")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Identification of a novel CACNA1A mutation in a Chinese family with autosomal recessive progressive myoclonic epilepsy
Authors Lv YD, Wang Z, Liu C, Cui L
Received 7 July 2017
Accepted for publication 21 August 2017
Published 19 October 2017 Volume 2017:13 Pages 2631—2636
DOI https://doi.org/10.2147/NDT.S145774
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
Yudan Lv, Zan Wang, Chang Liu, Li Cui
Department of Neurology, Department of Neurology and Neuroscience Center, The First Hospital of Jilin University, Changchun, People’s Republic of China
Background: Progressive myoclonic epilepsy (PME) is a heterogeneous neurodegenerative disorder, which is commonly manifested with refractory seizures and neurologic deterioration. The prognosis of PME is poor, so early diagnosis of PME is critical. The aim of our study is to identify the novel pathogenic gene in a Chinese family with PME, which may be helpful in future.
Subjects and methods: A three-generation consanguineous Chinese Han family with PME was recruited. A novel homozygous variant was identified by the genetic technique of exome sequencing and certificated by Sanger sequencing and functional prediction.
Results: A novel homozygous variant, c.6975_6976insCAG, in the CACNA1A was identified in the PME family. The novel variant encoding the alpha-1A subunit of the calcium channel Cav2.1 was found in two siblings in the Chinese family and was absent in 50 normal controls. Our research indicates that the homozygous c.6975_6976insCAG might be the pathogenic mutation for PME.
Conclusion: As a molecular diagnostic strategy, our research explores the mutation gene spectrum of PME and has resulted in significant predictions for genetic counseling.
Keywords: CACNA1A, PME, exome sequencing, myoclonus, muscular hypotonia
A Letter to the Editor has been received and published for this article.
Introduction
The CACNA1A gene has been reported to encode the pore-forming alpha-1A subunit of the calcium channel Cav2.1, which acts as a voltage sensor and an ion pore.1,2 Several mutations within this gene lead to variable clinical phenotypes such as familial hemiplegic migraine type 1, spinocerebellar ataxia type 6 and episodic ataxia type 2 (EA2), which have been recognized widely. There are only two reports on a CACNA1A-related generalized epileptic seizure3 and a photoparoxysmal response (PPR).4 Muscular hypotonia or muscular atrophy has also been reported to be associated with CACNA1A gene.5
Progressive myoclonic epilepsy (PME) is generally manifested with refractory myoclonus seizures, cognitive decline and neurologic deterioration, caused by metabolic, genetic and neurodegenerative diseases, which has an unfavorable course and poor prognosis. A wide range of specific etiology contributes to the PME, such as neuronal ceroid lipofuscinoses (NCLs), Tay–Sachs, myoclonic epilepsy with ragged red fibers (MERRF), POLG1 Mutation, Lafora Disease and Unverricht–Lundborg disease (ULD).
Herein, we describe two PME patients. Those gene mutations recognized widely in the PME were screened with negative results. On the other hand, a causative novel mutation in the CACNA1A gene was found in the PME by exome sequencing (ES), and then confirmed by Sanger sequencing subsequently.
Subjects and methods
Subjects
A three-generation consanguineous Chinese Han family with PME was studied, and four members of the family agreed to participate in our study. PME has been found in the two siblings (II:1 and II:3; Figure 1) with the same phenotype of refractory myoclonic seizures, neurologic deterioration and cognitive decline, who have the same problems of refractory myoclonic seizures, neurologic deterioration and cognitive decline. However, their parents (I:1 and I:2; Figure 1) and their children (III:1–3; Figure 1) were normal persons. Fifty healthy Chinese Han subjects (age 32.5±4.5 years) were enrolled as controls with neurologic physical and clinical assessments. After obtaining written informed consent from the participants, we collected the peripheral blood samples from April 19 to October 30, 2015.
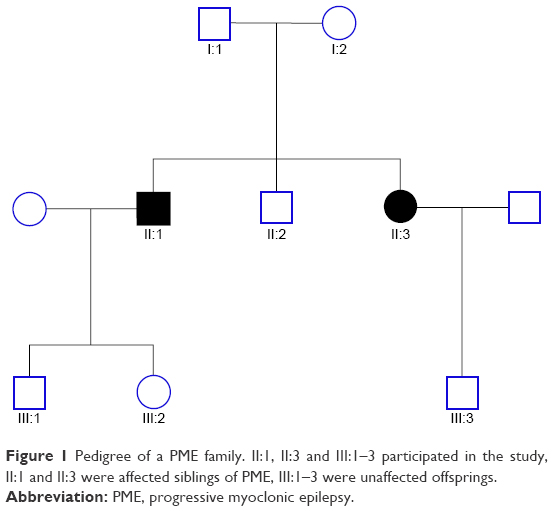 | Figure 1 Pedigree of a PME family. II:1, II:3 and III:1–3 participated in the study, II:1 and II:3 were affected siblings of PME, III:1–3 were unaffected offsprings. |
Clinical evaluations
Clinical and electrophysiologic assessments were performed on the PME family in the First Hospital of Jilin University. Both patients were afraid of light. Electroencephalogram (EEG) was conducted in both patients with PPR during the process of light stimulation frequently. Magnetic resonance imaging was also carried out.
Whole ES and relevant variant analysis
Genomic DNA was extracted from peripheral blood samples by using standard phenol–chloroform extraction method.6 ES was performed according to relevant guidelines and regulations. At least 1.5–2 μg of genomic DNA was obtained from the proband (II:1 and II:3; Figure 1), and then following the manufacturers’ procedures, the samples was sheared, enriched, hybridized and captured on the Agilent SureSelect. The captured library was enriched with Illumina HiSeq 2000 sequencing instruments and provided enough depth to identify variants at 97.4% of targeted exome exactly.7
All variants were screened with the single nucleotide polymorphism (SNP) database version 142 (dbSNP142), 1000 Genomes Project (version 2014 October) and NHLBI ES Project (ESP) 6500. The clean reads were mapped to the human reference genome (UCSC hg19, http://genome.ucsc.edu/) using Burrows–Wheeler Alignment tool.8,9 SNPs and insertions/deletions were identified by the Sequence Alignment/Map tools (SAMtools),10 and duplicate reads were marked by Picard tools. Functional prediction was carried out by Sorting Intolerant from Tolerant and Polymorphism Phenotyping version 2 (PolyPhen-2). Candidate variants were analyzed by the ANNOVAR (Annotate Variation) software.11
Sanger sequencing and functional prediction
Potential causative variants were confirmed by direct Sanger sequencing using the ABI3500 sequencer system (Thermo Fisher Scientific, Waltham, MA, USA).12 Primers for the mutation of RS753460234 in the CACNA1A gene were designed as follows: RS753460234-F: TCCCGTGTCTCCTTTGATTT; R: GGTAGTAGCCGTTGGGGAGT. MutationTaster was applied to predict the functional changes in the CACNA1A variant.13
Ethics approval and consent to participate
This study has been approved by the Research Ethics Board of Jilin University First Hospital. Written informed consents were obtained from the patients and patients’ family members for publication of this study and any accompanying images. A copy of the written consent is available for review by the editor of this journal.
Results
Clinical findings
Both patients (II:1 and II:3; Figure 1) presented with refractory myoclonus at around 50 years of age. Their previous history was noncontributory. The seizures could be described as symmetrical and rhythmic jerk. The frequency increased to dozens of times a day. They had much more myoclonus in day than at night. Besides this, they were afraid of light and sound before seizure onset. And 2 years later, they gradually presented with muscle weakness, muscular atrophy and cognitive decline. Neurologic examination showed both proximal and distal muscle atrophy of lower limbs, muscle weakness and progressive ataxia. Mini-Mental State Examination scores were 20/30 and 22/30, respectively, which revealed that they had trouble in memory and locating. EEG on admission showed generalized polyspike waves or generalized spike-slow complex wave without previous EEG available (Figure 2). Also, the electromyogram results were normal. Light stimulation caused photosensitivity frequently. Magnetic resonance imaging was normal. According to the clinical history, presentation and EEG features, we made a diagnosis of PME. After the administration of antiepileptic drug valproic acid, no improvement was seen. The clinical picture of PME family is summarized in Table 1.
 | Figure 2 EEG showed generalized polyspike-slow complex wave mixed with some muscle artifacts, which indicated a myoclonic seizure. |
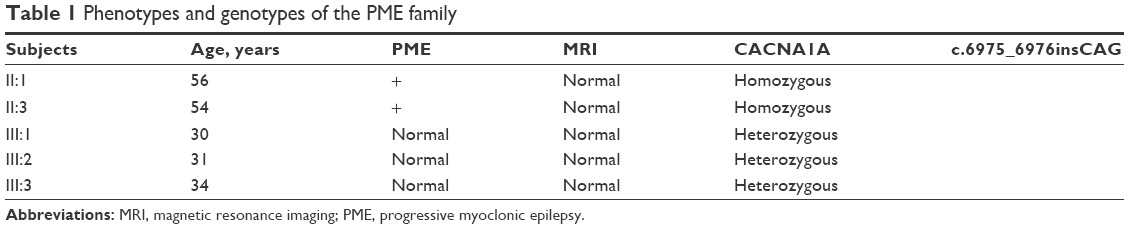 | Table 1 Phenotypes and genotypes of the PME family |
Common pathogenic gene of PME screening
A wide range of specific gene mutation contributes to the PME, such as NCLs, Tay–Sachs, MERRF, POLG1 mutation, Lafora Disease and ULD. However, we obtained negative results with the common pathogenic gene screening.
Exome sequencing
A total of 162.565 million reads were generated from ES with an average read length of 150 bp, and 162.308 million reads (99.84%) were mapped to the human genome. Also, 16,041.78 Mb of effective sequences was mapped to the target region. Moreover, 180.973 million bases covered the target region and occupied about 99.7% of the target region. The sequence at 10× or greater covered 98.7% of the target region. Also, 64,952 SNPs in the exon regions and 7,001 SNPs in the splicing sites were identified in total. Five hundred and sixty indels in the exon regions and 402 in the splicing sites were identified, respectively.7 We used a prioritization scheme to eliminate common known variants reported in public databases, such as dbSNP137 (Minor Allele Frequency >1%), 1,000 genomes project with a frequency >0.5% and NHLBI ESP6500, in order to detect the possible pathogenic variant. Sorting Intolerant from Tolerant and PolyPhen-2 analyses were utilized to predict the functional changes of the mutation. As a result, a homozygous CACNA1A c.6975_6976insCAG variant was observed in the proband (II:1 and II:3; Figure 1) as a possible pathogenic mutation.
Confirmation of pathogenic mutation
A homozygous CACNA1A c.6975_6976insCAG in the exon 47 identified in the proband was further confirmed by direct Sanger sequencing. The same homozygous variant was found in the affected sibling (II:1 and II:3; Figure 1), but not in both unaffected offsprings (III:1–3; Figure 1). The identical variant was absolutely absent in 50 ethnically matched normal controls (Figure 3). The homozygous CACNA1A c.6975_6976insCAG variant relevant with the PME phenotype, leading to an insertion at amino acid position 2326, was predicted to change alanine to another quisqualic acid, which may trigger neurotoxicity. CADD (http://cadd.gs.washington.edu/score) predicts the variant to be harmful with a functional CADD_Phred of 14.13 (≤10); the mutation may be the disease-causing mutation in the PME family.
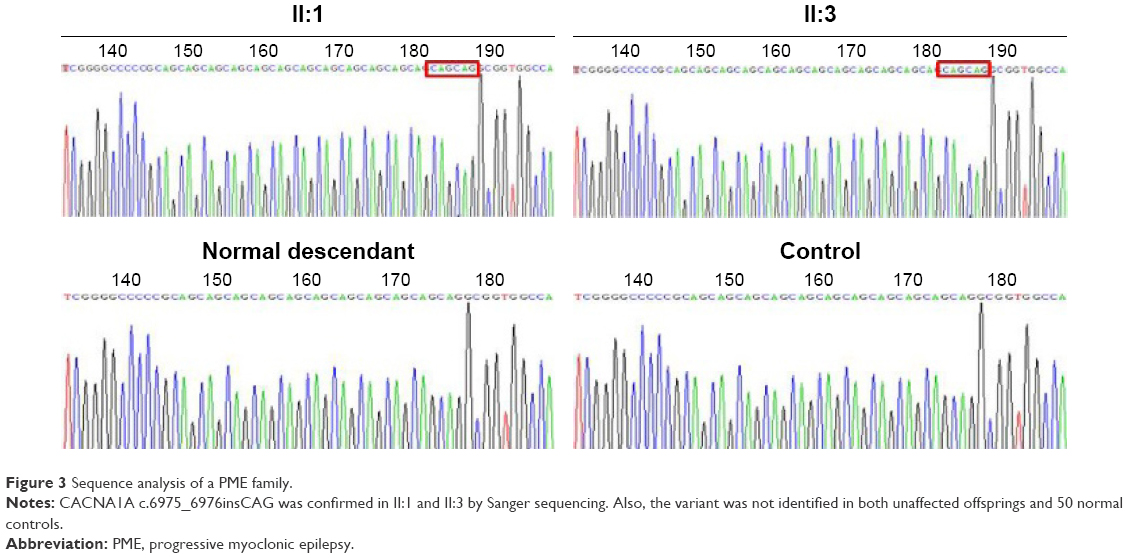 | Figure 3 Sequence analysis of a PME family. |
Discussion
PME is an epilepsy syndrome characterized by progressive myoclonus, cognitive impairment, ataxia as well as other neurologic deficits. It includes several specific etiologies. Cystatin B gene mutations cause ULD, a rare form of PME.14 Mutation of the nuclear lamin gene LMNB2 causes PME with early ataxia.15 CLN6 mutation is reported to cause NCLs, another form of PME.16 Also, PME encompasses a wide spectrum of mitochondrial and metabolic diseases such as MERRF and Lafora disease. MERRF is the common form with the underlying mutation in the tRNALys gene,17 and Lafora was reported to be associated with mutation in EPM2A gene.18 However, in our cases, none of the screened mutation genes are positive. By whole ES, we identified CACNA1A mutation in our subjects. This mutation cannot be seen in normal controls, but has been found in our PME family. Only homozygous mutation can cause PME, although the offsprings may show heterozygous mutation, while they are normal people without PME presentation. Therefore, one good allele is enough in our family.
The CACNA1A gene encodes the alpha-1A subunit of the Cav2.1 P/Q-type voltage-gated calcium channel, which is in connection with the transmembrane performing. Several heterozygous mutations in this gene have been reported to cause three allelic autosomal dominant diseases, such as EA2, familial hemiplegic migraine type 1 and spinocerebellar ataxia type 6. Among these mutations, nonsense, missense and cytosine–adenine–guanine repeats are commonly observed. However, an association with idiopathic epilepsy and a link with epileptic syndrome have only been recently recognized. Reinson et al5 describe in their report that two patients, a sister and brother, developed daily refractory seizures with extreme muscular hypotonia and global developmental delay. A missense mutation c.4315T>A in CACNA1A was identified. Similar clinical symptoms of refractory seizures, muscular hypotonia and progressive ataxia are seen in our cases. Kaido et al4 report two cases of EA2 with PPR in EEG. A novel single base deletion (c.3575delA) in CACNA1A was uncovered. Special and rare PPR response to light stimulation is also presented in our cases.
In consistent with our clinical findings, mice with loss of Cav2.1 channel exhibit a severe phenotype of chronic dystonia and ataxia.19 Inborn errors of CACNA1A, an early P/Q-type voltage-gated calcium release defect, produces a stable generalized epilepsy phenotype.20 The various phenotypes caused due to mutation in CACNA1A have been found in both human and mice. Thus, CACNA1A is likely a novel disease-causing gene for the PME.
The mutation of polymorphic CAG insertion in exon 47 was located in an important functional region of the human a1A subunit of the neuronal Cav2.1 (P/Q-type) Ca2+ channel, which is associated with dysfunction of the intracellular C-terminus21 closely (Figure 4). Furthermore, mutation p.A2326delinsQA in CACNA1A changes alanine to another quisqualic acid, which may contribute to convulsions.22 Chase reports that L-quisqualic acid is an excitatory amino acid agonist that may trigger neurotoxicity.23
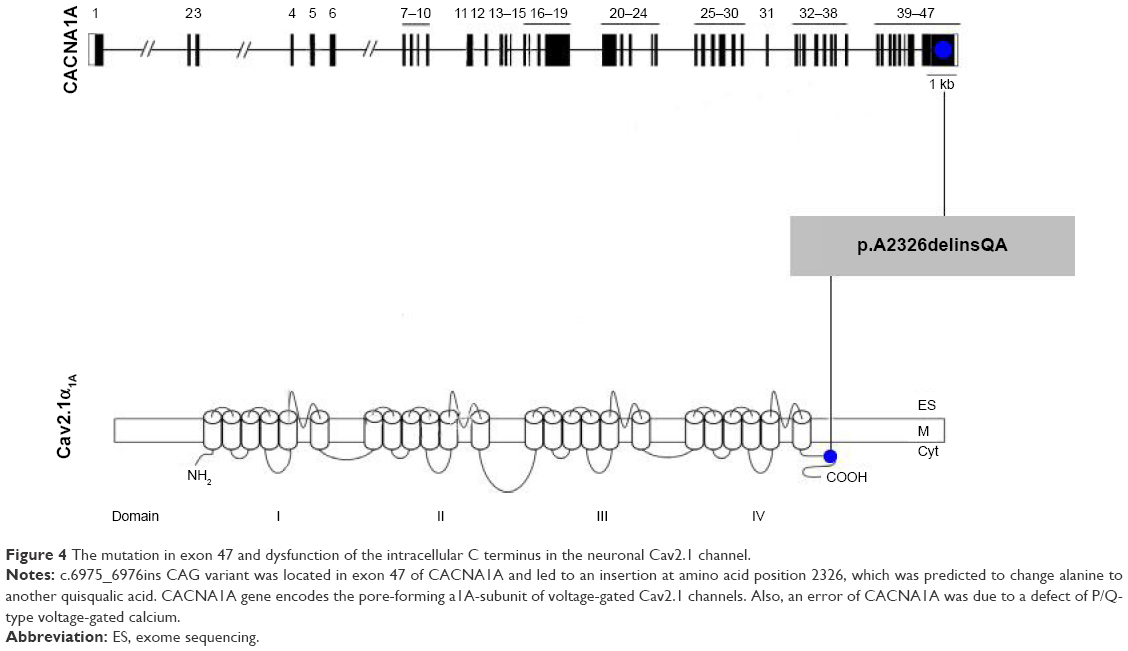 | Figure 4 The mutation in exon 47 and dysfunction of the intracellular C terminus in the neuronal Cav2.1 channel. |
Conclusion
In our study, the homozygous CACNA1A c.6975_6976insCAG mutation was identified in two affected siblings of PME family, but was absent in the unaffected children and 50 normal controls. The mutation is associated with PME phenotype closely and might be disease causing. The c.6975_6976insCAG mutation in CACNA1A in the PME family is a novel finding. This mutation of the base-insertion was responsible for the function of amino acid and protein significantly.
In summary, ES reveals a novel disease-causing CACNA1A gene for PME as a powerful molecular diagnostic technique.
Acknowledgment
The authors thank Dr Jun Lu who was involved in revising the manuscript critically for important intellectual content. The authors acknowledge assitance from National Natural Science Foundation of China (Grant no 81701270).
Author contributions
All authors participated in drafting the article or revising it critically for important intellectual content. All authors participated in conception and design, acquisition of data, or analysis and interpretation of data. All authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work. Also, all authors agreed and gave final approval of the version to be published.
Disclosure
The authors report no conflicts of interest in this work.
References
Vahedi K, Denier C, Ducros A, et al. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology. 2000;55(7):1040–1042. | ||
Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics. 2007;4(2):267–273. | ||
Damaj L, Lupien-Meilleur A, Lortie A, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism, and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. 2015;23(11):1505–1512. | ||
Kaido M, Furuta M, Nakamori M, Yuasa Y, Takahashi MP. Episodic ataxia type 2 manifests as epileptiform electroencephalographic activity with no epileptic attacks in two family members. Rinsho Shinkeigaku. 2016;56(4):260–264. | ||
Reinson K, Oiglane-Shlik E, Talvik I, et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am J Med Genet A. 2016;170(8):2173–2176. | ||
Zheng W, Deng X, Liang H, et al. Genetic analysis of the fused in sarcoma gene in Chinese Han patients with essential tremor. Neurobiol Aging. 2013;34(8):2078.e3–e4. | ||
Yuan L, Wu S, Xu H, et al. Identification of a novel PHEX mutation in a Chinese family with X-linked hypophosphatemic rickets using exome sequencing. Biol Chem. 2015;396(1):27–33. | ||
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. | ||
Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. | ||
Li H, Handsaker B, Wysoker A, et al; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. | ||
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. | ||
Guo Y, Yang H, Deng X, et al. Genetic analysis of the S100B gene in Chinese patients with parkinson disease. Neurosci Lett. 2013;555:134–136. | ||
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. | ||
Duarte AJ, Ribeiro D, Chaves J, Amaral O. Characterization of a rare Unverricht-Lundborg disease mutation. Mol Genet Metab Rep. 2015;4:68–71. | ||
Damiano JA, Afawi Z, Bahlo M, et al. Mutation of the nuclear lamin gene LMNB2 in progressive myoclonus epilepsy with early ataxia. Hum Mol Genet. 2015;24(16):4483–4490. | ||
Sato R, Inui T, Endo W, et al. First Japanese variant of late infantile neuronal ceroid lipofuscinosis caused by novel CLN6 mutations. Brain Dev. 2016;38(9):852–856. | ||
Zsurka G, Becker F, Heinen M, et al. Mutation in the mitochondrial t RNA(Ile) gene causes progressive myoclonus epilepsy. Seizure. 2013;22(6):483–486. | ||
Schorlemmer K, Bauer S, Belke M, et al. Sustained seizure remission on perampanel in progressive myoclonic epilepsy (Lafora disease). Epilepsy Behav Case Rep. 2013;1:118–121. | ||
Hoffman EP. Hemiplegic migraine-downstream of a single-base change. N Engl J Med. 2001;345(1):57–59. | ||
Bomben VC, Aiba I, Qian J, Mark MD, Herlitze S, Noebels JL. Isolated P/Q calcium channel deletion in layer VI corticothalamic neurons generates absence epilepsy. J Neurosci. 2016;36(2):405–418. | ||
Wan J, Mamsa H, Johnston JL, et al. Large genomic deletions in CACNA1A cause episodic ataxia type 2. Front Neurol. 2011;2:51. | ||
Verleye M, Andre N, Heulard I, Gillardin JM. Nefopam blocks voltage-sensitive sodium channels and modulates glutamatergic transmission in rodents. Brain Res. 2004;1013(2):249–255. | ||
Chase LA, Roon RJ, Wellman L, Beitz AJ, Koerner JF. L-Quisqualic acid transport into hippocampal neurons by a cystine-sensitive carrier is required for the induction of quisqualate sensitization. Neuroscience. 2001;106(2):287–301. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.