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177(4):1647–1656. doi:10.2353/ajpath.2010.090885
18. O’Brien NA, Browne BC, Chow L, et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9(6):1489–1502. doi:10.1158/1535-7163.MCT-09-1171
19. Ou SI, Schrock AB, Bocharov EV, et al. HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol. 2017;12(3):446–457. doi:10.1016/j.jtho.2016.11.2224
20. Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18(2):221–223. doi:10.1038/nm.2609
21. D’Amato V, Raimondo L, Formisano L, et al. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev. 2015;41(10):877–883. doi:10.1016/j.ctrv.2015.08.001
22. Kancha RK, Von BN, Bartosch N, et al. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS One. 2011;6(10):e26760. doi:10.1371/journal.pone.0026760
23. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237. doi:10.1158/2159-8290.CD-12-0349
24. Weng G, Wang E, Wang Z, et al. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019;47(W1):W322–W330. doi:10.1093/nar/gkz397
25. Chen F, Liu H, Sun H, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys Chem Chem Phys. 2016;18(32):22129–22139. doi:10.1039/C6CP03670H
26. Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51(1):69–82. doi:10.1021/ci100275a
27. Sun H, Li Y, Tian S, Xu L, Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys. 2014;16(31):16719–16729. doi:10.1039/C4CP01388C
28. Leung KM, Batey S, Rowlands R, et al. A HER2-specific modified Fc fragment (Fcab) induces antitumor effects through degradation of HER2 and apoptosis. Mol Ther. 2015;23(11):1722–1733. doi:10.1038/mt.2015.127
29. Agus DB, Akita RW, Fox WD, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2(2):127–137. doi:10.1016/S1535-6108(02)00097-1
30. Franklin MC, Carey KD, Vajdos FF, et al. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5(4):317–328. doi:10.1016/S1535-6108(04)00083-2
31. Greulich H, Kaplan B, Mertins P, et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A. 2012;109(36):14476–14481. doi:10.1073/pnas.1203201109
32. Kavuri SM, Jain N, Galimi F, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5(8):832–841. doi:10.1158/2159-8290.CD-14-1211
33. Zuo WJ, Jiang YZ, Wang YJ, et al. Dual characteristics of novel HER2 kinase domain mutations in response to HER2-targeted therapies in human breast cancer. Clin Cancer Res. 2016;22(19):4859–4869. doi:10.1158/1078-0432.CCR-15-3036
34. Subramanian J, Katta A, Masood A, Vudem DR, Kancha RK. Emergence of ERBB2 mutation as a biomarker and an actionable target in solid cancers. Oncologist. 2019;24(12):e1303–e1314. doi:10.1634/theoncologist.2018-0845
35. Britta W, Reis-Filho JS. Activating mutations in HER2: neu opportunities and neu challenges. Cancer Discov. 2013;3(2):145–147. doi:10.1158/2159-8290.CD-12-0585
36. Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–440. doi:10.1016/j.ccr.2009.03.020
37. Reynolds K, Sarangi S, Bardia A, Dizon DS. Precision medicine and personalized breast cancer: combination pertuzumab therapy. Pharmgenomics Pers Med. 2014;7:95–105. doi:10.2147/PGPM.S37100
38. Lua WH, Gan SK, Lane DP, Verma CS. A search for synergy in the binding kinetics of Trastuzumab and Pertuzumab whole and F(ab) to Her2. NPJ Breast Cancer. 2015;1:15012. doi:10.1038/npjbcancer.2015.12
39. Tabernero J, Hoff PM, Shen L, et al. Pertuzumab plus trastuzumab and chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer (JACOB): final analysis of a double-blind, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2018;19(10):1372–1384. doi:10.1016/S1470-2045(18)30481-9
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.