")
Back to Journals » OncoTargets and Therapy » Volume 12
Identification Of Actionable Genetic Targets In Primary Cardiac Sarcomas
Authors Salvador-Coloma C, Saigí M , Díaz-Beveridge R, Penín RM, Pané-Foix M, Mayordomo E , Melián M, Schuler M, García Del Muro X, Font de Mora J
Received 3 May 2019
Accepted for publication 9 August 2019
Published 7 November 2019 Volume 2019:12 Pages 9265—9275
DOI https://doi.org/10.2147/OTT.S214319
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Carmen Salvador-Coloma,1,2,* María Saigí,3,* Roberto Díaz-Beveridge,1 Rosa María Penín,4 María Pané-Foix,4 Empar Mayordomo,5 Marcos Melián,1 Mona Schuler,6 Xavier García Del Muro,3 Jaime Font de Mora2
1Department of Medical Oncology, Hospital Universitari i Politècnic La Fe, Valencia, Spain; 2Laboratory of Cellular and Molecular Biology, Clinical and Translational Research in Cancer, Instituto de Investigación Sanitaria La Fe, Valencia, Spain; 3Department of Medical Oncology, Institut Català Oncologia, IDIBELL, L’Hospitalet de Llobregat, Barcelona, Spain; 4Department of Pathology, Hospital Universitari de Bellvitge, Barcelona, Spain; 5Department of Pathology, Hospital Universitari i Politècnic La Fe, Valencia, Spain; 6Department of Cardiac Surgery, Hospital Universitari i Politècnic La Fe, Valencia, Spain
*These authors contributed equally to this work
Correspondence: Carmen Salvador-Coloma
Jaime Font de Mora Instituto de Investigación Sanitaria La Fe, Avenida Fernando Abril Martorell, 106, Torre A, 5-07, Valencia 46026 Spain
Tel +34-961246646
Email [email protected]
Background: Primary cardiac tumors are extremely rare; most are myxomas with a benign prognosis. However, primary sarcomas are highly aggressive and treatment options are limited. Radical surgery is often not feasible and conventional therapies provide only modest results. Due to the rare nature of primary cardiac tumors, there are no proper randomized studies to guide treatment. Their complexity requires alternative approaches in order to improve treatment efficacy.
Methods: We isolated DNA from 5 primary cardiac sarcomas; the quality of DNA from 3 of them was sufficient to perform high-resolution single nucleotide polymorphism (SNP) array analysis.
Results: In the present study, molecular karyotyping revealed numerous segmental chromosomal alterations and amplifications affecting actionable genes that may be involved in disease initiation and/or progression. These include chromosomal break flanking AKT2 in undifferentiated pleomorphic rhabdomyosarcoma, chromosomal break in promoter of TERT, and gain of CDK4 and amplification of MDM2 in inflammatory myofibroblastic tumor. We detected segmental break flanking MOS in high-grade myxofibrosarcoma. In addition, the high number of chromosomal aberrations in high-grade myxofibrosarcoma may cause multiple tumor-specific epitopes, supporting the study of immunotherapy treatment in this type of aggressive tumor.
Conclusion: Our results provide a genetic rationale that supports an alternative, personalized therapeutic management of primary cardiac sarcomas.
Keywords: primary cardiac sarcomas, molecular karyotype, actionable genes
Introduction
Primary cardiac tumors are extremely rare, with a global incidence of 0.0017–0.019% per year.1 Heart metastases are over 20 times more frequent than primary cardiac tumors.2 Benign primary cardiac tumors are the most prevalent (around 75%) of which are myxomas.3 Malignant primary neoplasms are uncommon and sarcomas represent 20% of all cases.4,5 The most common subtypes are angiosarcoma (37%), followed by MFH (24%), leiomyosarcoma (9%), rhabdomyosarcoma (7%), unclassified (7%) and others (16%).6 These malignant tumors rapidly invade all layers of the heart and metastasize, with over 80% of patients presenting metastasis at diagnosis.7 Prevalence between males and females has not been reported and the mean age of presentation is 40 years.8
The symptoms depend on the location and the extent of the tumor. In some patients, detection of tumors is incidental, although in most cases, due to the aggressive nature of the tumors, there are symptoms which are usually non-specific. Symptoms include those derived from an obstruction to blood flow and impairment of valvular function, embolism, local myocardial invasion causing arrhythmia or pericardial effusion, or systemic symptoms of dyspnea (the most common symptom 60%), fever, malaise and weight loss.6,9
In most cases, a transthoracic echocardiography is the method of initial evaluation. A transesophageal echocardiography can provide valuable information, especially in difficult locations where the transthoracic route may not be sensitive enough. Cardiac magnetic resonance imaging (MRI), and computed tomography (CT) scan provide complementary information. In selected cases, a coronary angiography is specifically required to assess the location of the tumor’s nutrient vessels. The CT scan and MRI of the chest and abdomen are complementary and can show the extracardiac extent of the tumor and presence of metastases.
We present a series of five primary cardiac sarcomas. We performed molecular karyotyping of three samples using high-resolution SNP arrays and found genetic alterations that may provide alternative treatment options to improve the outcome of cardiac sarcomas.
Materials And Methods
Human Subject Research
This study was carried out according to the code of ethics of the World Medical Association (Declaration of Helsinki). Informed consent was obtained, and the study was approved by the author’s institutional review board.
Immunohistochemistry
Immunohistochemistry was performed as described earlier in a two-step staining method by a Techmate 500 immunostainer (DAKO).10 Antibodies used from DAKO were as follows: cytokeratin proteins (clone AE1-AE3), vimentin (V9), desmin (D33), smooth muscle actin (1A4), muscle-specific actin (HHF35), S-100 (#GA504), melanosome (HMB45), CD31 (JC0A), CD34 (QBEnd), CD68 (KP1), CD3 (#GA503), CD20 (L26), CD45LCA (2B11+PD7/26), estrogen receptor (EP1), progesterone receptor (PgR636), factor VIII (#GA527), Ki67 (MIB-1), kappa chain (#GA506), gamma chain (#GA512), calretinin (DAK-Calret 1), WT1 (6F-H2), epithelial membrane antigen (E29), Myogenin (clone F5D), smooth muscle actin (1A4) and E-cadherin (NCH38).
Fluorescence In Situ Hybridization
Paraffin-embedded tissue sections were hybridized with FISH Break-apart probe (Vysis LSI SS18 Dual Color Break Apart Probe-Abbott Molecular Inc) following the manufacturer’s protocol.
DNA Extraction And Molecular Karyotyping
DNA from tumor-selected sections of paraffin-embedded tissues was isolated with QIAamp DNA Investigator Kit (QIAGEN). Integrity of the DNA was assessed by Qubit and further analyzed by molecular karyotyping with SNP arrays (Oncoscan, Affymetrix). SNP array results were analyzed with Chromosome Analysis Suite software (Affymetrix, ChAS; version 3.1). The annotation version used by the ChAS software is based on the February 2009 human reference sequence GRCh37 (hg19). SNP array data quality was assessed with the internal array quality control parameter “Median of the Absolute Values of all Pairwise Differences” (MAPD). SNP array data were plotted and interpreted as described previously.11
Results
Case Report 1
A 50-year-old woman attended the Emergency Department with palpitations and worsening dyspnea. Her previous medical history consisted of a ductal breast carcinoma treated 4 years before with chemotherapy followed by surgery and radiotherapy. The electrocardiogram showed a supraventricular tachycardia. CT scan revealed a mass in the right atrium. A transthoracic echocardiogram (TTE) showed an immobile, heterogeneous, intracavitary mass (41×34mm) in the right atrium. The initial presumptive diagnosis was an intracavitary thrombus (Figure 1). Anticoagulants and beta-blockers were given. However, the patient’s condition quickly worsened with persistent hypotension and tachycardia. She underwent emergency cardiac surgery and thrombectomy and incomplete resection of the mass was performed.
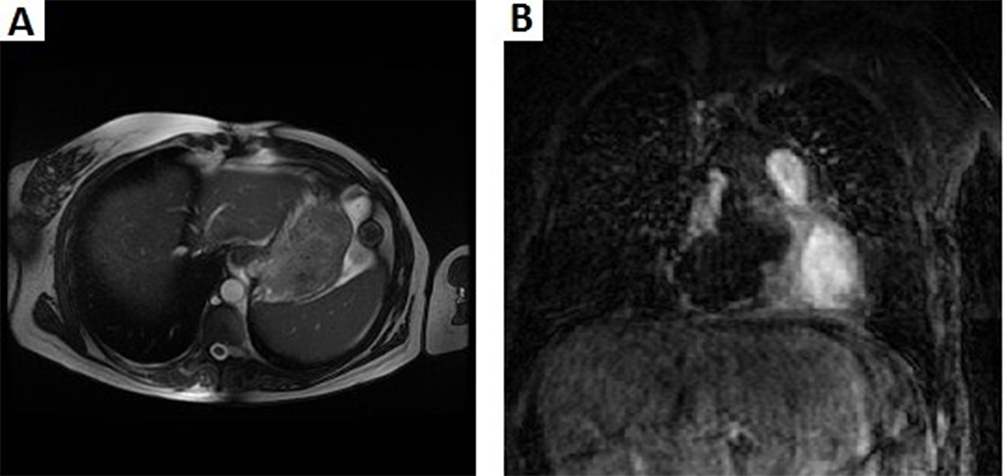 |
Figure 1 (A) CT scan of the chest and (B) CT scan of the abdomen reveal a hypodense and heterogeneous mass in the atrium. |
Histopathological examination revealed an undifferentiated pleomorphic sarcoma positive for smooth muscle actin (SMA) and vimentin expression, weak and focal staining for cytokeratin AE1-AE3, 90% Ki67 (Figure 2) and positive margins. Immunostaining was negative for epithelial membrane antigen, desmin, S-100, melanosome, CD31, CD34, factor VIII, estrogen and progesterone receptors, and CD45. Mesenchymal cells of medium and large size were very atypical and arranged in a vaguely fascicular pattern with marked pleomorphism. There was also a massive thrombosis on the right atrium with an extension to the inferior vena cava. The patient received palliative CHEMOTHERAPY with non-pegylated liposomal doxorubicin-ifosfamide, resulting in stable disease after 3 cycles. Due to high-grade hematological toxicity, treatment was changed to weekly paclitaxel and a partial response was obtained. Unfortunately, disease symptoms reappeared after 7 months of treatment, and her clinical condition quickly worsened. She died 19 months after the initial diagnosis.
 |
Figure 2 (A) Hematoxylin eosin staining at 10× magnification and (B) at 20× magnification showing atypical mesenchymal cell proliferation of fascicular pattern with numerous mitotic figures and necrosis areas. (C) Positive immunostaining for vimentin, (D) smooth muscle actin (SMA), (E) cytokeratins AE1/AE3 and F 90% Ki67 proliferation index. |
Molecular karyotype with SNPs array showed several segmental chromosomal aberrations (SCA, indicative of chromosomal break points) that led to 50% to 100% mosaic gains in 2p21p22.2, 4p14pter, 6p21.1pter (flanked by the osteoblast differentiation transcription factor RUNX2), 13q31.1qter, 17p11.1pter, 19q13.11qter (flanked by AKT2) and 22q11.1q13.1 (Figure 3). A mosaic loss in 10q11.23qter (50%) was also detected, but no whole numerical chromosomal alterations (NCA) were observed with this technique.
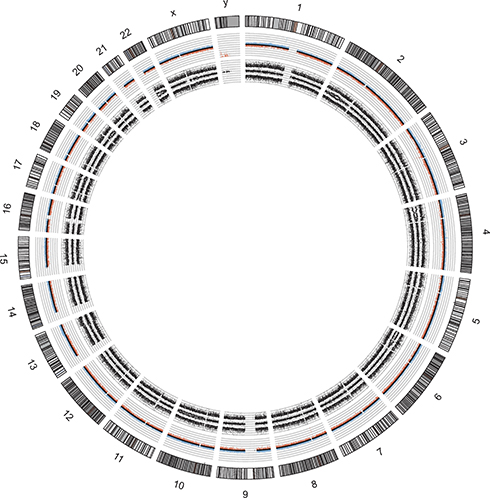 |
Figure 3 Molecular karyotype in circos plot of the undifferentiated pleomorphic rhabdomyosarcoma (case report 1). Allele peaks (inner plots) and weighted Log2ratio (middle plots) information were obtained from Affymetrix software ChAS and further used to generate circos plot. |
Case Report 2
A 72 years-old woman with unremarkable previous medical history was admitted to the hospital due to high fever and chest pain. CT scan revealed a cardiac mass in the right atrium with a myxoid compound, invading the inferior vena cava and suprahepatic veins. Study revealed a heart MRI with a multi-lobulated 60mm mass and associated thrombus (Figure 4A and B). Anticoagulant treatment was started and she underwent cardiac surgery with an incomplete resection. The pathological examination revealed a heterogeneous mesenchymal proliferation of large polygonal cells (40% Ki67 expression level) embedded in a collagenous stroma with extensive necrotic areas (Figure 5). Immunostaining for muscle-specific actin, desmin and myogenin were positive (data not shown). The final diagnosis was a high-grade pleomorphic rhabdomyosarcoma of the heart.
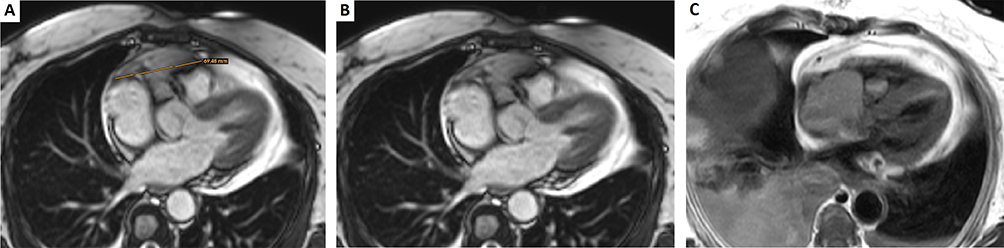 |
Figure 4 (A) Cardiac Magnetic Resonance Imaging of the pericardial tumor with a maximum diameter of 69.48 mm. (B)Large mass arising from the pericardium adjacent to the right atrium, which extends superiorly towards the left pulmonary artery. (C). Lesion relapsed arising from the pericardium in the right atrium which is invading the right ventricle. It is associated with extending thrombosis towards the suprahepatic veins and inferior cava vein. Bilateral pleural effusion. |
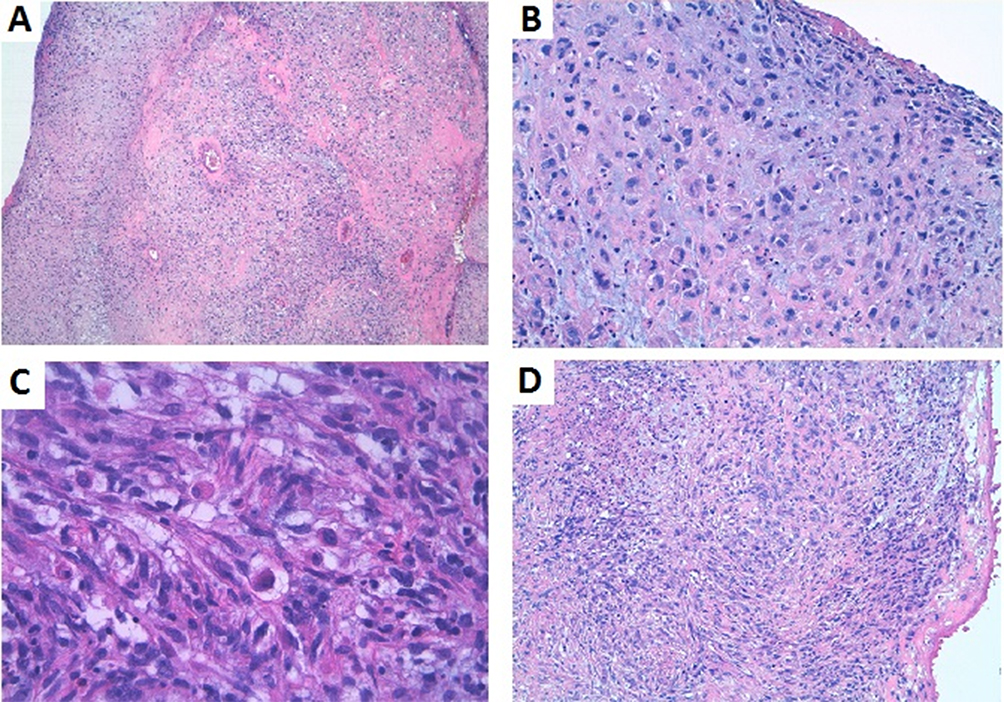 |
Figure 5 (A) Hypercellular areas with focal necrosis (H-E 10×). (B) The cells showed large, irregular nuclei and prominent nucleoli, with a wide, eosinophilic cytoplasm. (C) Cells with rhabdoid features and cytoplasmatic cross striations (H-E 40×). (D) Diffuse proliferation with a storiform growth pattern, embedded in a collagenous/myxoid stroma (H-E 10×). |
CT scan after surgery showed thrombosis of the pulmonary artery associated with pulmonary infarction and bilateral pleural effusion (Figure 4C). The patient presented congestive heart failure, and despite medical interventions, her clinical condition quickly deteriorated and she died 3 months after the initial diagnosis.
Case Report 3
A 66-year-old woman with no medical history entered the hospital with orthopnea and progressive dyspnea. The physical examination displayed signs of severe left heart failure and CT scan revealed an immobile, homogeneous, intracavitary mass in the septum of the left atrium (4×3cm) which extended into the right inferior pulmonary vein.
She underwent surgery with radical excision of the mass in February 2015 (Figure 6). The pathologist diagnosed an inflammatory myofibroblastic neoplasm. Stromal cells were positive for SMA, calponin, focal CD68, focal XIIa factor. Infiltrating inflammatory cells were polyclonal kappa and gamma chains positive (Figure 7). She recovered after surgery but had local relapse 37 months after the initial diagnosis, followed by a second surgical resection of the tumor. One year later she was diagnosed with two brain lesions and finally died of brain metastasis 56 months after initial diagnosis. SNP array analysis of the primary tumor revealed alterations in TERT, CDK4 and MDM2, among others (Figure 8). FISH analysis of the relapsed tumor confirmed MDM2 amplification (data not shown).
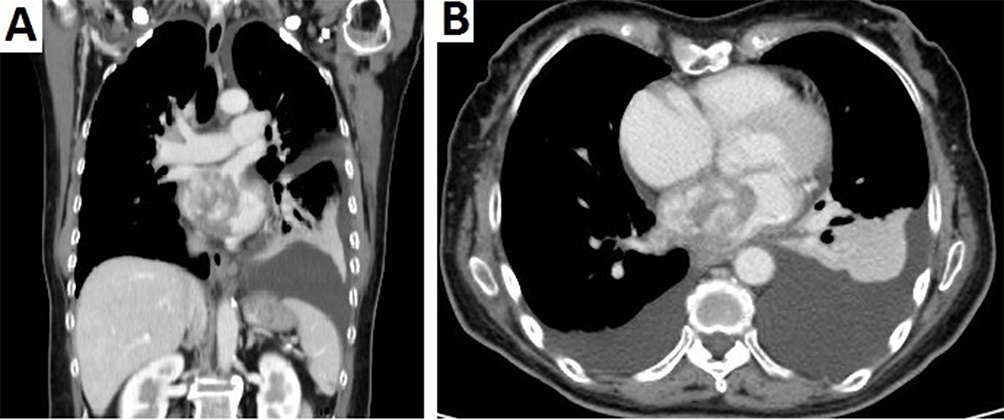 |
Figure 6 (A) CT scan of the abdomen and (B) CT scan of the chest reveal a large tumor in the left atrium extending to the lower pulmonary vein. |
 |
Figure 7 (A) Hematoxylin-eosin staining at 10× magnification and (B) at 20× magnification showing mixed inflammatory infiltrate, occasional plasma cells and mesenchymal spindle cells without atypia or mitotic figures. (C) Cells CD68+. (D) T-lymphocytes CD3+. (E) B-lymphocytes CD20+. (F) Smooth muscle actin. |
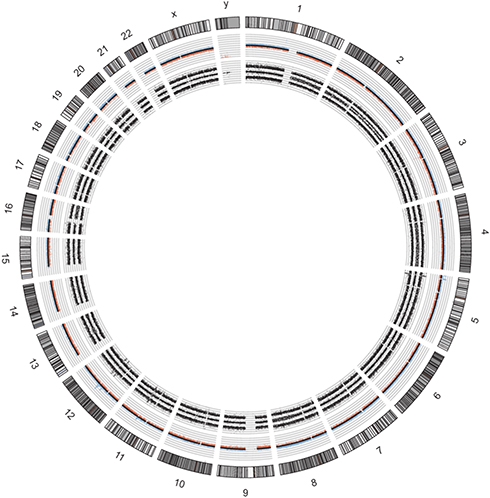 |
Figure 8 Molecular karyotype in circos plot of the inflammatory myofibroblastic tumor (case report 3). Allele peaks (inner plots) and weighted Log2ratio (middle plots) information were obtained from Affymetrix software ChAS and further used to generate circos plot. Paraffin-embedded tissue with a 50–60% tumor cell content was used for molecular karyotype analysis, revealing a 25% mosaic numerical chromosomal gain in whole chromosome 8 and numerous SCAs including 25–50% mosaic losses in 1q21.1qter, 2p25.3pter, 4q12.2qter, 9p13.2pter, 16q12.2q24.3; two sub-telomeric amplifications (4+1 allele copies) in 5p15.33 containing TERT promoter in the flanking breakpoint and in 5p15.2p15.31, gains (3+1 allele copies) in 2q11.1qter, 11q14.1, 12q13.2q13.3 and in 12q13.3q14.1 containing CDK4, and amplification (9 copies) in 12q14.3q21.1 region containing MDM2. |
Case Report 4
A 51-year-old man with no previous medical history arrived at the Emergency Department with shortness of breath, chest pain, and an episode of conscience loss. Physical examination revealed severe hypotension and tachycardia. The TTE revealed a large pericardial effusion consistent with a cardiac tamponade. A pericardiocentesis was performed and the patient was admitted for further analysis. A CT scan identified a 3×65 mm irregular mass in the pericardium of the right atrium (Figure 9). He underwent cardiac surgery and pathologic examination of biopsy revealed a heterogeneous cell proliferation with a biphasic pattern, composed of epithelial cells forming solid nests and glandular structures separated by spindle cells forming intersecting fascicles (Figure 10A). Mitotic rate was estimated as 4 mitotic cells per field using the 20X objective and resection margins were positive. Immunohistochemistry was positive for vimentin, BCL2 and epithelial markers (AE1/AE3 and CK7), epithelial membrane antigen, SMA, calretinin, and WT1 (Figure 8B and C and not shown). Fluorescent in-situ hybridization (FISH) showed SYT translocation (18q11.2) in 90% of tumor cell nuclei, further supporting the diagnosis of high-grade biphasic synovial sarcoma of the pericardium (Figure 10D).
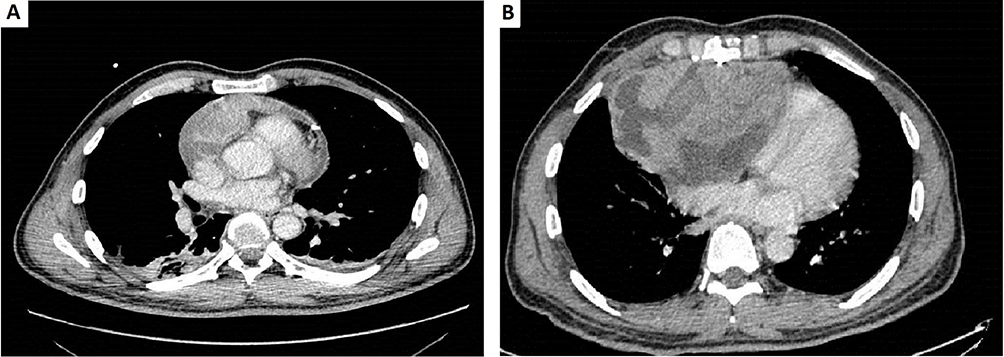 |
Figure 9 (A) Diagnostic computed tomography scan. Pericardial effusion with heterogenic areas suggesting recent bleeding. Pericardial tumor mass depending on the right atrium wall. (B) Computed tomography scan showing progression of the pericardial mass that measures 154×93.5 mm and presents mass effect on the right cavities. Composed of vascular structures and necroticoquistic areas. |
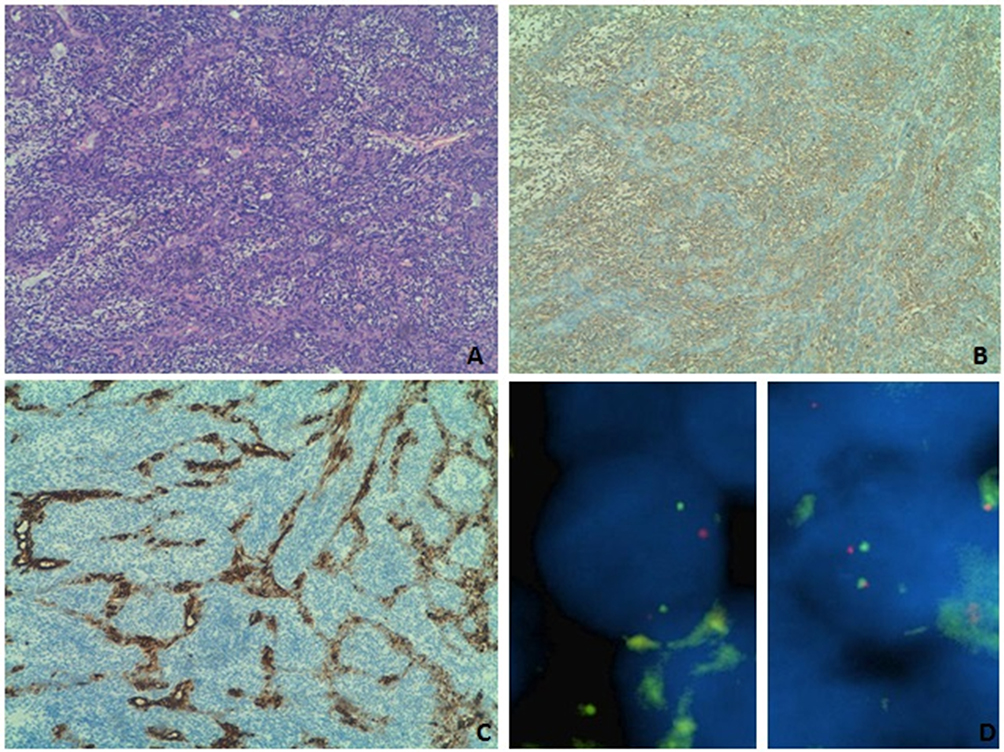 |
Figure 10 (A) Biphasic synovial sarcoma showing epithelial structures surrounded by spindle cells. (B) The epithelioid cells showed immunohistochemical expression of cytokeratin 7, (C) whereas the spindle cell component displayed a predominant vimentin expression. (D) FISH break-apart probe result was consistent with a translocation, yielded one yellow fusion, one red and one green pattern. |
Four cycles of adjuvant chemotherapy based on ifosfamide and epirubicin were administrated. A control CT scan in April 2015 suggested relapse. The patient was administered palliative chemotherapy with DTIC plus gemcitabine but the disease was continued to progress after 4 cycles. Therefore, a new line of chemotherapy was started with trabectedin but the patient was admitted to the hospital for congestive heart failure. He deteriorated rapidly and died due to congestive heart failure and tumor progression.
Case Report 5
A 27-year-old man with no previous medical history arrived at the Emergency Department with shortness of breath, orthopnea, tachypnea and cyanosis. He had a rhythmic tachycardia and 80% oxygen saturation. Chest radiography showed an alveolar pattern. Transthoracic echocardiogram revealed a mass on the left atrium protruding into the ventricle. Another extra-cardiac mass was detected on the right ventricle wall, and based on these findings, emergency surgery was performed (Figure 11). Pathologic examination revealed medium-sized spindle-shaped cells distributed in a myxoid stroma with focal necrosis in 20% of the sample. Cells of variable sizes, some of them enormous, displayed well-defined eosinophilic cytoplasm with pleomorphic nuclei. Immunohistochemical analysis revealed a very focal, cytoplasmic expression of desmin and myogenin and no expression of SMA, calponin, caldesmon or MyoD1, consistent with a high-grade myxofibrosarcoma (Figure 12). SYT translocation was also analyzed by FISH and was negative. Expression of the proliferation marker Ki67 was 35% and the resection edges were affected (not shown).
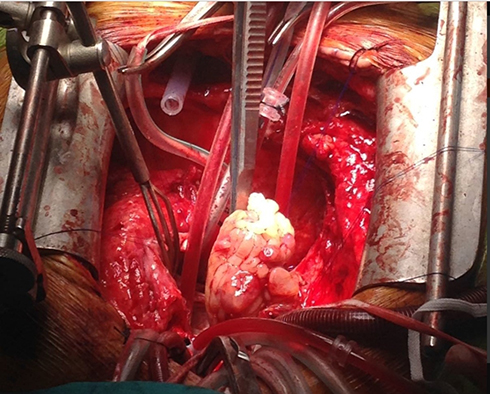 |
Figure 11 Surgical exposure of the heart confirmed an extra-cardiac mass on the left atrium protruding into the ventricle. |
 |
Figure 12 Hematoxylin and eosin staining in case report 5 revealed a myxoid variant of a high-grade undifferentiated pleomorphic sarcoma with moderate inflammatory infiltration. |
CT scan was consistent with incomplete tumor resection. After three chemotherapy cycles with epirubicin and ifosfamide, a cardiac MRI revealed the disappearance of the pericardial collection and intracardiac masses loss. Treatment was completed for a total of six cycles with an excellent tolerance. He currently maintains a complete response with no progression.
SNPs array analysis showed a high number of NCAs as well as SCAs, suggesting multiple rearrangements and a complex karyotype (Figure 13). Notably, chromothripsis was also detected in chromosomes 12q and 18q (Figure 13). Similarly to case report 3, CDK4 and MDM2 were also amplified (7 copies each).
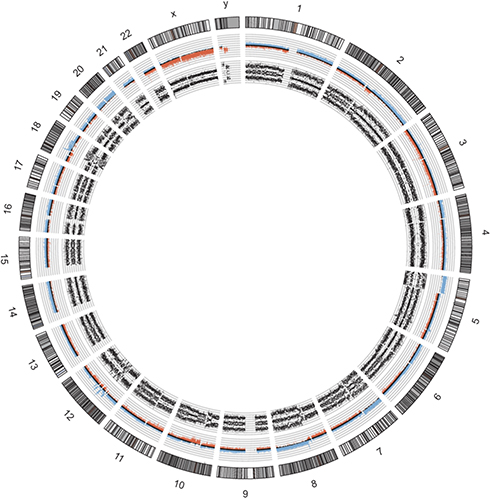 |
Figure 13 Molecular karyotype in circos plot of our reported high-grade myxofibrosarcoma (case report 5). Allele peaks (inner plots) and weighted Log2ratio (middle plots) information were obtained from Affymetrix software ChAS and further used to generate circos plot. NCA gains and losses were detected at variable mosaicisms in chromosomes −3, +9, +13, +14 (2+2 allele copies), +15, +16 (2+2), +17, +20 (2+2), +21 and −22. SCAs were detected in +1p21.1pter, −1p13.1p21.1, +1q21.1qter (2+2), +2q11.2pter, −2q11.2q14.1, +2q14.1qter, +4p12pter, +5p12pter (4–5 copies), +6q15pter (2+2), +6q15qter, +7q11.21pter (3+1), +7q11.21qter, −8q12.1pter, +8q12.1qter (2+2), −10p12.2pter, −10q11.21q21.3, +10q24.1qter, −11p11.2pter, +12p11.21pter, −12q21.2q23.1, +12q23.1q24.32, −12q24.32qter, −18q12.1pter, +18q21.32qter, +19q13.31pter (2+2), +19q13.31qter, +Xp11.21pter. An amplification in 8q (average of 7 copies) containing the Pleomorphic adenoma gene 1 (PLEG1) was flanked by MOS proto-oncogene promoter. Chromosomes 4, 12p, 12q23.3-12qter and 18q21.32qter displayed loss of heterozygosity (LOH). |
Discussion
Many types of sarcomas have been reported in the heart,5,7,8,12,13 although due to their rarity, most have been isolated case reports. Cardiac MRI provides useful information for characterizing cardiac masses and their anatomical localization. Thus, masses in the left atrium are more commonly benign, whereas masses in the right atrium and pericardium are more often malignant.14 However, three of the five cases we report originally affected the left atrium: primary pleomorphic rhabdomyosarcoma, inflammatory myofibroblastic tumor, and myxofibrosarcoma. Although benign myxomas are the most common tumors occurring in the left atrium, malignant tumors can simulate myxomas and should be considered in the differential diagnosis.15
Undifferentiated pleomorphic tumours are usually poorly differentiated, with atypical spindle cells, high mitotic activity, necrosis, and nuclear polymorphism.16,17 The prognosis of these aggressive tumours is generally poor. Although radical surgery offers an important means for alleviating symptoms, local recurrence and metastasis occur usually within 1 year. Chemotherapy and radiation therapy have limited benefit in most published cases.18 A recent study of an undifferentiated cardiac intimal sarcoma using next-generation sequencing reported an activating mutation in PDGFRB, suggesting a novel therapeutic target for this rare disease.19 Our molecular karyotype analysis identified numerous gains and losses with two well-defined segmental breaks flanked by AKT2 and RUNX2 genes, suggesting that deregulation of these two genes may also be involved in disease initiation and/or progression.
Primary pleomorphic rhabdomyosarcomas in the heart are extremely rare and the risk of hemodynamic complications and cardiac failure determine the generally poor prognosis of these tumors. In our case, the tumor rapidly progressed after an initial surgical resection and the patient developed rapidly progressive congestive heart failure. To our knowledge, only one other case of this pathology was been reported,20 which emphasizes the rarity of this condition and the relevance of reporting our own experience to improve the management of this disease.
The inflammatory myofibroblastic tumor is a very rare intracardiac neoplasia. It mainly occurs in children and young adults, and although the most common location is the lung, it has also been described in other sites (pelvis, head and neck, abdomen, liver, etc.) and exceptionally in the heart.21 The specificity of ALK may help in differential diagnosis and can be efficiently targeted with ALK inhibitors.22 Molecular karyotype analysis in our patient revealed a 50% mosaic NCA (2 to 3 chromosomes) for whole chromosome 2, where ALK is located. Taking into account that tumor cell content in the sample analyzed was around 60%, our results suggest that almost all tumor cells had an extra copy of chromosome 2. However, this technique is not adequate for the detection of ALK translocations if no copy number alterations are involved. Noteworthy, we identified two subtelomeric amplicons in 5p, one of them with the segmental break right at TERT promoter, suggesting a role for telomere maintenance in this disease. In addition, we detected a small segment 12q gain (4 copies) containing CDK4 gene and a nearby 9 copies-amplification containing MDM2 gene. Our results further support previous findings in an oral inflammatory myofibroblastic tumor23 and in two primary cardiac tumors24,25 and provide a rationale for applying precision medicine approaches to these types of tumors with TP53 therapy (PRIMA-1 or MDM2 inhibitors) and CDK4 inhibitors such as palbociclib. The neoplastic nature of inflammatory myofibroblastic tumors and the possible relation with inflammatory events are still debated: some exceptional spontaneous or steroid-induced regression has been described,26 but surgical resection remains the treatment of choice. Despite the infrequent relapse of inflammatory myofibroblastic tumors (25–35%), those arising within the heart are very rare and scant cases with incomplete resection may result in rapid local relapse.27,28 The case we report with brain metastasis showed the long-term poor outcome, suggesting that ensuring complete resection is not enough and supporting the use of alternative strategies in addition to surgical resection plus conventional therapy.
Primary synovial sarcoma arising from the pericardium is an extremely rare disease, as most of them arise from the extremities, displaying a preference for juxta-articular location. The mainstay of the diagnosis is the detection of the reciprocal translocation t(X; 18) (p11.2; q11.2) and SS18-SYT fusion gene by FISH, which is present in more than 80% of the cases.29 Although very little is known about the oncogenic pathways triggered by the chimeric protein SS18X-SYT, it is thought to play a role in the transcriptional deregulation of targeted genes.30 Cytogenetic analysis provides an added value in the confirmation of the diagnosis, being especially useful when they arise from atypical sites.
The management of synovial sarcoma is challenging and requires a personalized treatment strategy. A broad surgical resection is the cornerstone of therapy when feasible. The external beam radiation therapy (EBR) is used when the margins of resection are positive. Despite the initial radical management, the prognosis is poor with 12-month disease-free survival.31 Currently, new molecular-targeted therapies are under investigation, such as trastuzumab in those who express HER2.32
Myxofibrosarcoma is one of the most aggressive variants of soft tissue neoplasms. Although it usually involves the left atrium, in our case, the ventricles were affected. Consistent with being high-grade and aggressiveness, our reported myxofibrosarcoma displayed highly complex chromosomal rearrangements with NCAs, LOHs, SCA gains and losses, amplifications and chromothripsis. All these alterations reflect high chromosomal instability and suggest the role of genomic maintenance genes in this disease. In addition, these multiple rearrangements may result in multiple tumor-specific epitopes, and hence, immunotherapy would be an interesting treatment option to explore in these cases.
In conclusion, cardiac sarcomas proliferate rapidly and cause death in most cases with 6–12 months of median survival.13 The complexity of heart sarcomas requires personalized management of each case and alternative strategies in order to improve short and long-term outcome. They should be treated in specialized oncology centers, in order to obtain an accurate diagnosis and improve treatment efficacy. Observations from our study support the implementation of genetic analysis of primary cardiac sarcomas to identify actionable targets for a precision medicine strategy that may improve the prognosis of these rare oncologic diseases.
Ethics Approval And Informed Consent
This study was carried out according to the code of ethics of the World Medical Association (Declaration of Helsinki). The authors’ Institutional Review Board at La Fe Hospital approved the study. Patients provided written informed consent for participation in the study and for publication of case details.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748–756.
2. Silvestri F, Bussani R, Pavletic N, Mannone T. Metastases of the heart and pericardium. G Ital Cardiol. 1997;27(12):1252–1255.
3. Odim J, Reehal V, Laks H, Mehta U, Fishbein MC. Surgical pathology of cardiac tumors. Two decades at an urban institution. Cardiovasc Pathol. 2003;12(5):267–270.
4. Orlandi A, Ferlosio A, Roselli M, Chiariello L, Spagnoli LG. Cardiac sarcomas: an update. J Thor Oncol. 2010;5(9):1483–1489.
5. Truong PT, Jones SO, Martens B, et al. Treatment and outcomes in adult patients with primary cardiac sarcoma: the British Columbia cancer agency experience. Ann Surg Oncol. 2009;16(12):3358–3365.
6. Devbhandari MP, Meraj S, Jones MT, Kadir I, Bridgewater B. Primary cardiac sarcoma: reports of two cases and a review of current literature. J Cardiothorac Surg. 2007;2:34.
7. NA. S. Primary cardiac tumours. Ann Surg. 1980;19:127–131.
8. Burke AP, Cowan D, Virmani R. Primary sarcomas of the heart. Cancer. 1992;69(2):387–395.
9. Putnam JB
10. Berlanga P, Munoz L, Piqueras M, et al. miR-200c and phospho-AKT as prognostic factors and mediators of osteosarcoma progression and lung metastasis. Mol Oncol. 2016;10(7):1043–1053.
11. Sanmartin E, Yanez Y, Fornes-Ferrer V, et al. TIAM1 variants improve clinical outcome in neuroblastoma. Oncotarget. 2017;8(28):45286–45297.
12. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295–304.
13. Zhang PJ, Brooks JS, Goldblum JR, et al. Primary cardiac sarcomas: a clinicopathologic analysis of a series with follow-up information in 17 patients and emphasis on long-term survival. Hum Pathol. 2008;39(9):1385–1395.
14. Mousavi N, Cheezum MK, Aghayev A, et al. Assessment of cardiac masses by cardiac magnetic resonance imaging: histological correlation and clinical outcomes. J Am Heart Assoc. 2019;8(1):e007829.
15. Kumar S, Chaudhry MA, Khan I, Duthie DJ, Lindsay S, Kaul P. Metastatic left atrial synovial sarcoma mimicking a myxoma. J Thorac Cardiovasc Surg. 2004;128(5):756–758.
16. Gupta A. Primary cardiac sarcomas. Expert Rev Cardiovasc Ther. 2008;6(10):1295–1297.
17. Hottenrott G, Mentzel T, Peters A, Schroder A, Katenkamp D. Intravascular (“intimal”) epithelioid angiosarcoma: clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 1999;435(5):473–478.
18. Grebenc ML, Rosado de Christenson ML, Burke AP, Green CE, Galvin JR. Primary cardiac and pericardial neoplasms: radiologic-pathologic correlation. Radiographics. 2000;20(4):1073–1103. quiz 1110–1071, 1112.
19. Fu X, Niu W, Li J, et al. Activating mutation of PDGFRB gene in a rare cardiac undifferentiated intimal sarcoma of the left atrium: a case report. Oncotarget. 2017;8(46):81709–81716.
20. Chang YL, Lin CY, Wang SS, Kuo KT, Lee YC, Wu CT. Concomitant intramyocardial and epicardial vasculitis in an autopsied heart allograft for cardiac rhabdomyosarcoma. Clin Transplant. 2002;16(6):461–464.
21. Pucci A, Valori A, Muscio M, Garofalo L, Ferroni F, Abbruzzese PA. Asymptomatic inflammatory myofibroblastic tumor of the heart: immunohistochemical profile, differential diagnosis, and review of the literature. Cardiovasc Pathol. 2009;18(3):187–190.
22. Theilen TM, Soerensen J, Bochennek K, et al. Crizotinib in ALK(+) inflammatory myofibroblastic tumors-Current experience and future perspectives. Pediatr Blood Cancer. 2018;65:4.
23. Brooks JK, Nikitakis NG, Frankel BF, Papadimitriou JC, Sauk JJ. Oral inflammatory myofibroblastic tumor demonstrating ALK, p53, MDM2, CDK4, pRb, and Ki-67 immunoreactivity in an elderly patient. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99(6):716–726.
24. Crombe A, Lintingre PF, Le Loarer F, Lachatre D, Dallaudiere B. Multiple skeletal muscle metastases revealing a cardiac intimal sarcoma. Skeletal Radiol. 2018;47(1):125–130.
25. Watson R, Frye J, Trieu M, Yang MX. Primary undifferentiated pleomorphic cardiac sarcoma with MDM2 amplification presenting as acute left-sided heart failure. BMJ Case Rep. 2018;2018.
26. Ferbend P, Abramson LP, Backer CL, et al. Cardiac plasma cell granulomas: response to oral steroid treatment. Pediatr Cardiol. 2004;25(4):406–410.
27. Hartyanszky IL, Kadar K, Hubay M. Rapid recurrence of an inflammatory myofibroblastic tumor in the right ventricular outflow tract. Cardiol Young. 2000;10(3):271–274.
28. Yang X, Xiao C, Liu M, Wang Y. Cardiac inflammatory myofibroblastic tumor: does it recur after complete surgical resection in an adult? J Cardiothorac Surg. 2012;7:44.
29. Pearson PJ, Smithson WA, Driscoll DJ, Banks PM, Ehman RL. Inoperable plasma cell granuloma of the heart: spontaneous decrease in size during an 11-month period. Mayo Clin Proc. 1988;63(10):1022–1025.
30. Ladanyi M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001;20(40):5755–5762.
31. Yokouchi Y, Hiruta N, Oharaseki T, et al. Primary cardiac synovial sarcoma: a case report and literature review. Pathol Int. 2011;61(3):150–155.
32. Van der Mieren G, Willems S, Sciot R, et al. Pericardial synovial sarcoma: 14-year survival with multimodality therapy. Ann Thorac Surg. 2004;78(3):e41–42.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.